

Abstract
The recently emerged SARS-CoV2 caused a major pandemic of coronavirus disease (COVID-19). Non structural protein 1 (nsp1) is found in all beta coronavirus that cause severe respiratory disease. This protein is considered as a virulence factor and has an important role in pathogenesis. This study aims to elucidate the structural conformations of nsp1 to aid in the prediction of epitope sites and identification of important residues for targeted therapy against COVID-19. In this study, molecular modelling coupled with molecular dynamics simulations were performed to analyse the conformational landscape of nsp1 homologs of SARS-CoV1, SARS-CoV2 and MERS-CoV. Principal component analysis escorted by free energy landscape revealed that SARS-CoV2 nsp1 protein shows greater flexibility compared to SARS-CoV1 and MERS-CoV nsp1. Sequence comparison reveals that 28 mutations are present in SARS-CoV2 nsp1 protein compared to SARS-CoV1 nsp1. Several B-cell and T-cell epitopes were identified by an immunoinformatics approach. SARS-CoV2 nsp1 protein binds with the interface region of the palm and finger domain of POLA1 via hydrogen bonding and salt bridge interactions. Taken together, these in silico findings may help in the development of therapeutics specific against COVID-19.
Keywords: Non structural protein1, COVID-19, Protein modelling, Simulation, Immunoinformatics, Protein-protein docking
Graphical abstract
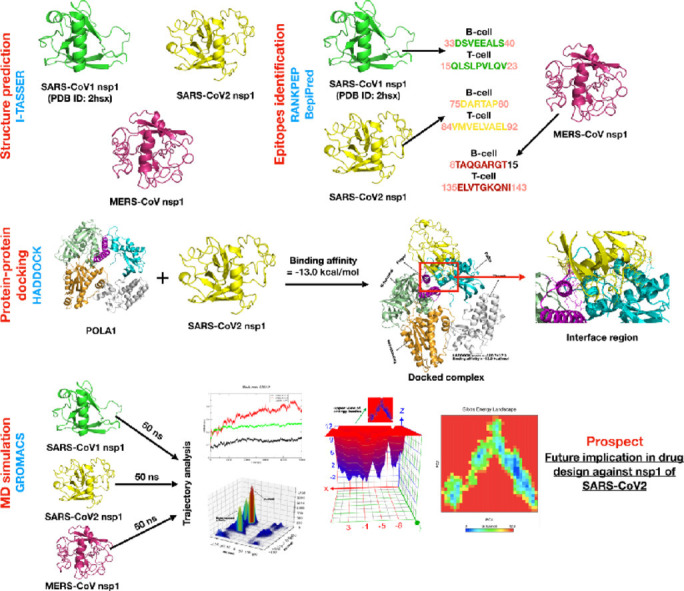
1. Introduction
In December 2019, the first epidemic novel coronavirus (SARS-CoV2) was identified in Wuhan city, China [1,2]. Since the initial outbreak, the WHO (World Health Organization) declared COVID-19 a pandemic level threat on March 11, 2020 given its global threat to human health and rapid spread. The causative agent of the COVID-19 disease is a severe acute respiratory syndrome coronavirus 2 (SARS-CoV2). The worldwide number of coronavirus cases reached 27,354,078 with a death toll of 8,94,005 as of Sep 07, 2020 (Last accessed Sep 07, 2020) [3]. Although it was first reported from China, the number of active cases in India, USA, Brazil, Russia, Spain, Italy, France, Germany, and UK have surpassed the cases identified in China (85,134) (Last accessed May 25, 2020) [3]. Almost all countries initiated social distancing and lockdown precautions to prevent and slow human to human transmission of the virus. Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses (ssRNA+) belonging to the Coronaviridae family. COVID-19 is a member of beta coronaviruses, like the other human coronaviruses SARS-CoV and MERS-CoV [2,4]. There are seven strains of human CoVs, which include NL63, 229E, HKU1, OC43, Middle East respiratory syndrome (MERS-CoV), severe acute respiratory syndrome (SARS-CoV or SARS-CoV1), and 2019-novel coronavirus (SARS-CoV2), responsible for the infection of the respiratory tract. Among these seven strains, three strains are highly pathogenic (SARS-CoV1, SARS-CoV2 and MERS-CoV) and are responsible for lower respiratory ailments like bronchiolitis, bronchitis, and pneumonia [5]. The genome of SARS-CoV2 is 30 kb and encodes structural and non-structural proteins like other CoVs. The two-thirds of the 5′ end of the CoV genome consists of two overlapping open reading frames (ORFs 1a and 1b) that encode non-structural proteins (nsps). The other one-third of the genome consists of ORFs that encode structural proteins. SARS-CoV2 structural proteins include S protein (Spike), E protein (Envelope), M protein (Membrane), and N protein (Nucleocapsid) [6,7]. The ORF1a and ORF1b encodes a polyprotein which is cleaved into sixteen non-structural proteins (nsp1–16) that form the replicase / transcriptase complex (RTC) [8]. Alpha and beta CoVs consists of 16 nsps, while gamma and delta CoVs, lacking nsp1, consists of 15 nsps (nsp2–16) [9]. The amino acid sequences of nsp1 homologs are highly divergent among CoVs [10]. It is among the least well-understood nsps, and other than in coronaviruses, no viral or cellular homologs are known. The nsp1 of SARS-CoV inhibits host gene expression by blocking the translation process through interaction with the 40S ribosomal subunit and degrades host mRNA via the recruitment of unidentified host nuclease(s) [11,12]. SARS-CoV nsp1 inhibits the expression of the IFN genes and the host antiviral signaling pathway in infected cells. The dysregulation of IFN genes is the key factor for inducing lethal pneumonia [13,14]. MERS-CoV nsp1 also induced mRNA degradation and translational suppression. SARS-CoV nsp1 also regulates the induction of cytokines and chemokines in human lung epithelial cells [15]. Thus, nsp1 is considered a major possible virulence factor for coronaviruses. SARS-CoV2 nsp1 antagonizes interferon induction to suppress the host antiviral response. The inflammatory phenotype of SARS-CoV and SARS-CoV2 pathology was also contributed by nsp1 protein [15]. The nsp1 protein of SARS-CoV2 interacts with six proteins of the infected host cells. They are POLA1, POLA2, PRIM1, PRIM2, PKP2 and COLGALT1. Four of these host proteins (POLA1, POLA2, PRIM1 and PRIM2) form DNA polymerase alpha complexes. These events raise the possibility that the nsp1 protein of SARS-CoV2 may interact with the DNA polymerase alpha complex and change its functional activity to antagonise the innate immune system [8].
In this study we propose the molecular model of the three nsp1 proteins and SARS-CoV2 nsp1-POLA1 complex. The epitope sites of SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 protein were identified by an immunoinformatics process. Molecular dynamics simulation, principal component analysis (PCA), and Gibbs free energy landscape (FEL) were performed to evaluate the structural flexibility and dynamic stability of the SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 protein. The mutated amino acids of SARS-CoV2 nsp1 protein were reported by using multiple sequence alignment. The binding interactions of SARS-CoV2 nsp1 protein with its host cell receptor POLA1 were determined by a protein-protein docking procedure to identify the important interacting residues at the interface region. The all-atom MD simulations for 50 ns on the three nsp1 proteins were performed to describe conformational changes under explicit solvent conditions. This study delivers an atomistic insight into the structure and conformation of nsp1 of SARS-CoV1, SARS-CoV2 and MERS-CoV by utilizing state-of-the-art computational approaches.
2. Materials and methods
2.1. Sequence retrieval
The protein sequences of nsp1 were retrieved from the curated NCBI database [16]. The accession numbers of the nsp1 of SARS-CoV1, SARS-CoV2 and MERS-CoV are NP_828860.2, YP_009725297.1 and YP_009047213.1, respectively. The pairwise sequence identity between COVID-19 nsp1 protein and each of the other HCoV nsp1 proteins (SARS-CoV1 and MERS-CoV) was calculated using BLASTp (basic local alignment tool) [17]. To check the conservation pattern, a multiple sequence alignment (MSA) of all of the nsp1 sequences was generated using the Clustal Omega program of the European Bioinformatics Institute (EMBL-EBI) [18].
2.2. Three dimensional structure prediction
The NMR structure of the non structural protein 1 (nsp1) of SARS-CoV1 (residues 13–127) was identified by Almeida et al. 2007 and deposited in Protein Data Bank (PDB) as ID no. 2hsx/2gdt [19]. Currently no crystallographic structures of nsp1 of SARS-CoV2 or MERS-CoV are available in Protein Data Bank (PDB). Thus, in silico modelling was employed to predict the three dimensional structure of nsp1 of SARS-CoV2 and MERS-CoV by using the I-TASSER web server [20]. I-TASSER (Iterative Threading Assembly Refinement) is a bioinformatics approach to predict the structure of proteins from their sequence. It first detects structural templates from the protein data bank database by fold recognition or multiple threading approach LOMETS [21]. The lowest energy predicted structures are subjected to a refinement process resulting in a final three dimensional protein structural model. I-TASSER (as 'Zhang-Server') has regularly been the top ranked server for prediction of protein structure in recent community-wide CASP (Critical Assessment of Protein Structure Prediction) method experiments [22]. The modelled nsp1 proteins were optimised to avoid any stereochemical restraints by steepest descent energy minimization method. The stereochemical quality of the nsp1 proteins was validated by Ramachandran plot using PROCHECK [23,24]. The models were further validated by ProSA [25], ProQ [26]. The ligand binding residues of nsp1 of SARS-CoV1, SARS-CoV2 and MERS-CoV were predicted by COACH meta server [27]. COACH generates complementary ligand binding sites of the target proteins by using two comparative processes, S-SITE and TM-SITE. These two methods recognize ligand-binding templates from the BioLiP database [28] by sequence profile comparisons and binding-specific substructure.
2.3. Prediction of T-cell (HLA class I and II) epitopes
The RANKPEP, sequence-based screening server was used to identify the T-cell epitopes [29] of the nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV. This server predicts the short peptide that binds to MHC molecules from protein sequences using the position-specific scoring matrix (PSSM). All the HLA class I alleles were selected for prediction of epitopes of HLA class I. For the prediction of epitopes of HLA class II, we selected some alleles such as DRB10101, DRB10301, DRB10401, DRB10701, DRB10801, DRB11101, DRB11301, and DRB11501 that cover HLA variability of over 95% of the human population worldwide [30].
2.4. B-cell epitopes (linear) identification
B-cell epitopes of the three nsp1 proteins were predicted by using BepiPred and Kolaskar and Tongaonkar Antigenicity (http://www.iedb.org/) servers [31]. BepiPred for linear epitope prediction uses both amino acid propensity scales and hidden Markov model methods. The cut off score for linear B-cell epitopes prediction is 0.50. Kolaskar and Tongaonkar evaluate the protein for B cell epitopes using the physicochemical properties of the amino acids and their frequencies of occurrence in recognized B cell epitopes [32,33].
2.5. Molecular dynamics simulation
Molecular dynamics simulations were used to predict the dynamic behaviour of the protein macromolecule. The nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV were subjected to MD simulation by using Gromacs v 2018.2 software suite [34] with OPLS-AA force field [35]. The three systems were solvated in a cubic box with SPC (simple point charge) water model [36] by maintaining periodic boundary condition (PBC) through the simulation process. Sodium and chloride ions were added to neutralize the three systems. Each system was energy minimized using the steepest descent algorithm until the maximum force was found to be smaller than 1000.0 kJ/mol/nm. This was done to remove any steric clashes on the system. Each system was equilibrated with 100 ps isothermal-isochoric ensemble, NVT (constant number of particles, volume, and temperature) followed by 100 ps isothermal-isobaric ensemble NPT (constant number of particles, pressure, and temperature). The two types of ensemble of equilibration methods stabilized the three systems at 310 K and 1 bar pressure. The Berendsen thermostat and Parrinello-Rahman were applied for temperature and pressure coupling methods, respectively [37]. Particle Mesh Ewald (PME) method [38] was used for the calculations of the long-range electrostatic interactions and the cut off radii for Van der Waals and coulombic short-range interactions were set to 0.9 nm. The Linear Constraint Solver (LINCS) constraints algorithm was used to fix the lengths of the peptide bonds and angles [39]. All the three systems were subjected to MD simulations for 50 ns. The resulting MD trajectories were utilized through the inbuilt tools of GROMACS for analysis purposes. The subsequent analyses were performed using VMD [40], USCF Chimera [41], Pymol [42], and plots were generated with xmgrace [43].
2.6. Principal components analysis (PCA) or essential dynamics
Principal components analysis or essential dynamics is a process which extracts the essential motions from the MD trajectory of the targeted protein molecule [44]. The nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV were analysed by PCA. The initial step of PCA analysis is to construct the covariance matrix which examines the linear relationship of atomic fluctuations for individual atomic pairs. The diagonalization of covariance matrix results in a matrix of eigenvectors and eigenvalues. The eigenvectors determine the movement of atoms having corresponding eigenvalues which represents the energetic contribution of an atom participating in motion. The covariance matrix and eigenvectors were analysed using the gmx cover and gmx anaeig tool respectively. Gibbs free-energy landscape (FEL) elucidates the protein dynamic processes by representing the conformational states and the energy barriers to those states [45]. The FEL of SARS-CoV1, SARS-CoV2 and MERS-CoV was constructed based on the first (PC1) and second (PC2) principal components, Rg and rmsd, and psi and phi angles. FEL was calculated and plotted by using gmx sham and gmx xpm2ps module of GROMACS.
2.7. Protein-protein docking
Nsp1 protein of SARS-CoV2 interacts with POLA1 and blocks the host cell replication process. [8]. The molecular interactions of nsp1 protein of SARS-CoV2 (COVID-19) with the catalytic subunit of human DNA polymerase alpha, POLA1 was analysed by using the HADDOCK (High Ambiguity Driven protein-protein DOCKing) program. It is a flexible docking approach for the modelling of biomolecular complexes. It encodes instruction from predicted or identified protein interfaces in ambiguous interaction restraints (AIRs) to drive the docking procedure [46]. The coordinates of the solved structure of the catalytic domain of DNA polymerase alpha, POLA1 was downloaded from PDB database (PDB ID: 6AS7) and prepared for the docking experiments by removing water, ions and the ligands. The interface residues were utilized for the docking procedure. The active residues of POLA1 (Asp860, Ser863, Leu864, Arg922, Lys926, Lys950, Asn954 and Asp1004) were retrieved from the literature [47]. The active residues (Leu16, Leu18, Phe31, Val35, Glu36, Leu39, Arg43, Leu46, Gly49, Iso71, Pro109, Arg119, Val121 and Leu123) of nsp1 of SARS-CoV2 were predicted from COACH server. The amino acids surrounding the active residues of both proteins were selected as passive in the docking procedure. Active residues are the amino acids from the interface region of the two proteins that take part in direct binding with the other protein partner while passive residues are the amino acids that can interact indirectly during the docking procedure. Approximately 163 structures in 8 clusters were obtained from HADDOCK server, which represented 81.5% of the water-refined models. PRODIGY software [48] was used to predict the binding affinity and dissociation constant for each SARS-CoV2 nsp1-POLA1 complex from the best three clusters. The generated model of the SARS-CoV2/nsp1-POLA1 complex was optimized to avoid any stereochemical restraints by steepest descent energy minimization method. The optimized complex was validated by Ramachandran plot analysis of ψ/φ angle from PROCHECK. PISA server (http://www.ebi.ac.uk/msd-srv/prot_int/, Last accessed May 25, 2020) was used to calculate total buried surface area, nature of interactions and amino acids involved in interactions at interface region.
3. Results and discussion
3.1. Sequence analysis and protein structure prediction
The sequence identity of nsp1 protein of SARS-CoV2 with SARS-CoV1 and MERS-CoV was 84.4% and 20.61%, respectively. A multiple sequence alignment (MSA) of nsp1 proteins was generated to identify the conserved residues. The amino acids marked as asterisk illustrate the positions of nsp1 protein that were highly conserved over the evolutionary time scale (Fig. 1 ). The differences in the amino acid changes were also recorded. It was observed that compared to SARS-CoV1, there were 28 mutations in the nsp1 protein of SARS-CoV2 (Fig. 1). The 3D structures of the nsp1 homologs of SARS-CoV2 and MERS-CoV were modelled using the I-TASSER web server (Fig. 2 ). The NMR structure of SARS-CoV1 nsp113–127 (PDB: 2hsx/2gdt) was identified as the template for three dimensional structure prediction of SARS-CoV2 nsp1 on the I-TASSER server. The highly significant templates used in the modelling of the nsp1 protein of SARS-CoV2 and MERS-CoV through I-TASSER are listed in Table 1 . High Z score (>1 means good alignment) and good coverage for most of the structural templates indicates confidence in the structural model in both cases. As shown from the topological analysis, SARS-CoV2 nsp1 13–127 exhibits similar α/β-folds with SARS-CoV nsp113–127 that consist of a characteristic six-stranded β-barrel and a long α-helix. It was also observed that a short α helix (163–168) is present in the C-terminal region of the of the SARS-CoV2 nsp1 protein. This α-helix may play an important role in the inhibition of the host protein synthesis process [49]. In the case of MERS-CoV nsp1, the structure consists of an eight-stranded β-barrel and four α-helices, which is different from the SARS-CoV1 and SARS-CoV2 nsp1 (Fig. S1). The stereochemical quality of the nsp1 models was validated on the basis of Ramachandran analysis of ψ/φ angle from PROCHECK. Examination of Ramachandran plot of nsp1 protein of SARS-CoV2 and MERS-CoV showed above 92% residues lie in the allowed regions (Table S1). From ProSA and ProQ analysis, it is clear that the overall model quality of the SARS-CoV2 and MERS-CoV nsp1s are within the range of scores typically found for proteins of similar size (Table S1). The important residues of the nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV that are involved in the ligand binding process are listed in Table 2 .
Fig. 1.

Multiple sequence alignment for nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV. The alignment is shown using the Clustal Omega web server. Asterisks represent conserved residues. Mutated residues of SARS-CoV2 nsp1 protein are highlight with a red box (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Fig. 2.

Prediction of three dimensional structure of nsp1 protein by I-TASSER program. A. Modelled structure of SARS-CoV2 nsp1 protein. B. Modelled structure of MERS-CoV nsp1 protein. C. Superimposition of SARS-CoV1 (PDB ID: 2hsx) with SARS-CoV2 and MERS-CoV nsp1 (Orange, blue and green colour represents the SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1, respectively) (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Table 1.
The highly significant structural templates for sequence alignment obtained from PDB library for modelling through I-TASSER.
| SARS-CoV2 | MERS-CoV | ||
|---|---|---|---|
| PDB ID | Normalized Z Score | PDB ID | Normalized Z Score |
| 2hsxA | 1.83 | 2hsx | 8.11 |
| 2 gdtA | 2.52 | 2hsx | 9.76 |
| 2 gdt | 1.70 | 2hsx | 5.19 |
| 2hsx | 10.03 | 5xbc | 1.43 |
| 2hsx | 4.59 | 3zbd | 1.14 |
| 2hsxA | 2.14 | 2v0gA | 0.72 |
| 2hsx | 7.61 | 2 gdtA | 0.95 |
| 2 gdtA | 3.07 | 2p97A | 0.63 |
| 2 gdtA | 2.75 | 1v8eA | 0.60 |
| 2hsxA | 2.34 | 3fdfA | 0.69 |
Table 2.
Active residues prediction by COACH server.
| Nsp1 protein | Active site residues |
|---|---|
| SARS-CoV1 | Leu16, Leu18, Phe31, Leu39, Arg43, Leu46, Gly49, Iso71, Pro109, Arg119, Val121, Leu123 |
| SARS-CoV2 | Leu16, Leu18, Phe31, Val35, Glu36, Leu39, Arg43, Leu46, Gly49, Iso71, Pro109, Arg119, Val121, Leu123 |
| MERS-CoV | Arg13 |
3.2. Defining T-cell and linear B-cell epitopes
Several studies revealed that a specific T-cell response is required for the elimination of several viral infections such as influenza A, SARS-CoV, MERS-CoV and para-influenza. These studies conclude that T-cell mediated response is essential for the development of specific vaccines [50,51]. CD8+ cytotoxic T-cells recognize the infected cells in the lungs whereas CD4+ helper T-cells are essential for the production of specific antibodies against viruses. Here we used the RankPep server to predict peptide binders to MHC class I and MHC class II alleles from nsp1 protein sequences by using Position Specific Scoring Matrices (PSSMs). The antigenic epitopes of three nsp1 proteins with high binding affinity were predicted and summarized in Tables 3 and 4 . Secreted neutralising antibodies play an important role to protect the body against viruses. The entry process of the viruses is blocked by the SARS-CoV specific neutralizing antibodies [52]. The Bepipred web server was employed for the linear B-cell epitope prediction study. SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 proteins were used for this purpose. The Kolaskar and Tongaonkar Antigenicity method was employed for cross-checking of the predicted epitopes. The linear B-cell epitopes of the three nsp1 proteins are depicted in Table 5 . Both humoral and cellular immune responses are important factors against coronavirus infection [52]. Finally, in SARS-CoV2 nsp1 protein, four epitope rich regions (15–27, 45–81, 121–140 and 147–178) that were shared between T-cell and B-cell were reported. This information will be helpful for vaccine design by targeting SARS-CoV2 nsp1 protein.
Table 3.
HLA I antigenic epitopes predicted using Rankpep.
| Sl. No | Alleles | SARS-CoV1 | SARS-CoV2 | MERS-CoV |
|---|---|---|---|---|
| 1 | HLA_A0201 | 15–23 QLSLPVLQV 84–92 KVVELVAEM 103–111 TLGVLVPHV 169–177 ALRELTREL |
84–92 VMVELVAEL 15–23 QLSLPVLQV 106–114 VLVPHVGEI |
135–143 ELVTGKQNI 89–97 YLVERLIAC 62–70 MLLKKEPLL 75–83 RLAGHTRHL |
| 2 | HLA_A0204 | 78–86 STNHGHKVV |
78–86 TAPHGHVMV 45–53 HLKDGTCGL |
80–88 TRHLPGPRV 3–11 FVAGVTAQC |
| 3 | HLA_A0206 | 103–111 TLGVLVPHV 38–46 ALSEAREHL |
106–114 VLVPHVGEI 103–111 TLGVLVPHV |
- |
| 4 | HLA_A0301 | 121–129 VLLRKNGNK |
121–129 VLLRKNGNK |
57–66 EVVKAMLLKK |
| 5 | HLA_A11 | - | - | 58–66 VVKAMLLKK |
| 6 | HLA_A1101 | 76–84 ALSTNHGHK |
3–11 SLVPGFNEK |
- |
| 7 | HLA_A2402 | 96–104 QYGRSGITL |
153–161 PYEDFQENW 96–104 QYGRSGETL |
- |
| 8 | HLA_A31 | 116–124 IAYRNVLLR |
116–124 VAYRKVLLR |
154–162 HYTPFHYER |
| 9 | HLA_A6801 | 116–124 IAYRNVLLR 76–84 ALSTNHGHK |
- | - |
| 10 | HLA_B0702 | - | 114–122 IPVAYRKVL |
- |
| 11 | HLA_B2705 | - | 76–84 ARTAPHGHV |
74–82 IRLAGHTRH |
| 12 | HLA_B35 | - | - | 124–132 KPIGMFFPV |
| 13 | HLA_B3501 | 61–69 LPQLEQPYV |
61–69 LPQLEQPYV |
33–42 VPLCGSGNLV 99–108 NPFMVNQLAY |
| 14 | HLA_B51 | 61–69 LPQLEQPYV 108–116 VPHVGETPI |
61–69 LPQLEQPYV |
83–91 LPGPRVYLV 33–41 VPLCGSGNL |
| 15 | HLA_B5101 | 108–116 VPHVGETPI 18–26 LPVLQVRDV |
108–116 VPHVGEIPV 114–122 IPVAYRKVL 18–26 LPVLQVRDV |
- |
| 16 | HLA_B5102 | 61–69 LPQLEQPYV 18–26 LPVLQVRDV |
- | - |
| 17 | HLA_B5103 | 108–116 VPHVGETPI 18–26 LPVLQVRDV |
18–26 LPVLQVRDV |
- |
| 18 | HLA_B5401 | - | - | 83–91 LPGPRVYLV |
| 19 | HLA_X | 169–177 ALRELTREL |
- | 82–90 HLPGPRVYL 124–132 KPIGMFFPY 128–136 MFFPYDIEL |
Table 4.
HLA II antigenic epitopes predicted using Rankpep.
| Sl. No | Alleles | SARS-CoV1 | SARS-CoV2 | MERS-CoV |
|---|---|---|---|---|
| 1 | HLADRB10101 | 68–76 YVFIKRSDA |
71–79 IKRSDARTA 68–76 YVFIKRSDA |
108–116 YSSSANGSL 99–107 NPFMVNQLA 3–11 FVAGVTAQG 80–88 TRHLPGPRV |
| 2 | HLADRB10401 | 157–165 YEQNWNTKH 97–105 YGRSGITLG 68–76 YVFIKRSDA 116–124 IAYRNVLLR |
97–105 YGRSGETLG 157–165 FQENWNTKH 68–76 YVFIKRSDA 55–63 EVEKGVLPQ |
3–11 FVAGVTAQG 2–10 SFVAGVTAQ 106–114 YERDNTSCP 114–122 GSLVGTTLQ |
| 3 | HLADRB10701 | - | - | 95–103 IACENPFMV |
| 4 | HLADRB11101 | 159–167 QNWNTKHGS 163–171 TKHGSGALR |
159–167 ENWNTKHSS 169–177 VTRELMREL |
59–67 VKAMLLKKE 182–190 YAQNLLKKL |
| 5 | HLADRB11501 | 54–62 VELEKGVLP 20–28 VLQVRDVLV 133–141 GHSYGIDLK |
54–62 VEVEKGVLP 20–28 VLQVRDVLV 133–141 GHSYGADLK 69–77 VFIKRSDAR |
126–134 IGMFFPYDI 56–64 YEVVKAMLL |
Table 5.
Predicted linear B-cell epitopes of SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 protein via Bepipred and Kolaskar and Tongaonkar antigenicity.
| Antigen | Server | Amino acid position | Sequece |
|---|---|---|---|
| SARS-CoV1 | Bepipred | 33–40 76–79 95–97 111–116 127–135 145–168 |
DSVEEALS ALST IQY VGETPI GNKGAGGHS LGDELGTDPIEDYEQNWNTKHGSG |
| Kolaskar and Tongaonkar | 13–29 51–72 83–89 103–114 118–124 137–143 |
HVQLSLPVLQVRDVLVR CGLVELEKGVLPQLEQPYVFIK HKVVELV TLGVLVPHVGET YRNVLLR GIDLKSY |
|
| SARS-CoV2 | Bepipred | 9–11 33–34 45–46 75–80 97–102 127–138 148–168 |
NEK DS HL DARTAP YGRSGE GNKGAGGHSYGA ELGTDPYEDFQENWNTKHSSG |
| Kolaskar and Tongaonkar | 13–29 51–72 81–92 104–124 |
HVQLSLPVLQVRDVLVR CGLVEVEKGVLPQLEQPYVFIK HGHVMVELVAEL LGVLVPHVGEIPVAYRKVLLR |
|
| MERS-CoV | Bepipred | 8–15 22–26 50 53–56 81–84 109–116 121–123 150–154 160–162 |
TAQGARGT SEKHQ M ENA RHLP SSSANGSL LQG RGGYH YERDNTSCPEWMDDFEADPKGKY |
| Kolaskar and Tongaonkar | 26–39 56–62 66–76 85–98 103–109 131–137 |
QDHVSLTVPLCGSG YEVVKAM KEPLLYVPIRL GPRVYLVERLIACE VNQLAYS PYDIELV |
3.3. MD simulation analysis
MD simulation methods have been utilized as a computational tool to explore the conformational behavior of proteins. Here, to understand the structural behaviour of three nsp1 proteins, we have performed MD simulation studies for 50 ns. To determine the equilibration and system stability during the simulations of three nsp1 proteins, the backbone RMSDs, radius of gyration (Rg), solvent accessible surface area (SASA), and root mean square fluctuations (RMSF) were calculated and monitored over the course of 50 ns simulations and are represented in Fig. 3 . Evaluation of the structural drift was provided by the analysis of the RMSDs from the initial structures as a function of simulation time. The resulting RMSD graph was explored to assess the conformational changes during simulation. The nsp1 of SARS-CoV1 shows the smallest deviation during the initial 0–15 ns and achieved equilibrium at ~15 ns and it remained stable for the period of 50 ns. This suggests no significant changes observed during simulation. The backbone RMSD of SARS-CoV2 nsp1 increases sharply till 14 ns and reaches a value from 0.2 to 0.7 nm. Thereafter, it showed flexible stability at ~0.7 to 0.85 nm during ~ 18 to 44 ns. After 44 ns RMSD drop down ~ 0.68 nm. This maximum RMSD fluctuation indicated that the SARS-CoV2 nsp1 was undergoing a large conformational change during the simulation. In case of MERS-CoV nsp1, the backbone RMSD deviates ~ 0.44 nm during first 8 ns and remained stable thereafter. (Fig. 3 A). The average backbone RMSDs of SARS-CoV1, SARS-CoV2 and MERS-CoV were 0.25 nm, 0.77 nm and 0.44 nm, respectively (Fig. 3 A). SASA analysis suggested that the exposure of the three nsp1 protein surfaces to the solvent and the changes in solvent accessibility could lead to conformational changes of the nsp1 proteins. Fig. 3 B shows the variations in SASA for the SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 protein with respect to simulation time. The average value of SASA of SARS-CoV1, SARS-CoV2 and MERS-COV was 68 nm2, 110 nm2, and 118 nm2, respectively. The SASA values for the SARS-CoV1 were reduced when compared with the case of SARS-CoV2 and MERS-CoV. Overall, the SASA of three systems seems to attain a stable equilibrium without any major peak during the simulation, signifying the stability of compactness. Root mean square fluctuations (RMSFs) of each amino acid highlights the flexible regions of the three nsp1 systems. RMSFs values higher than 0.25 nm are characteristic of amino acid residues belonging to flexible regions. The RMSF plot of SARS-CoV1 nsp1 showed less flexible sites (67, 70, 116) during the simulation. The most significant conformational shifts occurred in seven regions: 1–3, 6–11, 32–36, 58–60, 80–81, 99, 126–129, 130–138, 144–148, 150–160 and 165–180 of SARS-CoV2 nsp1 protein. The higher fluctuations are related to the residues of a loop and helix regions. The RMSF analysis of MERS-CoV1 nsp1 showed that residues:1, 18–21, 49–52, 98–99, 113–119, 193 exhibited fluctuations during simulation (Fig. 3 C). The high RMSF values of SARS-CoV2 indicated more flexibility in this protein compared with SARS-CoV1 and MERS-CoV (Fig. 3 C). To further understand the structural stability of the three nsp1 proteins, we studied the compactness parameter of the structure by determining the radius of gyration (Rg). Radius of gyration (Rg) is defined as the root mean square deviation between the center of gravity of the respected protein and its end. Rg defines the stability and firmness of the simulation system and can reveal protein folded-unfolded states [53]. The average Rg value was 1.34 nm, 1.69 nm and 1.61 nm for SARS-CoV1, SARS-CoV2 and MERS-CoV respectively. The Rg plot of SARS-CoV2 showed a smallest deviation till 14 ns. Thereafter it showed flexible stability. However, the Rg plot shows two small drifts during 50 ns simulation. The gyration curve showed a decrease in the overall Rg value of the SARS-CoV1 nsp1 protein compared with the SARS-CoV2 and MERS-CoV, indicating that nsp1 protein of SARS-CoV1 was in a compact, well folded state (Fig. 3 D). From RMSD, RMSF and Rg analysis, it was observed that SARS-CoV2 shows flexible stability during MD simulation. An overall trend of backbone RMSD, SASA, RMSF and radius of gyration indicated that all three nsp1 protein systems were well equilibrated and stable during the simulation run. Hydrogen bonds provide most of the directional interactions that represents protein structure, protein folding and molecular recognition. The formation of hydrogen bonds as a function of simulation time was analysed. The average number of hydrogen bonds of SARS-CoV1, SARS-CoV2 and MERS-CoV were 69±5, 92±10 and 90±11, respectively ( Fig. 4 ).
Fig. 3.

Analysis of MD simulation of three nsp1 proteins. A. Root mean square deviation (RMSD). B. Solvent accessible surface area (SASA). C. Root mean square fluctuations (RMSF). D. Radius of gyration. Black, red and green colour represents the SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1, respectively. Flexible regions of each nsp1 are highlighted with circles (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Fig. 4.

Trajectory analysis of hydrogen bonds. Hydrogen bonds are responsible for the stability of the protein molecules. Black, red and green colour depicts the number of hydrogen bonds of the SARS-CoV1, SARS-CoV2 and MERS-CoV, respectively throughout the simulation run (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.4. Ramachandran (ψ/Φ) space of the residues by molecular dynamics simulation
The frequency of dihedral angles phi (Φ) and psi (Ψ) was monitored during the 50 ns simulation time. The plot of the dihedral angle frequencies in a Ramachandran-like graph provides conformational preferences of the three nsp1 proteins. The allowed regions of the Ramachandran plot of three nsp1 proteins are delimited by green lines in Fig. 5 . High peaks mean that this combination of Ramachandran-angles is occupied often during simulation. In the case of SARS-CoV1, the major Ramachandran ψ/Φ angle distribution, as obtained by the MD analysis was found to peak at Ramachandran coordinates of −100° ≤ Φ ≤ −60° and −55° ≤ Ψ ≤ −40°. The other comparatively medium distributions were at −80° ≤ Φ ≤ −60° and 120° ≤ Ψ ≤ 150° in the Ramachandran (ψ, Φ) space. The highly populated Φ/Ψ values were close to the right-handed α-helical space and medium populated Φ/Ψ values were close to the β-sheet space (Fig. 5A and Table 6 ). In case of SARS-CoV2, it is worth noting that the distribution of β-sheets (−90° ≤ Φ ≤ −70°, 110° ≤ Ψ ≤ 150°) is more pronounced in comparison to that for α-helices (−95° ≤ Φ ≤ −65°, −40° ≤ Ψ ≤ −30°) (Fig. 5B and Table 6). The most highly populated (Φ, Ψ) angles for MERS-CoV were −90° ≤ Φ ≤ −65° and 120° ≤ Ψ ≤ 150°, which are by definition β-sheets. The other medium and weak distributions were at right-handed α-helix and left handed α-helix regions, respectively (Fig. 5C and Table 6). The analysis of the Ramachandran (ψ/Φ) space of three nsp1 proteins suggests that the distribution of the secondary structures spanned mainly swings between α-helices and β-sheets.
Fig. 5.

2D and 3D Ramachandran plots obtained by sampling the dihedrals of the three nsp1 proteins during the 50 ns simulations. A. SARS-CoV1. B. SARS-CoV2. C. MERS-CoV. In the 2D plots, the regions outlined by green lines are the most populated and are associated with a well-defined secondary structure. In the 3D plots, the most probable structures are colored in deep orange, while the least probable structures are coloured in blue. High peaks mean that this combination of Ramachandran-angles is assumed often during simulation (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Table 6.
Ramachandran space of three nsp1 proteins.
| Name of the protein | Dihedral angles | Frequency of population | Secondary structure |
|---|---|---|---|
| 1. SARS-CoV1 | −100° ≤ Φ ≤ −60°, −55° ≤ Ψ ≤ −40° |
Higher | Right-handed α-helix |
| −80° ≤ Φ ≤ −60°, 120° ≤ Ψ ≤ 150° |
Medium | β -sheet | |
| 2. SARS-CoV2 | −90° ≤ Φ ≤ −70°, 110° ≤ Ψ ≤ 150° |
Higher | β -sheet |
| −95° ≤ Φ ≤ −65°, −40° ≤ Ψ ≤ −30° |
Lower | Right-handed α-helix | |
| 3. MERS-CoV | −90° ≤ Φ ≤ −65°, 120° ≤ Ψ ≤ 150° |
Higher | β -sheet |
| −90° ≤ Φ ≤ −60°, −40° ≤ Ψ ≤ −30° |
Medium | Right-handed α-helix |
3.5. Principal component analysis (PCA) and Gibbs free energy landscape (FEL)
The overall pattern of motion of the atoms was monitored using the MD trajectories projected on the first (PC1) and second (PC2) principal components to gain a better understanding of the conformational changes in the nsp1 protein of SARS-CoV1, SARS-CoV2 and MERS-CoV. The eigen vectors described the collective motion of the atoms, while the eigenvalues signified the atomic influence in movement. A large distribution of lines indicated greater variance in accordance with more conformational changes in the SARS-CoV2 nsp1 protein compared with SARS-CoV1 and MERS-CoV nsp1 protein. The trajectories of SARS-CoV2 nsp1 protein covered a wider conformational space and showed higher space magnitudes. The trace values, which are the sums of the eigenvalues, were 4.71 nm2, 44.082 nm2, and 21.611 nm2 for SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1, respectively. It was suggested that SARS-CoV2 nsp1 protein appeared to cover a larger conformational space due to its greater flexibility when compared with the other two nsp1 proteins (Fig. 6 ). This observation correlates with the trajectory of the stability parameters such as RMSD, RMSF and Rg of SARS-CoV2 nsp1. It is evident from the PCA plot that the collective motions of the SARS-CoV1 nsp1 protein are localized in a small subspace compared to the SARS-CoV2 and MERS-CoV nsp1 (Fig. 6)
Fig. 6.

Projection of motion of nsp1 protein atoms of SARS-CoV1 (Black colour), SARS-CoV2 (red colour) and MERS-CoV (green colour) on PC1 and PC2 (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
The Gibbs free energy landscape (FEL) deliver a precise portrayal of a protein's most stable conformational space, which are important when analysing the conformational changes during simulation. Gibbs free energy landscape (FEL) was calculated by using PC1, PC2 coordinates and RMSD, Rg coordinates of the three nsp1 protein molecules. Both 2D and 3D graphs of the FEL were plotted using PC1, PC2 and RMSD, Rg coordinates. The ∆G values for SARS-CoV1, SARS-CoV2 and MERS-CoV nsp1 protein were 0 to 13.3, 12.2 and 12.9 kJ/mol, respectively. The broader, shallow and narrow energy basin were observed during the trajectory analysis of the simulation. Each nsp1 protein has a different pattern for the FEL. SARS-CoV1 nsp1 has less conformational mobility, restricted to a more confined conformational space within a single local basin with the lowest energy (Fig. 7A ). Extraction of snapshots from these deep energy wells represent the most native-like conformation with a RMSD of 0.2702 nm and Rg of 1.2807 nm (Fig. S2A). In the case of SARS-CoV2 nsp1, local minima distributed to about three to four energy basins within the energy landscape which indicates a wide range of conformations (Fig. 7 B). One of the well populated conformations was located at a RMSD of around 0.5433 nm and a Rg of around 1.7332 nm (Fig. S2B). MERS-CoV nsp1 has a broader and narrow with two to three energy basins (Fig. 7 C). The native-like conformation from the low energy basins was centerd at the coordinate (RMSD = 0.5724 nm, Rg = 1.6221 nm) (Fig. S2C). Fig. 7 displays the FELs of SARS-CoV1, SARS-CoV2 and MERS-COV nsp1 proteins where the deeper blue indicates the stable conformational states having lower energy.
Fig. 7.

2D and 3D plots of Gibbs free energy landscape (FEL) of three nsp1 proteins. A. SARS-CoV1. B. SARS-CoV2. C. MERS-CoV. The blue, cyan and green regions in the free energy landscape plot denotes low energy state with highly stable protein conformation while the red region signify high energy state with unstable protein conformation (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.6. Molecular interactions of SARS-CoV2 nsp1 with POLA1
The catalytic domain of DNA polymerase alpha, POLA1 is involved in the replication process. The molecular association of SARS-CoV2 nsp1 with POLA1 was predicted by the HADDOCK program. The nsp1-POLA1 docked complexes were analysed based on Z-score and HADDOCK score (Fig. 8 and Table S2). PRODIGY was used to predict the binding energy for each nsp1-POLA1 complex from the best three cluster. The three best docked complexes were selected on the basis of lowest binding energy. (Fig. S3). The best energy values obtained were −13.0 , −11.6 and −9.6 kcal/mol. (Table S2). Interface area, involvement of amino acids and molecular interactions were calculated by PISA server. The interface area of the best docked complex (ΔG = −13.0 kcal/mol) was 1260.9 Å2 and shown in Fig. 9 A. The best complex structure of SARS-CoV2 nsp1-POLA1 was evaluated by the Ramachandran plot which shows 98.4% of residues are present in the allowed region. From ProSA and ProQ analysis, it is clear that the overall model quality of the final protein–protein complex is within the range of scores typically found for proteins of similar size (Table S1). Interaction studies of this nsp1-POLA1 complex showed thirteen hydrogen bonds and eight salt bridge interactions at the interface region (Table 7 and Fig. 9 B). It was observed from the docking experiments that the residues of finger domain (Lys923, Gln927, Gln932) and palm domain (Glu1060, Lys1020, Lys1024, Asn1030, Lys1031 and Glu1037) of POLA1 are mainly involved in the binding process with SARS-CoV2 nsp1 protein (Fig. 8). The hydrogen bonds and salt bridge interactions play an important role in the stability of the SARS-CoV2 nsp1-POLA1 complex formation.
Fig. 8.

Model of docking complex between SARS-CoV2 nsp1 protein and POLA1. SARS-CoV2 nsp1 is represented by a yellow cartoon. POLA1 is composed of five domains. N-terminal (338–534 and 761–808), exonuclease (535–760), palm (834–908 and 968–1076), finger (909–967), and thumb (1077–1250) domain are represented by pale green, orange, cyan, magenta and grey colour respectively. The binding affinity of nsp1 is higher at the interface region of palm (cyan colour) and finger (magenta colour) domain of POLA1 (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Fig. 9.

The proposed binding mode of the host cell POLA1 and the COVID-19 nsp1 model. A. Nsp1 (yellow surface) interacts with the palm (cyan surface) and finger (magenta surface) domain. Interface region is represented by a red surface. B. Molecular interactions between SARS-CoV2 nsp1 and POLA1. Interface residues are represented as a line model. Several bonds are denoted by an orange dotted line (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Table 7.
Intermolecular interactions of the best docked complex of SARS-CoV2 nsp1-POLA1 predicted by PISA analysis.
| Sl.No | Amino acids of nsp1 | Amino acids of POLA1 | Distance (Å) | Interactions |
|---|---|---|---|---|
| 1 | Glu 2 [OE2] | Lys 923 [HZ2] | 1.63 | Hydrogen bond |
| 2 | Lys47 [O] | Lys 923 [HZ1] | 1.82 | Hydrogen bond |
| 3 | Glu 2 [OE1] | Gln 927 [HE21] | 1.81 | Hydrogen bond |
| 4 | Met 1 [O] | Gln 927 [HE22] | 2.17 | Hydrogen bond |
| 5 | Pro 115 [O] | Gln 932 [HE21] | 1.78 | Hydrogen bond |
| 6 | Asp 33 [OD2] | Lys 1020 [HZ1] | 1.62 | Hydrogen bond |
| 7 | Glu 41 [OE1] | Ans 1030 [HD21] | 1.78 | Hydrogen bond |
| 8 | Glu 36 [O] | Lys 1031 [HZ3] | 1.80 | Hydrogen bond |
| 9 | Met 1 [N] | Gln 927 [OE1] | 3.50 | Hydrogen bond |
| 10 | Arg 29 [HH21] | Glu 1016 [OE2] | 2.11 | Hydrogen bond |
| 11 | Arg 29 [HH11] | Glu 1016 [OE2] | 1.94 | Hydrogen bond |
| 12 | Lys47 [HZ3] | Glu 1037 [OE1] | 1.63 | Hydrogen bond |
| 13 | Lys47 [HZ1] | Glu 1037 [OE2] | 2.36 | Hydrogen bond |
| 14 | Glu 2 [OE2] | Lys 923 [NZ] | 2.57 | Salt bridge |
| 15 | Asp 33 [OD2] | Lys 1020 [NZ] | 2.64 | Salt bridge |
| 16 | Glu 37 [OE1] | Lys 1024 [NZ] | 2.88 | Salt bridge |
| 17 | Glu 37 [OE2] | Lys 1024 [NZ] | 2.91 | Salt bridge |
| 18 | Arg 29 [NH2] | Glu 1016 [OE2] | 2.97 | Salt bridge |
| 19 | Arg 29 [NH1] | Glu 1016 [OE2] | 2.85 | Salt bridge |
| 20 | Lys47 [NZ] | Glu 1037 [OE1] | 2.67 | Salt bridge |
| 21 | Lys47 [NZ] | Glu 1037 [OE2] | 2.70 | Salt bridge |
4. Conclusion
The COVID-19 pandemic has led to a health, economic, and social crisis in the world. The development of a specific targeted therapy could reduce the rate of infection. This comprehensive study represents an immunoinformatics approach towards the identification of specific B-cell and T-cell epitopes of three nsp1 proteins. Four epitope rich regions (15–27, 45–81, 121–140 and 147–178) that were shared between T-cell and B-cell were reported in SARS-CoV2 nsp1 protein. The in-depth structural elucidation of nsp1 proteins together with dynamic conformations showed that SARS-CoV2 nsp1 protein covers a large conformational space due to its greater flexibility compared with SARS-CoV1 and MERS-CoV nsp1. A three-dimensional structural model of the complex structure between SARS-CoV2 nsp1 protein and catalytic subunit of DNA polymerase alpha POLA1 was constructed using a protein-protein docking approach. During complex formation between SARS-CoV2 nsp1 and POLA1, salt bridge interactions help to bring the two proteins in close proximity and form 13 strong hydrogen bonds that contribute to the stability of the complex formation. Knowledge of this important binding site could enable further simulation and experimental studies on the mode of SARS-CoV2 nsp1 protein recognition by the catalytic site of DNA polymerase alpha POLA1. From FEL analysis, it was observed that SARS-CoV1 and MERS-CoV nsp1 show stable RMSD and Rg such that their energy minimas are tightly confined. Taken together, structural evaluation and immunological analysis suggest that the nsp1 protein could be considered as a possible drug target and candidate molecule for the vaccine development process against COVID-19.
Data availability
The modelled and docking structures are available upon request from the corresponding author.
CRediT authorship contribution statement
Ankur Chaudhuri: Supervision, Conceptualization, Formal analysis, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgment
The author wishes to express sincere thanks to Dr. Sibani Sen Chakraborty, West Bengal State University, Kolkata, India and Dr. Asim Kumar Bera, Institute for Protein Design, University of Washington, Seattle, USA for their useful suggestions in preparation of the manuscript. I must acknowledge Dr. Samuel Pellock, Institute for Protein Design, University of Washington, Seattle, USA, for graciously editing the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.130854.
Appendix. Supplementary materials
References
- 1.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/s0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.-.M., Wang W., Song Z.-.G., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(worldometers.info. (2020). COVID-19 coronavirus pandemic. https://www. worldometers.info/coronavirus/(Last accessed May 25, 2020)
- 4.Elfiky A.A., Mahdy S.M., Elshemey W.M. Quantitative structure-activity relationship and molecular docking revealed a potency of anti-hepatitis C virus drugs against human corona viruses. J. Med. Virol. 2017;89:1040–1047. doi: 10.1002/jmv.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bogoch I.I., Watts A., Thomas-Bachli A., Huber C., Kraemer M.U.G., Khan K. Potential for global spread of a novel coronavirus from China. J. Travel Med. 2020:27. doi: 10.1093/jtm/taaa011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woo P.C.Y., Huang Y., Lau S.K.P., Yuen K.-.Y. Coronavirus genomics and bioinformatics analysis. Viruses. 2010;2:1804–1820. doi: 10.3390/v2081803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed S.F., Quadeer A.A., Mckay M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses. 2020;12:254. doi: 10.3390/v12030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neuman B.W., Chamberlain P., Bowden F., Joseph J. Atlas of coronavirus replicase structure. Virus Res. 2014;194:49–66. doi: 10.1016/j.virusres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor R.F., Roper R.L. Unique SARS-CoV protein nsp1: bioinformatics, biochemistry and potential effects on virulence. Trends Microbiol. 2007;15:51–53. doi: 10.1016/j.tim.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamitani W., Huang C., Narayanan K., Lokugamage K.G., Makino S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009;16:1134–1140. doi: 10.1038/nsmb.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lokugamage K.G., Narayanan K., Huang C., Makino S. Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J. Virol. 2012;86:13598–13608. doi: 10.1128/jvi.01958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Channappanavar R., Fehr A.R., Vijay R., Mack M., Zhao J., Meyerholz D.K., et al. Dysregulated Type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-Infected mice. Cell Host Microbe. 2016;19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kindler E., Thiel V. SARS-CoV and IFN: too Little, Too Late. Cell Host Microbe. 2016;19:139–141. doi: 10.1016/j.chom.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Law A.H.Y., Lee D.C.W., Cheung B.K.W., Yim H.C.H., Lau A.S.Y. Role for nonstructural protein 1 of severe acute respiratory syndrome coronavirus in chemokine dysregulation. J. Virol. 2006;81:416–422. doi: 10.1128/jvi.02336-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.NCBI. National Center of Biotechnology Informatics (NCBI) database website http://www.ncbi.nlm.nih.gov/. 2020. http://www.ncbi.nlm.nih.gov/2020 (Last accessed May 25, 2020)
- 17.Gish W., States D.J. Identification of protein coding regions by database similarity search. Nat. Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 18.Notredame C., Higgins D.G., Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. 1 1Edited by J. Thornton. [DOI] [PubMed] [Google Scholar]
- 19.Almeida M.S., Johnson M.A., Herrmann T., Geralt M., Kurt Wüthrich. Novel β-Barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 2007;81:3151–3161. doi: 10.1128/jvi.01939-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2014;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng W., Zhang C., Wuyun Q., Pearce R., Li Y., Zhang Y. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 2019:47. doi: 10.1093/nar/gkz384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cozzetto D., Kryshtafovych A., Tramontano A. Evaluation of CASP8 model quality predictions. Proteins Struct. Funct. Bioinform. 2009;77:157–166. doi: 10.1002/prot.22534. [DOI] [PubMed] [Google Scholar]
- 23.Laskowski R.A., Macarthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. doi: 10.1107/s0021889892009944. [DOI] [Google Scholar]
- 24.Morris A.L., Macarthur M.W., Hutchinson E.G., Thornton J.M. Stereochemical quality of protein structure coordinates. Proteins Struct. Funct. Genet. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- 25.Wiederstein M., Sippl M.J. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007:35. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cristobal S., Zemla A., Fischer D., Rychlewski L., Elofsson A. A study of quality measures for protein threading models. BMC Bioinform. 2001;2:5. doi: 10.1186/1471-2105-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J., Roy A., Zhang Y. Protein–ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29:2588–2595. doi: 10.1093/bioinformatics/btt447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J., Roy A., Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 2012:41. doi: 10.1093/nar/gks966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reche P.A., Reinherz E.L. Prediction of peptide-MHC binding using profiles. Methods Mol. Biol. Immunoinform. 2007:185–200. doi: 10.1007/978-1-60327-118-9_13. [DOI] [PubMed] [Google Scholar]
- 30.Kruiswijk C., Richard G., Salverda M.L., Hindocha P., Martin W.D., Groot A.S.D., et al. In silico identification and modification of T cell epitopes in pertussis antigens associated with tolerance. Hum. Vaccines Immunother. 2020;16:277–285. doi: 10.1080/21645515.2019.1703453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2018:47. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kolaskar A., Tongaonkar P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276:172–174. doi: 10.1016/0014-5793(90)80535-q. [DOI] [PubMed] [Google Scholar]
- 33.Mirza M.U., Rafique S., Ali A., Munir M., Ikram N., Manan A., et al. Towards peptide vaccines against Zika virus: immunoinformatics combined with molecular dynamics simulations to predict antigenic epitopes of Zika viral proteins. Sci. Rep. 2016;6 doi: 10.1038/srep37313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berendsen H., Spoel D.V.D., Drunen R.V. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-e. [DOI] [Google Scholar]
- 35.Jorgensen W.L., Maxwell D.S., Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996;118:11225–11236. doi: 10.1021/ja9621760. [DOI] [Google Scholar]
- 36.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 37.Parrinello M., Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- 38.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 39.Hess B., Bekker H., Berendsen H.J.C., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(sici)1096-987x(199709)18. 12<1463::aid−jcc4>3.0.co;2-h. [DOI] [Google Scholar]
- 40.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 41.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., et al. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.DeLano W.L. PyMOL: an open-source molecular graphics tool. Ccp4. Newslett. Protein Crystallogr. 2002;40:11. [Google Scholar]
- 43.Turner P.J., XMGRACE, Version 5.1.19 . Beaverton, ORE, USA; 2005. Central For Costal and Land-Margin Research; Oregon Graduate Institute of Science and Technology. [Google Scholar]
- 44.Amadei A., Linssen A.B.M., Berendsen H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 45.Frauenfelder H., Sligar S., Wolynes P. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 46.Zundert G.V., Rodrigues J., Trellet M., Schmitz C., Kastritis P., Karaca E., et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016;428:720–725. doi: 10.1016/j.jmb.2015.09.014. [DOI] [PubMed] [Google Scholar]
- 47.T. Tahirov, A. Baranovskiy, N. Babayeva, Crystal structure of the catalytic core of human DNA polymerase alpha in ternary complex with an DNA-Primed DNA template and Dctp, (2018). doi:10.2210/pdb6as7/pdb
- 48.Xue L.C., Rodrigues J.P., Kastritis P.L., Bonvin A.M., Vangone A. PRODIGY: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics. 2016 doi: 10.1093/bioinformatics/btw514. [DOI] [PubMed] [Google Scholar]
- 49.Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Channappanavar R., Zhao J., Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol. Res. 2014;59:118–128. doi: 10.1007/s12026-014-8534-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh H.-L.J., Gan S.K.-E., Bertoletti A., Tan Y.-.J. Understanding the T cell immune response in SARS coronavirus infection, Emerg. Microbes Infect. 2012;1:1–6. doi: 10.1038/emi.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsueh P.-.R., Huang L.-.M., Chen P.-.J., Kao C.-.L., Yang P.-.C. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin. Microbiol. Infect. 2004;10:1062–1066. doi: 10.1111/j.1469-0691.2004.01009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivankov D.N., Bogatyreva N.S., Lobanov M.Y., Galzitskaya O.V. Coupling between properties of the protein shape and the rate of protein folding. PLoS ONE. 2009:4. doi: 10.1371/journal.pone.0006476. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The modelled and docking structures are available upon request from the corresponding author.