

Abstract
Tardive dyskinesia (TD) is a severe condition characterized by repetitive involuntary movement of orofacial regions and extremities. Patients treated with antipsychotics typically present with TD symptomatology. Here, we conducted the largest GWAS of TD to date, by meta-analyzing samples of East-Asian, European, and African American ancestry, followed by analyses of biological pathways and polygenic risk with related phenotypes. We identified a novel locus and three suggestive loci, implicating immune-related pathways. Through integrating trans-ethnic fine mapping, we identified putative credible causal variants for three of the loci. Post-hoc analysis revealed that SNPs harbored in TNFRSF1B and CALCOCO1 independently conferred three-fold increase in TD risk, beyond clinical risk factors like Age of onset and Duration of illness to schizophrenia. Further work is necessary to replicate loci that are reported in the study and evaluate the polygenic architecture underlying TD.
Subject terms: Pharmacogenomics, Predictive markers
Introduction
Tardive Dyskinesia (TD) is a persistent and potentially debilitating involuntary movement disorder characterized by choreiform, athetoid, and or dystonic movements1,2. TD is largely caused by antipsychotic treatment and was first described in 19573. Although commonly observed in patients with schizophrenia, TD can occur in individuals with other psychiatric disorders, as long as they have been similarly exposed to prolonged antipsychotic treatment. The prevalence of TD in schizophrenia is estimated to be between 15 and 30%, although rates of TD have reduced with prescription of second-generation antipsychotics4–6. Nevertheless, second-generation antipsychotics still carry with them a risk of developing TD, and TD remains a clinically relevant phenotype as it has been associated with more severe schizophrenia illness, cognitive impairments, lowered quality of life and increased mortality7–10.
The pathophysiology of TD is currently unknown. Theories pertaining to dopamine receptor hypersensitivity, serotonergic dysfunction, GABA insufficiency, and free radical damage have been put forth2,11,12. It is postulated that TD is related to the schizophrenia disease process; recent reports have linked basal ganglia (caudate nucleus) volume reductions to TD in schizophrenia; notably, these samples were on second-generation antipsychotic—suggesting the pathological process towards TD is beyond neurochemical properties13. As TD is potentially irreversible with no effective treatment, there needs to be an added emphasis on the prevention and identification of genetic risk factors. Several genes (e.g.-, DRD2, DRD3, MnSOD, CYP2D6, GRIN2A, and GRIN2B) have been implicated in candidate gene studies, but replication remains equivocal14–18. Genome-Wide Association Studies (GWAS) performed on the TD phenotype suggested that the GLI family zinc finger 2 (GLI2), heparan sulfate proteoglycan 2 (HSPG2), dipeptidyl-peptidase 6 (DPP6), and GABA pathway genes could putatively be considered susceptibility genes for TD19–23. Nonetheless, these studies are limited by small samples and require further replication. Here, we report the findings of the largest GWAS of TD to date. We identified a novel locus at 16q24.1 (P = 3.01 × 10−8) and three suggestive loci (1p36.22, 6q23.2, 12q13.13; P < 5 × 10−7) associated with TD.
Methodology
Participants from the Singapore Clinical and Translational Research program in Singapore, and the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE)24–26 study were included in the current report. All participants met criteria for DSM-IV diagnosis for schizophrenia. Tardive dyskinesia was ascertained via the Abnormal Involuntary Movement Scale (AIMS)27,28. After quality control procedures, 1406 participants (280 with TD), and 6,291,020 SNPs remained. Linear Mixed Models GWA was performed via GEMMA29, and independent cohorts across the two studies were meta-analyzed via fixed effects inverse variance approach in METAL30. GWAS summary statistics were then subjected to functional annotation for GWAS identified loci31, eQTL lookups, pathway analysis32, and transcriptome-wide analysis33, which were conducted to characterize potential biological mechanisms underlying TD. Further fine-mapping analysis was also carried out to identify credible causal variants34–36. A series of polygenic risk score analyses were conducted to compare the genetic architecture of TD with related phenotypes37. We also carried out a series of post-hoc multivariate logistic regression analysis to investigate joint clinical and genetic factors that predict the emergence of TD. Finally, variants previously associated with TD were compared with the current GWAS results11,18,22,23,38–46. Detailed methodological approaches are further reported in the Supplementary Information included with the current report.
Results
Demographics and assessment of tardive dyskinesia
Demographics are reported in Table 1 and Supplementary Table 1. There were 71.1% males and 28.9% females with TD. There was no significant difference in gender proportion between individuals with TD and those without, (1, n = 1406) = 0.142, P = 0.706. There were significant differences in age, t(1404) = −14.06, P = 4.0 × 10−42, age of illness onset, t(392.7) = −3.06, P = 0.0024, duration of illness, t(1390) = −11.28, P = 2.76 × 10−28, and antipsychotic dose measured in CPZ equivalent, t(516.3) = 3.73, P = 0.002 between individuals with and without TD. These differences in demographics and clinical characteristics were further modelled using a polygenic risk score approach that allows us to examine genetic effects for TD alongside demographics effects; results are reported in subsequent sections.
Table 1.
Characteristics of tardive dyskinesia versus non-tardive dyskinesia samples.
| TD samples (N = 280) | Non-TD samples (N = 1126) | |
|---|---|---|
| Gender (male/female) | 199/81 | 813/313 |
| Age (years) | 55.51 (11.38) | 44.23 (12.17) |
| Age of illness onset (years) | 28.71 (10.71) | 26.55 (9.50) |
| Duration of Illness (years) | 26.52 (12.55) | 17.22 (12.23) |
| PANSS (score) | ||
| Positive symptoms | 12.90 (6.01) | 13.51 (5.99) |
| Negative symptoms | 15.42 (6.89) | 15.69 (6.99) |
| General psychopathology | 27.44 (10.11) | 27.96 (10.12) |
| Total | 55.76 (20.67) | 57.17 (20.40) |
| Antipsychotics, n (%) | ||
| Typical only | 170 (60.7%) | 487 (43.3%) |
| Atypical only | 66 (23.6%) | 394 (35.0%) |
| Typical + atypical | 17 (6.1%) | 93 (8.3%) |
| None | 2 (0.7%) | 7 (0.6%) |
| Daily CPZ equivalent dose, mg (SD) | 494.7 (494.30) | 637.16 (679.07) |
| Total aims score | 11.09 (5.11) | 1.19 (2.02) |
Genome-wide association of tardive dyskinesia
Standard GWAS quality control procedures were carried out (Supplementary Figs. 1–5). We carried out Principal Components Analysis (PCA) on each ancestry group to identify overall population stratification across samples, and, within population PCA to identify fine-grain population outliers (See Supplementary Information). We removed population outliers detected by PCA via a series of k-means clustering. Notably, association analysis was carried out within each ancestry first and thereafter meta-analyzed. Linear mixed models conducted via the GEMMA29 package revealed significant genome-wide association of TD at the level of the CATIE cohorts, but not the STCRP cohort (Supplementary Fig. 6). Subsequent fixed-effect inverse variance meta-analysis30 (λGC = 1.02; Table 2, Figs. 1–3) across the STCRP and CATIE cohorts revealed one novel locus on chromosome 16 (rs11639774, downstream of GSE1) (P = 3.01 × 10−8). Three other suggestive independent genomic loci (P < 5 × 10−7) were identified on chromosome 1 (rs499646, P = 8.30 × 10−8, TNFRSF1B), chromosome 6 (rs6926250, P = 2.54 × 10−7, EPB41L2), and chromosome 12 (rs4237808, P = 1.08 × 10−7, CALCOCO1). Due to low minor allele frequencies in the STCRP sample, two of the SNPs were only present in the CATIE cohorts. Proxy SNPs in LD within the STCRP cohort were identified using the SNP Annotation and Proxy Search (SNAP)47. Further meta-analysis of association results from both proxy SNPs were carried out using Fisher’s p-value meta-analysis approach (https://CRAN.R-project.org/package=metap). Fisher’s meta-analysis for the top SNP (rs11639774) and proxy SNP (rs9928615) in the STCRP cohort was significant ( (4) = 40.28, P = 3.79 × 10−08). Similarly, results for SNP (rs499626) and proxy SNP (rs11569835) in the STCRP cohort reached suggestive significance ( (4) = 36.48, P = 2.31 × 10−07), with both primary variants with proxy SNP meta-analysis showing consistent effects (Supplementary Table 2). GWAS markers were further annotated via ANNOVAR, eQTL, Chromatin Interaction modules within the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA)31 (Supplementary Tables 3, 4; Supplementary Fig 7).
Fig. 2. QQ plot for meta-analysis of STCRP, CATIE-EUR, and CATIE-AFR.
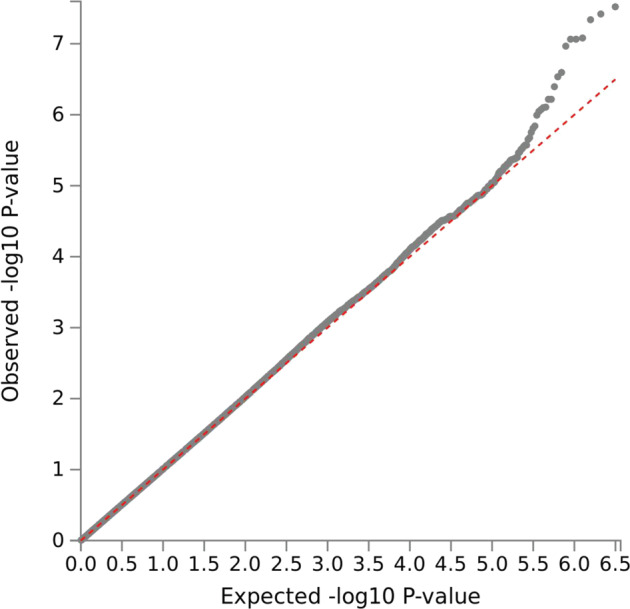
Note: Lambda = 1.02.
Table 2.
Top SNPs associated with tardive dyskinesia.
| SNP | Symbol | Location | Position | Variant | Effect allele | Other allele | EAF | Beta | SE | P-value | HetP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs11639774 | GSE1 | 16q24.1 | 85221868 | Intergenic | A | G | 0.168 | 0.151 | 0.0273 | 3.01E-08 | 0.067 |
| rs499646 | TNFRSF1B | 1p36.22 | 12239089 | Intronic | A | G | 0.12 | 0.1949 | 0.0364 | 8.30E-08 | 0.246 |
| rs4237808 | CALCOCO1 | 12q13.13 | 54093797 | Intergenic | T | C | 0.4914 | 0.0765 | 0.0144 | 1.08E-07 | 0.506 |
| rs6926250 | EPB41L2 | 6q23.2 | 131302473 | Intronic | T | C | 0.6872 | −0.0968 | 0.0188 | 2.54E-07 | 0.447 |
SNP single-nucleotide polymorphism, EAF effect allele frequency, SE standard error, HetP heterogenity P-value.
Fig. 1. Manhattan plot for GWAS fixed effect meta-analysis of TD.

Note: Three cohorts were included in the fixed-effect meta-analysis, STCRP, CATIE-EUR, and CATIE-AFR. A single GWAS signal was found at 16q24.1 for rs11639774.
Fig. 3. Regional loci and fine-mapping plots.

Top panel represents regional plots for a Chromosome 1, b Chromosome 12, and c Chromosome 16. Bottom panel represents the visualization of 99% credible SNP set for a Chromosome 1, b Chromosome 12, and c Chromosome 16, against location of SNP, with top annotation bars.
Gene-based and pathway analysis
MAGMA was used to conduct both the gene-based and geneset analysis32. Gene-based analysis tests for association of TD with markers within the gene by mapping the SNPs to gene level, while geneset analysis aggregates individual genes to a collection of genes with overlapping characteristics28. Results of MAGMA gene-based analysis on 18,259 mapped autosomal genes after Bonferroni correction are presented in Supplementary Table 5. None of the genes were significant after Bonferroni correction. MAGMA competitive geneset analysis32 was conducted using the most recent Molecular Signature Database version 6.148. Although none of the 17,199 genesets survived Bonferroni correction, top pathways implicated regulation of immunoglobulin production and immunoglobulin isotype switching, expression of chemokine receptors, and regulation of cell growth, and monocytes (Supplementary Table 6).
eQTL lookups/transcriptome-wide analysis
Expression quantitative trait loci (eQTL) were performed as part of FUMA31 GWAS pipeline. Notably, eQTL effect of ATP5G2 (Bonferroni corrected P = 1.24 × 10−14, Supplementary Table 7) and MAP3K12 (Bonferroni corrected p = 6.29 × 10−10, Supplementary Table 7) was observed for subthreshold genomic significance loci for 12q13.13, TNFRS1B (Bonferroni corrected p = 4.60 × 10−08, Supplementary Table 7) for 1p36.22, EPB41L2 (Bonferroni corrected p = 2.86 × 10−07, Supplementary Table 7) and AKAP7 (Bonferroni corrected p = 1.48 × 10−06, Supplementary Table 7) for 6q23.2, with expression in various tissues, including those related to motor functions (Table 3).
Table 3.
Summary of expression quantitative trait loci (eQTL) analysis.
| Symbol | Chr | Tissue | Database |
|---|---|---|---|
| TNFRSF1B | 1 | BIOS_eQTL_geneLevel | BIOSQTL |
| AKAP7 | 6 | BIOS_eQTL_geneLevel | BIOSQTL |
| AKAP7 | 6 | Muscle_Skeletal | GTEx_v7 |
| AKAP7 | 6 | Thyroid | GTEx_v7 |
| EPB41L2 | 6 | xQTLServer_eQTLs | xQTLServer |
| EPB41L2 | 6 | Skin_Sun_Exposed_Lower_leg | GTEx_v7 |
| EPB41L2 | 6 | Esophagus_Muscularis | GTEx_v7 |
| EPB41L2 | 6 | Esophagus_Mucosa | GTEx_v7 |
| EPB41L2 | 6 | Lung | GTEx_v7 |
| ATF7 | 12 | BIOS_eQTL_geneLevel | BIOSQTL |
| ATP5G2 | 12 | Esophagus_Mucosa | GTEx_v7 |
| ATP5G2 | 12 | BIOS_eQTL_geneLevel | BIOSQTL |
| ATP5G2 | 12 | Thyroid | GTEx_v7 |
| ATP5G2 | 12 | Nerve_Tibial | GTEx_v7 |
| ATP5G2 | 12 | Adipose_Subcutaneous | GTEx_v7 |
| ATP5G2 | 12 | Brain_Cerebellar_Hemisphere | GTEx_v7 |
| ATP5G2 | 12 | Brain_Cerebellum | GTEx_v7 |
| ATP5G2 | 12 | Testis | GTEx_v7 |
| ATP5G2 | 12 | Esophagus_Muscularis | GTEx_v7 |
| ATP5G2 | 12 | Adipose_Visceral_Omentum | GTEx_v7 |
| ATP5G2 | 12 | Small_Intestine_Terminal_Ileum | GTEx_v7 |
| ATP5G2 | 12 | Lung | GTEx_v7 |
| ATP5G2 | 12 | Skin_Not_Sun_Exposed_Suprapubic | GTEx_v7 |
| ATP5G2 | 12 | Artery_Tibial | GTEx_v7 |
| ATP5G2 | 12 | CRBL | BRAINEAC |
| ATP5G2 | 12 | MEDU | BRAINEAC |
| ATP5G2 | 12 | PUTM | BRAINEAC |
| ATP5G2 | 12 | SNIG | BRAINEAC |
| ATP5G2 | 12 | FCTX | BRAINEAC |
| ATP5G2 | 12 | aveALL | BRAINEAC |
| ATP5G2 | 12 | TCTX | BRAINEAC |
| ATP5G2 | 12 | WHMT | BRAINEAC |
| ATP5G2 | 12 | OCTX | BRAINEAC |
| ATP5G2 | 12 | THAL | BRAINEAC |
| ATP5G2 | 12 | HIPP | BRAINEAC |
| ESPL1 | 12 | Esophagus_Mucosa | GTEx_v7 |
| MAP3K12 | 12 | BIOS_eQTL_geneLevel | BIOSQTL |
| NPFF | 12 | BIOS_eQTL_geneLevel | BIOSQTL |
| SP1 | 12 | Esophagus_Mucosa | GTEx_v7 |
| SP7 | 12 | Thyroid | GTEx_v7 |
| SP7 | 12 | Testis | GTEx_v7 |
Transcriptome-wide analysis was implemented via MetaXcan33. Unlike eQTL lookups, the MetaXcan approach further incorporates evidence from GWAS summary statistics with genome-wide gene expression profiles from the GTex49 database. This provides information of potential functional enrichment within a genomic locus. Here, we found lower expression of a transcript, EPB41L2, in the esophagus muscularis at Bonferroni-corrected significance (P = 0.02, Supplementary Table 8).
Fine-mapping analysis
Cross-Ancestry fine mapping was performed on PAINTOR v3.134,35 to identify putative causal variants on the 4 loci (1p36.22, 6q23.2, 12q13.13, and 16q24.1). The 99% cumulative posterior probability identified a total of 231 putative credible causal SNPs across three loci (1p36.22, 12q13.13, and 16q24.1; Supplementary Table 9, Supplementary Fig. 8). From these, highly credible SNPs were defined as posterior probability > 0.8. This identified a putative causal variant for each locus: 1p36.22 (rs499646, posterior probability = 0.99), 12q13.13 (rs4237808, posterior probability = 0.863), and 16q24.1 (rs28468398, posterior probability = 0.952). Notably, for loci on 1p36.22 and 12q13.13, the index SNP identified in GWAS is also a putative causal SNP identified by PAINTOR (See Fig. 3). Further annotations via the Variant Effect Predictor50 revealed that (i) rs499646 is an intronic variant 3500 bp from the promotor of TNFRSF1B and is a known protein-coding variant and appears to be a loss-of-function intolerant variant, (ii) rs4237808 is an intergenic variant between ATP5G2 and CALCOCO1, but lies within 1000 bp of two CTCF binding sites, and 3200 bp from a known promotor flank, and (iii) rs28468398 is a regulatory region variant, which lies within a transcription factor binding site on CTC-786C10.1/GSE1 gene. Notably, additional joint finemap annotations revealed that all three variants were enriched by known brain level gene expression sites close by (See Fig. 3). GCTA-COJO36 revealed no further signals present in the loci beyond independent variants identified via LD clumping or fine mapping (Supplementary Fig. 9).
Polygenic risk modelling of TD with other diseases and traits
Polygenic risk score modelling via PRSice237 (Supplementary Fig. 10) revealed best polygenic association of TD with amyotrophic lateral Sclerosis (ALS), albeit only two PRS thresholds remained significant after Bonferroni correction (PT = 0.5; PT = 1.0). ALS PRS-based pathway analysis indicated “misfolded protein” as a top pathway shared between TD and ALS (Supplementary Fig. 11), although this remains a trend finding. We followed up on the possibility that there might be pleiotropic genetic effects of ALS and TD, given that both conditions implicated motor neurons. A post-hoc lookup of ALS variants and genes was carried out on GWAS catalog. We filtered GWAS SNPs previously reported to be associated with ALS based on p < 1e-6 and extracted corresponding mapped genes. These were then looked up in the MAGMA gene results reported earlier. We found that three previously known ALS genes, FBXO15, FAM19A1, and NP5 genes were nominally associated with TD genes (TD Gene P-values: FBXO15: 0.0210, FAM19A1: 0.0356 and NPS: 0.0455). Cross-Trait polygenic risk scores were also conducted on other psychopathological or autoimmune traits—such as schizophrenia, bipolar disorder, Alzheimer’s disease, Parkinson’s disease, depression, rheumatoid arthritis, and Crohn’s disease but were not significant after multiple testing correction. It is, however, notable that aspect of the schizophrenia and rheumatoid arthritis did appear to be close to the multiple correction threshold for PT < 0.05.
Post-hoc clinical analysis: effects of nedication type and SNP effects on TD
We performed post-hoc multivariate logistic regression analysis to investigate potential effects of clinical factors affecting TD beyond genetic effects. Preliminary linear-by-linear Chi-square analysis were significant (, p = 1.40 × 10−5, see Fig. 4a). Expectedly, there was a significant proportion of TD cases taking typical, compared to atypical antipsychotics. The Clinical Baseline model (Age of Onset, Duration of illness, Daily CPZ equivalents) was significantly associated with TD, for individuals who were taking either typical (, df = 25, p = 2.15 × 10−15) or atypical antipsychotics (, df = 25, p = 5.55 × 10−7). There were no significant model differences between the Clinical Baseline model nor Clinical + Genetic model (Clinical Baseline + rs6926250, rs4237808, rs499646, rs11639774) in predicting TD for individuals taking typical antipsychotics. However, for individuals taking atypical antipsychotics, there was a significant genetic contribution to predicting TD cases beyond clinical factors (See Fig. 4b, c, Δ = 38.96, df = 4, p = 7.09 × 10−8). It is notable that after adjusting for population stratification, database, and sex, Age of Onset (OR = 1.071, 95% CI = 1.029–1.114, p.adj = 1.96 × 10−2) and Duration of illness (OR = 1.105, 95% CI = 1.069–1.142, p.adj = 1.18 × 10−7) significantly predicted TD. Beyond clinical factors, two of the top SNPs rs499646 [TNFRSF1B] (OR = 2.957, 95% CI = 1.669–5.239, p.adj = 5.86 × 10−3) and rs4237808 [CALCOCO1] (OR = 2.937, 95% CI = 1.713–5.038, p.adj = 2.62 × 10−3) identified in the earlier GWAS significantly predicted the emergence of TD. Note that p.adj are Bonferroni adjusted p-values for 29 variables in the logistic prediction model (See Supplementary Information for further details).
Fig. 4. Clinical and genetic risk factors for TD.

a Stratified analysis for antipsychotic type and TD b Logistic regression model for Clinical Baseline versus Clinical + Genetic Model. Y-axis: Liability scale R2 for logistic regression model, case-control proportions adjusted for TD prevalence set at 15%. Clinical Baseline: Age of Onset, Duration of Illness, Daily CPZ equivalents. Clinical + Genetic: includes Clinical Baseline + rs6926250, rs4237808, rs499646, rs11639774. ***p = 7.09e-8. c Odds ratio for predictors in Clinical + Genetic model. Black colored bars are significant after Bonferroni correction for all variables entered in the logistic regression model.
Lookup of past tardive dyskinesia studies
Variants previously found to be associated with TD were meta-analyzed with results from the current GWAS (Supplementary Table 10)11,18,22,23,38–46. The following variants, independent from CATIE were replicated in the current study CYP1A2 (rs762551, N = 1725, P = 0.018), DRD1 (rs4532, N = 1788, P = 0.045), GRIN2A (rs1345423, N = 1837, P = 0.012), GRIN2B (rs2192970, N = 1057, P = 0.0018), HSPG2 (rs878949 (proxy), N = 1572, P = 0.0051), and DPP6 (rs6977820, N = 1701, P = 0.00068). These genes implicated pathways involving drug metabolism, dopamine, and glutamate.
Discussion
To our knowledge, here we report the largest GWAS for TD. A single novel locus at 16q24.1 was found to be associated with TD. We also identified three loci (1p36.22, 6q23.2, and 12q13.13) with suggestive evidence of association to TD. Of these, putative causal variants were identified for three of the loci. The top GWAS hit at 16q24.1, in the GSE1 coiled-coil protein gene, encodes for a proline rich protein which was reported to be a subunit of BRAF35-HDAC (BHC) histone deacetylase complex51. This gene has been known to be implicated in the proliferation, migration, and invasion of breast cancer cells52. A lookup in the GWAS catalog revealed GSE1 was also associated at GWAS significance with platelet count and distribution53, and suggestive GWAS significance with amyotrophic lateral sclerosis54 and sulfasalazine-induced agranulocytosis55. GSE1 contributes to a geneset that are predicted targets of a microRNA biomarker for schizophrenia56.
Other suggestive associations with TD included TNFRSF1B (1p36.22), EPB41L2 (6q23.2), and CALCOCO1 (12q13.13). The tumor necrosis factor receptor superfamily member 1B (TNFRSF1B) is a protein-coding gene that mediates antiapoptotic signaling. Expression of TNFRSF1B is specific to cells in the immune systems, specific neuronal subtypes, certain T-cells subtypes, and endothelial cells57. This appears to be supported by results from the competitive geneset enrichment analysis, albeit nonsignificant, revealed top pathways that implicated immunoglobulin production, chemokine receptors, and monocytes. These findings appear to support existing theories on the role of immune and inflammation in the pathogenesis of TD11,12.
The erythrocyte membrane protein band 4.1 like 2 (EPB41L2) is involved in actin and cytoskeletal protein binding. More recently, EPB41L2 deficiency was found to result in myelination abnormalities in the peripheral nervous system, leading to motor neuropathy in a mice study58. This putative association of EPB41L2 deficiency is consistent with the direction of effect found in our GWAS results, suggesting that the expression of EPB41L2 might confer a protective effect against motor neuropathy. Functional enrichment in and around the EPB41L2 based on the MetaXcan finding is intriguing, further research is needed to dissect the function of EPB41L2 in TD pathophysiology.
The calcium binding and coiled-coil domain 1 (CALCOCO1) is a protein-coding gene that is involved in the activation of transcriptional activities of targets genes in the Wnt signaling pathway, neuronal receptor and aryl hydrocarbon receptor (AhR)59. Notably, one function of the AhR, a ligand-based transcription factor, is the regulation of transcriptional activity for drug metabolizing enzymes, including the family of cytochrome P450 (CYP) genes60. The family of CYP has been postulated to be a candidate for TD susceptibility11. Specifically, CYP enzymes, such as CYP1A2, CYP2D6, and CYP3A4, metabolize antipsychotics (e.g., clozapine, olanzapine, and haloperidol) and antidepressants (e.g., fluvoxamine)60. Meta-analysis of CYP1A2 from past and current study revealed that this gene was significantly associated with TD at trend level (P < 0.05; Supplementary Table 10).
Follow-up post-hoc investigation showed that individuals that were on Typical antipsychotics were expectedly more likely to be a TD case. We also demonstrated that clinical factors such as Age of Onset and Duration of illness of schizophrenia also significantly predicted TD. However, beyond clinical factors, top SNPs within CALCOCO1 and TNFRSF1B were responsible for accounting for nearly three times the risk of having TD and that joint clinical and genetic model accounted for greater than 75% of variance for individuals having TD and were on atypical antipsychotics. Nevertheless, further investigation is required to confirm the replicability and generalizability of the observed phenomenon.
Polygenic risk score analysis on traits with polygenic architecture revealed overlapping genetic polygenic risk of TD and ALS. The shared protein misfolding pathway between TD and ALS is consistent with findings from our MAGMA pathway analysis. Post-hoc lookups of known ALS genes that might be associated with TD revealed that an F-Box protein-encoding gene FBXO15, chemo/neurokine encoding gene FAM19A1, and neuropeptide encoding gene, NPS. Proteinopathies commonly implicated in neurodegenerative conditions have been proposed to also underlie TD61,62. Prior reports have implicated the accumulative role of proteinopathies in neurotoxicity, synaptic dysfunction, and its bidirectional effect with oxidative stress, neuroinflammation, and its consequence on the immune system61,62. We do note, however, that genetic overlap between TD and other seemingly related traits is generally weak, such as neurodegenerative conditions and autoimmune conditions. Nevertheless, Schizophrenia and Rheumatoid Arthritis were close to the multiple correction threshold. Future work in more powered samples may be carried out to investigate how the potential biological mechanisms for ALS, rheumatoid arthritis, and schizophrenia might be related to TD. We note that potential ancestry differences in LD patterns and allele frequencies of the target and training dataset could result in poorer polygenic prediction. The generally small pharmacogenomic datasets utilized in the context of this report, and the use of multiple reference panels (i.e., HapMap and 1000 genomes project) could have contributed to the variability of the results. Nevertheless, pharmacogenomic datasets such as those reported here continue to allow more clues to be shed regarding the potential biological mechanisms that underlie complex clinical effects that arise from the prescription of psychotropic medication.
Emergent results from the current report demonstrate the polygenic architecture of TD. While the current study remains underpowered for GWAS analysis (Supplementary Fig. 12), we present the largest GWAS of TD to date. The findings reported here suggest that multiple overlapping biological systems might contribute to the etiopathogenesis of the condition. Taken together, results suggest that TD is associated with significant proteinopathies, disrupted neuronal function within the brain and potentially including muscular innervation, existing in a background of inflammation. Further work is necessary to identify the cascade of biological disruptions and events that trigger TD symptomatology. Further work is needed to replicate findings in the current report and further unravel the biology of these risk variants and pathways identified.
Web resources
FUMA, http://fuma.ctglab.nl/.
GAS Power Calculator, http://csg.sph.umich.edu/abecasis/cats/gas_power_calculator/index.html
GCTA, https://cnsgenomics.com/software/gcta/
GEMMA, http://www.xzlab.org/software.html
GWAS Catalog, https://www.ebi.ac.uk/gwas/
LD-HUB, http://ldsc.broadinstitute.org/
MAGMA, https://ctg.cncr.nl/software/magma
METAL, https://genome.sph.umich.edu/wiki/METAL_Documentation
MetaXcan, https://github.com/hakyimlab/MetaXcan
Michigan Imputation Server, https://imputationserver.sph.umich.edu/index.html
MSigDB, http://software.broadinstitute.org/gsea/msigdb
PAINTOR, https://github.com/gkichaev/PAINTOR_V3.0
PLINK1.9, https://www.cog-genomics.org/plink2
PRSice2, https://github.com/choishingwan/PRSice/wiki
PredictDB, http://predictdb.org/
SNAP, http://www.broad.mit.edu/mpg/snap/
Variant Effect Predictor, https://www.ensembl.org/vep
Supplementary information
Acknowledgements
This research is supported by grants from the Ministry of Health Singapore, National Medical Research Council (Grant No.: NMRC/TCR/003/2008, NMRC/CG/004/2013). M.L. is supported by the National Medical Research Council Research Training Fellowship (Grant No.: MH095:003/008-1014). The work was (partly) supported by an A*STAR SPF grant to the SAPhIRE program. J.J.L. acknowledges the BMRC Central Research Fund (UIBR), Agency for Science, Technology and Research, Singapore and the BMRC Strategic Position Fund (SPF2014/001), Agency for Science, Technology and Research, Singapore. We thank the NIH for providing limited access datasets for the NIMH CATIE (ClinicalTrials.gov identifier NCT00014001, NIMH contract #N01MH90001). We would like to express our gratitude to Professor Arinami Tadao for his valuable comments. We also thank all participants and researchers who contributed to the collection of this data.
Author contributions
M.L. and K.L. ran the analysis and drafted the manuscript, J.J.L. and J.L. designed the study and supervised preparation of the manuscript. All other authors were involved in the drafting and approved the final manuscript. J.L. takes responsibility for data access.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Keane Lim, Max Lam
These authors jointly supervised this work: Jianjun Liu, Jimmy Lee
Contributor Information
Jianjun Liu, Email: liuj3@gis.a-star.edu.sg.
Jimmy Lee, Email: Jimmy_lee@imh.com.sg.
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-021-01471-y.
References
- 1.Correll CU, Kane JM, Citrome LL. Epidemiology, prevention, and assessment of tardive dyskinesia and advances in treatment. J. Clin. Psychiatry. 2017;78:1136–1147. doi: 10.4088/JCP.tv17016ah4c. [DOI] [PubMed] [Google Scholar]
- 2.Zai, C. C. et al. Genetics of tardive dyskinesia: promising leads and ways forward. J. Neurol. Sci. 10.1016/j.jns.2018.02.011 (2018). [DOI] [PubMed]
- 3.Weiner, W. J. Drug-induced Movement Disorders. in Encyclopedia of Movement Disorders (ed. Metman, K. K. A. L.) 340–347 (Elsevier, 2010).
- 4.Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr. Opin. Psychiatry. 2008;21:151–156. doi: 10.1097/YCO.0b013e3282f53132. [DOI] [PubMed] [Google Scholar]
- 5.Lee J, Jiang J, Sim K, Chong S-A. The prevalence of tardive dyskinesia in Chinese Singaporean patients with schizophrenia: revisited. J. Clin. Psychopharmacol. 2010;30:333–335. doi: 10.1097/JCP.0b013e3181dcf1d7. [DOI] [PubMed] [Google Scholar]
- 6.Zhang XY, et al. Gender differences in the prevalence, risk and clinical correlates of tardive dyskinesia in Chinese schizophrenia. Psychopharmacology. 2009;205:647–654. doi: 10.1007/s00213-009-1590-8. [DOI] [PubMed] [Google Scholar]
- 7.Chong S-A, Tay JAM, Subramaniam M, Pek E, Machin D. Mortality rates among patients with schizophrenia and tardive dyskinesia. J. Clin. Psychopharmacol. 2009;29:5–8. doi: 10.1097/JCP.0b013e3181929f94. [DOI] [PubMed] [Google Scholar]
- 8.Wu JQ, et al. Cognition impairment in schizophrenia patients with tardive dyskinesia: association with plasma superoxide dismutase activity. Schizophr. Res. 2014;152:210–216. doi: 10.1016/j.schres.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Adrianzén C, et al. Relative association of treatment-emergent adverse events with quality of life of patients with schizophrenia: post hoc analysis from a 3-year observational study. Hum. Psychopharmacol. 2010;25:439–447. doi: 10.1002/hup.1143. [DOI] [PubMed] [Google Scholar]
- 10.Tenback DE, van Harten PN, Slooff CJ, van Os J. Evidence that early extrapyramidal symptoms predict later tardive dyskinesia: a prospective analysis of 10,000 patients in the European Schizophrenia Outpatient Health Outcomes (SOHO) study. Am. J. Psychiatry. 2006;163:1438–1440. doi: 10.1176/ajp.2006.163.8.1438. [DOI] [PubMed] [Google Scholar]
- 11.Lanning RK, Zai CC, Müller DJ. Pharmacogenetics of tardive dyskinesia: an updated review of the literature. Pharmacogenomics. 2016;17:1339–1351. doi: 10.2217/pgs.16.26. [DOI] [PubMed] [Google Scholar]
- 12.Lee H-J, Kang S-G. Genetics of tardive dyskinesia. Int. Rev. Neurobiol. 2011;98:231–264. doi: 10.1016/B978-0-12-381328-2.00010-9. [DOI] [PubMed] [Google Scholar]
- 13.Sarró S, et al. Structural brain changes associated with tardive dyskinesia in schizophrenia. Br. J. Psychiatry. 2013;203:51–57. doi: 10.1192/bjp.bp.112.114538. [DOI] [PubMed] [Google Scholar]
- 14.Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYP1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol. Psychiatry. 2008;13:544–556. doi: 10.1038/sj.mp.4002142. [DOI] [PubMed] [Google Scholar]
- 15.Tsai H-T, et al. A candidate gene study of tardive dyskinesia in the CATIE schizophrenia trial. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010;153B:336–340. doi: 10.1002/ajmg.b.30981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thelma BK, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9:1285–1306. doi: 10.2217/14622416.9.9.1285. [DOI] [PubMed] [Google Scholar]
- 17.Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and the Ser9Gly polymorphism in the DRD3 gene: a meta analysis. Schizophr. Res. 2006;83:185–192. doi: 10.1016/j.schres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Roberto Bakker P, et al. Antipsychotic-induced movement disorders in long-stay psychiatric patients and 45 Tag SNPs in 7 candidate genes: a prospective study. PLoS ONE. 2012;7:e50970. doi: 10.1371/journal.pone.0050970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenbaum L, Alkelai A, Rigbi A, Kohn Y, Lerer B. Evidence for association of the GLI2 gene with tardive dyskinesia in patients with chronic schizophrenia. Mov. Disord. 2010;25:2809–2817. doi: 10.1002/mds.23377. [DOI] [PubMed] [Google Scholar]
- 20.Inada T, et al. Pathway-based association analysis of genome-wide screening data suggest that genes associated with the gamma-aminobutyric acid receptor signaling pathway are involved in neuroleptic-induced, treatment-resistant tardive dyskinesia. Pharmacogenet. Genomics. 2008;18:317–323. doi: 10.1097/FPC.0b013e3282f70492. [DOI] [PubMed] [Google Scholar]
- 21.Syu A, et al. Association of the HSPG2 gene with neuroleptic-induced tardive dyskinesia. Neuropsychopharmacology. 2010;35:1155–1164. doi: 10.1038/npp.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka S, et al. DPP6 as a candidate gene for neuroleptic-induced tardive dyskinesia. Pharmacogenomics J. 2013;13:27–34. doi: 10.1038/tpj.2011.36. [DOI] [PubMed] [Google Scholar]
- 23.Greenbaum L, Alkelai A, Zozulinsky P, Kohn Y, Lerer B. Support for association of HSPG2 with tardive dyskinesia in Caucasian populations. Pharmacogenomics J. 2011;12:513. doi: 10.1038/tpj.2011.32. [DOI] [PubMed] [Google Scholar]
- 24.Lieberman JA, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N. Engl. J. Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 25.Stroup TS, et al. The National Institute of Mental Health Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) project: schizophrenia trial design and protocol development. Schizophr. Bull. 2003;29:15–31. doi: 10.1093/oxfordjournals.schbul.a006986. [DOI] [PubMed] [Google Scholar]
- 26.Sullivan PF, et al. Genomewide association for schizophrenia in the CATIE study: results of stage 1. Mol. Psychiatry. 2008;13:570–584. doi: 10.1038/mp.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guy. W. ECDEU Assessment Manual for Psychopharmacology. 534–537 (U.S. Dept. of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs, 1976).
- 28.Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch. Gen. Psychiatry. 1982;39:486–487. doi: 10.1001/archpsyc.1982.04290040080014. [DOI] [PubMed] [Google Scholar]
- 29.Zhou X, Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barbeira, A. et al. MetaXcan: Summary statistics based gene-level association method infers accurate PrediXcan results. bioRxiv10.1101/045260 (2016).
- 34.Kichaev G, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722. doi: 10.1371/journal.pgen.1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kichaev G, Pasaniuc B. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet. 2015;97:260–271. doi: 10.1016/j.ajhg.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Euesden J, Lewis CM, O’Reilly PF. PRSice: Polygenic Risk Score software. Bioinformatics. 2015;31:1466–1468. doi: 10.1093/bioinformatics/btu848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fedorenko OY, et al. Association study indicates a protective role of phosphatidylinositol-4-phosphate-5-kinase against tardive dyskinesia. Int. J. Neuropsychopharmacol. 2014;18:1–6. doi: 10.1093/ijnp/pyu098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hsieh C-J, Chen Y-C, Lai M-S, Hong C-J, Chien K-L. Genetic variability in serotonin receptor and transporter genes may influence risk for tardive dyskinesia in chronic schizophrenia. Psychiatry Res. 2011;188:175–176. doi: 10.1016/j.psychres.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 40.Ivanova SA, et al. Cytochrome P450 1A2 co-determines neuroleptic load and may diminish tardive dyskinesia by increased inducibility. World J. Biol. Psychiatry. 2015;16:200–205. doi: 10.3109/15622975.2014.995222. [DOI] [PubMed] [Google Scholar]
- 41.Ivanova SA, et al. Dehydroepiandrosterone sulphate as a putative protective factor against tardive dyskinesia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2014;50:172–177. doi: 10.1016/j.pnpbp.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 42.Ivanova SA, et al. NMDA receptor genotypes associated with the vulnerability to develop dyskinesia. Transl. Psychiatry. 2012;2:e67. doi: 10.1038/tp.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai I-C, et al. Analysis of genetic variations in the dopamine D1 receptor (DRD1) gene and antipsychotics-induced tardive dyskinesia in schizophrenia. Eur. J. Clin. Pharmacol. 2011;67:383–388. doi: 10.1007/s00228-010-0967-2. [DOI] [PubMed] [Google Scholar]
- 44.Son W-Y, et al. Gaba transporter SLC6A11 gene polymorphism associated with tardive dyskinesia. Nord. J. Psychiatry. 2014;68:123–128. doi: 10.3109/08039488.2013.780260. [DOI] [PubMed] [Google Scholar]
- 45.Tiwari AK, et al. Association study of cannabinoid receptor 1 (CNR1) gene in tardive dyskinesia. Pharmacogenomics J. 2012;12:260–266. doi: 10.1038/tpj.2010.93. [DOI] [PubMed] [Google Scholar]
- 46.Zai CC, et al. Association study of the vesicular monoamine transporter gene SLC18A2 with tardive dyskinesia. J. Psychiatr. Res. 2013;47:1760–1765. doi: 10.1016/j.jpsychires.2013.07.025. [DOI] [PubMed] [Google Scholar]
- 47.Johnson AD, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McLaren W, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hakimi M-A, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J. Biol. Chem. 2003;278:7234–7239. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- 52.Chai P, et al. GSE1 negative regulation by miR-489-5p promotes breast cancer cell proliferation and invasion. Biochem. Biophys. Res. Commun. 2016;471:123–128. doi: 10.1016/j.bbrc.2016.01.168. [DOI] [PubMed] [Google Scholar]
- 53.Astle WJ, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167:1415–1429.e19. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie T, et al. Genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging. 2014;35:1778.e9–1778.e23. doi: 10.1016/j.neurobiolaging.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 55.Wadelius, M. et al. Sulfasalazine-induced agranulocytosis is associated with the human leukocyte antigen locus. Clin. Pharmacol. Ther. 10.1002/cpt.805 (2017). [DOI] [PMC free article] [PubMed]
- 56.Hass J, et al. Associations between DNA methylation and schizophrenia-related intermediate phenotypes—a gene set enrichment analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2015;59:31–39. doi: 10.1016/j.pnpbp.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naudé PJW, den Boer JA, Luiten PGM, Eisel ULM. Tumor necrosis factor receptor cross-talk. FEBS J. 2011;278:888–898. doi: 10.1111/j.1742-4658.2011.08017.x. [DOI] [PubMed] [Google Scholar]
- 58.Saitoh Y, Ohno N, Yamauchi J, Sakamoto T, Terada N. Deficiency of a membrane skeletal protein, 4.1G, results in myelin abnormalities in the peripheral nervous system. Histochem. Cell Biol. 2017;148:597–606. doi: 10.1007/s00418-017-1600-6. [DOI] [PubMed] [Google Scholar]
- 59.Yang CK, Kim JH, Stallcup MR. Role of the N-terminal activation domain of the coiled-coil coactivator in mediating transcriptional activation by beta-catenin. Mol. Endocrinol. 2006;20:3251–3262. doi: 10.1210/me.2006-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramadoss P, Marcus C, Perdew GH. Role of the aryl hydrocarbon receptor in drug metabolism. Expert Opin. Drug Metab. Toxicol. 2005;1:9–21. doi: 10.1517/17425255.1.1.9. [DOI] [PubMed] [Google Scholar]
- 61.Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 2015;47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sweeney P, et al. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl. Neurodegener. 2017;6:6. doi: 10.1186/s40035-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.