

Abstract
Cyclin-dependent kinase 9 (CDK9) is a member of the cyclin-dependent kinase (CDK) family which is involved in transcriptional regulation of several genes, including the oncogene Myc, and is a validated target for pancreatic cancer. Here we report the development of an aminopyrazole based proteolysis targeting chimera (PROTAC 2) that selectively degrades CDK9 (DC50 = 158 ± 6 nM). Mass spectrometry-based kinome profiling shows PROTAC 2 selectively degrades CDK9 in MiaPaCa2 cells and sensitizes them to Venetoclax mediated growth inhibition.
Graphical Abstract
Cyclin-dependent kinase (CDK) 9 is a member of the CDK family of serine-threonine kinases that are involved in a variety of cellular functions such as the regulation of the cell cycle and transcription.1, 2 CDKs are activated through association with cyclins or activators that mimic cyclins.3 CDK9 interacts with Cyclin K and the CDK9/Cyclin K complex regulates the replication stress response.4, 5 CDK9 also interacts with cyclin T to form the catalytic subunit of the positive transcription elongation factor b (P-TEFb) that facilitates productive transcriptional elongation.6 Among others, CDK9/Cyclin T complex regulates transcription of anti-apoptotic protein Mcl-1 and the oncogene Myc.7, 8 More recently, CDK9 was implicated as a therapeutic target in KRAS-mutant driven pancreatic cancer using the MiaPaCa2 cell line.9
Analyses of data from large-scale kinome screens suggest that it is easy to develop inhibitors that are selective for CDKs; however, these inhibitors are seldom selective for a specific CDK/cyclin combination.1, 10 This lack of selectivity for various CDKs have been addressed by proteolysis targeting chimeras (PROTACs).11, 12 PROTACs are heterobifunctional molecules, wherein ligands that bind to two different proteins are conjugated via a linker. One end of the PROTAC binds to the protein of interest (POI) while the other binds to an E3-ligase to facilitate the formation of a ternary complex (POI:PROTAC:E3-ligase). The formation of a stable ternary complex allows the E3-ligase to ubiquitinate a proximal lysine on the POI to enable proteasomal degradation of POI.13–15 Since the distribution of surface-exposed lysine residues are different among the various CDKs, a PROTAC generated using a non-selective CDK inhibitor with an appropriate linker could lead to a selective CDK PROTAC. Consistently, others and we used non-selective CDK inhibitors with aminopyrazole,16 aminothiazole and 4H-chromen-4-one cores to generate the CDK9 selective PROTACs, Compound 3, THAL-SNS-032 and Wogonin-based 11c, respectively.17–19 A recent report used a CDK9 inhibitor BAY-1143572 as a warhead to develop a CDK9 PROTAC,20 which is similar to a CDK4/6 inhibitor Palbociclib-based CDK6 selective PROTAC.21–23
The goal here was to improve upon our previously reported aminopyrazole based CDK9 degrader and identify one with sub-μM potency.17 Toward this, we explored linker length and linker composition by synthesis of a focused library of aminopyrazole-based PROTACs and evaluated them as CDK9 degraders (Figure 1). The PROTAC panel was screened in a time-course (Figure S1) and a dose-response study (Figure 2) to identify an aminopyrazole based CDK9 degrader with sub-μM potency. Briefly, HEK293 cells were subjected to increasing concentrations (0.01 – 10 μM) of the PROTACs for 24 hr and the lysates were subjected to western blot analysis to assess CDK9 degradation (Figure 2).
Figure 1.
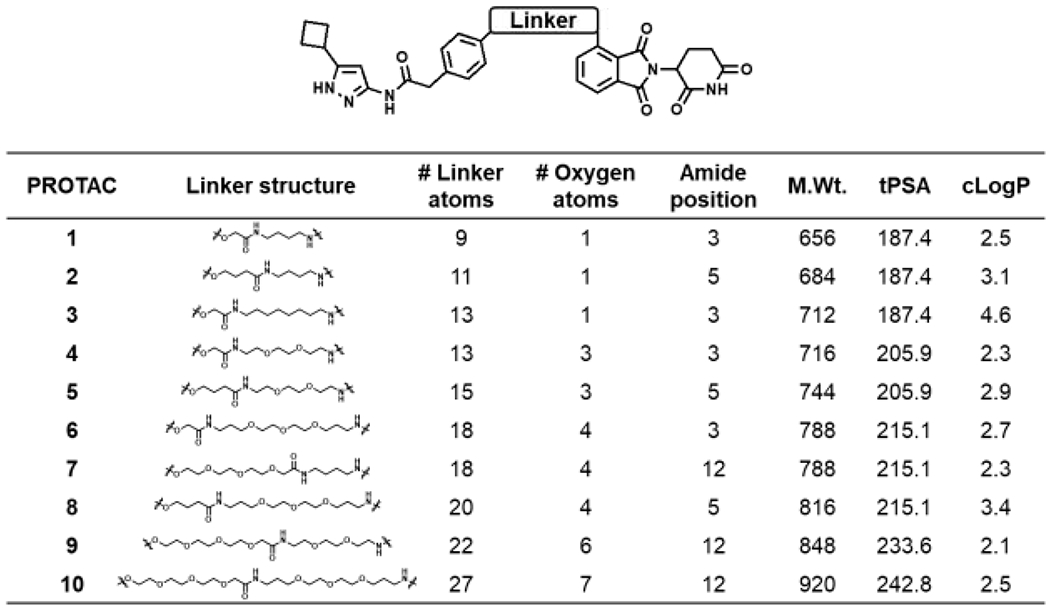
Aminopyrazole based PROTACs.
Figure 2.
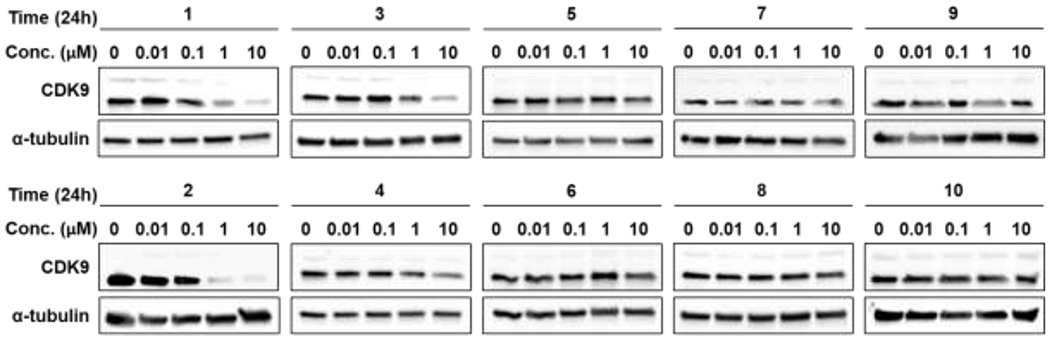
Screening of aminopyrazole based PROTACs for CDK9 degradation.
The screen identified PROTAC 2 as a CDK9 degrader with sub-μM DC50 value (158 ± 6 nM), which induced complete CDK9 degradation at 1 μM (Figure 2). We also determined that PROTAC 2 does not degrade the CDK9 binding partner Cyclin K (Figure S2). Decreasing the linker length by 2-carbon atoms (PROTAC 1), resulted in partial loss of CDK9 degradation. Increasing the linker length by two additional carbon atoms in PROTAC 3 resulted in partial CDK9 degradation with DC50 ∼ 1 μM (Figure 2). Surprisingly, replacement of alkyl linkers with PEG linker (PROTAC 3 vs PROTAC 4) exhibited little to no CDK9 degradation (Figure 2). We also varied PEG linker lengths on either side of amide (PROTAC 5 - 9) and observed no CDK9 degradation. These studies suggest that linker length and linker composition could be important for efficient CDK9 degradation.
We next evaluated the selectivity of PROTAC 2 across other CDK family members in dose-dependent (Figure 3A) and time-dependent (Figure 3B) studies. HEK293 cells were treated with indicated concentrations of PROTAC 2 for 24 h and the lysates were subjected to western blot analyses and probed for the different CDKs (Figure 3A). In the dose-dependent study, PROTAC 2 selectively degraded CDK9 without affecting the levels of other CDK family members. Next, we subjected HEK293 cells to 1 μM of PROTAC 2 and probed the lysates for various CDKs at indicated time points (Figure 3B). PROTAC 2 degraded CDK9 as early as the 4 h time point and the degradation was sustained for 24 h. It is important to note that PROTAC 2 did not degrade other CDKs probed (Figure 3B). CDK9 is a validated target for pancreatic cancer and we observed PROTAC 2 induced CDK9 degradation in pancreatic cancer cell lines (MiaPaCa2 and S2013) (Figure S3A and S3B). Together these studies show that PROTAC 2 selectively degrades CDK9 in dose- and time-dependent manner.
Figure 3.
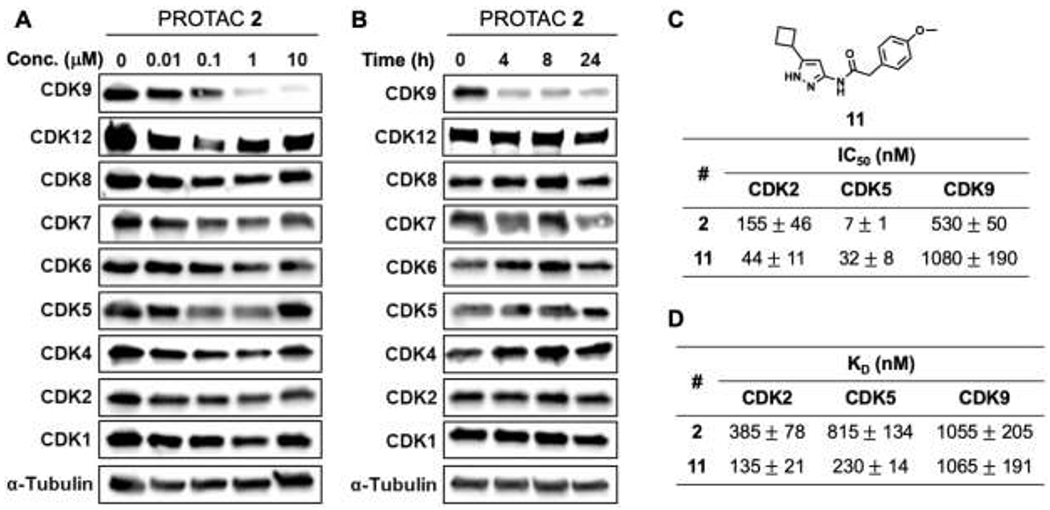
Selectivity profile of CDK9 PROTAC 2. Western blot analysis showing dose-dependent (24 h) (A) and time-dependent (1 μM) (B) effects of PROTAC 2 in HEK293 cells. In vitro cell-free IC50 (C) and KD (D) profiling of inhibitor 11 and PROTAC 2, respectively.
Previous reports from our group and others showed that aminopyrazole analogs are CDK 2/5 inhibitors.16, 24, 25 Remarkably, an aminopyrazole based PROTAC selectively degraded CDK9.17 To determine if the selective degradation of CDK9 is a result of loss of binding to CDK2/5 due to the modifications of the aminopyrazole inhibitor to convert it into a PROTAC, we evaluated PROTAC 2 and CDK inhibitor 1116, 17 in in vitro cell-free kinase assays. Inhibitor 11 and PROTAC 2 exhibited nM potency (IC50) in blocking the kinase activities of CDK2 and CDK5. The CDK2, CDK5 and CDK9 kinase activity inhibition profiles of Inhibitor 11 and PROTAC 2 were similar (Figure 3C). Likewise, the binding affinity (KD) profiles of Inhibitor 11 and PROTAC 2 for CDK2, CDK5 and CDK9 were also similar (Figure 3D). Since the binding affinity profile and the kinase activity inhibitory profile of analog 11 and PROTAC 2 are similar, the selective degradation of CDK9 by PROTAC 2 could be attributed to the differential topographical distributions of lysine residues among CDK2, CDK5 and CDK9.
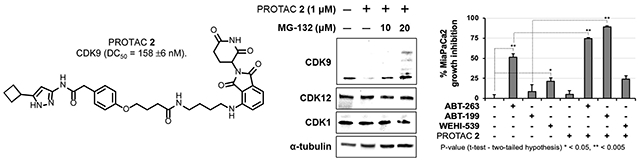
Next, we conducted a series of competition experiments to evaluate the mechanism of action of PROTAC 2. To determine if degradation of CDK9 by PROTAC 2 followed the formation of a ternary complex between CDK9:PROTAC 2:CRBN E3-ligase, we conducted two sets of competition experiments with PROTAC 2 and pomalidomide (a CRBN ligand), and PROTAC 2 and flavopiridol (a CDK9 inhibitor). HEK293 cells were treated with either 10 μM of pomalidomide (Figure 4A) or flavopiridol (Figure 4B) alone and in combination with 1 μM of PROTAC 2 for 24 h and 8 h, respectively, and the lysates were subjected to western blot analysis. The results show that pomalidomide or flavopiridol by itself did not affect CDK9 levels but was able to block PROTAC 2 mediated degradation of CDK9 when treated in combination (Figure 4A and 4B). No such changes were observed in CDK7 levels, another CDK involved in transcriptional regulation was used as a control. These competition studies demonstrate the need for simultaneous engagement of CDK9 and a CRBN E3 ligase (ternary complex) by PROTAC 2 to facilitate CDK9 degradation. Since the PROTAC-based strategy involves the ubiquitination of target protein followed by its proteasomal degradation, we also subjected HEK293 cells to increasing concentrations of proteasome inhibitor MG132 and PROTAC 2 for 24 h. CDK9 degradation was abrogated in the presence of MG132. The membranes were also probed for CDK1 and CDK12 and no such changes were observed in their levels (Figure 4C). Collectively, these studies confirmed the formation of the ternary complex between CDK9:PROTAC 2:CRBN, followed by CDK9 ubiquitination and subsequent proteasomal degradation as the mechanism of action of PROTAC 2, which is similar to our previous report of a CDK6 selective PROTAC.22
Figure 4.
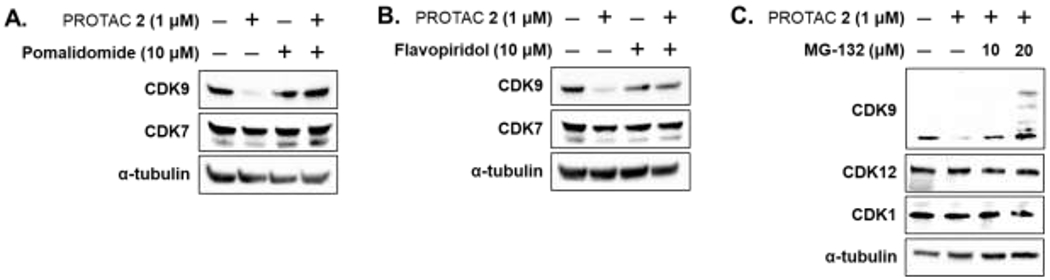
Mechanism of action of CDK9 PROTAC 2. (A) Western blot analysis showing inhibition of CDK9 degradation upon simultaneous treatment of Pomalidomide and PROTAC 2. (B) Western blot analysis showing inhibition of CDK9 degradation upon simultaneous treatment of Flavopiridol and PROTAC 2. (C) Western blot analysis showing inhibition of CDK9 degradation upon simultaneous treatment of MG132 and PROTAC 2.
To assess the kinome and proteome selectivity of PROTAC 2, we performed quantitative mass spectrometry with lysates from HEK293 and MiaPaCa2 cell lines treated with PROTAC 2 for 24 h (Figure 5). MiaPaCa2 cells were selected for the proteome-wide profiling as they were used to validate CDK9 as a therapeutic target for pancreatic cancers.9 We were able to quantify 229 unique kinases, including 13 CDKs in the HEK293 lysate and CDK9 was the only kinase identified as a hit with a 1.5-fold change and a significance threshold of P < 0.001 (Figure 5A). We were able to quantify 3433 proteins in the MiaPaCa2 lysate and three kinases CDK9, CDK2 and RPS6KA1 were identified as hits with a 1.5-fold change and a significance threshold of P < 0.001 (Figure 5B).
Figure 5.
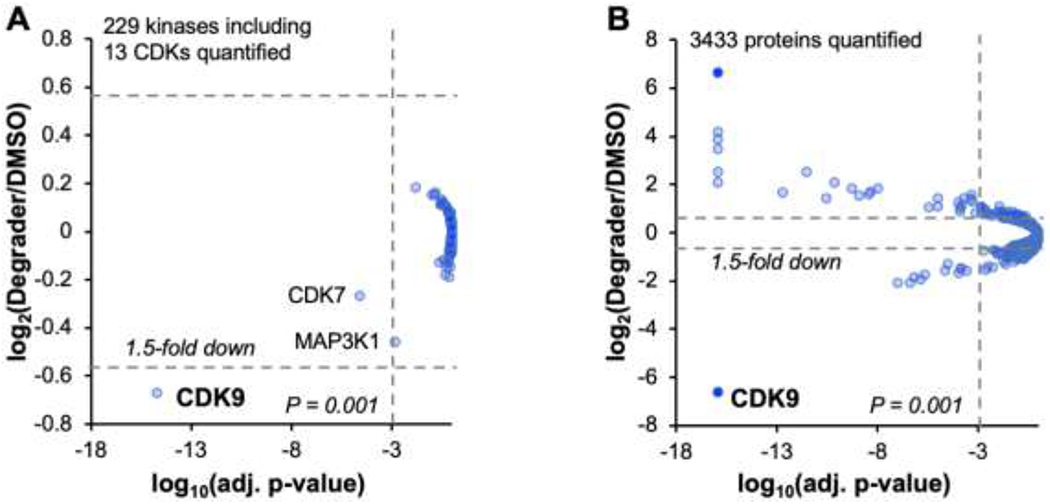
Kinome and proteome-wide profile of PROTAC 2. (A) Volcano plot of the kinome in HEK293 cells treated with 1μM of PROTAC 2 and incubated for 24 h. (B) Volcano plot of the proteome in MiaPaCa2 cells treated with 1 μM of PROTAC 2 and incubated for 24 h.
Recent studies demonstrated that concurrent inactivation of Mcl-1 and Bcl-xL resulted in robust induction of apoptosis.16, 17, 25–30 Consequently, targeting Mcl-1 and Bcl-xL is considered a therapeutic strategy for pancreatic cancer therapy.28 Abbott Laboratories (ABT) successfully developed direct inhibitors of Bcl-xL/Bcl2/Bcl-w (Figure 6A).31, 32 However, resistance to Bcl-xL/Bcl2/Bcl-w inhibition has been attributed to compensatory activity by Mcl-133 and Mcl-1 inactivation sensitized cancer cells to Bcl2 inhibitors.34,35 Since CDK9 activity regulates the levels of pro-survival protein Mcl-1,34, 36 we tested whether the selective CDK9 PROTAC 2 sensitizes MiaPaCa2 to Bcl2 inhibitors.
Figure 6.
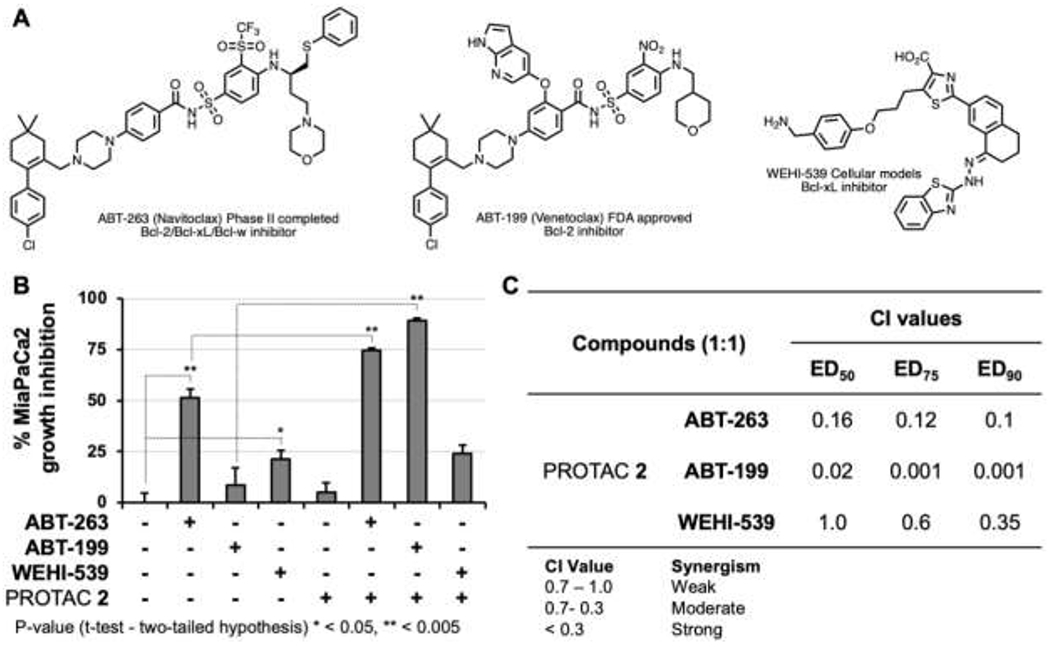
Synergism studies using BCL inhibitors and PROTAC 2. (A) Structure of Bcl2 inhibitors Navitoclax (ABT-263); Venetoclax (ABT-199); WEHI-539; (B) Growth inhibitory effects of different inhibitors combinations at 5 μM, (C) Combination index (Cl) values for the three Bcl2 inhibitors and PROTAC 2 combinations.
To test the above, we selected three Bcl2 inhibitors with varying Bcl2 selectivity profiles: ABT-263 (Navitoclax) is a clinical candidate that underwent a phase II trial and is a Bcl2/Bcl-xL/Bcl-w inhibitor, ABT-199 (Venetoclax) is a FDA approved Bcl2 inhibitor, and WEHI-539 is an experimental Bcl-xL inhibitor (Figure 6A). We conducted growth inhibition studies to determine if PROTAC 2 sensitizes MiaPaCa2 cells to the above Bcl2 inhibitors. Briefly, MiaPaCa2 cells were treated individually with ABT-263 (Bcl2/Bcl-xL/Bcl-w), ABT-199 (Bcl2), WEHI-537 (Bcl-xL), PROTAC 2 (CDK9), and the combination of Bcl2 inhibitors and PROTAC 2, respectively. Following a 3-day incubation, cell viability was measured using a PrestoBlue assay. The selective compounds PROTAC 2, ABT199 and WEHI-539 showed minimal growth inhibitory effects individually at equimolar concentration, while the non-selective inhibitor ABT-263 induced ∼50% growth inhibition. However, PROTAC 2 sensitized MiPaCa2 cells to ABT-263 or ABT-199. Interestingly, PROTAC 2 did not sensitize MiaPaCa2 to the Bcl-xL selective inhibitor WEHI-539 (Figure 6B). Consistently, the combination index (Cl) values determined using CalcuSyn at effective doses (ED) of 50, 75 and 90 showed that PROTAC 2 exhibited strong synergism with ABT-263 and ABT-199 but not with WEHI-539 (Figure 6C). Together, these data show that PROTAC 2 potently sensitizes MiaPaCa2 cells to the Bcl2 inhibitor Venetoclax.
In conclusion, we report the development of an aminopyrazole based CDK9 degrader (PROTAC 2) with a DC50 value of ∼150 nM. In cells, PROTAC 2 selectively degrades CDK9 while sparing other CDK family members, which can be attributed to the differential topological arrangement of surface-exposed lysine residues among the CDK family members. Using competition studies with a CDK9 inhibitor, a CRBN binder and a proteasome inhibitor, we established that PROTAC 2 forms a ternary complex with CDK9 and CRBN to induce ubiquitination mediated proteasomal degradation of CDK9. The cellular selectivity of PROTAC 2 for CDK9 was also established using kinome- and proteome-wide mass spectrometry-based profiling studies. We also showed that PROTAC 2 sensitizes pancreatic cancer cells to the Bcl2 selective FDA approved inhibitor Venetoclax. Synergism studies to validate the above findings in vivo are currently underway and will be reported in due course.
Supplementary Material
Acknowledgments:
This work was supported in part by NIH grants CA197999, CA251151, GM121316, and CA036727. HMK was supported by a UNMC graduate fellowship. We would like to thank the Natarajan lab members for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Supplementary data associated with this article can be found in the online version.
References
- 1.Sonawane YA, Taylor MA, Napoleon JV, Rana S, Contreras JI, Natarajan A. Cyclin Dependent Kinase 9 Inhibitors for Cancer Therapy. J Med Chem. 2016;59(19): 8667–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peyressatre M, Prevel C, Pellerano M, Morris MC. Targeting cyclin-dependent kinases in human cancers: from small molecules to Peptide inhibitors. Cancers (Basel). 2015;7(1): 179–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wood DJ, Endicott JA. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018;8(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu DS, Zhao R, Hsu EL, et al. Cyclin-dependent kinase 9-cyclin K functions in the replication stress response. EMBO Rep. 2010;11(11): 876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu DS, Cortez D. A role for CDK9-cyclin K in maintaining genome integrity. Cell Cycle. 2011;10(1): 28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majello B, Napolitano G, Giordano A, Lania L. Transcriptional regulation by targeted recruitment of cyclin-dependent CDK9 kinase in vivo. Oncogene. 1999;18(32): 4598–4605. [DOI] [PubMed] [Google Scholar]
- 7.Manohar SM, Rathos MJ, Sonawane V, Rao SV, Joshi KS. Cyclin-dependent kinase inhibitor, P276-00 induces apoptosis in multiple myeloma cells by inhibition of Cdk9-T1 and RNA polymerase II-dependent transcription. Leuk Res. 2011;35(6): 821–830. [DOI] [PubMed] [Google Scholar]
- 8.Huang CH, Lujambio A, Zuber J, et al. CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 2014;28(16): 1800–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blake DR, Vaseva AV, Hodge RG, et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci Signal. 2019;12(590). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11): 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15): 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chembiochem. 2007;8(17): 2058–2062. [DOI] [PubMed] [Google Scholar]
- 13.Zengerle M, Chan KH, Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10(8): 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Qian Y, Altieri M, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem Biol. 2015;22(6): 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winter GE, Buckley DL, Paulk J, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241): 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rana S, Sonawane YA, Taylor MA, Kizhake S, Zahid M, Natarajan A. Synthesis of aminopyrazole analogs and their evaluation as CDK inhibitors for cancer therapy. Bioorg Med Chem Lett. 2018;28(23–24): 3736–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robb CM, Contreras JI, Kour S, et al. Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC). Chem Commun (Camb). 2017;53(54): 7577–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bian J, Ren J, Li Y, et al. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg Chem. 2018;81: 373–381. [DOI] [PubMed] [Google Scholar]
- 19.Olson CM, Jiang B, Erb MA, et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat Chem Biol. 2018;14(2): 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu X, Li Y, Yu B, et al. Discovery of selective CDK9 degraders with enhancing antiproliferative activity through PROTAC conversion. Eur J Med Chem. 2020;211: 113091. [DOI] [PubMed] [Google Scholar]
- 21.Su S, Yang Z, Gao H, et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J Med Chem. 2019;62(16): 7575–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rana S, Bendjennat M, Kour S, et al. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg Med Chem Lett. 2019;29(11): 1375–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brand M, Jiang B, Bauer S, et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem Biol. 2019;26(2): 300–306 e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pevarello P, Brasca MG, Amici R, et al. 3-Aminopyrazole inhibitors of CDK2/cyclin A as antitumor agents. 1. Lead finding. J Med Chem. 2004;47(13): 3367–3380. [DOI] [PubMed] [Google Scholar]
- 25.Robb CM, Kour S, Contreras JI, et al. Characterization of CDK(5) inhibitor, 20-223 (aka CP668863) for colorectal cancer therapy. Oncotarget. 2018;9(4): 5216–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez H, Zhang L, George NM, et al. Perturbation of the Bcl-2 network and an induced Noxa/Bcl-xL interaction trigger mitochondrial dysfunction after DNA damage. J Biol Chem. 2010;285(20): 15016–15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rajule R, Bryant VC, Lopez H, Luo X, Natarajan A. Perturbing pro-survival proteins using quinoxaline derivatives: a structure-activity relationship study. Bioorg Med Chem. 2012;20(7): 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Contreras JI, Robb CM, King HM, et al. Chemical Genetic Screens Identify Kinase Inhibitor Combinations that Target Anti-Apoptotic Proteins for Cancer Therapy. ACS Chem Biol. 2018;13(5): 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kour S, Rana S, Contreras JI, et al. CDK5 Inhibitor Downregulates Mcl-1 and Sensitizes Pancreatic Cancer Cell Lines to Navitoclax. Mol Pharmacol. 2019;96(4): 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rana S, Kour S, Sonawane YA, et al. Symbiotic prodrugs (SymProDs) dual targeting of NFkappaB and CDK. Chem Biol Drug Des. 2020;96(2): 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9): 3421–3428. [DOI] [PubMed] [Google Scholar]
- 32.Touzeau C, Dousset C, Bodet L, et al. ABT-737 induces apoptosis in mantle cell lymphoma cells with a Bcl-2high/Mcl-1low profile and synergizes with other antineoplastic agents. Clin Cancer Res. 2011;17(18): 5973–5981. [DOI] [PubMed] [Google Scholar]
- 33.Konopleva M, Contractor R, Tsao T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5): 375–388. [DOI] [PubMed] [Google Scholar]
- 34.Abid M, Sonawane YA, Contreras JI, Rana S, Natarajan A. Recent Advances in Cancer Drug Development: Targeting Induced Myeloid Cell Leukemia-1 (Mcl-1) Differentiation Protein. Curr Med Chem. 2017;24(40): 4488–4514. [DOI] [PubMed] [Google Scholar]
- 35.Peddaboina C, Jupiter D, Fletcher S, et al. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer. 2012;12: 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacCallum DE, Melville J, Frame S, et al. Seliciclib (CYC202, R-Roscovitine) induces cell death in multiple myeloma cells by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65(12): 5399–5407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.