

Abstract
Oxidative and endoplasmic reticulum (ER) stress are involved in mediating high-fat diet (HFD)-induced insulin resistance. As the ER-localized methionine sulfoxide reductase B3 (MsrB3) protects cells against oxidative and ER stress, we hypothesized that MsrB3 might be associated with HFD-induced insulin resistance. To test this hypothesis, we examined the effect of MsrB3 deficiency on HFD-induced insulin resistance using MsrB3 knockout (KO) mice. Mice were fed a control diet or HFD for 12 weeks and insulin sensitivity was measured using a hyperinsulinemic-euglycemic clamp. HFD consumption increased the body weight of both wild-type and MsrB3 KO mice, and no significant difference was observed between the genotypes. The HFD increased oxidative stress and induced insulin resistance in the skeletal muscle of wild-type mice, but did not affect either in MsrB3 KO mice. The unfolded protein response (UPR) was increased in MsrB3 KO mice upon consumption of HFD, but not in wild-type mice. Mitochondrial oxidative phosphorylation proteins and the levels of superoxide dismutase 2 and glutathione peroxidase 1 were increased in MsrB3 KO mice upon HFD consumption. The respiratory control ratio was reduced in wild-type mice consuming HFD but not in MsrB3 KO mice. The levels of calcium/calmodulin-dependent protein kinase kinase β, phosphorylated AMP-activated protein kinase, and peroxisome proliferator-activated receptor gamma coactivator 1α were increased in MsrB3 KO mice following HFD consumption. These results suggest that MsrB3 deficiency inhibits HFD-induced insulin resistance, and the increased mitochondrial biogenesis and antioxidant induction might be the mechanisms underlying this phenomenon.
Keywords: Methionine sulfoxide reductase B3, Insulin resistance, High-fat diet, Oxidative stress, Unfolded protein response, Mitochondrial oxidative phosphorylation
Abbreviations: AMPK, AMP-activated protein kinase; ER, Endoplasmic reticulum; ERAD, ER-associated protein degradation; GIR, Glucose infusion rate; HFD, High-fat diet; HGP, Hepatic glucose production; UPR, Unfolded protein response
Graphical abstract
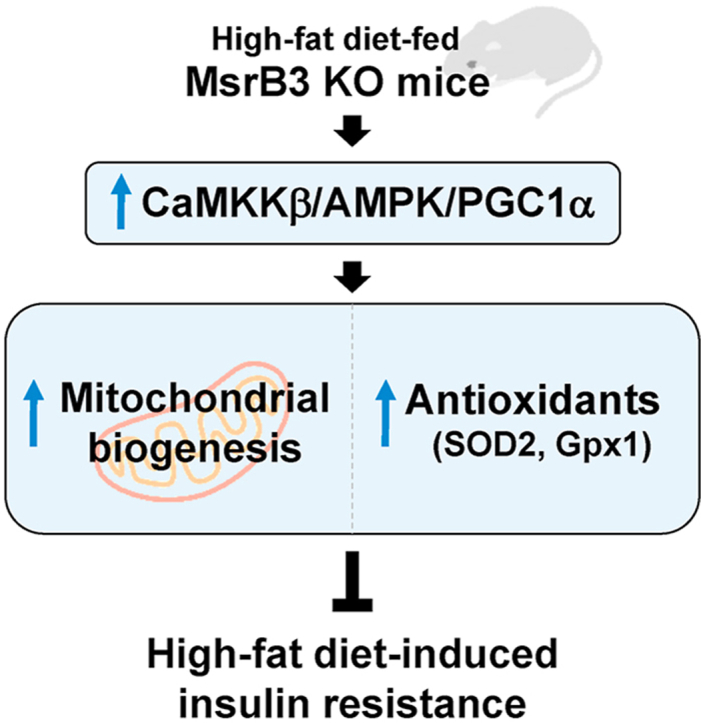
Highlights
-
•
High-fat diet increases CaMKKβ/AMPK/PGC1α in MsrB3-deficient mice.
-
•
MsrB3 deficiency increases mitochondrial oxidative phosphorylation proteins in response to a high-fat diet.
-
•
MsrB3 deficiency increases antioxidant expression in response to high-fat diet.
-
•
MsrB3 deficiency inhibits the development of high-fat diet-induced insulin resistance.
1. Introduction
Obesity is a pandemic due to an increasingly sedentary lifestyle and dietary changes. The increased prevalence of chronic conditions such as type 2 diabetes and cardiovascular diseases is closely associated with the obesity pandemic [1]. Increased fat mass in obese subjects induces insulin resistance, which is an independent risk factor for obesity-related chronic diseases [1]. The accumulation of fat mass increases reactive oxygen species (ROS) production, which is a key factor in inducing insulin resistance in obese subjects [2].
ROS are produced as a byproduct of normal cellular metabolism, and physiological ROS levels are maintained by a balance between ROS production and a detoxifying antioxidant system. Increased ROS production or reduced antioxidant capacity results in ROS accumulation, which causes tissue damage, called oxidative stress. A large body of evidence suggests that oxidative stress contributes to the development of insulin resistance by interfering with insulin signaling pathways [3]. In addition, oxidative stress induces endoplasmic reticulum (ER) stress, which is closely linked to insulin resistance [4].
The ER is a cellular organelle involved in post-translational protein modifications, such as folding, glycosylation, and oligomerization [5]. Misfolded proteins are retro-translocated to the cytoplasm for proteasomal degradation, i.e., ER-associated protein degradation (ERAD) [6]. The accumulation of unfolded or misfolded proteins in the ER induces the generation of ER stress, which activates signaling pathways to sustain ER homeostasis, called unfolded protein response (UPR) [7]. The UPR results in reduced protein synthesis but increases chaperone production and ERAD, leading to the re-establishment of ER function [7]. When the UPR is overwhelmed by persistent ER stress, cells undergo apoptosis [7]. Because redox status is a critical factor for proper protein folding, increased oxidative stress can induce ER stress [8]. Many previous studies have reported positive correlations between UPR and insulin resistance. Glucose-regulated protein 78 (GRP78) is an ER chaperone that is involved in protein folding and initiation of the UPR by binding to misfolded proteins. GRP78 expression is known to be increased in the skeletal muscle of glucose-intolerant patients with obesity and was positively correlated with fasting glucose levels [9]. The expression of ER stress markers in adipocytes is higher in obese human subjects than that in lean controls, and is correlated with body mass index and fat mass [10].
Methionine sulfoxide reductases, which catalyze the reduction of methionine sulfoxide to methionine, function as antioxidant enzymes; they consist of MsrA and MsrB in mammals [11]. While MsrA, specific for methionine-S-sulfoxide, is coded a single gene, MsrB, specific for methionine-R-sulfoxide, is coded by three genes whose products localize in different subcellular compartments (MsrB1 in the nucleus and cytoplasm, MsrB2 in the mitochondria, and MsrB3 in the mitochondria and ER) [11,12]. In rodents, MsrB3 might localize only to the ER [11,13]. MsrA-deficient mice show aggravated oxidative stress and insulin resistance following high-fat diet (HFD) consumption [14], whereas MsrB1 deficiency does not influence oxidative stress or insulin resistance in HFD-fed mice [15]. However, there are no reports regarding the roles of MsrB2 or MsrB3 in HFD-induced insulin resistance.
We had previously reported that MsrB3 protects the cells from ER stress as well as oxidative stress [[16], [17], [18]]. Because the dysregulation of ER is closely associated with insulin resistance, it is possible that the modulation of MsrB3 would affect insulin sensitivity. We hypothesized that MsrB3 deficiency might accelerate insulin resistance by increasing oxidative stress and ER stress. To examine the role of MsrB3 in HFD-induced insulin resistance, we fed MsrB3 knockout (KO) and wild-type mice with HFD for 12 weeks and measured the insulin sensitivity using the hyperinsulinemic-euglycemic clamp technique. In contrast to our hypothesis, MsrB3 deficiency inhibited the development of HFD-induced insulin resistance.
2. Materials and methods
2.1. Animals
The generation of MsrB3 KO mice has been described previously [19]. Eight-week-old male C57BL/6 mice were housed in a controlled environment with a 12:12-h light:dark cycle. The mice were fed a control diet (16% of kcal from fat; AIN-93G, Research Diets, New Brunswick, NJ, USA) or HFD (60% of kcal from fat, Research Diets) for 12 weeks. After the completion of all experiments, the mice were euthanized by intraperitoneal injection of 2,2,2,-tribromoethanol and tert-amyl alcohol (375 mg/kg) and cervical dislocation. Blood was collected from the retro-orbital plexus using heparin-coated capillary tubes, and the plasma was stored at −80 °C until analysis. Skeletal muscles and adipose tissue were excised, weighed, and stored at −80 °C. For ER stress induction, mice were injected intraperitoneally with tunicamycin (1 mg/kg), fasted for 24 h, and then anesthetized. This study was conducted in strict accordance with guidelines and protocols approved by the Institutional Animal Care and Use Committee of Yeungnam University College of Medicine (YUMC-AEC2016-015).
2.2. Hyperinsulinemic-euglycemic clamp
The hyperinsulinemic-euglycemic clamp technique was performed as described previously [20]. During the clamp test, human insulin (Lilly, USA) was infused at a dose of 24 pmol kg−1·min−1 and plasma glucose levels were maintained at approximately 6 mM by infusion with 20% glucose. Radiolabeled [3-3H] glucose (0.1 μCi/min; PerkinElmer, Waltham, MA, USA) was infused to assess whole-body glucose turnover, and 2-deoxy-D-[1–14C] glucose (10 μCi, PerkinElmer) was injected as a bolus to measure the tissue glucose uptake. Hepatic glucose production (HGP) following insulin stimulation was estimated by subtracting the glucose infusion rate (GIR) from the whole-body glucose uptake rate. Plasma glucose levels were measured using a GM9 glucose analyzer (Analox, Stourbridge, UK), and plasma insulin concentrations were measured using an enzyme-linked immunosorbent assay (Merck, Kenilworth, NJ, USA).
2.3. Homeostatic model assessment-insulin resistance (HOMA-IR)
HOMA-IR was calculated based on the values of fasting glucose and insulin as follows: [fasting glucose (mg/dL) × fasting insulin (μU/mL)]/405.
2.4. H2O2 levels
H2O2 levels in the gastrocnemius muscle were determined using the ferric-sensitive dye xylenol orange (Sigma-Aldrich, St. Louis, MO, USA) as previously described [21].
2.5. Western blotting
The antibody against MsrB3 has been described previously [19]. The antibodies against oxidative phosphorylation (OXPHOS) proteins, peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α), and 4-hydroxynonenal (4HNE) were obtained from Abcam (Cambridge, UK). The antibody against Gpx1 was obtained from AbFrontier (Seoul, Korea). The antibodies against activating transcription factor 4 (ATF4), superoxide dismutase 2 (SOD2), ubiquitin (Ub), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The antibody against Grp78 was obtained from Enzo (Farmingdale, NY, USA). The antibodies against phosphorylated Akt (pAkt), Akt, phosphorylated AS160 (pAS160), phosphorylated GSK3β (pGSK3β), GSK3β, phosphorylated eIF2α (p-eIF2α), eIF2α, calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ), phosphorylated liver kinase B1 (pLKB1), LKB1, phosphorylated AMP-activated protein kinase (pAMPK), and AMPK were obtained from Cell Signaling (Danvers, MA, USA). Western blotting was performed as described previously [21]. Skeletal muscle samples were homogenized in lysis buffer and then 30 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The resolved proteins were transferred onto PVDF membranes (Merck), and then the membranes were blocked and probed with primary antibodies. After probing with the appropriate secondary antibodies, protein presence was detected with chemiluminescence detection reagent (Merck) and then quantified using a LAS-4000 image analyzer and Multi Gauge 3.0 software (Fujifilm, Japan).
2.6. Quantitative real time polymerase chain reaction (qRT-PCR)
The gene expression of MsrB3 was measured by qRT-PCR using a Real-Time PCR 7500 System and Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA), as previously described [20]. The gene expression was normalized by ribosomal protein lateral stalk subunit P0 (36B4). The following primer sequences were used: 36B4 (forward, 5′-CACTGGTCTAGGACCCGAGAA-3′; reverse, 5′-GGTGCCTCTGGAGATTTTCG-3′) and MsrB3 (forward, 5′-GGTGGAAACCAGCTGTTCTC-3′; reverse, 5′-GGATGCTGAGTTGATGCAGTA -3′).
2.7. Mitochondrial respiration
Mitochondrial function was measured using an Oxytherm oxygen electrode (Hansatech Instruments, Norfolk, UK), as previously described [22]. Briefly, skeletal muscle samples were minced with scissors and homogenized using a motorized Glass/Teflon Potter Elvehjem homogenizer. The homogenates were centrifuged at 700×g for 10 min at 4 °C, and the supernatant was removed and centrifuged at 10 500×g for 10 min at 4 °C. The pellets were suspended and used to measure mitochondrial oxygen consumption and protein concentration. The mitochondrial suspension (50 μL) was added to the Oxytherm chamber containing 500 μL of respiration buffer at 37 °C under conditions of constant stirring. Pyruvate and malate (5 mM and 2.5 mM, respectively) were added to aid complex I-mediated oxygen consumption, which was followed by the addition of ADP (250 μM) for state 3 respiration. Then, 5 μM oligomycin was added to inhibit ATP synthase activity (state 4 respiration). The oxygen consumption rate was standardized based on protein levels. The respiratory control ratio was determined by dividing state 3 consumption by state 4 consumption.
2.8. Statistical analysis
The values are presented as the mean ± SE. Statistical analyses were performed using Prism version 7 (Graph Pad Software, Inc., La Jolla, CA, USA). Differences among the four groups were analyzed via two-way analysis of variance (ANOVA) followed by Tukey's HSD post hoc test unless otherwise specified. Differences between the two groups were assessed using an unpaired Student's t-test. A p-value less than 0.05 (p < 0.05) was considered significant.
3. Results
3.1. MsrB3 deficiency decreases susceptibility to diet-induced insulin resistance
Feeding HFD for 12 weeks resulted in increased body weight and reduced skeletal muscle mass in both wild-type and MsrB3 KO mice. However, no significant differences were observed between the two genotypes in either the control or HFD groups (Fig. 1A and B). HFD tended to increase retroperitoneal fat mass in both genotypes (Fig. 1C). The levels of plasma glucose were not significantly altered in response to HFD or MsrB3 deficiency (Fig. 1D). The HFD significantly increased plasma insulin levels in wild-type mice but not in MsrB3 KO mice. Accordingly, HOMA-IR was increased by HFD in wild-type mice, showed no significant difference with respect to that in MsrB3 KO mice. Plasma insulin levels and HOMA-IR were not significantly different between the two genotypes in either the control or HFD groups (Fig. 1E–F).
Fig. 1.

Body weight, skeletal muscle weight, adipose tissue mass, and plasma levels of glucose and insulin in wild-type and MsrB3 knockout (KO) mice following consumption of a control diet or high-fat diet for 12 weeks. (A) Body weight. (B) The weights of gastrocnemius and soleus muscles. (C) Retroperitoneal fat mass. (D) Plasma glucose levels. (E) Plasma insulin levels. (F) Homeostatic model assessment-insulin resistance (HOMA-IR). Skeletal muscle and fat masses are standardized based on the body weight. Results are presented as means ± SE (n = 7–9 in each group). *p < 0.05 and **p < 0.01.
Fig. 3.

The levels of oxidative stress markers and antioxidant enzyme in the gastrocnemius muscle of wild-type (WT) and MsrB3 knockout (KO) mice following the consumption of a control diet or high-fat diet for 12 weeks. (A) MsrB3 protein levels. (B) Hydrogen peroxide (H2O2) levels. (C) 4-hydroxy-2-nonenal (4HNE). (D) Superoxide dismutase 2 (SOD2) protein levels. (E) Glutathione peroxidase 1 (Gpx1) protein levels. Protein levels were normalized with those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results are presented as means ± SE (n = 6–11 in each group). *p < 0.05, **p < 0.01, and ***p < 0.001.
Next, a hyperinsulinemic-euglycemic clamp was used to identify the tissue responsible for the development of insulin resistance. During the clamp test, plasma glucose levels were maintained at ~6.1 mM in the control diet and HFD-fed mice. Plasma insulin levels were 1105 ± 43.84 and 1088 ± 99.20 pM in wild-type and Msr3 KO mice, respectively, in the control diet group, and 1351 ± 122.89 and 1399 ± 109.59 pM, respectively, in wild-type and MsrB3 KO mice in the HFD group. GIR was significantly reduced in wild-type mice following HFD consumption, whereas it was not significantly decreased by HFD in MsrB3 KO mice. No significant difference was observed between wild-type and MsrB3 KO mice in either the control or HFD groups (Fig. 2A). The HFD also significantly reduced whole-body glucose turnover in wild-type mice but not in MsrB3 KO mice. Whole-body glucose turnover was not significantly different between the two genotypes in either the control or HFD groups (Fig. 2B). HFD significantly reduced skeletal muscle glucose uptake in wild-type mice but not in MsrB3 KO mice. No significant difference was observed between the two genotypes in either the control or HFD groups (Fig. 2C). Hepatic glucose production was not significantly different between the groups (Fig. 2D). These data suggest that HFD consumption for 12 weeks induces insulin resistance in the skeletal muscle of wild-type mice but not in that of MsrB3 KO mice.
Fig. 2.

Glucose metabolism during the hyperinsulinemic-euglycemic clamp test in wild-type and MsrB3 knockout (KO) mice following the consumption of a control diet or high-fat diet for 12 weeks. (A) Glucose infusion rate (GIR). (B) Whole-body glucose turnover. (C) Soleus muscle glucose uptake. (D) Insulin-stimulated hepatic glucose production (HGP). Results are presented as means ± SE (n = 7–9 in each group). *p < 0.05 and **p < 0.01.
3.2. MsrB3 deficiency increases resistance against oxidative stress due to HFD
HFD consumption for 12 weeks did not alter the protein levels of MsrB3 in the skeletal muscle of wild-type mice (Fig. 3A). Further, HFD increased hydrogen peroxide (H2O2) levels in the skeletal muscle of wild-type mice (p = 0.0545), but did not affect hydrogen peroxide levels in the skeletal muscle of MsrB3 KO mice. Hydrogen peroxide levels were not significantly different between wild-type and MsrB3 KO mice in either the control diet or HFD group (Fig. 3B).
The levels of 4HNE, an oxidative stress marker, were also increased by HFD in wild-type mice, whereas they were not significantly altered in MsrB3 KO mice. The 4HNE levels did not significantly differ between the two genotypes in either the control or HFD groups (Fig. 3C). The protein levels of SOD2 and Gpx1 were significantly increased by HFD in MsrB3 KO mice but not in wild-type mice. The Gpx1 protein levels were significantly higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice. There was no difference in SOD2 protein levels between the two genotypes (Fig. 3D and E). These results suggest that HFD consumption for 12 weeks increases oxidative stress in wild-type mice but not in MsrB3 KO mice.
3.3. MsrB3 deficiency enhances UPR following HFD consumption
Next, we assessed UPR by measuring the expression of Grp78, p-eIF2α, and ATF4, and levels of ubiquitination. HFD did not significantly alter Grp78 protein levels in wild-type mice, but it significantly increased Grp78 protein levels in MsrB3 KO mice. Grp78 protein levels were significantly higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice (Fig. 4A). HFD seems to increase the levels of p-eIF2α in MsrB3 KO mice (Fig. 4B). HFD also increased the ATF4 protein levels in MsrB3 KO mice but not in wild-type mice (Fig. 4C). The levels of ubiquitinated protein were increased by HFD in MsrB3 KO mice but not in wild-type mice. The ubiquitinated protein levels were significantly higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice (Fig. 4D). These results suggest that MsrB3 deficiency enhances UPR following HFD consumption.
Fig. 4.

The expression of proteins involved in unfolded protein response in the gastrocnemius muscle of wild-type (WT) and MsrB3 knockout (KO) mice following consumption of a control diet or high-fat diet for 12 weeks. (A) Glucose-regulated protein 78 (GRP78). (B) Phosphorylated eukaryotic translation initiation factor 2α (p-eIF2α). (C) Activating transcription factor 4 (ATF4). (D) Ubiquitinated (Ub) protein levels. Protein levels were normalized with those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results are presented as means ± SE (n = 9–11 in each group). *p < 0.05 and **p < 0.01.
3.4. MsrB3 deficiency increases mitochondrial OXPHOS proteins following HFD consumption
ER stress is closely linked to changes in mitochondrial functions [23]. We measured the levels of proteins involved in mitochondrial OXPHOS. The levels of complex I and II proteins were similar in wild-type mice following HFD consumption, while they were increased upon HFD consumption in MsrB3 KO mice. The levels of complex I and II proteins were significantly higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice (Fig. 5A and B). The levels of complex III, IV, and V proteins tended to increase in response to HFD consumption in MsrB3 KO mice but not in wild-type mice (Fig. 5C–E). The mitochondrial respiratory control ratio was reduced upon HFD consumption in wild-type mice, but was not affected in MsrB3 KO mice (Fig. 5F). These results suggest that MsrB3 deficiency increases the expression of mitochondrial OXPHOS proteins and sustains mitochondrial function following HFD consumption.
Fig. 5.

The levels of proteins involved in mitochondrial oxidative phosphorylation and respiratory control ratio in the gastrocnemius muscle of wild-type and MsrB3 knockout (KO) mice following consumption of a control diet or high-fat diet for 12 weeks. (A) Complex I. (B) Complex II. (C) Complex III. (D) Complex IV. (E) Complex V. (F) Respiratory control ratio. Protein levels were normalized with those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results are presented as means ± SE (n = 6–12). *p < 0.05 and **p < 0.01.
To examine the mechanism by which HFD increased mitochondrial OXPHOS proteins in MsrB3 KO mice, we measured the protein levels of CaMKKβ, pAMPK and PGC1α as AMPK/PGC1α is known to regulate mitochondrial biogenesis. While the levels of CaMKKβ were not significantly altered upon HFD consumption in wild-type mice, they were significantly increased upon HFD consumption in MsrB3 KO mice. The protein levels of CaMKKβ were significantly higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice (Fig. 6A). The levels of pAMPK protein were increased in MsrB3 KO mice but not in wild-type mice upon consumption of HFD (Fig. 6B). The protein levels of PGC1α were higher in HFD-fed MsrB3 KO mice than those in HFD-fed wild-type mice (Fig. 6C). These results suggest that MsrB3 deficiency enhances CaMKKβ/AMPK/PGC1α signaling in response to the HFD.
Fig. 6.

The protein levels of CaMKKβ, pAMPK, and PGC1α in the gastrocnemius muscle of wild-type (WT) and MsrB3 knockout (KO) mice following consumption of a control diet or high-fat diet for 12 weeks. (A) Calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ). (B) Phosphorylated AMP-activated protein kinase (pAMPK). (C) Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α). Protein levels of CaMKK and PGC1α were normalized with those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Results are presented as means ± SE (n = 9–11 in each group). *p < 0.05, ***p < 0.001, and ****p < 0.0001.
3.5. MsrB3 deficiency has no effect on tunicamycin-induced insulin resistance
Tunicamycin is known to induce UPR by inhibiting glycosylation, leading to the development of insulin resistance in cell culture and ex vivo [24]. In contrast, administration of tunicamycin to mice results in reduced blood glucose levels in vivo [25]. Therefore, to confirm the development of insulin resistance in response to tunicamycin, we injected tunicamycin intraperitoneally in wild-type mice and measured the fasting plasma glucose and insulin levels to calculate HOMA-IR. Although tunicamycin did not affect plasma glucose levels, it significantly increased plasma insulin levels. Accordingly, HOMA-IR was significantly increased in response to tunicamycin administration. Tunicamycin reduced the levels of phosphorylated AS160 in the skeletal muscle and those of phosphorylated GSK3β in the liver (Supplementary Fig. S1). These results suggest that tunicamycin induces insulin resistance in the liver and skeletal muscle in mice.
Tunicamycin administration in wild-type mice significantly reduced both mRNA and protein levels of MsrB3 in the skeletal muscle (Fig. 7A and B), suggesting that tunicamycin inhibits MsrB3 expression at the transcriptional level. Tunicamycin was administered to wild-type and MsrB3 KO mice after they were fed a control diet or HFD for 12 weeks. The HFD increased fasting plasma glucose and insulin levels in both wild-type and MsrB3 KO mice, but no difference was observed between the two genotypes in the control and HFD groups (Fig. 7C and D). Accordingly, HOMA-IR was increased in both groups upon HFD consumption, but it was not affected by MsrB3 deficiency (Fig. 7E). Tunicamycin did not affect the levels of antioxidant enzymes or mitochondrial OXPHOS proteins in either genotype following HFD consumption (Fig. 8). These results suggest that MsrB3 deficiency does not affect tunicamycin-induced insulin resistance.
Fig. 7.

Effect of tunicamycin on the plasma levels of glucose and insulin in wild-type (WT) and MsrB3 knockout (KO) mice following consumption of a control diet or high-fat diet for 12 weeks. (A) MsrB3 mRNA and (B) protein levels in the gastrocnemius muscle. (C) Plasma glucose levels. (D) Plasma insulin levels. (E) Homeostatic model assessment-insulin resistance (HOMA-IR). Results are presented as means ± SE (n = 3–5 in each group). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Fig. 8.

Effect of tunicamycin on the levels of antioxidant enzymes and proteins involved in mitochondrial oxidative phosphorylation in wild-type and MsrB3 knockout (KO) mice following a high-fat diet for 12 weeks. (A) Superoxide dismutase 2 (SOD2). (B) Glutathione peroxidase 1 (Gpx1). (C) Complex I. (D) Complex II. (E) Complex III. (F) Complex V. Results are presented as means ± SE (n = 4–5 in each group).
4. Discussion
In previous studies, MsrA deficiency aggravated HFD-induced insulin resistance, whereas MsrB1 deficiency had no effect on diet-induced insulin resistance. In the present study, the effect of MsrB3 deficiency on HFD-induced insulin resistance was examined. HFD consumption for 12 weeks in wild-type mice resulted in the development of insulin resistance and increased oxidative stress, but no difference in UPR was observed in the skeletal muscle. In contrast, MsrB3 deficiency resulted in inhibition of HFD-induced insulin resistance development and ROS accumulation in the skeletal muscle. Furthermore, MsrB3 deficiency increased UPR and the protein levels of mitochondrial OXPHOS and antioxidant enzymes. These results suggest that MsrB3 deficiency reduces susceptibility to diet-induced insulin resistance, which may be mediated by increased mitochondrial biogenesis and antioxidant induction.
High levels of oxidative stress are observed in the plasma and insulin-sensitive tissues of patients with obesity and type 2 diabetes. In experimental animals, catalase, glutaredoxin, peroxiredoxins [[2], [3], [6]], and MsrA knockout increases oxidative stress and insulin resistance [26], whereas overexpression of SOD2 and peroxiredoxin 4 reduces oxidative stress and prevents the development of HFD-induced insulin resistance [27,28]. Treatment with antioxidants, such as tempol, hemin, and vitamin C, improves insulin resistance [26]. However, the modulation of antioxidant enzymes is not always followed by altered levels of oxidative stress. Loss of MsrB1 and selenoprotein W does not affect oxidative stress or insulin sensitivity in either control diet- or HFD-fed mice [15,21]. In the present study, MsrB3 deficiency did not significantly alter oxidative stress compared with that in wild-type mice following consumption of a control diet. However, interestingly, while the HFD increased oxidative stress in the wild-type mice, it failed to increase oxidative stress in the MsrB3-deficient mice compared with that in the respective control diet-fed mice. Accordingly, the HFD did not significantly alter insulin sensitivity in MsrB3-deficient mice. Increased expression of antioxidant enzymes, Gpx1 and SOD2, may counteract oxidative stress in MsrB3 knockout mice. Compensatory overexpression of other antioxidant enzymes has been observed in MsrB1 knockout and Gpx1 knockout mice [29].
AMPK is involved in a variety of physiological functions, including metabolism, inflammation, redox regulation, and mitochondrial biogenesis [30]. We observed increased levels of pAMPK in HFD-fed MsrB3 KO mice. Thus, activation of AMPK in HFD-fed MsrB3 KO mice increased the expression of the antioxidant enzymes Gpx1 and SOD2, which in turn may inhibit the accumulation of hydrogen peroxide. Accordingly, AMPK activation induces the expression of antioxidant enzymes, including Gpx1 and SOD2, in human mesenchymal stem cells [31], in human umbilical vein endothelial cells [32], and in the lungs and kidneys of mice [33,34]. Nrf2 plays an important role in the induction of antioxidant enzymes by AMPK [34,35]. We previously observed that deficiency of MsrB3 induces Nrf2 activation in human dermal fibroblasts [36]. AMPK is activated in response to increased AMP levels and increased expression of upstream kinases such as CaMKKβ and LKB1 [37]. MsrB3-deficient mice exhibited increased CaMKKβ protein levels following HFD consumption in this study. Previously, we had also observed increased intracellular calcium levels in MsrB3-deficient cancer cells [38]. Therefore, we assumed that HFD consumption would increase intracellular calcium levels, leading to the activation of CaMKKβ and AMPK in MsrB3-deficient mice.
We observed increased levels of PGC1α and mitochondrial OXPHOS proteins in the HFD-fed MsrB3 KO mice. AMPK has been shown to enhance mitochondrial biogenesis by regulating PGC1α expression in a variety of cells and tissues, including the skeletal muscle [33,39,40]. Numerous studies suggest that mitochondrial abnormalities are closely associated with insulin resistance [41]. In patients with obesity and type 2 diabetes, reductions in mitochondrial number and size are accompanied by reduced electron transport chain activity in the skeletal muscle [42]. Lipid infusion or HFD reduces the expression of genes required for mitochondrial oxidative phosphorylation and oxygen consumption in the skeletal muscle of humans [43,44]. Functional and morphological abnormalities of the mitochondria result in increased ROS generation and reduced beta-oxidation, leading to the development of insulin resistance [45]. Enhanced mitochondrial OXPHOS protein levels or activities are associated with improved insulin resistance [46,47]. Therefore, it is possible that enhanced mitochondrial biogenesis mediated by AMPK/PGC1α involves in the inhibition of HFD-induced insulin resistance in MsrB3 KO mice.
ER-stress has been proposed as a mechanism for inducing insulin resistance, which has mostly been demonstrated in cells in vitro. Many previous studies using experimental animals have shown a positive relationship between insulin resistance and UPR. Insulin resistance in obese rodents is accompanied by increased UPR, whereas ameliorated insulin resistance is followed by reduced UPR. In the present study, we showed that a chemical ER stress inducer, tunicamycin, induces insulin resistance in the skeletal muscle and liver of mice. These data indicate a causal relationship between ER stress and insulin resistance in vivo.
Previously, both HFD and MsrB3 deficiency have been reported to induce ER stress [38]. However, we did not observe an alteration in UPR upon HFD consumption or in response to MsrB3 deficiency; however, HFD consumption in the background of MsrB3 deficiency increased the UPR. Interestingly, UPR and insulin resistance were not positively correlated in wild-type and MsrB3-deficient mice in this study. In addition, MsrB3 deficiency resulted in inhibition of diet-induced insulin resistance development, whereas it increased UPR following HFD consumption. ER stress is closely linked to mitochondrial alterations, including increases in biogenesis and oxidative phosphorylation, and these mitochondrial changes lead to adaptive survival of cells [23]. This interaction between the ER and mitochondria is mediated by calcium [48]. Consistent with these previous results, increased ER stress in HFD-fed MsrB3-deficient mice was accompanied by increases in CaMKKβ/AMPK/PGC1α and mitochondrial biogenesis. Therefore, although severe ER stress induced by tunicamycin results in the development of insulin resistance, mild ER stress due to the HFD in MsrB3 deficiency seems to reduce susceptibility to insulin resistance. Consistent with our results, some previous studies have reported a beneficial effect of UPR on insulin sensitivity. Lipid infusion induces insulin resistance and decreases insulin-induced UPR in the adipose tissue of healthy human subjects [49]. In patients with type 2 diabetes, insulin-mediated UPR is absent in the adipose tissue [49]. HFD-induced steatosis and insulin resistance are improved by flaxseed, and are accompanied by increased UPR [50]. However, the underlying mechanisms of these inconsistent results regarding the relationship between UPR and insulin resistance remain to be explored.
Although MsrB3 deficiency reduced the susceptibility to HFD-induced insulin resistance, MsrB3 deficiency had no effect on tunicamycin-induced insulin resistance, suggesting that MsrB3 deficiency does not have a beneficial effect on insulin resistance caused by severe ER stress. Furthermore, tunicamycin abolished the increase in antioxidant expression and mitochondrial OXPHOS proteins following HFD consumption in MsrB3 KO mice. These data indirectly suggest that the positive effect of MsrB3 deficiency on diet-induced insulin resistance is mediated by mild ER stress, and that severe tunicamycin-induced ER stress overwhelms these adaptive changes in MsrB3 KO mice.
In summary, MsrB3 deficiency inhibits the development of HFD-induced insulin resistance in the skeletal muscle, which might be mediated by increased mitochondrial biogenesis and antioxidant induction via the CaMKKβ/AMPK/PGC1α pathway. Although the activation of CaMKKβ/AMPK/PGC1α is possibly linked to ER stress, further studies are needed to clarify these mechanisms.
Funding
This research was supported by grants from the Medical Research Center Program (2015R1A5A2009124) and the Basic Science Research Program (2019R1A2C1088730) through the National Research Foundation of Korea (NRF), funded by the Korean government. The funding source played no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or the decision to submit the article for publication.
Declaration of competing interest
There is no conflict of interest.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101823.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Multimedia component 1
References
- 1.Saltiel A.R., Olefsky J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 2017;127(1):1–4. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Di Meo S., Iossa S., Venditti P. Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J. Endocrinol. 2017;233(1):R15–R42. doi: 10.1530/JOE-16-0598. [DOI] [PubMed] [Google Scholar]
- 3.Barazzoni R., Zanetti M., Gortan Cappellari G., Semolic A., Boschelle M., Codarin E. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-kappaB inhibitor (IkappaB)-nuclear factor-kappaB (NFkappaB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia. 2012;55(3):773–782. doi: 10.1007/s00125-011-2396-x. [DOI] [PubMed] [Google Scholar]
- 4.Khan S., Wang C.H. ER stress in adipocytes and insulin resistance: mechanisms and significance (Review) Mol. Med. Rep. 2014;10(5):2234–2240. doi: 10.3892/mmr.2014.2532. [DOI] [PubMed] [Google Scholar]
- 5.Braakman I., Hebert D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5(5):a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samant R.S., Livingston C.M., Sontag E.M., Frydman J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature. 2018;563(7731):407–411. doi: 10.1038/s41586-018-0678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012;13(2):89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 8.Cao S.S., Kaufman R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants Redox Signal. 2014;21(3):396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marciniak C., Duhem C., Boulinguiez A., Raverdy V., Baud G., Verkindt H. Differential unfolded protein response in skeletal muscle from non-diabetic glucose tolerant or intolerant patients with obesity before and after bariatric surgery. Acta Diabetol. 2020;57(7):819–826. doi: 10.1007/s00592-020-01490-z. [DOI] [PubMed] [Google Scholar]
- 10.Sharma N.K., Das S.K., Mondal A.K., Hackney O.G., Chu W.S., Kern P.A. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J. Clin. Endocrinol. Metab. 2008;93(11):4532–4541. doi: 10.1210/jc.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H.Y. The methionine sulfoxide reduction system: selenium utilization and methionine sulfoxide reductase enzymes and their functions. Antioxidants Redox Signal. 2013;19(9):958–969. doi: 10.1089/ars.2012.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H.Y., Gladyshev V.N. Methionine sulfoxide reduction in mammals: characterization of methionine-R-sulfoxide reductases. Mol. Biol. Cell. 2004;15(3):1055–1064. doi: 10.1091/mbc.E03-08-0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H.Y., Gladyshev V.N. Characterization of mouse endoplasmic reticulum methionine-R-sulfoxide reductase. Biochem. Biophys. Res. Commun. 2004;320(4):1277–1283. doi: 10.1016/j.bbrc.2004.06.078. [DOI] [PubMed] [Google Scholar]
- 14.Styskal J., Nwagwu F.A., Watkins Y.N., Liang H., Richardson A., Musi N. Methionine sulfoxide reductase A affects insulin resistance by protecting insulin receptor function. Free Radic. Biol. Med. 2013;56:123–132. doi: 10.1016/j.freeradbiomed.2012.10.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heo J.Y., Cha H.N., Kim K.Y., Lee E., Kim S.J., Kim Y.W. Methionine sulfoxide reductase B1 deficiency does not increase high-fat diet-induced insulin resistance in mice. Free Radic. Res. 2017;51(1):24–37. doi: 10.1080/10715762.2016.1261133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwak G.H., Lim D.H., Han J.Y., Lee Y.S., Kim H.Y. Methionine sulfoxide reductase B3 protects from endoplasmic reticulum stress in Drosophila and in mammalian cells. Biochem. Biophys. Res. Commun. 2012;420(1):130–135. doi: 10.1016/j.bbrc.2012.02.128. [DOI] [PubMed] [Google Scholar]
- 17.Lee E., Kwak G.H., Kamble K., Kim H.Y. Methionine sulfoxide reductase B3 deficiency inhibits cell growth through the activation of p53-p21 and p27 pathways. Arch. Biochem. Biophys. 2014;547:1–5. doi: 10.1016/j.abb.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 18.Kwak G.H., Kim T.H., Kim H.Y. Down-regulation of MsrB3 induces cancer cell apoptosis through reactive oxygen species production and intrinsic mitochondrial pathway activation. Biochem. Biophys. Res. Commun. 2017;483(1):468–474. doi: 10.1016/j.bbrc.2016.12.120. [DOI] [PubMed] [Google Scholar]
- 19.Kwon T.J., Cho H.J., Kim U.K., Lee E., Oh S.K., Bok J. Methionine sulfoxide reductase B3 deficiency causes hearing loss due to stereocilia degeneration and apoptotic cell death in cochlear hair cells. Hum. Mol. Genet. 2014;23(6):1591–1601. doi: 10.1093/hmg/ddt549. [DOI] [PubMed] [Google Scholar]
- 20.Cha H.N., Park S., Dan Y., Kim J.R., Park S.Y. Peroxiredoxin2 deficiency aggravates aging-induced insulin resistance and declines muscle strength. J Gerontol A Biol Sci Med Sci. 2019;74(2):147–154. doi: 10.1093/gerona/gly113. [DOI] [PubMed] [Google Scholar]
- 21.Shin M.G., Cha H.N., Park S., Kim Y.W., Kim J.Y., Park S.Y. Selenoprotein W deficiency does not affect oxidative stress and insulin sensitivity in the skeletal muscle of high-fat diet-fed obese mice. Am. J. Physiol. Cell Physiol. 2019;317(6):C1172–C1182. doi: 10.1152/ajpcell.00064.2019. [DOI] [PubMed] [Google Scholar]
- 22.Finlin B.S., Memetimin H., Confides A.L., Kasza I., Zhu B., Vekaria H.J. Human adipose beiging in response to cold and mirabegron. JCI Insight. 2018;3(15) doi: 10.1172/jci.insight.121510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knupp J., Arvan P., Chang A. Increased mitochondrial respiration promotes survival from endoplasmic reticulum stress. Cell Death Differ. 2019;26(3):487–501. doi: 10.1038/s41418-018-0133-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh H.J., Toyoda T., Didesch M.M., Lee M.Y., Sleeman M.W., Kulkarni R.N. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat. Commun. 2013;4:1871. doi: 10.1038/ncomms2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng B., Huang X., Jiang D., Hua L., Zhuo Y., Wu Endoplasmic reticulum stress inducer tunicamycin alters hepatic energy homeostasis in mice. Int. J. Mol. Sci. 2017;18(8) doi: 10.3390/ijms18081710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park S., Park S.Y. Can antioxidants be effective therapeutics for type 2 diabetes? Yeungnam Univ J Med. 2020;38(2) doi: 10.12701/yujm.2020.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boden M.J., Brandon A.E., Tid-Ang J.D., Preston E., Wilks D., Stuart E. Overexpression of manganese superoxide dismutase ameliorates high-fat diet-induced insulin resistance in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2012;303(6):E798–E805. doi: 10.1152/ajpendo.00577.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nabeshima A., Yamada S., Guo X., Tanimoto A., Wang K.Y., Shimajiri S. Peroxiredoxin 4 protects against nonalcoholic steatohepatitis and type 2 diabetes in a nongenetic mouse model. Antioxidants Redox Signal. 2013;19(17):1983–1998. doi: 10.1089/ars.2012.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis P., Stefanovic N., Pete J., Calkin A.C., Giunti S., Thallas-Bonke V. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation. 2007;115(16):2178–2187. doi: 10.1161/CIRCULATIONAHA.106.664250. [DOI] [PubMed] [Google Scholar]
- 30.Jeon S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016;48(7):e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X., Yan J., He F., Zhong D., Yang H., Pei M. Mechanical stretch induces antioxidant responses and osteogenic differentiation in human mesenchymal stem cells through activation of the AMPK-SIRT1 signaling pathway. Free Radic. Biol. Med. 2018;126:187–201. doi: 10.1016/j.freeradbiomed.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kukidome D., Nishikawa T., Sonoda K., Imoto K., Fujisawa K., Yano M. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55(1):120–127. [PubMed] [Google Scholar]
- 33.Wang G., Song Y., Feng W., Liu L., Zhu Y., Xie X. Activation of AMPK attenuates LPS-induced acute lung injury by upregulation of PGC1alpha and SOD1. Exp Ther Med. 2016;12(3):1551–1555. doi: 10.3892/etm.2016.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong Y.A., Lim J.H., Kim M.Y., Kim Y., Park H.S., Kim H.W. Extracellular superoxide dismutase attenuates renal oxidative stress through the activation of adenosine monophosphate-activated protein kinase in diabetic nephropathy. Antioxidants Redox Signal. 2018;28(17):1543–1561. doi: 10.1089/ars.2017.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joo M.S., Kim W.D., Lee K.Y., Kim J.H., Koo J.H., Kim S.G. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol. Cell Biol. 2016;36(14):1931–1942. doi: 10.1128/MCB.00118-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak G.H., Kim K.Y., Kim H.Y. Methionine sulfoxide reductase B3 deficiency stimulates heme oxygenase-1 expression via ROS-dependent and Nrf2 activation pathways. Biochem. Biophys. Res. Commun. 2016;473(4):1033–1038. doi: 10.1016/j.bbrc.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Auciello F.R., Ross F.A., Ikematsu N., Hardie D.G. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014;588(18):3361–3366. doi: 10.1016/j.febslet.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwak G.H., Kim H.Y. MsrB3 deficiency induces cancer cell apoptosis through p53-independent and ER stress-dependent pathways. Arch. Biochem. Biophys. 2017;621:1–5. doi: 10.1016/j.abb.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Wan Z., Root-McCaig J., Castellani L., Kemp B.E., Steinberg G.R., Wright D.C. Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity. 2014;22(3):730–738. doi: 10.1002/oby.20605. [DOI] [PubMed] [Google Scholar]
- 40.Jager S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. U. S. A. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bournat J.C., Brown C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010;17(5):446–452. doi: 10.1097/MED.0b013e32833c3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ritov V.B., Menshikova E.V., He J., Ferrell R.E., Goodpaster B.H., Kelley D.E. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54(1):8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 43.Richardson D.K., Kashyap S., Bajaj M., Cusi K., Mandarino S.J., Finlayson J. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem. 2005;280(11):10290–10297. doi: 10.1074/jbc.M408985200. [DOI] [PubMed] [Google Scholar]
- 44.Sparks L.M., Xie H., Koza R.A., Mynatt R., Hulver M.W., Bray G.A. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54(7):1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 45.Kim J.A., Wei Y., Sowers J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008;102(4):401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ngo D.T.M., Sverdlov A.L., Karki S., Macartney-Coxson D., Stubbs R.S., Farb M.G. Oxidative modifications of mitochondrial complex II are associated with insulin resistance of visceral fat in obesity. Am. J. Physiol. Endocrinol. Metab. 2019;316(2):E168–E177. doi: 10.1152/ajpendo.00227.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H.H., Qin G.J., Li X.L., Zhang Y.H., Du P.J., Zhang P.Y. SIRT1 overexpression in skeletal muscle in vivo induces increased insulin sensitivity and enhanced complex I but not complex II-V functions in individual subsarcolemmal and intermyofibrillar mitochondria. J. Physiol. Biochem. 2015;71(2):177–190. doi: 10.1007/s13105-015-0396-x. [DOI] [PubMed] [Google Scholar]
- 48.Kaufman R.J., Malhotra J.D. Calcium trafficking integrates endoplasmic reticulum function with mitochondrial bioenergetics. Biochim. Biophys. Acta. 2014;1843(10):2233–2239. doi: 10.1016/j.bbamcr.2014.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boden G., Cheung P., Kresge K., Homko C., Powers B., Ferrer L. Insulin resistance is associated with diminished endoplasmic reticulum stress responses in adipose tissue of healthy and diabetic subjects. Diabetes. 2014;63(9):2977–2983. doi: 10.2337/db14-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu X., Deng Q., Tang Y., Xiao L., Liu L., Yao P. Flaxseed oil attenuates hepatic steatosis and insulin resistance in mice by rescuing the adaption to ER stress. J. Agric. Food Chem. 2018;66(41):10729–10740. doi: 10.1021/acs.jafc.8b03906. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multimedia component 1