

Abstract
The eligibility for kidney donation and long-term post-donation renal prognosis of patients with Gitelman syndrome (GS) are unknown. We herein report a 44-year-old woman with GS who donated her kidney for transplant. A gene sequence analysis revealed compound heterozygous mutations of T180K and L858H in the SLC12A3 gene. Since transplantation, the renal function and serum potassium and magnesium levels of the donor and recipient have remained stable for seven years with careful monitoring and supplementation. Patients with asymptomatic GS who have no complications can be considered eligible to donate their kidney for transplant with proper monitoring after transplantation.
Keywords: Gitelman syndrome, kidney transplantation, hypokalemia, the SLC12A3 gene
Introduction
Gitelman syndrome (GS) (OMIM: 263800) is an autosomal recessive disorder caused by inactivating mutations in the solute carrier family 12 member 3 gene (SLC12A3), which encodes the thiazide-sensitive Na-Cl cotransporter (NCC) located on the apical surface of distal convoluted tubular (DCT) cells (1-4). GS is a rare salt-losing tubulopathy characterized by hypokalemic metabolic alkalosis with hypomagnesemia and hypocalciuria (3,5) and has been widely described as a benign or mild variant of Bartter's syndrome (6). Clinical symptoms are often unremarkable or relatively mild and include muscular weakness, fatigue, salt craving, thirst, nocturia, and cramps. However, recent reports have emphasized the phenotypic variability and potential severity in patients with GS (7,8). Patients with GS are at risk of developing ventricular arrhythmias, diabetes, and chronic kidney disease (CKD).
Living kidney transplantation from GS donors is rare; only three cases have been reported to date (9-11). In addition, the kidney donation eligibility and long-term post-donation renal prognosis of patients with GS are unknown. We herein report the clinical course of a 44-year-old woman who was diagnosed with GS by a genetic analysis during a preoperative donor evaluation for kidney transplantation. We previously reported the clinical course of the recipient who received her kidney (12).
Case Report
A 44-year-old Japanese woman underwent a preoperative evaluation for kidney transplantation donation to her husband, who was a patient with type 2 diabetes receiving peritoneal dialysis. She showed no clinical signs or symptoms and had no significant medical or family history. Her blood pressure, heart rate, and body mass index were 110/70 mmHg, 68 beats/min, and 18.3 kg/m2, respectively. She had no muscle weakness or sensory abnormalities, and her deep tendon reflexes were normal. Biochemical analyses showed mild hypokalemia, hypomagnesemia, hyperreninemia, and metabolic alkalosis. Thyroid function testing was normal (Table). Her urinary calcium-to-creatinine ratio was 0.012, indicating hypocalciuria. Based on these findings, GS was suspected, and direct DNA sequencing of the SLC12A3 gene was performed for the patient and her parents, who also had no obvious clinical symptoms. Sequencing using DNA samples derived from white blood cells showed that the patient had compound heterozygous missense mutations in the SLC12A3 gene with single-base substitutions at nucleotides 539 of exon 4 (ACG to AAG, T180K) and 2573 of exon 22 (CTC to CAC, L858H). A linkage analysis confirmed that T180K was the maternal allele, while L858H was the paternal allele (Fig. 1).
Table.
Biochemical Data.
| Parameter | Measured value |
Reference range |
|---|---|---|
| Hemoglobin (g/dL) | 11.6 | 11.5–14.5 |
| White blood cell count (/mm3) | 5,400 | 3,300-8,600 |
| Albumin (g/dL) | 4.3 | 3.5–5.2 |
| Creatinine (mg/dL) | 0.5 | 0.4–0.8 |
| Urea (mg/dL) | 13 | 8.0-20.0 |
| Sodium (mEq/L) | 139 | 136–146 |
| Potassium (mEq/L) | 3.1 | 3.6–4.8 |
| Chloride (mEq/L) | 101 | 98–109 |
| Calcium (mg/dL) | 9.5 | 8.6–10.2 |
| Phosphate (mg/dL) | 2.5 | 2.6–4.6 |
| Magnesium (mg/dL) | 1.7 | 1.8–2.6 |
| Bicarbonate (mEq/L) | 27.3 | |
| TSH (μIU/mL) | 2.28 | 0.34–4.04 |
| FT3 (pg/mL) | 2.60 | 2.36–5.00 |
| FT4 (ng/dL) | 1.12 | 0.88–1.67 |
| PRA (ng/mL/hr) | 4.0 | |
| Aldsterone (pg/mL) | 88.6 | |
| FBG (mg/dL) | 97 | 65–109 |
| HbA1c (%) | 5.3 | 4.6–6.2 |
| Total urinary potassium (mEq/24h) | 46.6 | |
| Total urinary calcium (mg/24h) | 1.89 | |
| Urinary calcium/Urinary creatinine | 0.012 | |
| Total urinary sodium (mEq/24h) | 281.4 | |
| Total urinary magnesium (mg/24h) | 11.97 | |
| Total urinary chloride (mEq/24h) | 256.1 | |
| Creatinine clearance (mL/min) | 268 |
FBG: fasting blood glucose, TP: total protein, TSH: thyroid-stimulating hormone, PRA: plasma renin activity
Figure 1.

An SLC12A3 gene sequence analysis of the patient showed a heterozygous transition (C-to-A) at nucleotide 539 in exon 4, resulting in a Thr-to-Lys substitution at amino acid 180, and a heterozygous transition (T-to-A) at nucleotide 2573 in exon 22, resulting in a Leu-to-His substitution at amino acid 858.
As the patient met all other donation criteria for kidney transplantation and showed no significant symptoms or complications of GS, kidney explantation was performed. To determine the distribution of SLC12A3 protein expression, we performed immunohistological staining with anti-SLC12A3 antibodies. Immunostaining was performed on formalin-fixed paraffin-embedded sections treated by heating in 0.01 mol/L citrate buffer at pH 6.0 using an electric kettle for antigen retrieval. Tissue sections were incubated with anti-SLC12A3 antibodies (HPA028748; Atlas Antibodies, Bromma, Sweden) overnight at 4℃, according to the manufacturer's instructions. The secondary antibody was N-HistofineⓇ Simple Stain MAX PO (G) (Nichirei Biosciences, Tokyo, Japan). Tissue sections were stained using DAB-H2O2 as a substrate. We found immunoreactivity in the apical surface of the DCT cells of the transplanted kidney, similar to the kidney of a healthy individual (Fig. 2A-D). Renal fibrosis in the patient was evaluated using Masson's trichrome stain; nonspecific vacuolar lesions in the epithelial cells in the proximal tubule and very mild tubulointerstitial fibrosis were observed. The interstitial damage of the kidney was not noticeable, and the renal tissue showed an almost normal structure (Fig. 2E, F).
Figure 2.
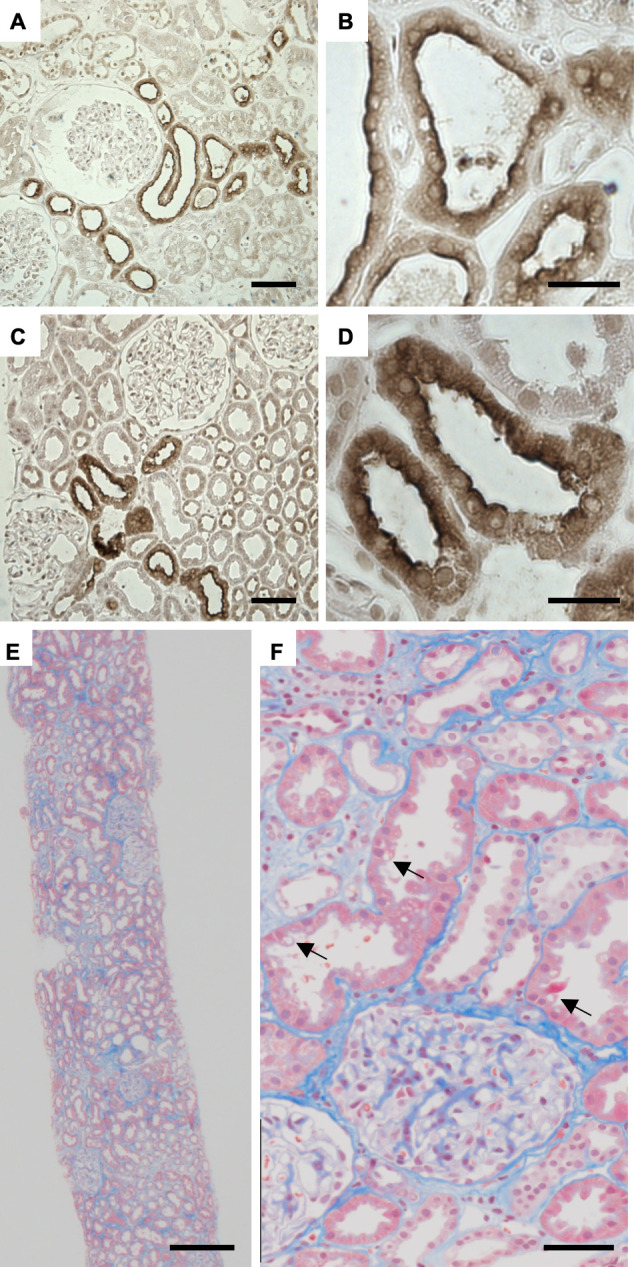
Immunohistochemical staining of kidney specimens with antibodies to SLC12A3, the sodium-chloride cotransporter (NCC) (A-D). Staining of the membrane surface of the distal convoluted tubule was similar between (A, B) a healthy control and (C, D) the patient with Gitelman syndrome. Masson’s trichrome staining of the patient’s kidney (E, F). A light microscopic examination showed nonspecific vacuolar lesions in the epithelial cells of the proximal tubule (arrows) and mild tubulointerstitial fibrosis. The right-side images (B, D, F) are enlarged forms of the left-side images (A, C, E), respectively. Scale bars=50 μm (A, C, F), 20 μm (B, D), and 250 μm (E).
After transplantation, the patient was treated with potassium supplementation using potassium gluconate (8 mEq/day). At the 7-year follow-up evaluation, her serum electrolytes and renal function were maintained (serum magnesium 1.7-2.1 mg/dL, potassium 3.3-4.1 mg/dL, and creatinine 0.67-0.83 mg/dL) without any adverse events. Fig. 3 shows the clinical course of the donor's renal function and electrolytes before and after transplantation. Unilateral kidney donation did not worsen the donor's GS symptoms.
Figure 3.

Clinical course of the donor's renal function and electrolytes before and after transplantation.
During the early postoperative period, the recipient suffered from acute antibody-mediated rejection. The response to the intensive immunosuppressive therapy [3 consecutive administrations of methylprednisolone (mPSL) 500 mg/body, intravenous immunoglobulin (IVIG) 40 g/body, rituximab 200 mg/body] was satisfactory, and the renal function improved promptly within 2 months with a creatinine level of 1.5 mg/dL. At present, the graft function is stable at 7 years after transplantation [sCr 1.1 mg/dL, estimated glomerular filtration rate (eGFR) 54 mL/min/1.73 m2, and proteinuria 0.04 g/g·Cr]. The electrolyte levels were abnormal immediately following transplantation due to the acute antibody-mediated rejection; however, at the long-term follow-up, the serum potassium level was normal (3.7-4.5 mEq/mL), and mild hypomagnesemia approaching a normal magnesium level was present (1.7-1.9 mg/dL) without supplementation.
Discussion
We herein report a patient with asymptomatic GS who served as a kidney transplantation donor. The diagnosis was made by biochemical and genetic analyses. Since kidney transplantation, the correction of hypokalemia with low-dose potassium supplementation has maintained her renal function for seven years.
Only three cases of successful living kidney transplantation from donors with GS have been reported (9-11). In one of these, the donor was diagnosed after transplantation; in two of these, the diagnosis was based on biochemical parameters alone without genetic analyses. Although the clinical courses of all kidney recipients have been reported, those of the donors are not well described, and data regarding the long-term post-donation renal prognosis and donor eligibility of patients with GS have been lacking.
Although GS is considered a benign tubulopathy, recent reports have shown that GS is associated with a significant reduction in the quality of life. Severe manifestations, such as growth retardation, chondrocalcinosis, tetany, rhabdomyolysis, seizures, and ventricular arrhythmias, as well as early-onset disease (before six years old) have been described (6,8,13). A careful evaluation of kidney transplant donors with GS is necessary to ensure that they do not develop CKD. Most clinical problems in GS are linked to electrolyte disturbances, including chronic salt loss, hypokalemia, and hypomagnesemia. Due to hypokalemia, GS patients may experience mild renal impairment, impaired glucose tolerance, and prolonged QT syndrome, which leads to an increased risk of ventricular arrhythmias (14-16). CKD may develop in patients with GS due to either chronic hypokalemia, which is associated with tubulointerstitial nephritis, tubule vacuolization, and cystic changes known as hypokalemic nephropathy, or volume depletion and activation of the renin-angiotensin-aldosterone system, which contributes to renal damage and fibrosis (17-19). In the present case, the patient had only mild hypokalemia (serum potassium level 3.0-3.5 mEq/L) and mild hypomagnesemia (magnesium level 1.5-2.0 mg/dL) without clinically remarkable symptoms. Abnormal electrocardiography and impaired glucose tolerance were not observed, and the renal function was maintained (eGFR 103.3 mL/min/1.73 m2). However, continuous follow-up and management of the potassium and magnesium levels and renal function will be necessary in the future. Therefore, kidney donation by patients with hypokalemia due to GS should be undertaken with care.
However, the electrolyte levels were abnormal in the recipient immediately following transplantation because of kidney injury caused by acute antibody-mediated rejection; therefore, it was difficult to accurately evaluate the presence of laboratory findings suggestive of GS (e.g. hypokalemia, hypomagnesemia). In 3 previous reports of renal transplantation for GS (9-11), 2 recipients had hypokalemia and hypomagnesemia, which are laboratory findings suggestive of GS (10,11), and the third had a serum potassium level of 3.7 mEq/L and a magnesium level of 1.8 mg/dL at 1 year after transplantation (9). These levels were around the lower limits of the normal range, similar to those of the present case.
Our case report provides at least four explanations as to why our recipient did not show severe hypokalemia or hypomagnesemia: (1) a reduced kidney function (CKD3T), (2) the effect of the calcineurin inhibitor, (3) no evidence of proteinuria-associated renal magnesium wasting, and (4) no evidence of hyperaldosteronism. First, the risk of hypokalemia is decreased because most patients become CKD stage 2 or 3 after kidney transplantation (20-23), and our patient also showed CKD stage 3. Second, the calcineurin inhibitor induces the side-effect of hyperkalemia due to NCC overactivation while causing hypomagnesemia via renal magnesium wasting (24). Third, no evidence of proteinuria-associated renal magnesium wasting arose in our case. In general, 15% of patients with CKD stage 3 show hypomagnesemia, and this effect is associated with proteinuria. Proteinuria-associated renal magnesium wasting causes hypomagnesemia (25). Fourth, hyperaldosteronism was not observed in our case (serum aldosterone 88.6 pg/mL). A previous report showed that a high aldosterone level during GS is likely to cause hypomagnesemia due to TRPM6 downregulation (26). Therefore, in the post-transplant management for recipients transplanted from a donor with GS, it is necessary to follow the progress with special attention to electrolyte imbalances. It is important to note the degree of electrolyte imbalance in GS cases and the risk of hyperkalemia and hypomagnesemia due to the use of calcineurin inhibitors.
GS is caused by biallelic inactivating mutations in the SLC12A3 gene, which encodes a NCC that is expressed on the apical membrane of cells lining the DCT (1). The nature of homozygous and intronic or nonframeshift mutations in the SLC12A3 gene may influence the phenotype and cause more severe hypokalemia (18,27). In the present case, the compound heterozygous missense mutations of T180K and L858H in the SLC12A3 gene were detected by a genetic analysis. While some reports have shown that the T180K mutation is pathogenic (28), one previous report suggested that the T180K variant is simply a polymorphism and not functionally responsible for GS-related salt loss (29). Therefore, the pathogenicity of the T180K mutation is controversial. The L858H mutation is a missense mutation, and although unlike nonsense mutations it produces protein, it may be pathogenic and may cause a functional abnormality. In our patient, immunostaining showed immunoreactivity for the NCC along the apical membrane of the DCT. Previous reports have suggested a correlation between clinical functional defects and tissue NCC expression, indicating that patients with NCC expression defects along the apical membrane of the DCT exhibit more severe clinical symptoms (7,30). Women are less severely affected than men because of the estrogen-induced up-regulation of NCC in the DCT (8). Thus, the present patient here may have had a mild phenotype of GS.
Although our patient was initially asymptomatic and had mild hypokalemia, we were able to confirm GS by a genetic analysis. Furthermore, we confirmed kidney transplantation eligibility and conducted long-term follow-up. Strict monitoring of the potassium and magnesium levels after kidney transplantation showed that the renal function was maintained even seven years later. In general, the long-term prognosis of GS is good with proper treatment (31). Therefore, we anticipate excellent results, provided kidney donors with GS have their potassium and magnesium levels monitored and properly managed.
In conclusion, we herein report a case of successful living kidney transplant donation by an asymptomatic patient with GS who harbored a compound heterozygous mutation of the SLC12A3 gene. This is the first report of a patient with GS who has maintained a good long-term renal function after serving as a kidney transplant donor. It is important to carefully monitor the renal function and serum potassium and magnesium levels in donors with GS, taking the potential severity of GS into account.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
The authors thank Teruko Tamatsukuri for her excellent technical assistance during direct DNA sequencing studies.
References
- 1.Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24-30, 1996. [DOI] [PubMed] [Google Scholar]
- 2.Bhandari S, Turney JH. The molecular basis of hypokalaemic alkalosis: Bartter's and Gitelman's syndromes. Nephron 80: 373-379, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr 120: 38-43, 1992. [DOI] [PubMed] [Google Scholar]
- 4.Mastroianni N, De Fusco M, Zollo M, et al. Molecular cloning, expression pattern, and chromosomal localization of the human Na-Cl thiazide-sensitive cotransporter (SLC12A3). Genomics 35: 486-493, 1996. [DOI] [PubMed] [Google Scholar]
- 5.Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians 79: 221-235, 1966. [PubMed] [Google Scholar]
- 6.Pachulski RT, Lopez F, Sharaf R. Gitelman's not-so-benign syndrome. N Engl J Med 353: 850-851, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Riveira-Munoz E, Chang Q, Godefroid N, et al. Transcriptional and functional analyses of SLC12A3 mutations: new clues for the pathogenesis of Gitelman syndrome. J Am Soc Nephrol 18: 1271-1283, 2007. [DOI] [PubMed] [Google Scholar]
- 8. Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB; Yale Gitelman's and Bartter's Syndrome Collaborative Study Group. Gitelman's syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int 59: 710-717, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Hu DC, Burtner C, Hong A, Lobo PI, Okusa MD. Correction of renal hypertension after kidney transplantation from a donor with Gitelman syndrome. Am J Med Sci 331: 105-109, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Bansal R, Ranga VK. Acquired Gitelman's syndrome: an oxymoron? Int Urol Nephrol 43: 233-236, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Stewart D, Iancu D, Ashton E, Courtney AE, Connor A, Walsh SB. Transplantation of a Gitelman syndrome kidney ameliorates hypertension: a case report. Am J Kidney Dis 73: 421-424, 2019. [DOI] [PubMed] [Google Scholar]
- 12.Niikura T, Yamamoto I, Nakada Y, et al. Probable C4d-negative accelerated acute antibody-mediated rejection due to non-HLA antibodies. Nephrology (Carlton) 20(Suppl 2): 75-78, 2015. [DOI] [PubMed] [Google Scholar]
- 13.Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med 112: 183-190, 2002. [DOI] [PubMed] [Google Scholar]
- 14.Bettinelli A, Tosetto C, Colussi G, Tommasini G, Edefonti A, Bianchetti MG. Electrocardiogram with prolonged QT interval in Gitelman disease. Kidney Int 62: 580-584, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Foglia PEG, Bettinelli A, Tosetto C, et al. Cardiac work up in primary renal hypokalaemia-hypomagnesaemia (Gitelman syndrome). Nephrol Dial Transplant 19: 1398-1402, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Ren H, Qin L, Wang W, et al. Abnormal glucose metabolism and insulin sensitivity in Chinese patients with Gitelman syndrome. Am J Nephrol 37: 152-157, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Balavoine AS, Bataille P, Vanhille P, et al. Phenotype-genotype correlation and follow-up in adult patients with hypokalaemia of renal origin suggesting Gitelman syndrome. Eur J Endocrinol 165: 665-673, 2011. [DOI] [PubMed] [Google Scholar]
- 18.Tseng M-H, Yang S-S, Hsu Y-J, et al. Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation. J Clin Endocrinol Metab 97: E1478-E1482, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM 104: 939-944, 2011. [DOI] [PubMed] [Google Scholar]
- 20.Ansell D, Udayaraj UP, Steenkamp R, Dudley CRK. Chronic renal failure in kidney transplant recipients. Do they receive optimum care?: data from the UK renal registry. Am J Transplant 7: 1167-1176, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Djamali A, Kendziorski C, Brazy PC, Becker BN. Disease progression and outcomes in chronic kidney disease and renal transplantation. Kidney Int 64: 1800-1807, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Kukla A, Adulla M, Pascual J, et al. CKD stage-to-stage progression in native and transplant kidney disease. Nephrol Dial Transplant 23: 693-700, 2008. [DOI] [PubMed] [Google Scholar]
- 23.Karthikeyan V, Karpinski J, Nair RC, Knoll G. The burden of chronic kidney disease in renal transplant recipients. Am J Transplant 4: 262-269, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Lee CH, Kim G-H. Electrolyte and acid-base disturbances induced by clacineurin inhibitors. Electrolyte Blood Press 5: 126-130, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oka T, Hamano T, Sakaguchi Y, et al. Proteinuria-associated renal magnesium wasting leads to hypomagnesemia: a common electrolyte abnormality in chronic kidney disease. Nephrol Dial Transplant 34: 1154-1162, 2019. [DOI] [PubMed] [Google Scholar]
- 26.Filippatos TD, Rizos CV, Tzavella E, Elisaf MS. Gitelman syndrome: an analysis of the underlying pathophysiologic mechanisms of acid-base and electrolyte abnormalities. Int Urol Nephrol 50: 91-96, 2018. [DOI] [PubMed] [Google Scholar]
- 27.Zhong F, Ying H, Jia W, et al. Characteristics and follow-up of 13 pedigrees with Gitelman syndrome. J Endocrinol Invest 42: 653-665, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monkawa T, Kurihara I, Kobayashi K, Hayashi M, Saruta T. Novel mutations in thiazide-sensitive Na-Cl cotransporter gene of patients with Gitelman's syndrome. J Am Soc Nephrol 11: 65-70, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Naraba H, Kokubo Y, Tomoike H, Iwai N. Functional confirmation of Gitelman's syndrome mutations in Japanese. Hypertens Res 28: 805-809, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Jang HR, Lee JW, Oh YK, et al. From bench to bedside: diagnosis of Gitelman's syndrome - defect of sodium-chloride cotransporter in renal tissue. Kidney Int 70: 813-817, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Dimitrijevic Z, Salinger-Martinovic S, Mitic B, Nikolic V. Gitelman's syndrome as a cause of poorly controlled hypokalemia. Clin Ter 166: e173-e176, 2015. [DOI] [PubMed] [Google Scholar]