

Abstract
TAFRO syndrome and POEMS syndrome are lymphoproliferative disorders with elevated interleukin-6 and vascular endothelial growth factor (VEGF) levels; however, their underlying pathogenic mechanisms remain unclear. Similarities have been reported in the pathological findings of the lymph nodes of TAFRO syndrome, Multicentric Castleman disease (MCD), and some cases of POEMS syndrome. However, there is no consensus on the relationship among them. We encountered a case of lymphoproliferative disorder that presented with manifestations of both TAFRO syndrome and POEMS syndrome. This case may be a subtype of idiopathic MCD and will be very important for establishing the disease concept of TAFRO syndrome and POEMS syndrome.
Keywords: TAFRO syndrome, POEMS syndrome, Castleman disease, lymphoproliferative disorder
Introduction
Multicentric Castleman disease (MCD) is a rare lymphoproliferative disorder that is characterized by systemic inflammatory symptoms, lymph node proliferation, and organ dysfunction. MCD is subclassified into human herpesvirus 8 (HHV-8)-associated MCD and HHV-8-negative/idiopathic MCD (iMCD).
TAFRO syndrome is a recently recognized systemic inflammatory disease that was initially described by Takai et al. in 2010 (1). Several cases of TAFRO syndrome have since been reported in the US, Europe, and other countries. TAFRO syndrome is characterized by thrombocytopenia (T), anasarca (A), fever (F), reticulin fibrosis (R), and organomegaly (O). In TAFRO syndrome, serum interleukin-6 (IL-6) levels are elevated, and IL-6 and other proinflammatory cytokines have been implicated in its pathogenesis. Elevated alkaline phosphatase (ALP) levels and renal impairment are also characteristics of TAFRO syndrome. The pathological characteristics of the lymph nodes of TAFRO syndrome are similar to those of iMCD. Therefore, TAFRO syndrome may be a variant type of iMCD (2). However, since the clinical manifestations of TAFRO syndrome differ from those of MCD, it may be regarded as a distinct clinical entity (3).
POEMS syndrome is a plasma cell dyscrasia that is characterized by polyneuropathy (P), organomegaly (O), endocrinopathy (E), monoclonal gammopathy (M), and skin changes (S). In POEMS syndrome, vascular endothelial growth factor (VEGF) levels are markedly elevated and considered to be an important factor in its pathogenesis (4). The pathological findings of the lymph nodes of POEMS syndrome are sometimes similar to those of MCD, some cases of POEMS syndrome are associated with MCD, and the diagnostic criteria of POEMS syndrome include MCD (5).
The pathogenesis of both of these syndromes remains unclear, and there is currently no consensus on the relationship among TAFRO syndrome, POEMS syndrome, and MCD. We herein report a case of a lymphoproliferative disorder presenting with manifestations of both TAFRO syndrome and POEMS syndrome. A more detailed understanding of the pathophysiology of this case will contribute to the establishment of the disease concept of TAFRO syndrome and POEMS syndrome.
Case Report
A 49-year-old man was admitted to our hospital with a fever and anasarca. He had had muscle weakness and fatigue for approximately one year. He developed lower leg edema one month previously with progressive weakness in the lower limbs in addition to anorexia and nausea, and a fever had followed two weeks later.
His medical history and family history were unremarkable. A physical examination revealed a temperature of 38.3 °C, blood pressure 124/86 mmHg, heart rate 136/min, respiratory rate 16/min, and oxygen saturation of 94% on room air. He was wheelchair-bound and ambulated with aids because of weakness in the lower limbs and fatigue. He had severe lower leg edema and distal dominant dysesthesia of all four limbs with muscle weakness (proximal power of grade 4/5 and distal power of grade 4/5). The deep tendon reflexes were weak in the upper limbs and absent in the lower limbs. Skin lesions that resembled hemangioma were detected on his trunk (Fig. 1A). The lesion had been around for quite some time. He had gynecomastia but not hyperpigmentation or hypertrichosis.
Figure 1.

A: Gross features of hemangioma. B: The M protein was identified by immunofixation electrophoresis as κ-type Bence Jones protein. C: Computed tomography revealed abnormal body fluid retention, gynecomastia, and systemic lymphadenopathy.
Laboratory data (Table) showed a white blood cell count of 21,000 cells/μL, hemoglobin level 12.4 g/dL, platelet count 67,000 cells/μL, lactate dehydrogenase level 376 IU/L, interleukin-2 receptor level 3,111 U/mL, C-reactive protein (CRP) level 26.85 mg/dL, creatinine level 1.22 mg/dL, and alkaline phosphatase level 835 IU/L. His serum IL-6 level was elevated to 13.7 pg/mL, serum VEGF level to 2,370 pg/mL, and plasma VEGF level to 291 pg/mL. His thyroid-stimulating hormone (TSH), free T3 (FT3), and free T4 (FT4) levels had decreased to 0.245 μIU/mL, 0.41 ng/dL, and 0.58 pg/mL, respectively. Antinuclear antibody (ANA) and antineutrophil cytoplasmic antibodies (ANCAs) were negative. Blood, urine, and sputum cultures were negative. A polymerase chain reaction (PCR) analysis of serum did not detect human immunodeficiency virus (HIV), Epstein-Barr virus (EBV), or human herpesvirus 8 (HHV-8). Serum immunoelectrophoresis showed the absence of the M protein, whereas urine immunoelectrophoresis detected κ-type Bence Jones protein (Fig. 1B). Computed tomography (CT) revealed bilateral pleural effusion, ascites, hepatomegaly, enlarged breast glandular tissue, and multiple small lymphadenopathies in the cervical, axillary, mediastinal, mesenteric, and inguinal regions (Fig. 1C).
Table.
Laboratory Data on Admission.
| Blood cell count | Biochemistry | Serological test | ||||||
| WBC | 21,000 | /μL | AST | 36 | IU/L | CRP | 26.85 | mg/dL |
| My. | 0.7 | % | ALT | 33 | IU/L | IgG | 1,120 | mg/dL |
| Stab. | 1.7 | % | LDH | 376 | IU/L | IgG4 | 35.1 | mg/dL |
| Seg. | 82.9 | % | ALP | 835 | IU/L | IgA | 100 | mg/dL |
| Lymph. | 4.4 | % | γ-GTP | 106 | IU/L | IgM | 33 | mg/dL |
| Mono. | 10 | % | T-bil | 1.2 | mg/dL | freeκchain | 130 | mg/dL |
| Eos. | 0 | % | D-bil | 0.7 | mg/dL | freeλchain | 68.7 | mg/dL |
| Baso. | 0.3 | % | TP | 4.7 | g/dL | k/λ ratio | 1.89 | |
| RBC | 436×104 | /μL | Alb | 2.3 | g/dL | C3 | 120 | mg/dL |
| Hb | 12.4 | g/dL | CK | 19 | IU/L | C4 | 24 | mg/dL |
| Ht | 38.1 | % | UA | 8 | mg/dL | CH50 | 59 | U/mL |
| MCV | 87.4 | fL | BUN | 23.7 | mg/dL | ANA | negative | |
| Retic. | 15 | ‰ | Cre | 1.22 | mg/dL | PR3-ANCA | negative | |
| Plt | 6.7×104 | /μL | Na | 132 | mEq/L | MPO-ANCA | negative | |
| K | 4.3 | mEq/L | ||||||
| Urinalysis | Cl | 99 | mEq/L | Coagulation | ||||
| Protein | 0.32 | g/gCre | Ca | 7.6 | mEq/L | APTT | 42.1 | sec. |
| RBC | <1 | HPF | sIL-2R | 3,111 | U/mL | PT | 15 | sec. |
| WBC | <1 | HPF | IL-6 | 13.7 | pg/mL | Fib. | 509 | mg/dL |
| plasma VEGF | 291 | pg/mL | ||||||
| serum VEGF | 2,370 | pg/mL | ||||||
| TSH | 0.245 | μIU/mL | ||||||
| freeT3 | 0.41 | ng/dL | ||||||
| freeT4 | 0.58 | pg/mL | ||||||
WBC: white blood cell, My: myelocyte, Seg: segment, Lymph: lymphocytosis, Mono: mononucleosis, Eos: eosinophil, Baso: basophils, RBC: red blood cell, Hb: hemoglobin, Ht: hematocrit, MCV: mean corpuscular volume, Retic: reticulocyte, Plt: platelet, RBC: red blood cell, AST: aspartate transaminase, ALT: alanine aminotransferase, LDH: lactate dehydrogenase, ALP: alkaline phosphatase, γ-GTP: γ-glutamyl transpeptidase, T-bill: total bilirubin, D-bill: direct bilirubin, TP: total protein, Alb: albumin, CK: creatine kinase, UA: uric acid, BUN: blood urea nitrogen, Cre: creatinine, Na: natrium, K: kalium, Cl: chlorine, Ca: calcium, sIL-2R: soluble interleukin-2 receptor, IL-6: interleukin-6, VEGF: vascular endothelial growth factor, TSH: thyroid-stimulating hormone, freeT3: free triiodothyronine, freeT4: free thyroxin, CRP: C-reactive protein, IgG: immunoglobulin G, IgA: immunoglobulin A, IgM: immunoglobulin M, C3: complement component 3, C4: complement component 4, CH50: total complement activity test, ANA: antinuclear antibody, PR3-ANCA: proteinase-anti-neutrophil cytoplasmic antibody, MPO-ANCA: myeloperoxidase anti-neutrophil cytoplasmic antibody, APTT: activated partial thromboplastin time, PT: prothrombin time, Fib: fibrinogen
Sclerotic bone lesions were not observed on CT. A bone marrow biopsy revealed hyperplasia of megakaryocytes and reticulin fibrosis (Fig. 2A). There were no abnormal plasma cells in the bone marrow. A lymph node biopsy of the inguinal region revealed that the germinal centers were atrophic and surrounded by a mantle zone, which exhibited layering of lymphocytes, and plasma cell infiltration without light-chain restriction was noted in the interfollicular zone around high endothelial venules (Fig. 2B, C). These findings were consistent with MCD. The expression of latency-associated nuclear antigen 1 (LANA-1) was not detected in the lesion by immunohistochemistry.
Figure 2.
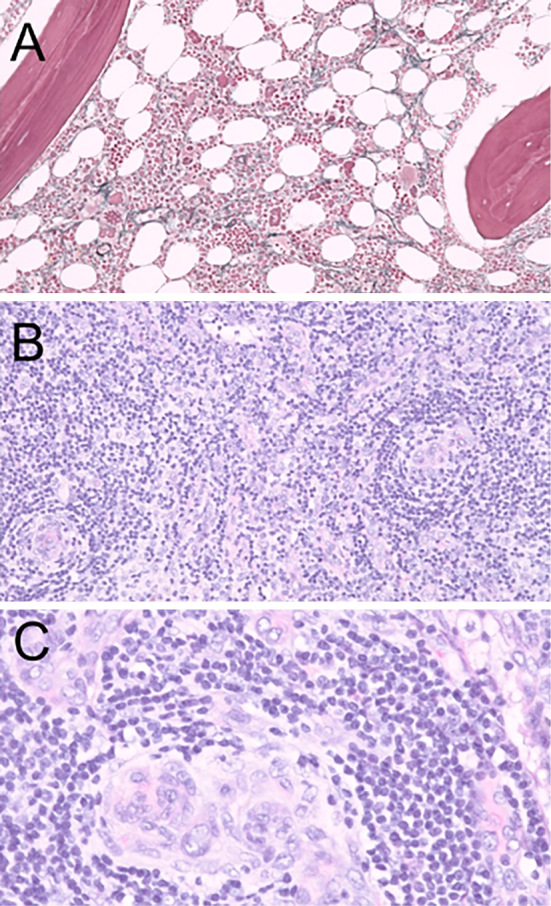
Histopathological findings of the bone marrow (A) and lymph nodes of the inguinal region (B, C). A: A bone marrow biopsy showed hyperplasia of megakaryocytes and reticulin fibrosis (Gitter staining). B, C: A lymph node biopsy of the inguinal region revealed that the germinal centers were atrophic and surrounded by a mantle zone that exhibited layering of lymphocytes, and plasma cell infiltration was observed in the interfollicular zone around high endothelial venules (Hematoxylin and Eosin staining).
As an initial evaluation, we conducted a neve conduction study of the left extremity. We were unable to perform a study of the right extremity because of his aggressive clinical course. In the left median nerve and ulnar nerve, an F-wave could not be elicited, and the distal latencies were prolonged, so the sensory nerve conduction velocities (SCVs) were assumed to be diminished. In the left tibial nerve, an F-wave could not be elicited, and the distal latency was prolonged. The sensory nerve action potential (SNAP) could not be elicited in the left sural nerve. These results suggested neuropathy mainly with sensory impairment and demyelination predominantly in the lower extremities and proximal areas. A diagnosis of peripheral neuropathy was reached based on the symptoms and electrophysiological findings, which were consistent with the features of POEMS syndrome (6).
Based on these findings, the differential diagnosis was TAFRO syndrome and POEMS syndrome.
The pathological findings of the lymph nodes and bone marrow, thrombocytopenia, anasarca, fever, organomegaly (hepatomegaly and splenomegaly), and elevated serum ALP levels were consistent with TAFRO syndrome, whereas the M protein (κ-type Bence Jones protein), polyneuropathy, endocrinopathy (hypothyroidism and gynecomastia), and skin changes (hemangioma) were not. However, the findings of M protein, polyneuropathy, organomegaly, endocrinopathy, and skin changes were compatible with POEMS syndrome, whereas the pathological findings of bone marrow and elevated serum ALP levels were not. Therefore, the present case was diagnosed as a borderline case of TAFRO syndrome and POEMS syndrome.
We initiated treatment for TAFRO syndrome because of the severity of inflammatory findings and administered prednisolone at 1 mg/kg/day. This treatment approach reduced the level of CRP but did not attenuate the other clinical manifestations. Therefore, we changed our treatment strategy to that for POEMS syndrome and initiated VRD therapy (bortezomib 1.3 mg/m2/day on days 1, 4, 8, and 11 of a 21-day cycle, lenalidomide 25 mg/day on days 1-14 of a 21-day cycle, dexamethasone 20 mg/day on days 1, 2, 4, 5, 8, 9, 11, and 12 of a 21-day cycle) after informed consent was obtained. After the initiation of VRD therapy, his CRP level decreased further; however, cerebral infarction occurred on day 5 of the first cycle of VRD therapy (Fig. 3). Cerebral lesion progression was noted two days later. Since the amount of ascites and pleural effusion increased, we performed plasma exchange; however, the patient ultimately died due to hypovolemic shock.
Figure 3.

On brain magnetic resonance imaging, multiple lesions with a high signal intensity on diffusion-weighted imaging (DWI) were observed in the right genu corpus callosum and bilateral cerebral white matter. On magnetic resonance angiography, neither stenosis nor occlusion was detected in the aorta or its main branches.
Autopsy findings revealed pleural effusion (right: 1,000 mL, left: 1,000 mL), ascites (1,800 mL), hepatomegaly, and splenomegaly. Renal histopathology showed some characteristic findings that suggested vascular endothelial injury, which was compatible with thrombotic microangiopathy (Fig. 4). Among 208 glomeruli examined, 6 were globally sclerosed. Most glomeruli showed swelling, and the diameter of 1 glomerulus was approximately 180-200 μm. Neither mesangial matrix expansion nor proliferation was observed. Arterioles showed endothelial swelling, and the capillary lumens were narrowed. A double-contoured glomerular basement membrane due to prominent endothelial swelling and a widened subendothelial space were detected. Neither lobulation, crescents, glomerular thrombosis, nor mesangiolysis was detected.
Figure 4.

Autopsy kidney. A, B: Endothelial swelling and a double contour were observed (A: PAM, B: PAS staining). C: Most glomeruli were swollen. The interstitium was not edematous, and cellular infiltration was only observed in limited areas (Hematoxylin and Eosin staining).
The interstitium was not edematous, and cellular infiltration was observed in a limited area. Tubular cells were detached from tubular basement membrane, but were suspected to be mainly due to postmortem changes. Arteriosclerotic changes were very mild. There was no lymphoma, myeloma, or other specific findings.
Discussion
POEMS syndrome is a lymphoproliferative disorder caused by an underlying plasma cell neoplasm. The mandatory criteria for a diagnosis of POEMS syndrome are polyneuropathy and clonal plasma cell disorder (almost always λ). Other criteria include MCD, sclerotic bone lesions, and elevated VEGF levels. Minor criteria are organomegaly, extravascular volume overload, endocrinopathy, skin changes, papilledema, and thrombocytosis or polycythemia (5). The M protein of POEMS syndrome is generally the λ type; however, POEMS syndrome with κ-type M protein has been reported (7-9).
The present case presented with polyneuropathy (P), organomegaly (O), endocrinopathy (E), monoclonal gammopathy (M), skin changes (S), and elevated VEGF and thus met the diagnosis criteria for POEMS syndrome. However, the characteristic symptoms of TAFRO syndrome were also detected, namely, thrombocytopenia (T), anasarca (A), fever (F), reticulin fibrosis (R), and organomegaly (O).
Although Masaki's criteria require the exclusion of POEMS syndrome for the diagnosis of TAFRO syndrome (3) and thrombocytosis and anasarca are also manifestations of POEMS syndrome, reticulin myelofibrosis in bone marrow and an elevated serum ALP level are not compatible with POEMS syndrome.
Difficulties are associated with performing a kidney biopsy for TAFRO syndrome because of thrombocytopenia, and only approximately 30 cases have been reported as of August 2020. Based on the findings obtained to date, local glomerular microangiopathy is a characteristic finding on a renal biopsy in cases of TAFRO syndrome. Other findings include membranoproliferative glomerulonephritis (MPGN)-like changes with endothelial injury and a lack of fibrin thrombi in glomerular capillaries or arterioles (10,11). An autopsy was conducted about one month from the disease onset in our patient, so the renal findings in our patient support the report of Nagayama et al. that thrombotic microangiopathy (TMA)-like lesions tend to appear in the acute phase of TAFRO syndrome (10).
The renal histology of POEMS syndrome is reported to be similar to that of TAFRO syndrome, and MPGN-like lesions and evidence of endothelial injury are the most common findings in POEMS syndrome (5). Some experts have said that DM nephropathy-like nodules are observed in POEMS syndrome, but this phenomenon is not widely accepted. Few reports have described the renal histology in patients with TAFRO or POEMS syndrome. Our report including autopsy findings may provide some clues concerning the pathogenesis of these diseases.
We considered the present case to be a borderline case of POEMS syndrome and TAFRO syndrome. Although TAFRO syndrome and POEMS syndrome are generally regarded as separate disease entities, a previously reported case of TAFRO syndrome presented with all of the major manifestations of POEMS syndrome, similar to the present case (12). Furthermore, cases of TAFRO syndrome complicated by endocrinopathy and hemangioma have been reported (13,14).
TAFRO syndrome and POEMS syndrome may be associated with MCD, and the pathological findings of the lymph nodes of the inguinal region of this case were similar to those of MCD; therefore, the present case may be a subtype of iMCD. A commonality in both diseases is that they were associated with IL-6, VEGF, and other proinflammatory cytokines; however, the underlying mechanisms have not yet been elucidated in detail. In both cases, the serum IL-6 and VEGF levels were elevated. We concluded that the manifestations of TAFRO syndrome and POEMS syndrome may overlap, and the findings obtained in the present patient will contribute to the establishment of the disease concept for TAFRO syndrome and POEMS syndrome.
Since the inflammatory findings of the present case were severe, we mainly suspected TAFRO syndrome and initiated treatment with prednisolone. The CRP level decreased after the administration of prednisolone without the attenuation of the other clinical manifestations, so we switched our treatment strategy. There is currently no standard treatment for TAFRO syndrome; however, some cases have been successfully treated with the immunosuppressant cyclosporine (15), the anti-IL-6 receptor antibody tocilizumab (16), and the anti-CD20 antibody rituximab (17). Furthermore, the effectiveness of thalidomide, an immunomodulatory drug, was demonstrated in one case (18). Similarly, no standard treatment has yet been established for POEMS syndrome. However, myeloma-type therapy, such as high-dose chemotherapy with autologous stem cell transplantation, alkylator-based therapy, the proteasome inhibitor bortezomib, and the immunomodulatory drug lenalidomide have been reported to be effective (5,19).
Since the present case exhibited manifestations of both TAFRO syndrome and POEMS syndrome, we needed to carefully consider our next approach. We suspected that the M protein indicated the presence of plasma cell dyscrasia in the background of this case and therefore selected a treatment strategy for POEMS syndrome involving bortezomib and lenalidomide. We expected immunomodulatory drugs to be effective against the symptoms of TAFRO syndrome as well as POEMS syndrome. As the patient's condition deteriorated because of cerebral infarction and he died, we were unable to accurately assess the effects of VRD therapy.
The patient developed cerebral infarction in the treatment course. Cases of cerebral infarction in patients with POEMS syndrome have been reported (20,21), and the 5-year risk of cerebral infarction in POEMS syndrome is 13.4% (22). Cerebral infarction also occurs in TAFRO syndrome (23). Lenalidomide plus dexamethasone is associated with an increased risk of thrombosis, and a treatment strategy to prevent thrombosis is recommended (24). Therefore, we administered aspirin to prevent thrombosis, but aspirin alone was not effective in preventing thrombotic events in this patient, who had multiple risk factors for thrombosis.
In conclusion, we encountered a case of a lymphoproliferative disorder that presented with manifestations of both TAFRO syndrome and POEMS syndrome, and the results obtained may help establish the disease concept of TAFRO syndrome and POEMS syndrome.
The authors state that they have no Conflict of Interest (COI).
Acknowledgement
We are grateful to Dr. Masayoshi YAMAMOTO and Dr. Kan NIIMI (Department of Neurology, Tazuke Kofukai, Medical Research Institute, Kitano Hospital) for their assistance in this study.
We are also grateful to the Department of Pathology, National Institute of Infectious Diseases for LANA-1 staining of the lymph node biopsy.
References
- 1.Takai K, Nikkuni K, Shibuya H, Hashidate H. Thrombocytopenia with mild bone marrow fibrosis accompanied by fever, pleural effusion, ascites and hepatosplenomegaly. Rinsho Ketsueki 51: 320-325, 2010. (in Japanese, Abstract in English). [PubMed] [Google Scholar]
- 2.Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV-8-negative multicentric Castleman disease. Am J Hematol 91: 220-226, 2016. [DOI] [PubMed] [Google Scholar]
- 3.Masaki Y, Kawabata H, Takai K, et al. 2019 Updated diagnostic criteria and disease severity classification for TAFRO syndrome. Int J Hematol 111: 155-158, 2020. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe O, Maruyama I, Arimura K, et al. Overproduction of vascular endothelial growth factor/vascular permeability factor is causative in Crow-Fukase (POEMS) syndrome. Muscle Nerve 21: 1390-1397, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Dispenzieri A. POEMS Syndrome: 2019 Update on diagnosis, risk-stratification, and management. Am J Hematol 94: 812-827, 2019. [DOI] [PubMed] [Google Scholar]
- 6.Min JH, Hong YH, Lee KW. Electrophysiological features of patients with POEMS syndrome. Clin Neurophysiol 116: 965-968, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Dao LN, Hanson CA, Dispenzieri A, Morice WG, Kurtin PJ, Hoyer JD. Bone marrow histopathology in POEMS syndrome: a distinctive combination of plasma cell, lymphoid, and myeloid findings in 87 patients. Blood 117: 6438-6444, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda S, Kobayashi T, Saito M, et al. Multiparameter flow cytometry for the identification of neoplastic plasma cells in POEMS syndrome with IgG-kappa gammopathy: successful treatment using lenalidomide and dexamethasone. Intern Med 58: 3461-3468, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romas E, Storey E, Ayers M, Byrne E. Polyneuropathy, organomegaly, endocrinopathy, M-protein and skin change (POEMS) syndrome with IgG kappa paraproteinemia. Pathology 24: 217-220, 1992. [DOI] [PubMed] [Google Scholar]
- 10.Nagayama Y, Yamano M, Yagame M, et al. TAFRO syndrome as a cause of glomerular microangiopathy: a case report and literature review. BMC Nephrol 20: 375, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leurs A, Gnemmi V, Lionet A, et al. Renal pathologic findings in TAFRO syndrome: is there a continuum between thrombotic microangiopathy and membranoproliferative glomerulonephritis? A case report and literature review. Front Immunol 10: 1489, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noda-Narita S, Sumida K, Sekine A, et al. TAFRO syndrome with refractory thrombocytopenia responding to tocilizumab and romiplostim: a case report. CEN Case Reports 7: 162-168, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miatech JL, Patel NR, Latuso NQ, Ellipeddi PK. TAFRO syndrome: a case of significant endocrinopathy in a Caucasian patient. Cureus 11: e4946, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujita K, Hatta K. Tufted-angioma-like lesion associated with vascular endothelial growth factor and interleukin-6 in TAFRO syndrome: is it a common histological feature of multicentric Castleman disease/POEMS syndrome? J Cutan Pathol 46: 280-284, 2019. [DOI] [PubMed] [Google Scholar]
- 15.Yamaga Y, Tokuyama K, Kato T, et al. Successful treatment with cyclosporin a in Tocilizumab-resistant TAFRO syndrome. Intern Med 55: 185-190, 2016. [DOI] [PubMed] [Google Scholar]
- 16.Kawabata H, Kotani SI, Matsumura Y, et al. Successful treatment of a patient with multicentric castleman's disease who presented with thrombocytopenia, ascites, renal failure and myelofibrosis using tocilizumab, an anti-interleukin-6 receptor antibody. Intern Med 52: 1503-1507, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Ozawa T, Kosugi S, Kito M, et al. Efficacy of rituximab for TAFRO syndrome, a variant type of multicentric Castleman's disease. Rinsho Ketsueki 55: 350-355, 2014. (in Japanese, Abstract in English). [PubMed] [Google Scholar]
- 18.Tatekawa S, Umemura K, Fukuyama R, et al. Thalidomide for tocilizumab-resistant ascites with TAFRO syndrome. Clinical Case Reports 3: 472-478, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao H, Huang X-fei, Gao X-min, et al. What is the best first-line treatment for POEMS syndrome: autologous transplantation, melphalan and dexamethasone, or lenalidomide and dexamethasone? Leukemia 33: 1023-1029, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang K, Chu K, Kim DE, Jeong SW, Lee JW, Roh JK. POEMS syndrome associated with ischemic stroke. Arch Neurol 60: 745-749, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, Wang L, Zhou W, Jin J. Hyaline vascular Castleman disease associated with POEMS syndrome and cerebral infarction. Ann Hematol 86: 59-61, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Dupont SA, Dispenzieri A, Mauermann ML, Rabinstein AA, Brown RD Jr. Cerebral infarction in POEMS syndrome: incidence, risk factors, and imaging characteristics. Neurology 73: 1308-1312, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goda T, Oyama N, Iwamoto T, et al. A case of cerebral infarction in a patient with TAFRO syndrome. J Neurol Sci 400: 21-22, 2019. [DOI] [PubMed] [Google Scholar]
- 24.Larocca A, Cavallo F, Bringhen S, et al. Aspirin or enoxaparin thromboprophylaxis for patients with newly diagnosed multiple myeloma treated with lenalidomide. Blood 119: 933-939, 2012. [DOI] [PubMed] [Google Scholar]