

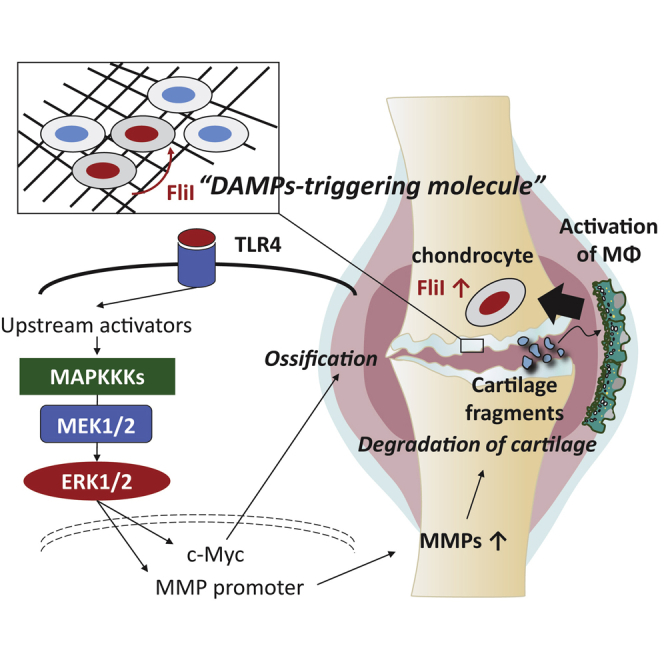
Summary
Synovial macrophages that are activated by cartilage fragments initiate synovitis, a condition that promotes hypertrophic changes in chondrocytes leading to cartilage degeneration in OA. In this study, we analyzed the molecular response of chondrocytes under condition of this type of stimulation to identify a molecular therapeutic target. Stimulated macrophages promoted hypertrophic changes in chondrocytes resulting in production of matrix-degrading enzymes of cartilage. Among the top-upregulated genes, FliI was found to be released from activated chondrocytes and exerted autocrine/paracrine effects on chondrocytes leading to an increase in expression of catabolic and hypertrophic factors. Silencing FliI in stimulated cells significantly reduced expression of catabolic and hypertrophic factors in cocultured chondrocytes. Our further results demonstrated that the FliI-TLR4-ERK1/2 axis is involved in the hypertrophic signaling of chondrocytes and catabolism of cartilage. Our findings provide a new insight into the pathogenesis of OA and identify a potentially new molecular target for diagnostics and therapeutics.
Subject areas: molecular physiology, immunology, cell biology
Graphical abstract
Highlights
-
•
Activated macrophages promote the secretion of FliI from chondrocytes
-
•
FliI acts as a DAMP-triggering molecule in cartilage
-
•
FliI promotes chondrocyte hypertrophy and cartilage catabolism
-
•
FliI represents attractive target for therapeutic intervention
Molecular physiology; Immunology; Cell biology
Introduction
Osteoarthritis (OA), a major, world-wide joint disease, frequently leads to the development of disabilities and long-term hospitalization and medication for people. The number of patients with OA has been reported to be increasing globally and it is estimated that 303 million people suffered from OA in 2017 (Abramson et al., 2006; James et al., 2018). The disease is characterized by progressive degeneration of cartilage, osteophyte formation, subchondral bone sclerosis, and synovial inflammation (synovitis). These issues limit mobility, physical functioning and dexterity of the affected population, leading to premature aging and, in many cases, early retirement. Therefore, OA is considered to be one of the most devastating diseases, in that it influences functionality and productivity in society with significant economic impact on individuals and medical services (Glyn-Jones et al., 2015; Loeser et al., 2012). To date, there are no specific treatments available for OA and current therapies, such as the use of acetaminophen and nonsteroidal anti-inflammatory drugs are given just to relieve pain in the joint. In late-stage OA, patients frequently need surgical intervention and joint arthroplasty to relieve pain and restore the function of the joint (Abramson et al., 2006; Glyn-Jones et al., 2015; Loeser et al., 2012).
The inability to develop a specific drug to inhibit OA pathology is most likely due to our lack of fundamental knowledge regarding the molecular pathogenesis of the disease. However, there is a consensus that mechanical force/loads, trauma, and aging that are associated with the breakage of cartilage and cartilage wear serve to initiate subsequent events leading to the impairment of joint function (Pap and Korb-Pap, 2015, De Lange-Brokaar et al., 2012). Synovitis is one of these primary events in OA and it becomes more prominent in the middle and late stages of OA. Macrophages are constitutively resident in the synovium in both heathy and pathological conditions and are believed to play the key role in the development of synovitis through promoting the infiltration of immune cells and the production of proinflammatory cytokines (De Lange-Brokaar et al., 2012; Robinson et al., 2016). Macrophages are generally classified on basis of the activation into proinflammatory M1 and anti-inflammatory M2. The macrophage M2 phenotype is the most dominant under normal conditions in joints, where they serve to maintain tissue homeostasis. However, upon exposure to microenvironmental stimuli, macrophages are polarized to the M1 phenotype which is responsible for the production of nitric oxide and proinflammatory cytokines that function to mediate tissue damage. A growing body of evidence suggests that an association exists between synovitis and increased cartilage damage. In fact, the presence of inflammatory cytokines in the synovium provokes cartilage breakage and the resulting fragments/tears can then cause further pathological changes (Macfarlane et al., 2019; Hamasaki et al., 2019). Consistent with these findings, our earlier studies demonstrated that cartilage fragments elucidate a robust inflammatory response in macrophages that are associated with increased catabolism in cartilages (Hamasaki et al., 2019, Hamasaki et al., 2020). Therefore, investigating the intercellular communication within the joint microenvironment namely between cells that reside in the synovium and cartilage would be expected to provide clues for development of a novel treatment.
Cartilage is an avascular non-self-renewing hyaline tissue that connects the surface of the bones and acts as a shock absorber in the joint. Specialized cells called chondrocytes, which are sparsely distributed in the cartilage, produce the extracellular matrix which is essential for cartilage homeostasis and function. Chondrocytes in the articular cartilage exhibit phenotypic heterogeneity including developing into proliferative, prehypertrophic, and hypertrophic chondrocytes, depending on their developmental origin, localization, and the stage of the disease (Ji et al., 2019). Physiologically, chondrocytes maintain metabolism in cartilage through regulating anabolic and catabolic activities in the cartilage. In progressive OA, metabolism is drastically switched toward catabolism, which is characterized by phenotypic changes in chondrocytes, including hypertrophy and apoptosis resulting in matrix calcification (Ji et al., 2019). Hypertrophic chondrocytes become the source of catabolic factors such as matrix metalloproteinases (MMPs) and aggrecanases (ADAMTS), and proinflammatory mediators that ultimately lead to the degradation of cartilage (Loeser et al., 2012; Goldring and Otero, 2011; Aigner et al., 2002). These findings indicate that chondrocytes play a crucial role in the progression of OA and our understanding of their pathophysiological function is essential for discovering new therapeutic interventions for preventing/modifying OA progression.
Given the fact that synovitis mediated by inflammatory macrophages alters the functional and phenotypic properties of chondrocytes, we analyzed the gene profile of chondrocytes that had been activated by coculturing with inflammatory macrophages using a transwell system. Macrophages were stimulated with cartilage fragments to mimic the environment in the synovium allowing the activation of macrophages. Bioinformatic analyses revealed that genes associated with hypertrophy and ossification in cocultured chondrocytes were upregulated. Our further data identified a novel chondrocyte catabolic factor, flightless I (FliI), that stimulates chondrocyte hypertrophy and cartilage catabolism associated with progression of OA.
Results
Gene profile of chondrocytes stimulated by coculturing with inflammatory macrophages
To fully exploit the molecular and biological characteristics of chondrocytes upon the onset of synovitis that is initiated by cartilage breakage, murine chondrocytes were cultured with bone marrow-derived macrophages that had been stimulated with cartilage fragments and their response was analyzed using an RNA-seq approach. A total 328 genes were upregulated in chondrocytes cultured with the cartilage fragment-stimulated macrophages as compared to those cultured with mock macrophages (Figure 1A and Table S3). A Venn diagram analysis demonstrated that of the regulated genes, 78 genes were present in the gene profile of the OA cartilage and 106 genes were present in the gene profile of the chondrocytes that had been stimulated with IL-1β (Figures 1B and 1C). These results revealed that inflammatory macrophages stimulated by cartilage fragments activate chondrocytes, causing them to express a gene profile that shares a certain degree of similarity to the gene expression signature of OA. To further understand the regulatory networks and functional relevance of this molecular response, differentially expressed genes (p < 0.05) were subjected to GO and pathway enrichment analyses. The top enriched terms included ossification for a biological process, heparin binding for molecular function, extracellular matrix for cellular component, and chondrocytes for overrepresented tissues (Figure 1D and Table S4). It is noteworthy that the upregulated genes in stimulated chondrocytes included number of molecular markers for cartilage catabolism, including Mmp13, Vegfa, and Col10a1. Consistent with these findings, a KEGG pathway database mapping analysis revealed that ECM-receptor interaction and protein digestion and absorption, which are involved in the OA process, were included in this response (Figure 1E and Table S4). Similarly, this response included the negative regulators of transcription factor SOX9 and the positive regulators RUNX2 that play essential roles in chondrocyte hypertrophy (Figure 1F and Table S4). Considering these collective findings, we conclude that the presence of cartilage fragments in the synovium promotes a molecular response in macrophages that mediates hypertrophic changes in chondrocytes and the catabolism of cartilage.
Figure 1.

Gene profile of murine chondrocytes cocultured with murine macrophages stimulated by cartilage fragments
For stimulation condition, chondrocytes seeded onto cell culture transwell insert were cocultured with macrophages activated by cartilage fragments with sizes of 0.5–55 μm at a ratio of 1:5 for 48 hr.
(A) Volcano plots represents molecular response of chondrocytes stimulated by coculturing with macrophages (n = 3). Top-regulated genes are indicated. Red plots represent upregulated genes, blue plots represent downregulated (p < 0.05) and black plots represent genes that are not significantly regulated.
(B) Venn diagram analysis for the numbers of genes which are significantly upregulated in OA cartilage and these reported in the current study.
(C) Venn diagram analysis for the numbers of genes which are significantly upregulated in chondrocytes stimulated with IL-1β and these reported in the current study. The significantly upregulated genes identified in the current study were compared to gene profiles of OA cartilage and chondrocytes stimulated with IL-1β and the results are shown as Venn diagrams.
(D) Gene enrichment analysis for biological process, molecular function, cellular component, and overrepresented tissue terms of the differentially expressed genes in chondrocytes cocultured with macrophages stimulated by cartilage fragments.
(E) KEGG pathways enrichment analysis.
(F) Enriched transcriptional factors of differentially expressed genes in chondrocytes that were cocultured with macrophages stimulated by cartilage fragments.
Identification of FliI as catabolic factor associated with chondrocyte hypertrophy
Our results prompted us to speculate that the synovitis initiated by cartilage fragments might elicit the production of autocrine/paracrine signaling molecules in chondrocytes that would then mediate hypertrophic differentiation. To further identify the target molecules involved in this process, the differentially expressed genes of chondrocytes were filtered to match p < 0.01 with FC ≥ 1.0 and this criterion led to the identification of 37 genes. These genes were functionally enriched, and included secreted, signal, developmental protein, and phosphoproteins terms (Figure 2A). The potential involvement of molecules enriched in the developmental protein term in cell maturation/development caused us to speculate that they may contribute to hypertrophic differentiation of chondrocytes. Of these genes, FliI which was found in the top-regulated genes (Figure 2B and Table S3) and structurally contains a functional domain involved in cell differentiation, was further characterized as a potential molecular target involved in hypertrophic changes in chondrocytes and the catabolism of cartilage. The upregulation of FliI expression in cocultured chondrocytes with macrophages was confirmed using qRT-PCR (Figure 2C). Next, OA cartilage samples were immunostained for the detection of FliI using IHC and IFA assays. Intracellular FliI was detected in chondrocytes that were located in the superficial and middle zones of OA cartilage and in isolated chondrocytes derived from OA cartilage (Figures 2D and 2E). These results suggest that FliI is expressed in a certain subpopulation of chondrocytes in damaged cartilage and under specific stimulation. Likewise, secreted FliI was detected in synovial fluids of the OA joint and chondrocyte cultures that had been stimulated with IL-1β (Figures 2F and 2G), suggesting a potential role of FliI in the OA process. To gain an insight into the pathological role of FliI in OA, recombinant FliI encoding a conserved repeated region of the Gelsolin-related domain was used to stimulate chondrocytes and cartilage in vitro. Remarkably, stimulation by recombinant FliI resulted in a significant elevation in the expression of catabolic factors, including Mmp3, Mmp13, and Adamts5 in murine primary chondrocytes (Figures 3A–3C), and a decrease in the expression of anabolic factors, including Col2a1 and aggrecan (Figure 3D) in a dose-dependent manner. Moreover, an increase in the expression of hypertrophic markers, including RUNX2, collagen X (COL X) and c-Myc but not VEGFA was observed in chondrocytes that had been stimulated by FliI (Figures 3E and 3F). In line with these results, cartilage stimulated with FliI exhibited a significant decrease in safranin O staining in the superficial zone of cartilage along with an increased concentration of sGAG in the medium (Figures 4A–4C). These collective results revealed that the release of FliI in cartilage promotes the production of catabolic factors and hypertrophic changes in chondrocytes thus leading to the degradation of extracellular matrix and the degeneration of cartilage in OA. To obtain further evidence on the paracrine effects of FliI and its pathological role in OA, a cocultured model containing naive chondrocytes and FliI-knocked down chondrocytes was developed (Figure 4D). The transfected chondrocytes with FliI siRNA (test) or with negative control siRNA (control) were stimulated with IL-1β for 24 hr and then added to cultures of naive chondrocytes (Figure 4E). Of note, the cocultured chondrocytes with FliI-knocked down cells exhibited a reduction in the expression of Mmp3 and the expression of Mmp13 and Runx2 tended to be lower than that for the control (Figure 4F), suggesting that FliI has pathological effects on chondrocytes in cartilage.
Figure 2.

Identification of FliI as a potential molecular target involved in OA process
(A) Gene enrichment analysis for functional categories of top-regulated genes in murine chondrocytes cocultured with murine macrophages stimulated by cartilage fragments.
(B) Violin plots showing the fold change of top-regulated genes in chondrocytes.
(C) Expression of FliI in chondrocytes as analyzed by qRT-PCR. Results represent means ± SEM for triplicates and significant difference was determined by the student t-test.
(D) Detection of FliI in OA cartilage using IHC. Scale bars are indicated and represent 500 μm for low magnification image and 100 μm for higher magnification images.
(E) Detection of FliI in chondrocytes isolated from OA cartilage using IFA. Scale bars represent 50 μm.
(F) Detection of FliI in synovial fluids by ELISA (left panel) and Western blotting (right panel).
(G) Detection of FliI in supernatant of chondrocyte cultures stimulated with IL-1β or TNF-α using ELISA (upper panel) and Western blotting (lower panel). Results represent means ± SEM for triplicates and significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
Figure 3.

In vitro stimulation of murine chondrocytes by FliI
(A) Dose-dependent manner of FliI in stimulating chondrocytes and promoting the expression of catabolic factors.
(B) Gene expression of catabolic factors in chondrocytes stimulated with FliI (5μg/mL) or IL-1β (positive control) as assayed by qRT-PCR. Results represent means ± SEM for 8 samples and significant difference was determined by the one-way ANOVA, followed by Tukey's multiple-comparison procedure.
(C) Protein levels of catabolic factors in stimulated chondrocytes as assayed by Western blotting.
(D) Gene expression of anabolic factors in stimulated chondrocytes with FliI (5μg/mL) (n = 8). Significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
(E) Protein levels of hypertrophic factors in stimulated chondrocytes with FliI (5μg/mL) as assayed by Western blotting.
(F) Quantification of the relative densities of the detected bands of RUNX2, c-Myc and COL X. Results represent means ± SEM for triplicates and significant difference was determined by the student t-test. Results from in vitro experiments are representative of at least two independent experiments.
Figure 4.

Pathological role of FliI in OA process
(A) Catabolic effect of FliI on cartilage (Ex-vivo stimulation). Representative images for safranin O staining of sectioned cartilage after Ex-vivo stimulation with FliI (10 μg/mL) or IL-1β. Scale bar represents 50 μm.
(B) Quantification of the safranin O staining of sectioned cartilage after stimulation with FliI or IL-1β.
(C) Detection of sGAG in supernatant of stimulated cartilage cultures. Results represent means of 9 samples ± SEM and significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
(D) Experimental design of knockdown experiment.
(E) Detection of FliI in transfected chondrocytes.
(F) Expression of catabolic factors in naive chondrocytes stimulated by culturing with IL-1β-stimulated-siRNA-transfected chondrocytes. Results represent means ± SEM for 6 samples and significant difference was determined by the student t-test.
Involvement of MAPKs signaling in chondrocytes activation by FliI
To understand the molecular mechanism associated with FliI activation, we evaluated the expression of ERK and p38 MAP kinases, and JNK, all of which are essential for the differentiation of hypertrophic chondrocytes and the development of endochondral ossification. It is noteworthy that the expression of pERK1/2 but not p38 and JNK were significantly increased in chondrocytes stimulated with FliI (Figure 5A). This elevation was coincident with an increase in the expression of pMEK1/2, which is known to be a factor involved in upper stream signaling of ERK1/2 (Figures 5B and 5C). Furthermore, the pretreatment of chondrocytes with the MEK1/2 inhibitor U0126 diminished the ability of FliI to increase the expression of pERK1/2, c-Myc, MMP13, and MMP3 in chondrocytes and this decrease was dose-dependent (Figure 5D). In fact, pretreatment with 10 μM U0126 significant abolished the effects of FliI, leading to an increase in the expression of pERK1/2 and MMP3 in chondrocytes (Figures 5E and 5F). These results reveal that the catabolic effects of FliI are probably mediated by MEK1/2-ERK1/2 axis signaling. Given the fact that FliI has the ability to interact with toll/interleukin-1 receptor (TIR) domains (Lei et al., 2012), we further investigated the potential involvement of TLR4 in FliI signaling. A pull-down assay showed a direct interaction between recombinant FliI and TLR4 (Figure 5G), suggesting that the changes in chondrocytes due to FliI activation are mediated by the TLR4-ERK1/2 axis. To test this supposition, chondrocytes were pretreated with the TLR4 signaling inhibitor TAK-242 after which, the cells were activated by FliI. This treatment resulted in a significant reduction in the increased expression of ERK1/2 as well as MMP3 and MMP13 (Figures 5H and 5I). From these results, we inferred that FliI promotes chondrocyte hypertrophy and cartilage catabolism though activation of the TLR4-ERK1/2 axis.
Figure 5.

Signaling pathway involved in FliI activation
(A) FliI activated ERK1/2 in chondrocytes stimulated with FliI (5μg/mL) as evidenced by increased expression of pERK1/2.
(B) Dose-dependent manner of FliI in activating ERK1/2 and MEK1/2.
(C) Quantification of the relative densities of the detected bands of pERK1/2 and pMEK1/2 for triplicates and significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
(D) Effects of different doses of MEK1/2 inhibitor U0126 on expression of to ERK1/2, MMP13, MMP3, c-Myc and RUNX2 after FliI activation.
(E) Western blot analysis showing that deduction of ERK1/2 expression due to U0126 is associated with reduction of expression of MMP3.
(F) Quantification of the relative densities of the detected bands of pERK1/2 and MMP3 for triplicates and significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
(G) Pull-down assay showing the ability of FliI to bind TLR4 of chondrocytes.
(H) Involvement of TLR4 in signaling mediated by FliI activation as analyzed by western blotting.
(I) Gene expression of MMP3 and MMP13 in FliI-stimulated chondrocytes after treatment with TAK-242. Results for triplicates and significant difference was determined by the One-way ANOVA, followed by Tukey's multiple-comparison procedure.
Discussion
There has been growing interest in the potential role of cartilage fragments in initiating synovitis in progressive OA. Under this condition, the production of proinflammatory cytokines in the synovium alter the chondrogenic phenotype and induce differentiation, hypertrophy and apoptosis leading to cartilage degeneration (Abramson et al., 2006, De Lange-Brokaar et al., 2012). In fact, our earlier findings, using cocultured chondrocytes model, demonstrated that cartilage fragments activate NF-κB signaling in macrophages resulting in the production of inflammatory cytokines that mediate the production of catabolic factors in chondrocytes (Hamasaki et al., 2019, 2020). Therefore, it is very important to study the molecular changes in chondrocytes that are cocultured with cartilage fragment-stimulated macrophages as a step toward developing a deeper understanding of synovium-cartilage crosstalk in the OA process. Hence, our data showed that chondrocytes cultured with cartilage fragment-stimulated macrophages expressed the gene signatures of ossification, differentiation, hypertrophy, and ECM degradation. An increased hypertrophic differentiation of chondrocytes and the degradation of the cartilage matrix are known to be associated with vascular invasion, endochondral ossification and calcification of cartilage (De Lange-Brokaar et al., 2012; Robinson et al., 2016). These results suggest that there is a link between endochondral ossification signaling and inflammation that occurs in the synovium due to the presence of cartilage fragments.
Our data support the conclusion that synovitis mediated by macrophages is an important switch for the development of progressive OA and cartilage degeneration. In fact, articular cartilage is a type of avascular tissue with no intrinsic vasculature or lymphatic supply, which makes it dependent on surrounding tissues including the synovium and subchondral bone for nutrition and signaling. Therefore, once damage occurs, by mechanical force for example, matrix degradation and cartilage wear activate synovial macrophages that initiate changes in cartilage metabolism, thus the perpetuation of the cycle of degradation. Components from the damaged extracellular matrix, such as fibronectin, aggrecan, and intracellular proteins derived from necrotic cells are recognized by macrophage receptors as danger-associated molecular patterns (DAMPs) that activate NLRP3 inflammasomes that are associated with the production of proinflammatory cytokines and MMP in macrophages (Blom et al., 2007; Nakayama, 2018). Upon the stimulation of cartilage, an extensive autocrine/paracrine communication occurs between different regions of the cartilage that regulates chondrocyte differentiation and endochondral ossification, as well as the production of matrix-degrading enzymes (Little et al., 2009; Goldring, 2000). Generally, cartilage structure, architecture, and function are regulated by a complex molecular network of balanced anabolic and catabolic activities that are mediated by chondrocytes. However, the increase in catabolic activities of cartilage accompanied by changes in chondrocyte phenotype impairs the architecture and function of cartilage which then exaggerates articular cartilage degeneration. Therefore, the identification of molecules that are shifting in tissue toward catabolic activities may aid in the discovery of disease modification agents for potential therapeutic applications.
Our further findings identified FliI as a catabolic factor of chondrocytes that exerted pathological effects on cartilage through promoting chondrocyte differentiation and cartilage catabolism. FliI was found to be highly expressed in chondrocytes of OA cartilage mainly in damaged superficial zones. On the other hand, high levels of extracellular FliI were detected in synovial fluids from patients with OA and the supernatant of chondrocytes that had been stimulated with inflammatory cytokines. The stimulation of chondrocytes with FliI appeared to activate the TLR4 and MAPKs-ERK1/2 signaling pathway resulting in an increase in the expression of hypertrophic and catabolic factors. These findings were further supported by results demonstrating that the pretreatment the cells with a MEK kinase inhibitor, upstream of ERKs signaling, and a TLR4 antagonist diminished the upregulation in the expression of catabolic factors induced by FliI stimulation. Together, the findings that FliI is associated with cartilage damage and the activation of TLR4-ERKs signaling in chondrocytes suggest its potential function as a DAMPs-triggering molecule in cartilage mediating hypertrophy and catabolism.
FliI is a member of the gelsolin family of proteins that is predominately expressed as intracellular protein and solely as secreted protein and functions as an actin-remodeling protein, and a nuclear receptor co-activator with the ability to interact with various other proteins that are important in cellular signaling (Kopecki and Cowin, 2008). Secreted FliI has been detected in fluids around damaged skin extracellular matrix, including blisters and wounds of patients undergoing abdominoplasty, as well as tumor microenvironments. The hypertrophic effects of FliI are evident by the facts that FliI impairs skin barrier development and promotes hypertrophic scarring through elevating collagen-Ι mediating hypertrophy and scar formation in skin (Cowin et al., 2007; Cameron et al., 2016). In the tumor microenvironment, FliI enhances tumor growth and spread through promoting tumor cell invasion and repressing apoptosis (Kopecki et al., 2015). Moreover, a recent study showed that FliI is expressed at high levels in damaged colonic mucosal matrix of patients with inflammatory bowel disease. The overexpression of FliI adversely affects mucosal healing and enhances the local production of inflammatory cytokines and immune cell infiltration (Kopecki et al., 2019). On the other hand, FliI exerts immunomodulatory functions on immune cells through interaction with TLR4 and the regulation of TLR-4/MyD88 signaling (Wang et al., 2006). These collective findings suggest that FliI might be a molecule that is associated with tissue damage and functions to promote tissue hypertrophy and catabolism.
Our data are consistent with earlier studies highlighting the association between the expression of members of the gelsolin family, such as villin and adseverin in the OA process via regulating chondrocyte differentiation and hypertrophy (Bucki et al., 2008). In essence, adseverin has been documented to regulate the onset of hypertrophy in a MAPK-dependent manner, and adseverin that is overexpressed in chondrocytes causes an increase in the expression of genes that are involved in differentiation and catabolism, as well as ERK1/2 and p38 signaling (Nurminsky et al., 2007). Generally, members of the gelsolin family including CapG, FliI, villin, and supervillin, are intracellular molecules that are involved in the regulation of actin dynamics. Extracellular forms of gelsolin proteins are released upon cell damage and act as DAMPs-triggering molecules that participate in the signaling of several signal transduction pathways that regulate essential functional processes, including inflammation, tumor growth, differentiation, and apoptosis (Bucki et al., 2008; Ahrens et al., 2012).
Hypertrophy of chondrocytes, the terminal stage of endochondral ossification, is characterized as a process where bone is replaced with cartilage. Hypertrophic chondrocytes express high levels of RUNX2, COL10A1 and MMPs and eventually undergo apoptosis, followed by vascular invasion that facilitates osteoclast invasion into cartilage and bone. Moreover, recently acuminating evidence suggests that the transdifferentiation of hypertrophic chondrocytes to osteoblasts occurs during the process of endochondral ossification (Aghajanian and Mohan, 2018). In this process, MAPK signaling pathways namely p38 and ERK-1/2 play a prominent role through regulating chondrocyte differentiation and the expression of matrix-degrading enzymes (Prasadam et al., 2010). It is interesting to note that the activation of TLRs in chondrocytes initiates hypertrophic/apoptotic changes in chondrocytes through the activation of MAPK signaling pathways. The contents of cartilage matrix that accumulate in joints due to traumatization and degeneration act as DAMPs that are recognized by TLRs that are expressed in chondrocytes. For example, a high-mobility group box 1 protein (HMGB-1) and proteoglycans such as biglycan and decorin activate chondrocytes via TLR4, resulting in the upregulation of catabolic factors (Kim et al., 2006). Generally, extracellular DAMPs, including ECM components and necrotic and apoptotic cells bind TLRs, NOD-like receptors (NLRs), and receptors for advanced glycosylation end products that are located on the surface of immune cells, chondrocytes, osteoblasts, and synoviocytes where they initiate downstream signaling cascades leading to the activation of several transcription factors that are associated with the production of catabolic factors. The essential role of DAMP in pathogenesis of OA has been demonstrated in experimental arthritis models as function blocking antibodies to HMGB1, carboxylate glycans or Coll2-1 to reduce catabolism and inflammation in the joint (Lambert et al., 2020). In line with these findings, our study identified FliI as a DAMPs-triggering molecule that mediates the activation of TLR4-ERK1/2 signaling in chondrocytes, thus leading to hypertrophy and cartilage catabolism. FliI appears to have the typical characteristics of DAMPs-triggering molecules, including a high expression in damaged tissues, acting as an endogenous molecule for pattern recognition receptors such as TLRs, can be passively released or actively secreted from stressed cells and, at high dosage, activates signal transduction and various signaling cascades that are associated with pathogenesis (Blom et al., 2007; Nakayama, 2018).
In conclusion, our findings suggest that macrophages that are activated by cartilage fragments in the synovium promote the release/secretion of FliI from chondrocytes which then mediates hypertrophic changes in chondrocytes and the catabolism of cartilage via TLR4-ERK1/2 signaling. This is the first study highlighting FliI as a DAMPs-triggering molecule that is involved in the pathogenesis of OA. Our further efforts will be directed at investigating FliI as a potential therapeutic target for OA in experimental models.
Limitation of the study
The major limitation of this study includes the lack of in vivo evidence supporting the pathological role of FliI in OA progression. Our future research aims at the therapeutic effects of FliI blocking antibody in OA animal models. Another limitation is the effective dose of FliI in vitro was relatively high as compared to the detected level of protein in OA joint. This is most likely due to the reason that recombinant FliI used in this study is expressing single gelsolin-related domain, while full protein contains 6 gelsolin-related domains. One additional limitation is that the macrophages used in the current study are derived from bone marrow cells, which might exhibit a different phenotype from synovial resident macrophages.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Flightless I | GeneTex | RRID: AB_10727618 |
| Anti-MMP13 antibody | Abcam | RRID: AB_776416 |
| ADAMTS5 | GeneTex | RRID: AB_1240417 |
| Anti-MMP3 antibody | Abcam | RRID: AB_881281 |
| RUNX2 | BioLegend | RRID: AB_2632769 |
| VEGFa | BioLegend | RRID: AB_2212504 |
| c-Myc antibody | Cell signaling | RRID: AB_2151827 |
| Phospho-c-Myc antibody | Cell signaling | RRID: AB_2151830 |
| Anti-Collagen X antibody | Abcam | RRID: AB_879742 |
| p44/42 MAPK (Erk1/2) antibody | Cell signaling | RRID: AB_390779 |
| Phosphor-p44/42 MAPK (Erk1/2) antibody | Cell signaling | RRID: AB_2315112 |
| p38 MAPK antibody | Cell signaling | RRID: AB_10999090 |
| Phospho-p38 MAPK antibody | Cell signaling | RRID: AB_2139682 |
| SAPK-JNK antibody | Cell signaling | RRID: AB_2250373 |
| Phospho-SAPK-JNK antibody | Cell signaling | RRID: AB_823588 |
| MEK1/2 antibody | Cell signaling | RRID: AB_10829473 |
| Phospho-MEK1/2 antibody | Cell signaling | RRID: AB_331648 |
| TLR4/MD2 | BioLegend | RRID: AB_313789 |
| Anti-β actin antibody | Applied Biological Materials | RRID: AB_2631287 |
| Biological samples | ||
| Human OA cartilage and synovial fluid from the patients undergoing total knee arthroplasty | Hokkaido University Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Flightless I Recombinant Protein Antigen | Novus Biologicals | NBP1-87925PEP |
| Recombinant Human IL-1 beta Protein | Novus Biologicals | NBP2-35895 |
| Recombinant Human TNF-a | Peprotech | Cat #. 300-01A |
| Recombinant Mouse IL-1β | BioLegend | Cat #. 575102 |
| Critical commercial assays | ||
| Human Flightless I Homolog ELISA Kit | MyBioSource, Huissen, Netherlands | catalog No. MBS459349 |
| Deposited data | ||
| Raw and Analysis data | This paper | GSE171420 |
| Raw and Analysis data of articular cartilage | Soul et al., 2018 | E-MTAB-6266 |
| Raw and Analysis data of articular cartilage | Ramos et al., 2014 | GSE57218 |
| Raw and Analysis data of articular cartilage | Dunn et al., 2016 | E-MTAB-4304 |
| Raw and Analysis data of human chondrocyte stimulated with IL-1β | Comblain et al., 2016 | GSE75181 |
| Raw and Analysis data of mouse chondrocyte stimulated with IL-1β | Son et al., 2017 | GSE104793 |
| Raw and Analysis data of bovine chondrocyte stimulated with IL-1β | Lv et al., 2019 | https://www.nature.com/articles/s41598-018-36500-2 |
| Experimental models: Organisms/strains | ||
| Mouse C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Software and algorithms | ||
| Image J | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Cufflinks and Cuffdiff | Hamasaki et al., 2020 | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/index.html |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, M Alaa Terkawi (materkawi@med.hokudai.ac.jp).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. The RNA-seq data generated during this study were deposited at the Gene Expression Omnibus (GEO) database (http: www.ncbi.nlm.nih.gov/geo/) and are publicly available under the accession number (GSE171420). Other bioinformatics data are publicly available under the accession numbers (E-MTAB-6266, GSE57218, E-MTAB-4304, GSE75181, GSE104793). Additional data related to this study might be requested from corresponding author.
Experimental model and subject details
Animal study
This study was performed to explore the molecular mechanism by which stimulated macrophages impair the function of chondrocytes in OA cartilage. Animal experiments were performed based on the ethical guidelines approved by the Institute of Animal Care and Use Committee of the Hokkaido University Graduate School of Medicine (approval ID: 19-0158). Specific pathogen-free C57BL/6 mice were kept in SPF facility and both male and female were used in all experiments. Primary articular chondrocytes were isolated from cartilages of 5-day-old C57BL/6 mice. Primary cartilages from femoral head were isolated from 6-weeks-old C57BL/6 mice. Chondrocytes were cocultured with stimulated macrophages in a transwell culture system and their response was analyzed RNA-Seq. Murine chondrocytes and femoral head cartilage were stimulated with the target molecule, and expression of catabolic/hypertrophic factors and histological changes were evaluated. Signaling pathway induced by the target molecule in chondrocytes was characterized in vitro using specific inhibitors.
Human study
Human samples including cartilage and synovial fluid samples were obtained from OA patients undergoing total knee arthroplasty under protocols approved by Hokkaido University Institutional Review Board and with the patients’ informed consent (ID: 018-0215). Tissues and synovial fluids were obtained from 4 OA patients including 1 man and 3 women with mean age of 74 years (59-83 years). Formalin-fixed human cartilage was immunostained by specific antibody for detecting the target molecule. On the other hand, human chondrocytes were cultured in presence or absence of stimuli for genes and proteins expression analyses.
Method details
In vitro coculture model of chondrocytes with macrophages
Macrophages, chondrocytes and cartilage fragments were isolated from C57BL/6 mice and cultured as earlier described (Hamasaki et al., 2019, Hamasaki et al., 2020). Briefly, bone marrow cells isolated using MACS beads (Monocyte isolation kit BM, Miltenyi Biotec) were differentiated into macrophages by stimulation with 50 ng/mL mouse recombinant macrophage colony-stimulating factor (MCSF; Peprotech, Tokyo, Japan) for 7 days. Primary articular chondrocytes were isolated from cartilages of 5-day-old C57BL/6 mice by enzymatic digestion with collagenase D (Sigma) in DMEM medium (DMEM, Sigma- Aldrich) at 37 °C overnight (Gosset et al., 2008). Cartilages were isolated from the femoral condyles and tibial plateau. To prepare the cartilage fragments, the femoral head cartilages of 4-week-old SPF C57BL/6 mice were crushed by Multi Beads Shocker (Yasui Kikai, Osaka, Japan) for 1 minute at 2500 rpm. Fragments with sizes ranged between 0.5 and 55 μm were kept at −80°C for further use. Thereafter, cartilage fragments at ratio of 5:1 were added to differentiated macrophages seeded onto 24-well plates at ratio 2 × 105. In parallel, chondrocytes were seeded (2 × 105) onto cell culture transwell insert (1.0 μm pore size; Falcon cell culture inserts, BD, Franklin Lakes, NJ, USA) and cultured in DMEM/F12 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich), 25 mg/L penicillin/streptomycin for 24h. The transwell inserts were next added to differentiated macrophages in 24-well plates and cultured in a 37 °C humidified atmosphere containing 5% CO2 for 48h. Control group was co-culture cells in absence of cartilage fragments.
Human samples
Human samples including cartilage and synovial fluid samples were obtained from OA patients undergoing total knee arthroplasty. Tissues and synovial fluids were obtained from 4 OA patients including 1 man and 3 women with mean age of 74 years (59-83 years) who underwent total knee arthroplasty. Obtained cartilage were fixed by 10% formalin for 24 h, decalcified by EDTA for 1 week at 4°C and then embedded frontally in paraffin. To isolate OA chondrocytes, cartilage was cut into small pieces and digested with 0.15% collagenase II solution (Gibco) in DMEM/F12 medium (Sigma) overnight at 37°C-water bath with shaking (Oseni et al., 2013). Cells were suspended in DMEM/F12 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich), 25 mg/L penicillin/streptomycin and cultured in a 37°C humidified atmosphere containing 5% CO2. After 5 days, chondrocytes were detached using 1% trypsin-EDTA solution (Wako), washed by PBS, seeded at 1X105 and stimulated with recombinant human IL-1β or TNF-α (Peprotech) for 24 h. Synovial fluids of OA knee samples were centrifuged at 2000 x g at 4°C for 10 min to remove cells/debris and preserved at -80°C until further analysis.
RNA sequencing and bioinformatics analysis
Cocultured chondrocytes were lysed with TRIzol Reagent (Invitrogen carlsbad, CA, USA) and RNA was purified using RNeasy Mini kit columns (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. High-quality DNA-free 1 μg RNA with integrity score > 8.0 was further used for generating libraries (TruSeq Stranded mRNA LT Sample Prep Kit). Only high-quality libraries were subjected to Illumina HiSeq 2500 (Illumina sequencing platforms). The paired-end reads (100 bp) were trimmed by fastp, aligned, normalized, and then annotated using Cufflinks and Cuffdiff (Hamasaki et al., 2020). Significantly expressed genes with p-value < 0.05 were subjected to gene ontology (GO) enrichment and pathway enrichment analyses of KEGG (Kyoto Encyclopedia of Genes and Genome). Analyses were performed using the Genomatix Gene Ranker (http://www.genomatix.de/), database for annotation visualization and integrated discovery online tools (DAVID: https://david.abcc.ncifcrf.gov/). Moreover, the gene profile of chondrocytes cultured with macrophages was compared to the available database for the gene profiles of OA-cartilage (E-MTAB-6266 (Soul et al., 2018), GSE57218 (Ramos et al., 2014), E-MTAB-4304 (Dunn et al., 2016)) and chondrocytes stimulated with IL-1β (GSE75181 (Comblain et al., 2016), GSE104793 (Son et al., 2017), and the dataset (Lv et al., 2019). Results were visualized as Venn diagram.
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
RNA was purified using kit columns (NucleoSpin® RNA, TAKARA, Tokyo, Japan) according to the manufacturer’s instructions. DNA-free RNA was reverse transcripted using GoScript TM reverse transcriptase and random primer (Promega, Madison, USA), according to manufacturer’s instructions. The qRT-PCR was performed by SYBR Premix Ex TaqTM II (Takara, Shiga, Japan) and the specific primers (Table S1) on a Thermal Cycler Dice System 2 (Takara). Gene expression of each target gene was calculated using the 2ΔΔCt method (Hamasaki et al., 2019, Hamasaki et al., 2020).
Immunohistochemistry (IHC) for human OA cartilage
Human OA cartilage were fixed by 10% formalin for 24 h, decalcified in EDTA solution (Wako, Japan) for 1 week and then embedded frontally in paraffin. Sections with of 3-μm thickness were prepared, deparaffinized, and treated for 5 min with proteinase K (Dako, CA, USA) for antigen retrieval. The slides were blocked with PBS-containing 1% BSA and 5% horse serum and incubated with FliI antibody (GeneTex) for 1 h. Signals was detected by horseradish peroxidase (HRP)-conjugated streptavidin using a Vectastain Elite ABC kit (Vector Laboratories, Burlingame, USA) followed by counterstaining with hematoxylin for nuclei detection.
Immunofluorescence (IFA) for human OA chondrocytes
Isolated chondrocytes from OA cartilage were seeded onto 96-well plate, fixed with 10% formalin, and then subjected to IFA. Briefly, fixed cells were treated with 0.1% triton X100 for 3 min and incubated with 5% FBS (Sigma)-PBS buffer for 1 h. Cells were incubated with primary rabbit antibody to Flightless I (GeneTex) diluted in blocking buffer at 1:200 for 1 h at 37°C. Bound antibody was detected by specific secondary antibody conjugated with Alexa Fluor®594 (Jackson ImmunoResearch, West Grove, PA, USA). The cellular nuclei were stained with DAPI (Dojindo Molecular Technologies, Kumamoto, Japan).
Detection of FliI by ELISA
FliI was detected in chondrocyte cultures and synovial fluid by Human Flightless I Homolog ELISA Kit (MyBioSource, Huissen, Netherlands, catalog No. MBS459349) according to the manufacturer's instructions.
Stimulation of primary murine chondrocytes
Murine chondrocytes were isolated from femoral condyles and tibial plateau using enzymatic digestion method. This method allows collecting all types of chondrocytes from different zones of cartilage including, superficial, transitional and deep zones (Gosset et al., 2008). Cells were seeded on 24-well plates at ratio 2×105 and cultured in DMEM medium in a 37°C humidified atmosphere containing 5% CO2 for 24 h. Cells were stimulated with murine IL-1β (10 ng/ml, Biolegend) or different concentrations of recombinant FliI, including 0.5, 1 , 5 or 10 μg/ml (Novus Biologicals) for 24 h. Recombinant FliI encoding of conserved repeated region of gelsolin-related domain, which may play a role in cell differentiation (Kopecki and Cowin, 2008). For knockdown experiment using small interfering RNA (siRNA), cells were seeded at 2 × 105 and cultured in presence of siRNA solution including murine FliI and negative control (Thermo Fisher, Silencer Select, Tokyo, Japan). Optimal concentration of siRNA was diluted by Opti-MEM (Gibco, Thermo Fisher) and then mixed with 25 μL Lipofectamine RNAiMAX Reagent (Thermo Fisher) diluted by Opti-MEM and incubated for 5 min as recommended by the manufacturer’s instructions. Silencing FliI was confirmed after 48 h by Western blotting. Transfected cells were cultured onto 1.0 μm pore size-transwell insert (Falcon, NC, USA) and stimulated with IL-1β (10 ng/ml) for 24 h. After 2 times washing with PBS, inserts were added to naïve chondrocytes 2 × 105 and cultured for 48 h. For inhibition assay, U0126, MEK1/2 inhibitor (Cell signaling, USA) and TAK-242 (Cayman, Canada), TLR4 signaling inhibitor were used. Chondrocytes were seeded onto 24-well plates at ratio 2.0×105 and then pretreated using different concentration of TAK-242 or U0126 for 2 h. Thereafter, cells were washed twice with PBS and stimulated with FliI (5 μg/ml) and cultured in a 37 °C humidified atmosphere containing 5% CO2 for 24 h. Finally, the cells were harvested for further gene expression and protein analyses.
Ex-vivo culture model
Primary cartilages from femoral head were isolated from 6-weeks-old C57BL/6 mice, washed twice by PBS, placed into 96-well plates (1 in each well) and cultured in 200 μl of pre-warmed medium (DMEM low-glucose supplemented with 10% FBS and 25 mg/L penicillin/streptomycin) for 24 h (Stanton et al., 2011; Marino et al., 2016). Cartilages were stimulated with 10 ng/ml murine recombinant IL-1β (Biolegend) or recombinant 10 μg/ml FliI (Novus Biologicals) for 72 h. The culture supernatant was harvested for measuring the release of glycosaminoglycan (GAG) using 1,9-Dimethylmethylene blue (DMMB) dye binding assay (Stanton et al., 2011; Terkeltaub et al., 2011). Cartilages were fixed using 4% PFA (Wako, Japan) for 48 h, decalcified by EDTA solution (Wako) for 2 weeks and then embedded in paraffin. Sections with 5 μm thickness were prepared and stained with hematoxylin and eosin and Safranin O. Safranin O staining indicating pathological changes was quantified using ImageJ (Schneider et al., 2012) (National Institutes of Health; NIH, USA) in the superficial area of articular cartilage and subtracted from the total area (Headland et al., 2015).
Western blot analysis
Chondrocytes were lysed in RIPA lysis buffer (ATTO corporation, Tokyo, Japan) and protein lysate was subjected to standard procedure for SDS-PAGE and Western blot analysis (ATTO). Targeted proteins were detected with specific antibodies listed in Table S2. Bands were visualized using a Quantity One v. 4.6.9 (Bio-Rad) software and their densities were quantified using ImageJ (NIH).
Pull-down assay
Chondrocytes were lysed in RIPA lysis buffer (ATTO corporation) and the concentration of protein was measured using BCA protein assay (Thermo Scientific). In parallel, 10 μg recombinant FliI (Novus biologicals) was incubated with his-tag beads (His60 Ni Superflow Resin, TAKARA, Japan) with low speed-rotation for 2 h at RT. FliI-binding His-tag beads were washed 3 time with PBST, and then chondrocytes lysate containing 500 μg protein were added to his-tag beads and incubated at low speed-rotation for 2 h at RT. Thereafter, the beads were washed 3 times with PBS-T and bound proteins were eluted for Western blotting.
Quantification and statistical analysis
Statistical comparisons were performed with mean ± SEM for continuous variables of experiments conducted at least two times. Data were analyzed using either unpaired Student’s t-test or One-way ANOVA multiple comparison (GraphPad Software Inc., La Jolla, CA, USA), and differences were considered statistically significant ∗p < 0.05, ∗∗ p < 0.01, and ∗∗∗ p < 0.001 and ∗∗∗∗ p < 0.0001.
Acknowledgments
We thank orthopedic surgeons who helped us to obtain samples from patients with OA. This work was supported by research grants from Kobayashi Foundation and Japan Agency for Medical Research and Development (JP20gm6210004).
Author contributions
Study designs were done by T.E., M.A.T., and N.I. In vitro and in vivo experiments were performed by T.E., M.H., G.M., T.O., M.K.A., S.Y., and H.A. Data acquisition and statistical analysis were done by T..E, T.S., D.T., K.H., and K.K. Drafting of manuscript was performed by T.E., M.A.T. Critical review of manuscript was done by T.O., N.I., K.K., and D.T. All authors have read and approved the submitted manuscript.
Declarations of interests
The authors declare no competing interests.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102643.
Contributor Information
Mohamad Alaa Terkawi, Email: materkawi@med.hokudai.ac.jp.
Tomohiro Onodera, Email: tomozou@med.hokudai.ac.jp.
Supplemental information
References
- Abramson S.B., Attur M., Yazici Y. Prospects for disease modification in osteoarthritis. Nat. Clin. Pract. Rheumatol. 2006;2:304–312. doi: 10.1038/ncprheum0193. [DOI] [PubMed] [Google Scholar]
- Aghajanian P., Mohan S. The art of building bone: emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018;6:19. doi: 10.1038/s41413-018-0021-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrens S., Zelenay S., Sancho D., Hanc P., Kjær S., Feest C., Fletcher G., Durkin C., Postigo A., Skehel M. F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity. 2012;36:635–645. doi: 10.1016/j.immuni.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Aigner T., Kurz B., Fukui N., Sandell L. Roles of chondrocytes in the pathogenesis of osteoarthritis. Curr. Opin. Rheumatol. 2002;14:578–584. doi: 10.1097/00002281-200209000-00018. [DOI] [PubMed] [Google Scholar]
- Blom A.B., Van Lent P.L., Libregts S., Holthuysen A.E., van der Kraan P.M., van Rooijen N., van den Berg W.B. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum. 2007;56:147–157. doi: 10.1002/art.22337. [DOI] [PubMed] [Google Scholar]
- Bucki R., Byfield F.J., Kulakowska A., Mccormick M.E., Drozdowski W., Namiot Z., Hartung T., Janmey P.A. Extracellular gelsolin binds lipoteichoic acid and modulates cellular response to proinflammatory bacterial wall components. J. Immunol. 2008;181:4936–4944. doi: 10.4049/jimmunol.181.7.4936. [DOI] [PubMed] [Google Scholar]
- Cameron A.M., Turner C.T., Adams D.H., Jackson J.E., Melville E., Arkell R.M., Anderson P.J., Cowin A.J. Flightless I is a key regulator of the fibroproliferative process in hypertrophic scarring and a target for a novel antiscarring therapy. Br. J. Dermatol. 2016;174:786–794. doi: 10.1111/bjd.14263. [DOI] [PubMed] [Google Scholar]
- Comblain F., Dubuc J.E., Lambert C., Sanchez C., Lesponne I., Serisier S., Henrotin Y. Identification of targets of a new nutritional mixture for osteoarthritis management composed by curcuminoids extract, hydrolyzed collagen and green tea extract. PLoS One. 2016;11:e0156902. doi: 10.1371/journal.pone.0156902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowin A.J., Adams D.H., Strudwick X.L., Chan H., Hooper J.A., Sander G.R., Rayner T.E., Matthaei K.I., Powell B.C., Campbell H.D. Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J. Pathol. 2007;211:572–581. doi: 10.1002/path.2143. [DOI] [PubMed] [Google Scholar]
- De Lange-Brokaar B.J., Ioan-Facsinay A., Van Osch G.J., Zuurmond A.M., Schoones J., Toes R.E., Huizinga T.W., Kloppenburg M. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis Cartilage. 2012;20:1484–1499. doi: 10.1016/j.joca.2012.08.027. [DOI] [PubMed] [Google Scholar]
- Dunn S.L., Soul J., Anand S., Schwartz J.M., Boot-Handford R.P., Hardingham T.E. Gene expression changes in damaged osteoarthritic cartilage identify a signature of non-chondrogenic and mechanical responses. Osteoarthritis Cartilage. 2016;24:1431–1440. doi: 10.1016/j.joca.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glyn-Jones S., Palmer A.J.R., Agricola R., Price A.J., Vincent T.L., Weinans H., Carr A.J. Osteoarthritis. The Lancet. 2015;386:376–387. doi: 10.1016/S0140-6736(14)60802-3. [DOI] [PubMed] [Google Scholar]
- Goldring M.B. The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000;43:1916–1926. doi: 10.1002/1529-0131(200009)43:9<1916::AID-ANR2>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Goldring M.B., Otero M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011;23:471–478. doi: 10.1097/BOR.0b013e328349c2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset M., Berenbaum F., Thirion S., Jacques C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008;3:1253–1260. doi: 10.1038/nprot.2008.95. [DOI] [PubMed] [Google Scholar]
- Hamasaki M., Terkawi M.A., Onodera T., Homan K., Iwasaki N. A novel cartilage fragments stimulation model revealed that macrophage inflammatory response causes an upregulation of catabolic factors of chondrocytes in vitro. Cartilage. 2019 doi: 10.1177/1947603519828426. 1947603519828426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M., Terkawi M.A., Onodera T., Tian Y., Ebata T., Matsumae G., Alhasan H., Takahashi D., Iwasaki N. Transcriptional profiling of murine macrophages stimulated with cartilage fragments revealed a strategy for treatment of progressive osteoarthritis. Sci. Rep. 2020;10:7558. doi: 10.1038/s41598-020-64515-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headland S.E., Jones H.R., Norling L.V., Kim A., Souza P.R., Corsiero E., Gil C.D., Nerviani A., Dell’Accio F., Pitzalis C. Neutrophil-derived microvesicles enter cartilage and protect the joint in inflammatory arthritis. Sci. Transl Med. 2015;7:315ra190. doi: 10.1126/scitranslmed.aac5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James S.L., Abate D., Abate K.H., Abay S.M., Abbafati C., Abbasi N., Abbastabar H., Abd-Allah F., Abdela J., Abdelalim A. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet. 2018;392:1789–1858. doi: 10.1016/S0140-6736(18)32279-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Q., Zheng Y., Zhang G., Hu Y., Fan X., Hou Y., Wen L., Li L., Xu Y., Wang Y. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 2019;78:100–110. doi: 10.1136/annrheumdis-2017-212863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.A., Cho M.L., Choi H.Y., Yoon C.S., Jhun J.Y., Oh H.J., Kim H.Y. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006;54:2152–2163. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- Kopecki Z., Cowin A.J. Flightless I: an actin-remodelling protein and an important negative regulator of wound repair. Int. J. Biochem. Cell Biol. 2008;40:1415–1419. doi: 10.1016/j.biocel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Kopecki Z., Yang G.N., Jackson J.E., Melville E.L., Calley M.P., Murrell D.F., Darby I.A., O’Toole E.A., Samuel M.S., Cowin A.J. Cytoskeletal protein Flightless I inhibits apoptosis, enhances tumor cell invasion and promotes cutaneous squamous cell carcinoma progression. Oncotarget. 2015;6:36426–36440. doi: 10.18632/oncotarget.5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecki Z., Yang G., Treloar S., Mashtoub S., Howarth G.S., Cummins A.G., Cowin A.J. Flightless I exacerbation of inflammatory responses contributes to increased colonic damage in a mouse model of dextran sulphate sodium-induced ulcerative colitis. Sci. Rep. 2019;9:12792. doi: 10.1038/s41598-019-49129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C., Zappia J., Sanchez C., Florin A., Dubuc J.-E., Henrotin Y. The Damage-Associated Molecular Patterns (DAMPs) as Potential Targets to Treat Osteoarthritis: Perspectives From a Review of the Literature. frontiers in Medicine. 2020;7:918. doi: 10.3389/fmed.2020.607186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei N., Franken L., Ruzehaji N., Offenhauser C., Cowin A.J., Murray R.Z. Flightless, secreted through a late endosome/lysosome pathway, binds LPS and dampens cytokine secretion. J. Cell Sci. 2012;125:4288–4296. doi: 10.1242/jcs.099507. [DOI] [PubMed] [Google Scholar]
- Little C.B., Barai A., Burkhardt D., Smith S.M., Fosang A.J., Werb Z., Shah M., Thompson E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009;60:3723–3733. doi: 10.1002/art.25002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser R.F., Goldring S.R., Scanzello C.R., Goldring M.B. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 2012;64:1697–1707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv M., Zhou Y., Polson S.W., Wan L.Q., Wang M., Han L., Wang L., Lu X.L. Identification of chondrocyte genes and signaling pathways in response to acute joint inflammation. Sci. Rep. 2019;9:93. doi: 10.1038/s41598-018-36500-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane L.A., Yang H., Collins J.E., Jarraya M., Guermazi A., Mandl L.A., Martin S.D., Wright J., Losina E., Katz J.N. Association of changes in effusion-synovitis with progression of cartilage damage over eighteen months in patients with osteoarthritis and meniscal tear. Arthritis Rheumatol. 2019;71:73–81. doi: 10.1002/art.40660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S., Staines K.A., Brown G., Howard-Jones R.A., Adamczyk M. Models of ex vivo explant cultures: applications in bone research. Bonekey Rep. 2016;5:818. doi: 10.1038/bonekey.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M. Macrophage recognition of crystals and nanoparticles. Front. Immunol. 2018;9:103. doi: 10.3389/fimmu.2018.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminsky D., Magee C., Faverman L., Nurminskaya M. Regulation of chondrocyte differentiation by actin-severing protein adseverin. Dev. Biol. 2007;302:427–437. doi: 10.1016/j.ydbio.2006.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oseni A.O., Butler P.E., Seifalian A.M. Optimization of chondrocyte isolation and characterization for large-scale cartilage tissue engineering. J. Surg. Res. 2013;181:41–48. doi: 10.1016/j.jss.2012.05.087. [DOI] [PubMed] [Google Scholar]
- Pap T., Korb-Pap A. Cartilage damage in osteoarthritis and rheumatoid arthritis--two unequal siblings. Nat. Rev. Rheumatol. 2015;11:606–615. doi: 10.1038/nrrheum.2015.95. [DOI] [PubMed] [Google Scholar]
- Prasadam I., Van Gennip S., Friis T., Shi W., Crawford R., Xiao Y. ERK-1/2 and p38 in the regulation of hypertrophic changes of normal articular cartilage chondrocytes induced by osteoarthritic subchondral osteoblasts. Arthritis Rheum. 2010;62:1349–1360. doi: 10.1002/art.27397. [DOI] [PubMed] [Google Scholar]
- Ramos Y.F., Den Hollander W., Bovee J.V., Bomer N., van der Breggen R., Lakenberg N., Keurentjes J.C., Goeman J.J., Slagboom P.E., Nelissen R.G. Genes involved in the osteoarthritis process identified through genome wide expression analysis in articular cartilage; the RAAK study. PLoS One. 2014;9:e103056. doi: 10.1371/journal.pone.0103056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson W.H., Lepus C.M., Wang Q., Raghu H., Mao R., Lindstrom T.M., Sokolove J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016;12:580–592. doi: 10.1038/nrrheum.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y.O., Park S., Kwak J.S., Won Y., Choi W.S., Rhee J., Chun C.H., Ryu J.H., Kim D.K., Choi H.S. Estrogen-related receptor gamma causes osteoarthritis by upregulating extracellular matrix-degrading enzymes. Nat. Commun. 2017;8:2133. doi: 10.1038/s41467-017-01868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soul J., Dunn S.L., Anand S., Serracino-Inglott F., Schwartz J.M., Boot-Handford R.P., Hardingham T.E. Stratification of knee osteoarthritis: two major patient subgroups identified by genome-wide expression analysis of articular cartilage. Ann. Rheum. Dis. 2018;77:423. doi: 10.1136/annrheumdis-2017-212603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton H., Golub S.B., Rogerson F.M., Last K., Little C.B., Fosang A.J. Investigating ADAMTS-mediated aggrecanolysis in mouse cartilage. Nat. Protoc. 2011;6:388–404. doi: 10.1038/nprot.2010.179. [DOI] [PubMed] [Google Scholar]
- Terkeltaub R., Yang B., Lotz M., Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011;63:1928–1937. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Chuang T.H., Ronni T., Gu S., Du Y.C., Cai H., Sun H.Q., Yin H.L., Chen X. Flightless I homolog negatively modulates the TLR pathway. J. Immunol. 2006;176:1355–1362. doi: 10.4049/jimmunol.176.3.1355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplemental information. The RNA-seq data generated during this study were deposited at the Gene Expression Omnibus (GEO) database (http: www.ncbi.nlm.nih.gov/geo/) and are publicly available under the accession number (GSE171420). Other bioinformatics data are publicly available under the accession numbers (E-MTAB-6266, GSE57218, E-MTAB-4304, GSE75181, GSE104793). Additional data related to this study might be requested from corresponding author.