

ABSTRACT
Anaplasma phagocytophilum (Ap), agent of human anaplasmosis, is an intracellular bacterium that causes the second most common tick-borne illness in North America. To address the lack of a genetic system for these pathogens, we used random Himar1 transposon mutagenesis to generate a library of Ap mutants capable of replicating in human promyelocytes (HL-60 cells). Illumina sequencing identified 1195 non-randomly distributed insertions. As the density of mutants was non-saturating, genes without insertions were either essential for Ap, or spared randomly. To resolve this question, we applied a biostatistical method for prediction of essential genes. Since the chances that a transposon was inserted into genomic TA dinucleotide sites should be the same for all loci, we used a Markov chain Monte Carlo model to estimate the probability that a non-mutated gene was essential for Ap. Predicted essential genes included those coding for structural ribosomal proteins, enzymes involved in metabolism, components of the type IV secretion system, antioxidant defense molecules and hypothetical proteins. We have used an in silico post-genomic approach to predict genes with high probability of being essential for replication of Ap in HL-60 cells. These results will help target genes to investigate their role in the pathogenesis of human anaplasmosis.
Keywords: tick-borne pathogen, Anaplasma phagocytophilum, transposon, mutagenesis, essential genes, virulence factor
A library of a mutant tick-borne bacterium, Anaplasma phagocytophilum, was developed that will facilitate identification of virulence factors, vaccine targets and drugs, and identification of essential genes.
INTRODUCTION
In North America, Europe and other temperate climate regions, ticks are the most important vectors of emerging zoonotic pathogens. One of these agents that can cause a potentially life-threatening illness in humans and domestic animals is Anaplasma phagocytophilum (Ap), an obligate intracellular tick-borne bacterium in the order Rickettsiales (Bakken et al. 1994; Chen et al. 1994; Dunning Hotopp et al. 2006). This pathogen is maintained in the environment by cycling through ticks and wildlife species that do not develop disease (Pancholi et al. 1995; Levin et al. 2002; Johnson et al. 2011; Massung et al. 2002). Incidental hosts such as humans, dogs, sheep and horses experience symptomatic infections following the bite of an infected tick. Illness manifests as fever, lethargy and reduced resistance to opportunistic pathogens such as Staphylococcus aureus, presumably as a consequence of infection of neutrophil granulocytes that are important members of the innate immune system (Stuen 2007). In North America, A. phagocytophilum falls into two genetically distinct groups that preferentially infect either small rodents, humans and dogs, or ruminants (Barbet et al. 2013). Despite their small genome of ∼1.5 Mb, the bacteria express host-specific sets of genes depending on whether they reside in a tick vector cell or a human cell (Nelson et al. 2008, 2020). In order to decipher their role, we used random transposon mutagenesis to facilitate functional characterization of the Ap genome, which is complicated by a sizable proportion (∼ 29%) of hypothetical genes with no known or predicted function due to a lack of homology to sequences in the databases (Nelson et al. 2020). To overcome these difficulties, we attempted large scale genetic manipulation to aid in functional characterization, which is generally difficult in obligate intracellular bacteria (Felsheim et al.2010; McClure et al. 2017; Oliva Chávez et al. 2019). The exogenous, transforming DNA must be delivered to purified bacteria while they are extracellular, and the transformants must remain infectious to be recovered in cell culture. The Himar1 mariner transposase has been used successfully for random mutagenesis in a variety of organisms, including Ap (Lampe, Churchill and Robertson 1996; Pelicic et al. 2000; Felsheim et al. 2006; Ashour and Hondalus 2003, Maier et al. 2006; Bilyk et al. 2013; Cartman and Minton 2010, Slamti and Picardeau 2012, Maglennon et al. 2013; Ichimura et al. 2014), Anaplasma marginale (Crosby et al. 2014, 2015) and Ehrlichia spp. (Cheng et al. 2013; Bekebrede et al. 2020). The transposase function of Himar1 is not dependent on specific host factors other than DNA repair enzymes, and randomly inserts transposons into the genome at ‘TA’ dinucleotide sites (Lampe, Churchill and Robertson 1996) that are readily available in the AT-rich (58%) genome of Ap.
Here, we describe the generation of a library of Ap mutants using the Himar1 system to introduce a transposon encoding the mCherry red fluorescent protein (Shaner, Steinbach and Tsien 2005) and spectinomycin resistance genes (aadA; Prentki and Krisch 1984) randomly into the genome of the Ap HGE1 isolate (GenBank accession number APHH01000000). The non-saturated library consisted of 857 isolates recovered in the human promyelocyte cell line HL-60, and contained 1195 mappable insertions. In a library with a subsaturation level of mutagenesis such as this, the location of insertions within genes of recovered mutants (i.e. those that sustained a non-lethal hit) can be used in a Bayesian model to predict which genes are likely to be essential rather than spared by chance alone (Lamichhane et al. 2003). In order to determine which genes among those that were spared from mutation might be essential for growth of Ap in HL-60 cells, the distribution of the recovered mutations was used in a hidden Monte Carlo–Markov scheme to predict the likelihood of a gene to be required for Ap survival in this cell line. This approach is particularly useful when saturating mutagenesis cannot be achieved, such as in our study involving an obligate intracellular bacterium. Beyond this caveat, the Himar1 transposable element is well suited for AT-rich Rickettsiales genomes, since it only requires a TA dinucleotide for insertion, which are ubiquitously present. Therefore, every Ap ORF contains Himar1 transposon target sites, making the mutant library amenable to Bayesian analysis. Lamichhane et al. (2003) used this method to predict essential genes in the M. tuberculosis genome that is 4.4 Mb in size with ∼4 250 ORFs based on 1425 defined insertions. Here, we present our prediction of essential genes based on 1 195 defined Himar1 insertions in the Ap genome that is 1.47 Mb with 1 369 ORFs (Dunning Hotopp et al.2006), which compares well, and highlights the usefulness of this approach.
In the text and tables, we reference both the APH and HGE1 locus tags to facilitate data base searches, because the HGE1 locus tags are not yet widely used. However, these two systems are not perfectly matched, since they were applied to the genome sequence of two different Ap strains, and because different sequencing methods were used, yielding different assemblies.
METHODS
Cell and bacterial culture
The human promyelocytic leukemia cell line HL-60 (ATCC, Manassas, VA, CCL-240) was used to culture the Ap HGE1 strain (Goodman et al. 1996). Cells were maintained in RPMI1640 medium with 10% heat-inactivated fetal bovine serum (FBS, BenchMark, Gemini Bio-Products, West Sacramento, CA), 25 mM HEPES buffer and 2 mM glutamine at 37°C with 5% CO2 in humidified air. HL-60 cells were maintained at a density of 1 × 105–1 × 106 cells/mL. For routine maintenance of Ap, infected cultures were monitored by examining Giemsa-stained cells centrifuged onto glass microscope slides using a Cytospin centrifuge (Thermo Scientific, Waltham, MA). When ≥ 90% of cells contained cytoplasmic inclusions containing numerous Ap bacteria (morulae), 100 µL of infected cell suspension was added to a 10-mL culture of 1 × 105 HL-60 cells/mL for continuous propagation.
Plasmid construction
The plasmid pCis Cherry Himar1 A7 was constructed and produced as previously described (Felsheim et al. 2006; Munderloh et al. 2012; Cheng et al. 2013), suspended in sterile DNase-free ultrapure water at a concentration of 1 µg/µL and stored at –20 °C. The plasmid combines the coding sequences for both the transposase and the transposon, each under control of the A. marginale Am tr promoter (Felsheim et al. 2006; Munderloh et al. 2012). The visible marker, a red fluorescent protein, was encoded by mcherry (Shaner, Steinbach and Tsien 2005), and for antibiotic-based selection, aadA, encoding an Escherichia coli adenylyl transferase that confers resistance to spectinomycin and streptomycin (Prentki and Krisch 1984), was included.
Library construction
Bacterial transformation, HL-60 cell infection, and selection
HL-60 cells were used when ≥ 90% of the cells were infected with A. phagocytophilum. Infected cells from a total volume of ∼100 mL of cultures were collected by centrifugation at 300 x g at room temperature, and resuspended in 3 mL of ice-cold, sterile 300 mM sucrose. Host cell free Ap were obtained by mechanical disruption of HL-60 cells using three passes through a 25 ga hypodermic needle attached to a 3-mL syringe with a Luer-lok hub. The lysate was filtered through a sterile 2 μm pore size filter (Whatman Puradisc 25 GD 2.0 µm GMF-150 25mm syringe filter), centrifuged at 9300 x g at 4°C for 5 min and the pellet containing bacteria was resuspended in 50 µL of ice cold 300 mM sucrose. The suspension was transferred to a 0.2 mm gap electroporation cuvette (BioRad, Hercules, CA), and incubated with 1 μg of the plasmid pCis Cherry Himar1 A7 (Lampe, Churchill and Robertson 1996; Munderloh et al. 2012) for 15 min on ice before being pulsed once (1.7 kV, 400 Ohms and 25 μF) using a BioRad Gene Pulser II. Electroporated bacteria were immediately added to 1.2 × 107 HL-60 cells in 0.5 mL RPMI1640 with 30% FBS and incubated for 60 min at 37°C with gentle agitation every 5 min. After 1 hour incubation, the electroporated bacteria and cells were diluted in 100 mL of RPMI1640 with 30% FBS, and 100 μL of this suspension was added to each well of ten 96-well plates. After resting overnight, an additional 100 μL of RPMI1640 with 20% FBS plus 200 μg/mL spectinomycin was added to each well to begin selection for transformed bacteria.
Isolation of mutant A. phagocytophilum
After 6 days, wells were examined for the presence of mCherry-expressing bacteria using a Nikon Eclipse TE2000 inverted microscope (Nikon Instruments Inc., Melville, NY) enclosed in an incubator at 37°C with an atmosphere of 5% CO2 in air, and equipped with an X-cite EXFO fluorescent light excitation source (Lumen Dynamics, Mississauga, Ontario, Canada) and a Rhodamine (TRITC) filter (544 nm excitation/570 emission wave length). Images of wells were captured using a Photometrics Cascade EMCCD camera (Photometrics, Tucson, AZ) and Metamorph software (Molecular Devices, Sunnyvale, CA). The contents of positive wells were added to HL-60 cell cultures in 12-well plates with 10% FBS supplemented RPMI1640 medium for propagation. When the proportion of infected cells reached ≥ 90%, transformed Ap from individual wells were frozen in RPMI1640 medium with 50% FBS and 10% dimethyl sulfoxide (DMSO) using a Mr. Frosty freezing container (Sigma, St. Louis, MO), and then stored in liquid nitrogen.
Identification of transposon insertion sites in the genome
Genomic DNA was prepared from an aliquot of each of the cultures from positive wells as described (Felsheim et al. 2006), stored individually and pooled into groups of 25 for sequencing. Each pool received a separate Illumina barcode. Libraries for sequencing were prepared at the University of Minnesota Genomics Center (UMGC) facilities and sequenced using a full lane of the Illumina HiSeq 2000 instrument with TruSeq Nano DNA reagents. Subsequently, files were sorted by barcode into 96 discrete fastq files using Illumina software and uploaded to the GALAXY suite (http://wiki.galaxyproject.org/; Blankenberg et al. 2010; Giardine et al. 2005; Goecks et al. 2010) at the high-performance computing center of the University of Florida (Gainesville). Sequence reads were processed to remove barcode sequences and filtered for quality and length. Reads containing genome-transposon junctions were identified using the NCBI BLAST tool with the A. phagocytophilum isolate HGE1 genome (GenBank accession number APHH01000000) and the Himar1 inverted-repeat sequences as references. Excel was used to sort 35 467 reads containing transposon sequences to determine the location of insertions. Insertions within repetitive regions were included only if three or fewer insertion locations were possible. For visualization, transposon insertion sites were mapped to the genome using the genome browser and annotation tool Artemis (Carver et al. 2012; Welcome Sanger Institute, UK), which can be downloaded from http://www.sanger-pathogens.github.io/Artemis/Artemis/. Under the menu item ‘File’ select ‘Open,’ and then the file ‘HGE1 all one contig w/insertions.gbk’ from the dialogue box (Nelson et al. 2008). The Artemis window will open with the annotation for the Ap genome loaded. Red boxes represent unique insertions, whereas orange boxes indicate insertions that could be in one, two or three different locations.
Identification of isolates containing insertions of interest
Using available sequence data (Dunning Hotopp et al. 2006) to identify the location and orientation of the insertion of interest in the HGE1 genome (GenBank accession number APHH0100000), PCR primers were designed to amplify across the junctions between genomic DNA and the transposon (Table 1). The individual DNA samples representing the pool of 25 sequenced isolates containing an insertion in a location of interest were pooled into groups of 5–6 isolates each, and PCR was performed using primers designed for the insertion of interest. Figure 1 is a schematic representation illustrating the general strategy for verification of insertions. For example, to identify the isolate number 100 containing the transposon in the HGE1_00035 locus (encoding a putative isoprenoid biosynthesis protein), the primers 00035 at 7443 and ChUp&Out were used (Table 1). DNA samples from Ap isolates in the pool that yielded the band of expected size (783 bp; Fig. 2A) were individually amplified by PCR to identify the population containing the mutant of interest (Fig. 2B). The second primer pair was subsequently used for confirmation (e.g. HGE1_01752 REV and cherry out; Table 1). A PCR reaction using the two flanking primers (e.g. 00035 at 7443 and 00035 at 7443 ID, expected size 2367 bp; Fig. 2C) was used to verify the absence (or detect presence) of any wild type DNA at that site in the isolate. PCR was performed using GoTaq DNA polymerase (Promega, Madison, WI), and cycling conditions were as follows: One initial denaturation step at 95°C for 2 min; and subsequently 95°C for 30 s, 53°C for 30 s and 72°C for 1 min (with ChUp&Out) or 3 min (with flanking primers) for 40 cycles, followed by a final extension at 72°C for 5 min. Amplification products were electrophoresed on a 1% agarose gel and stained with GelGreen (MilliporeSigma, St. Louis, MO).
Table 1.
Examples of primers used to identify and confirm the presence of the insertion of interest (Table 1). Primers were designed based on sequences of the transposon (encoding mCherry or aadA) and flanking genome regions identified by NextGen sequencing (Illumina HighSeq). Primer pairs to facilitate PCR amplification of DNA spanning the 5’ or 3’ transposon termini and adjacent genome regions were used to identify specific mutants in samples pooled for Illumina sequencing. Primer pairs complementary to genomic regions flanking the transposon were used to verify that a transposon was present at the target site.
| Primer List for selected mutants | Pairs with | |
|---|---|---|
| Spec out | GTATCAGCCCGTCATACTTGAA | - |
| Cherry out | GACCTTAAGTTTAGCCGTCTGT | - |
| ChUp&Out | ATTATCTTCCTCTCCCTTGCTGACC | - |
| 00035 at 7443 (isoprenoid biosynthesis protein) | ACTAAGCACTCATGACGTAAGGTCT | ChUp& out |
| 00035 at 7443 ID | ACACTACTAAGAGAAGCAGCAACGA | 00 035 at 7443 |
| HGE1 00140 FOR (hypothetical protein) | TGACGTTACCGTGCTCGAAG | Cherry out |
| HGE1 00140 REV | GTAATCCAGTCCCTGCCGAG | Spec out |
| HGE1 00255 FOR (hypothetical protein) | GAGCATGAGTCCGTGGGTAG | Spec out |
| HGE1 00255 REV | ATATGCTGAAACGTGCGCTG | Cherry out |
| HGE1 01752 FOR (HGE-14 protein) | ATGCCGTGGGTTCTTACGAG | Spec out |
| HGE1 01752 REV | GCATCCAAGCATAGCTGCAC | Cherry out |
| HGE1 05312 FOR (DNA-binding protein) | TGCGCAACAACTTTAAGCCC | Cherry out |
| HGE1 05312 REV | AGCGTAGAATAGGCGAACCG | Spec out |
| HGE1 05592 FOR (MerR family transcriptional regulator) | AAAAAGGAGAGGAAGGCGCT | Spec out |
| HGE1 05592 REV | GTCTCAACAGAGTTGCGCTG | Cherry out |
Figure 1.
Schematic illustrating the strategy used to identify individual mutants in pooled DNA samples. Primers were designed using sequence data for the A. phagocytophilum strain HGE1 genome, and information from Illumina sequencing to identify transposon insertion sites. Two sets of primers were used to amplify across the 5’ and 3’ junctions between the transposon and genomic sequences, and a third set was used to amplify DNA across both flanking regions and the transposon.
Figure 2.

Gel electrophoresis images depicting the process for identification of mutant numbers 99–122. A total of six individual mutant DNA samples of the 25 that had been pooled for sequencing, were pooled, and subjected to amplification using primers designed for the mutant of interest (Fig. 2A). Individual DNA samples for reactions that yielded a product of the expected size were separately amplified with the same primers (Fig. 2B). To verify the correct insertion locus, primers that amplified across the entire insert and flanking regions were used (Fig. 2C). Stars in panels A, B indicate the isolate with the mutation of interest. Primer combinations are indicated above the black bars, and mutant isolates are listed below the black bars. In panel C, mutant 100–1 to 100–8 and 116–1 to 116–8 denotes populations of that mutant isolate obtained by limiting dilution. The presence of a single high molecular weight amplification product indicates that there are no wild-type or other mutant A. phagocytophilum in these populations.
Culture of Ap containing an insertion of interest
Once a desired isolate was identified, it was retrieved from frozen stock and grown in HL-60 cells at a density of 1 × 105/mL with the addition of 100 µg/mL spectinomycin. Infected cultures were visually monitored for the presence of fluorescent bacteria producing mCherry and aliquots frozen in liquid nitrogen to maintain the library. DNA was isolated from infected cells to confirm the presence of the insertion in the gene of interest using the same primers as those for initial identification. Table 1 provides examples of primers used to identify mutants of HGE1_00035 (encoding an isoprenoid biosynthesis protein), HGE_00140 and HGE_00255 (encoding hypothetical proteins), HGE1_01752 (encoding a putative HGE-14 family effector), HGE1_05312 (encoding a DNA-binding protein) and HGE1_05592 (encoding a MerR family transcriptional regulator).
Purification of mutants from isolates
Sequence data showed that there were more insertions than isolated cultures of red-fluorescent and antibiotic-resistant cultures, suggesting that some of the isolated cultures contained more than one mutant population. In order to separate and recover individual mutants, infected cells from one well were serially diluted 2-fold across 96-well plates (eight replicate wells/dilution) pre-seeded with 104 HL-60 cells per well. Wells that were positive for fluorescent bacteria in the highest dilution were subcultured to propagate Ap bacteria for DNA extraction to test for purity by PCR using insertion-site specific primers.
Prediction of essential genes
In a library with a subsaturation level of mutagenesis, the location of insertions within genes of recovered mutants (i.e. those that did not sustain a lethal hit) can be used in a Bayesian model to predict which genes are likely to be essential rather than spared by chance alone (Lamichhane et al. 2003). A Markov chain Monte Carlo analysis was performed as described in Blades and Broman (2002) and using the package ‘negenes’ (2015) for R statistical software. The initial assumption was that essential genes were randomly distributed, and that only insertions within 80% of the 5’ region of the gene would effectively disrupt gene function. Because the Himar1 transposon was inserted randomly into ‘TA’ sites within the genome, the model used the number of ‘TA’ sites in all genes (the possible insertion sites) as well as the number of observed transposon insertion sites in all genes to predict which genes were likely essential and therefore intolerant of a disruptive transposon insertion.
RESULTS
Library generation
We generated a subsaturating library of Ap Himar1 transposon mutants of the HGE1 strain grown in HL-60 cells. The library contained 857 recovered isolates, however, after sequencing, 1195 mappable transposon insertion sites were identified (Table 2 and Table S1, Supporting Information; ‘HGE1 all one contig w/insertions.gbk’). Thus, up to 40% of isolates included more than one transposon mutant. The mapped insertions affected 267 predicted coding sequences (CDSs), representing approximately 23% of HGE1 genes. The library included isolates with insertions in 101 hypothetical genes, accounting for 38% of the recovered isolates. This indicated that the hypothetical genes that make up about 29% of the Ap genome did not differ in mutability from genes with known function. The repetitive nature of the Ap genome, illustrated by the p44/msp2 gene family and the duplication of genes encoding HGE-14 effectors, VirB2 and VirB6, vitamin and cofactor systems, added to the technical challenge of resolving repetitive sequences with the short length Illumina reads. We found ambiguous insertions that could be located in one or more different locations in the Ap genome that needed to be confirmed by the PCR screening scheme described above (supplementary file ‘HGE1 all in one contig w/insertions.gbk’). For example, 55 of the transposon insertions were within P44 outer membrane protein genes (none located in the expression site) or gene fragments. Figure 3 shows the distribution of the recovered mutants within the HGE1 genome.
Table 2.
Summary of mapped insertions in the A. phagocytophilum Himar1 transposon mutant library.
| Sequenced Insertions | 1195 |
| Insertions in open reading frames (ORFs)* | 522 |
| Insertions in intergenic spaces | 673 |
| Number of ORFs with insertions | 267 |
| ORFs with one insertion | 161 (60%) |
| ORFs with two insertions | 53 (20%) |
| ORFs with three insertions | 22 (8.2%) |
| ORFs with four insertions | 11 (4.1%) |
| ORFs with 5 or more insertions | 20 (7.5%) |
| ORFs without mapped insertions | 884 |
| % ORFs with recovered insertions | 23% |
For a complete list of open reading frames containing at least one insertion see Table S1 (Supporting Information). *This number includes inserts into the p44/msp2 pseudogenes, some of which are difficult to resolve due to their repeats.
Figure 3.
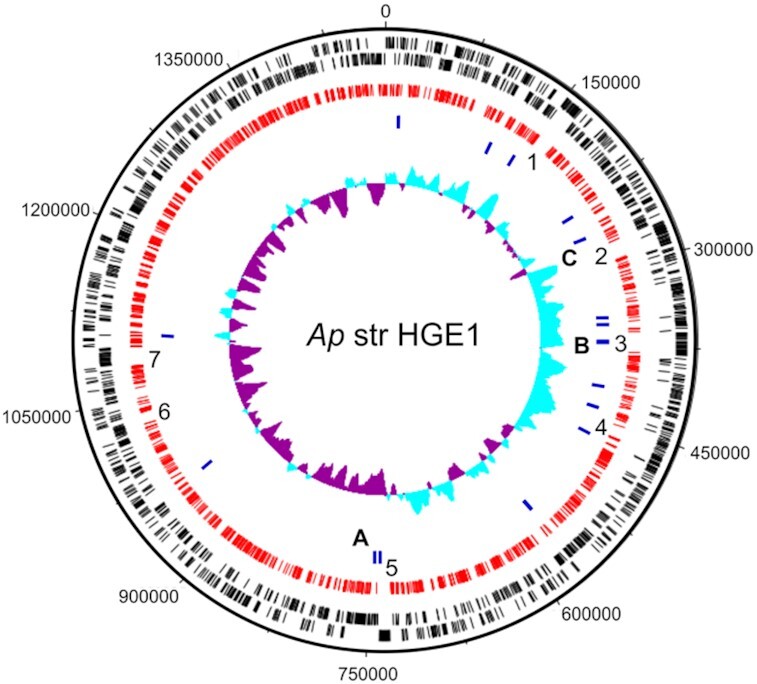
Representation of the sequenced genome of A. phagocytophilum strain HGE1 showing the distribution of transposon insertions. Black bars in the outer rings represent open reading frames on the plus and minus strands, respectively. Red bars in the innermost ring represent transposon insertion sites, while the blue bars indicate the positions of the 16 genes most highly predicted as essential. The innermost ring shows the G + C skew. The seven regions examined for TA content are noted as 1–7, while the components of Fig. 4 are noted as A, B and C. Region 2 corresponds to panel C of Fig. 4, while region 3 corresponds to panel B of Fig. 4. The image was generated using DNAPlotter, which is part of the Artemis software package (Carver et al. 2009; http://sanger-pathogens.github.io/Artemis/DNAPlotter/).
Isolation of specific mutants
Since DNA from 25 isolates was pooled for sequencing to determine insertion sites, each isolate had to be identified with primers targeting the junction between the transposon and the genomic insertion site using the PCR scheme described. In this way, specific isolates containing mutants of interest were identified. For example, the HGE1_05 592/APH_1282 mutant (disrupting transcription of the MerR transcriptional regulator, a helix-turn-helix DNA binding protein primarily found in bacteria; Esna Ashari, Brayton and Broschat 2019) was determined to be isolate 382 using primers listed in Table 1.
Prediction of essential genes
Because the library was developed in HL-60 cells, any mutants with insertions into genes essential for growth in HL-60 cells were not recovered. The 884 ORFs (77%) without a recovered insertion could be truly essential for growth or may have been spared by chance in a subsaturating library. We used a Bayesian model previously applied to a Himar1 transposon mutant library of Mycobacterium tuberculosis to predict essential genes based on our Ap library. The model predicted 16 genes to be more than 99% likely to have essential status for Ap survival in HL-60 cells, and 53 genes as more than 95% likely to be essential (Table S2, Supporting Information). The gene with the highest likelihood of essentiality was HGE1_03127 (APH_0711), encoding an NADH:ubiquinone oxidoreductase subunit H protein, belonging to complex I, which is a major source of reactive oxygen species (ROS) that are predominantly formed by electron transfer from FMNH(2). A total of six genes involved in the type IV secretion system were predicted to be essential, including HGE1_03857 (APH_0906) encoding a predicted effector (Meyer et al. 2013; Esna Ashari, Brayton and Broschat 2019). Indeed, a mutant with a transposon insertion into this gene that was previously recovered in ISE6 tick cells demonstrated a lack of growth in HL-60 cells (Oliva Chávez et al. 2019), corroborating the prediction. There were also stretches with a notable paucity of insertions. For example, the region from ∼153 kbp to 167 kbp on the first contig corresponding to HGE1_0645/APH-0149 to HGE1_0720/APH_0157, contained numerous genes that were not mutated. A total of five of these were assigned a > 90% likelihood of essentiality. The first in that stretch is HGE1_0645/APH_0149, annotated as a Tripartite ATP-independent periplasmic (TRAP) transporter (solute binding protein) that in other bacteria has been linked to pathogenicity (reviewed in Rosa et al. 2018), and HGE1_0680/APH_0157 is a putative ATP-binding cassette (ABC) transporter, highlighting the need of Ap to acquire nutrients from the host. HGE1_0685/APH_0158 and HGE1_0700/APH_0161 (tandemly repeated succinate dehydrogenase flavoprotein subunits, sdhA-1 and sdhA-2, with important function in the Krebs cycle that facilitates aerobic respiration, supporting their essential status) were assigned 90–95% essentiality. They are located distant from the sdhC (HGE1_04 282/APH_0999) gene that is co-transcribed with the 16S rRNA gene, an arrangement unique to Ap (Massung et al. 2008). However, it is likely that expression of both the sdhA and sdhB (HGE1_0690/APH_0159 to HGE1_0705/APH_0159) tandem repeat units is coordinated with transcription of sdhC. HGE1_0715/APH_0164 (90% essentiality) is annotated as a bicyclomycin resistance protein; its interactions, as identified by STRING analysis, with a protein of the type I secretion system and an outer membrane efflux protein indicate that it may be involved in substrate transport. Similarly, genes coding for structural ribosomal proteins were spared, as was most of the T4SS apparatus (Fig. 4A and B). A large, hypothetical protein coding gene (HGE_03162/APH_0720) that was expressed in Ap replicating in ISE6 tick cells sustained multiple transposon insertions (Fig. 4C).
Figure 4.

Screen shots from the Artemis genome browser, depicting selected genomic regions of A. phagocytophilum strain HGE1. (A) A large hypothetical gene (HGE1_03162/APH_0720) with multiple transposon insertions, encoding a 3038 amino acid protein that is expressed in tick cell culture. Red boxes indicate unique insert sites, and yellow boxes indicate insertions that may be located in other genomic regions with identical flanking sequences. (B) Components of the Type IV secretion system (T4SS) are spared from insertions with one exception of an insertion into the 3’ region of the virB6-4 gene (HGE1_01722/APH_0377; not shown, see text). The ORFs at HGE1_01665/APH_0371 encoding superoxide dismutase, HGE1_01675/APH_0373 encoding type IV secretion system ATPase VirB4-1 and HGE1_01680/APH_0374 to HGE1_01690/APH_0376 encoding type IV secretion system proteins VirB6-1 to VirB6-3 are depicted. C. The operon encoding multiple ribosomal proteins from HGE1_01235/APH_0281 encoding rplD 50S ribosomal protein L4 to HGE1_01330/APH_0299 secY preprotein translocase subunit SecY are devoid of transposon insertions. Shown are the reading frames on separate lines, with black vertical bars indicating stop codons. In panel A, only the forward reading frame is shown. In panels B and C, the two center horizontal bars contain the annotation for the forward or the reverse strand. Transposon insertion sites are annotated in red or yellow on the forward annotation track (in A), while protein coding sequences (CDS) are depicted in blue on the reading frame track and the gene is annotated in white on the annotation track.
DISCUSSION
We describe the first use of a Himar1 transposon library, coupled with high-throughput genome sequencing, to identify putative essential genes that are required for the viability of an intracellular, tick-borne bacterial pathogen, Ap, in mammalian cells in vitro. Our work represents the most extensive mutagenesis of A. phagocytophilum to date, and was obtained with 10 rounds of transformations and subsequent mutant recovery over the course of about a year. This library is a powerful tool to assist in the characterization of gene function in Ap (Akerley et al. 1998) as further phenotypic characterization will provide a unique catalogue of functionally annotated Ap genes with potential to significantly improve our understanding of the molecular strategies employed by this and related bacteria to subvert host cell function. Moreover, the methods described here are applicable to other bacteria that are difficult to manipulate genetically, and for which saturating mutations cannot be obtained. Two characteristics of the Himar1 transposase are likely to have contributed to its successful application, i.e. it does not depend on host factors to function, and selectively pastes sequences into TA dinucleotide sites that are abundant in the AT-rich Anaplasmataceae genomes. Himar1 functions in virtually any genome, including eukaryotes (Lampe, Churchill and Robertson 1996).
Although we did not obtain saturating coverage of the genome (meaning we did not recover mutants with insertions in every gene), identification of 1195 mapped insertions facilitated the in silico prediction of essential genes in a difficult to mutagenize obligate intracellular bacterium, which would otherwise not be possible.
When working with bacteria that do not require a host cell for survival, a population of genomes can theoretically be mutated so the bacterial chromosome is saturated with transposon insertions (meaning that each potential insertion site has at least one transposon insertion). To identify regions of the bacterial chromosome that are essential for viability under specific environmental conditions, bacteria are then tested by subjecting them to these conditions. However, when working with obligate intracellular bacteria, there is an overriding requirement that they must remain capable of invading host cells, and mutants with knock-outs in genes necessary for host cell invasion cannot be recovered for further testing. This may partially explain why we recovered a subsaturated transposon mutant library (1% saturation coverage), despite the overall high A + T content in Rickettsiales genomes, including that of Ap. Moreover, the numerous pseudogenes, duplicated genes and repetitive regions in the Ap genome make it difficult to map transposon insertions, and our model did not include insertions that could not be assigned with certainty to a specific location due to presence of identical sequences in more than two regions. Additionally, transposon insertions could have been missed due to differential mutant abundance. Mutants with attenuated growth (transposon insertions within quasi-essential genes) that are mixed with mutants that have no growth defect may be outcompeted. These types of mutants could be missed during library amplification and sequencing, or subsequently lost from the pools during propagation.
Nevertheless, we noted that many genes lacked insertions whereas others had one or multiple insertions. Therefore, we performed statistical analysis to estimate the probability that the low proportion of insertions in the Ap genome was due to biological selection and not attributable to chance. While it can reasonably be assumed that genes suffering hits predicted to result in gene knock-out would be non-essential for growth under conditions used for mutant recovery, genes that were spared could either be essential, i.e. would have resulted in death of the mutant and therefore not have been recovered, or could have been missed by chance.
Predicting which genes encoded by bacteria in the order Rickettsiales are essential is difficult, in part because approximately 30% or more of Rickettsiales genes have no matches in the data bases and are therefore designated hypothetical. Although bioinformatics may identify putative structural features and functions of hypothetical proteins, results remain tentative until proven (Oliva Chàvez et al. 2015, 2018). Hotopp et al. (2006) reported 12 ortholog clusters including 14 Ap genes that were present in all organisms in the order Rickettsiales. From these clusters, we recovered Ap mutants with insertions in six genes (HGE1_00350/APH_0077, HGE1_00805/APH_0187, HGE1_01120/APH_0256, HGE1_03207/APH_0734, HGE1_04477/APH_1042, HGE1_06102/APH_1392) and the model predicted one hypothetical protein gene with two insertions (HGE1_01872/APH_0406) to be likely essential for growth in HL-60 cells. In Ap, an additional 14 hypothetical genes and one GNAT family acetyltransferase gene (encoding a potential transcriptional activator) distinguish Anaplasmataceae from other Rickettsiales. We recovered six mutants from this group (HGE1_00050/APH_0011, HGE1_00890/APH_0206, HGE1_01575/APH_0351, HGE1_02747/APH_0615, HGE1_02827/APH_0615, HGE1_04987/APH_1153), demonstrating the encoded proteins were not essential for growth in HL-60 cells.
We recovered 14 mutants with transposon insertions in genes coding for HGE-14 proteins with a putative type IV effector motif (Hotopp et al. 2006), and which should thus be deemed non-essential. Nevertheless, our analysis predicted two of them (HGE1_02100/APH_0385 and HGE1_01782/APH_0387) to be essential at greater than 93% probability. The motif resembled, but differed from, the consensus sequence in Agrobacterium tumefaciens (R-X7-R-X-R-X-R-X-Xn; Vergunst et al. 2005). Given the importance of the T4SS and its effectors for Ap virulence and pathogenicity (Esna Ashari, Brayton and Broschat 2019), it would be important to further characterize these mutants under different biological conditions, as this could provide insights into the potential role of these genes for the growth and survival of Ap in mammals or ticks. Interestingly, the 16 genes that were predicted to be > 99% likely to be essential were found primarily, but not exclusively, on the leading strand of the genome (Fig. 3). Only two of these genes were associated with regions that were devoid of transposons. To understand whether there was a bias in the ability of a transposon to insert in these regions, we analyzed the prevalence of TA sites in these regions. We examined seven regions that ranged in size from 5 kb to 18 kb and found that the dinucleotide TA was present between 6.69 and 8.09% in these regions, while in the genome as a whole TA sites were represented at 8.28%. Region 5, which was 12 kb, contained just two genes, including HGE1_03122, an Ankyrin domain containing gene that is 9.9 kb. Several alternative explanations exist: (1) the mutants occur in one of two pseudogenes for this gene family, (2) the fact that these proteins are encoded by a gene family may mean that if one is rendered non-functional, another may compensate or (3) some members of the family were spared from mutagenesis and these could be the essential members. Finally, the mutation could impart an inability to infect tick cells, which can be tested.
We identified 55 insertions in p44/msp2 outer membrane protein pseudogene loci, but none in the expression site (ES). Ap genomes encode nearly 100 or more p44/msp2 pseudogenes that are recombined into the ES using conserved flanking sequences (Barbet et al. 2006; Wang et al. 2007), which results in antigenic variation of the immunodominant major surface protein 2 (Msp2), a mechanism postulated to allow escape from the host's immune response. In Anaplasma marginale, certain p44/msp2 variants have also been linked to infection of particular host cells (Chávez et al. 2012). Transposon insertion into a p44 pseudogene would presumably affect outer membrane proteins by reducing the number of recombination events that allow for antigen switching. Whether this would significantly reduce fitness of mutants may depend on the frequency with which the affected p44 donor sequence is used for recombination events, and whether removing it from that process would be deleterious. These outcomes are unknown at this time. In cell culture, antigenic variation is probably not necessary for survival (unless specific variants mediate specific cell invasion), however, some of these mutants may be less fit during mammalian infection in vivo, and be cleared more readily by the host's immune system.
Analysis of global gene expression in Ap growing in HL-60, neutrophil granulocytes, or HMEC-1 human cells versus ISE6 tick cells demonstrated that subsets of genes were preferentially expressed during replication in only one of these host systems (Nelson et al. 2008, 2020). We found that genes actively transcribed during Ap growth in tick cells sustained multiple mutations, which indicated that their gene products were dispensable for bacteria growing in HL-60 cells. For example, several hypothetical protein genes located in an Ap genome region primarily expressed during growth in tick cells (HGE1_06057/APH_1380–HGE1_06082/APH_1386) were mutated, further suggesting they were not used by bacteria residing in human host cells. By contrast, genes highly transcribed in HL-60 cells were less likely to be mutated. For example, HGE1_01595/APH_0355, predicted to be essential (Table S2, Supporting Information) is highly expressed in HL-60 cells (Nelson et al. 2008, 2020). The same is true for HGE1_05792/APH_1325 encoding an Msp2 family outer membrane protein, and virB2 (HGE1_04867/APH_1130). Likewise, a T4SS operon encoding sod and virB genes (HGE1_01665-HGE1_01695; Fig. 4B) remained nearly untouched except for a single insertion into the terminal gene encoding virB6-4 (locus tag HGE1_01695/APH_0377), just a few hundred base pairs upstream of the 3’ tandem repeats. Transposon insertions within a gene may also alter the expression of adjacent genes (polar effect). Characterization of the virB6-4 mutant indicated that the insertion not only disrupted expression of virB6-4 but also had a polar effect on the expression of upstream genes that resulted in an impaired growth phenotype in both human and tick cell cultures (Crosby et al. 2020). Our analysis to predict essential genes corroborated the importance of the sod and virB gene products for Ap replicating in HL-60 cells. Although HL-60 promyelocytes are not functionally identical to neutrophil granulocytes, they retain many of their defense features (Mark Welch et al. 2017), and we expect that many of the Ap genes predicted to be essential in HL-60 cells will also be essential in neutrophils. Additional transcript analysis (RNA-seq) of mutants will provide more detailed knowledge about transcription regulatory elements, small non-coding RNAs and polar effects of transposon insertions on the translation of proteins from multigene transcripts (operons). Future phenotypical characterization of this mutant library in different in vitro and in vivo environments will significantly improve functional annotation of the Ap genome, which is currently not well characterized. This will provide key insights on genes required for invasion and intracellular survival and ultimately identify potential vaccine targets and drugs.
Supplementary Material
Contributor Information
M Catherine O'Conor, College of Veterinary Medicine, 1365 Gortner Avenue St. Paul, MN 55108, USA.
Michael J Herron, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
Curtis M Nelson, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
Anthony F Barbet, Department of Infectious Diseases and Immunology, College of Veterinary Medicine, University of Florida, Academic Building 1017, room V2-200, 1945 SW 16th Ave. Gainesville Fl, 32608, USA.
F Liliana Crosby, Department of Infectious Diseases and Immunology, College of Veterinary Medicine, University of Florida, Academic Building 1017, room V2-200, 1945 SW 16th Ave. Gainesville Fl, 32608, USA.
Nicole Y Burkhardt, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
Lisa D Price, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
Kelly A Brayton, Department of Veterinary Microbiology and Pathology, Washington State University, Grimes Way, Bustad Hall, room 402, P.O. Box 647040 Pullman, WA 99164-7040, USA.
Timothy J Kurtti, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
Ulrike G Munderloh, Department of Entomology, University of Minnesota, UGM, 219 Hodson Hall, 1980 Folwell Avenue, Saint Paul, MN 55108, USA.
FUNDING
This work was supported by generous funding from the National Institutes of Health (NIH), grant number R01AI042792 to UGM, FLC and KAB; by a grant from the NIH, Office of the Director (T32OD010993; KO) and by a grant from the Minnesota Agricultural Experiment Station, project MIN-17-078.
Conflicts of interest
None declared.
REFERENCES
- Akerley BJ, Rubin EJ, Camilli Aet al. Systematic identification of essential genes by in vitro mariner mutagenesis. Proc Natl Acad Sci. 1998;95:8927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour J, Hondalus MK. Phenotypic mutants of the intracellular actinomycete Rhodococcusequi created by in vivo Himar1 transposon mutagenesis. J Bacteriol. 2003;185:2644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken JS, Dumler JS, Chen SMet al. Human granulocytic ehrlichiosis in the upper Midwest United States. A new species emerging?. JAMA. 1994;272:212–8. [PubMed] [Google Scholar]
- Barbet AF, Al-Khedery B, Stuen Set al. An emerging tick-borne disease of humans is caused by a subset of strains with conserved genome structure. Pathogens. 2013;2:544–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbet AF, Lundgren AM, Alleman ARet al. Structure of the expression site reveals global diversity in MSP2 (P44) variants in Anaplasmaphagocytophilum. Infect Immun. 2006;74:6429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekebrede H, Lin M, Teymournejad Oet al. Discovery of in vivo virulence genes of obligatory intracellular bacteria by random mutagenesis. Front Cell Infect Microbiol. 2020;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilyk B, Weber S, Myronovskyi Met al. In vivo random mutagenesis of streptomycetes using mariner-based transposon Himar1. Appl Microbiol Biotechnol. 2013;97:351–9. [DOI] [PubMed] [Google Scholar]
- Blades NJ, Broman KW. Estimating the number of essential genes in a genome by random transposon mutagenesis. 2002; Technical Report MS02-20, Department of Biostatistics, Johns Hopkins University, Baltimore, MD. https://www.biostat.wisc.edu/∼kbroman/publications/ms0220.pdf [Google Scholar]
- Blankenberg D, Von Kuster G, Coraor Net al. Galaxy: A web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010,Chapter 19:Unit 19.10.1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartman ST, Minton NP. A mariner-based transposon system for in vivo random mutagenesis of Clostridiumdifficile. Appl Environ Microbiol. 2010;76:1103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman Met al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Thomson N, Bleasby Aet al. DNAPlotter: circular and linear interactive genome visualization. Bioinformatics. 2009;25:119–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez AS, Felsheim RF, Kurtti TJet al. Expression patterns of Anaplasma marginale Msp2 variants change in response to growth in cattle, and tick cells versus mammalian cells. PLoS One. 2012;7:e36012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Nair AD, Indukuri VVet al. Targeted and random mutagenesis of Ehrlichiachaffeensis for the identification of genes required for in vivo infection. PLoS Pathog. 2013;9:e1003171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SM, Dumler JS, Bakken JSet al. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J Clin Microbiol. 1994;32:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby FL, Brayton KA, Magunda Fet al. Reduced Infectivity in cattle for an outer membrane protein mutant of Anaplasmamarginale. Appl Environ Microbiol. 2015;81:2206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby FL, Munderloh UG, Nelson CMet al. Disruption of VirB6 Paralogs inAnaplasmaphagocytophilum Attenuates Its Growth. J Bacteriol. 2020;202:e00301–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby FL, Wamsley HL, Pate MGet al. Knockout of an outer membrane protein operon of Anaplasmamarginale by transposon mutagenesis. BMC Genomics. 2014;15:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC, Lin M, Madupu Ret al. Comparative genomics of emerging human ehrlichiosis agents. PLos Genet. 2006;2:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esna Ashari Z, Brayton KA, Broschat SL. Prediction of T4SS effector proteins for Anaplasmaphagocytophilum using OPT4e, a new software tool. Front Microbiol. 2019;10:1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsheim RF, Chávez AS, Palmer GHet al. Transformation of Anaplasmamarginale. Vet Parasitol. 2010;167:167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsheim RF, Herron MJ, Nelson CMet al. Transformation of Anaplasmaphagocytophilum. BMC Biotech. 2006;6:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Riemer C, Hardison RCet al. Galaxy: A platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor Jet al. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman JL, Nelson C, Vitale Bet al. Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N Engl J Med. 1996;334:209–15. [DOI] [PubMed] [Google Scholar]
- Ichimura M, Uchida K, Nakayama-Imaohji Het al. Mariner-based transposon mutagenesis for Bacteroides species. J Basic Microbiol. 2014;54:558–67. [DOI] [PubMed] [Google Scholar]
- Johnson RC, Kodner C, Jarnefeld Jet al. Agents of human anaplasmosis and Lyme disease at Camp Ripley, Minnesota. Vector Borne Zoonotic Dis. 2011;11:1529–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane G, Zignol M, Blades NJet al. A post genomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc Natl Acad Sci. 2003;100:7213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;15:5470–9. [PMC free article] [PubMed] [Google Scholar]
- Levin ML, Nicholson WL, Massung RFet al. Comparison of the reservoir competence of medium-sized mammals and Peromyscusleucopus for Anaplasmaphagocytophilum in Connecticut. Vector Borne Zoonotic Dis. 2002;2:125–36. [DOI] [PubMed] [Google Scholar]
- Maglennon GA, Cook BS, Deeney ASet al. Transposon mutagenesis in Mycoplasmahyopneumoniae using a novel mariner-based system for generating random mutations. Vet Res. 2013;44:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier TM, Pechous R, Casey Met al. In vivo Himar1-based transposon mutagenesis of Francisellatularensis. Appl Environ Microbiol. 2006;72:1878–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch DB, Jauch A, Langowski Jet al. Transcriptomes reflect the phenotypes of undifferentiated, granulocyte and macrophage forms of HL-60/S4 cells. Nucleus. 2017;8: 222–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massung RF, Hiratzka SL, Brayton KAet al. Succinate dehydrogenase gene arrangement and expression in Anaplasmaphagocytophilum. Gene. 2008;414:41–8. [DOI] [PubMed] [Google Scholar]
- Massung RF, Lee K, Mauel Met al. Characterization of the rRNA genes of Ehrlichiachaffeensis andAnaplasmaphagocytophila. DNA Cell Biol. 2002;21:587–96. [DOI] [PubMed] [Google Scholar]
- McClure EE, Chávez ASO, Shaw DKet al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol. 2017;15:544–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer DF, Noroy C, Moumène Aet al. Searching algorithm for type IV secretion system effectors 1.0: a tool for predicting type IV effectors and exploring their genomic context. Nucleic Acids Res. 2013;41:9218–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munderloh UG, Felsheim RF, Burkhardt NYet al. The way forward: improving genetic systems. In: Palmer GH, Azad AF (eds). Intracellular Pathogens II: Rickettsiales. chapter 14ASM Press, 2012, 416–32. [Google Scholar]
- Nelson CM, Herron MJ, Wang XRet al. Global transcription profiles of Anaplasmaphagocytophilum at key stages of infection in tick and human cell lines and granulocytes. Front Vet Sci. 2020;7:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Herron MJ, Felsheim RFet al. Whole genome transcription profiling of Anaplasmaphagocytophilum in human and tick host cells by tiling array analysis. BMC Genomics. 2008;9:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva Chávez AS, Herron MJ, Nelson CMet al. Mutational analysis of gene function in the Anaplasmataceae: Challenges and perspectives. Ticks Tick Borne Dis. 2019;10:482–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva Chávez AS, Fairman JW, Felsheim RFet al. An O-methyltransferase is required for infection of tick cells by Anaplasmaphagocytophilum. PLoS Pathog. 2015;11:e1005248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancholi P, Kolbert CP, Mitchell PDet al. Ixodes dammini as a potential vector of human granulocytic ehrlichiosis. J Infect Dis. 1995;172:1007–12. [DOI] [PubMed] [Google Scholar]
- Pelicic V, Morelle S, Lampe Det al. Mutagenesis of Neisseriameningitidis by in vitro transposition of Himar1 mariner. J Bacteriol. 2000;182:5391–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki P, Krisch HM. In vitro insertional mutagenesis with a selectable DNA fragment. Gene. 1984;29:303–13. [DOI] [PubMed] [Google Scholar]
- Rosa LT, Bianconi ME, Thomas GHet al. Tripartite ATP-Independent Periplasmic (TRAP) Transporters and Tripartite Tricarboxylate Transporters (TTT): from Uptake to Pathogenicity. Front Cell Infect Microbiol. 2018;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–9. [DOI] [PubMed] [Google Scholar]
- Slamti L, Picardeau M. Construction of a library of random mutants in the spirochete Leptospirabiflexa using a mariner transposon. Methods Mol Biol. 2012;859:169–76. [DOI] [PubMed] [Google Scholar]
- Stuen S. Anaplasmaphagocytophilum - the most widespread tick-borne infection in animals in Europe. Vet Res Commun. 2007;31:79–84. [DOI] [PubMed] [Google Scholar]
- Vergunst AC, van Lier MC, den Dulk-Ras Aet al. Positive charge is an important feature of the C-terminal transport signal of the VirB/D4-translocated proteins of Agrobacterium. Proc Natl Acad Sci. 2005;102:832–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cheng Z, Zhang Cet al. Anaplasma phagocytophilum p44 mRNA expression is differentially regulated in mammalian and tick host cells: involvement of the DNA binding protein ApxR. J Bacteriol. 2007;189:8651659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.