

Abstract
Genetic testing for cardiac disorders continues to change. Our objective was to assess trends in variant classification in pediatric arrhythmia and cardiomyopathy. We conducted a retrospective review of patients tested for genetic arrhythmia and cardiomyopathy disorders from 2006–17. Variants were classified by CLIA labs. Trends were assessed by Spearman correlation. There were 914 variants in 583 patients from 337 families. The total number of tests ordered increased over time, accelerating after 2012. There was a strong positive correlation between the average number of genes tested per panel and year of testing (r=0.97, p <0.001) and a weak correlation between the year and a decrease in the percentage of clinically actionable variants (r=−0.20, p=0.005). By 2011, VUS represented >50% of variants reported on panels. Over 12 years, 203 genes were interrogated; one or more variants were reported in 91/203 genes (45%). 32% of patients had at least one clinically actionable variant; 28% had at least one VUS. Reclassification is an important long-term issue, with 21.5% variants changing clinical interpretation. We observed an increase over time in three areas: total number of tests ordered, average number of genes/panel, and percentage of VUS. Providers may need to interpret results from 90+ genes and ongoing education is critical. Due to their specific training in test result interpretation, we recommend the inclusion of a genetic counselor in pediatric electrophysiology and cardiomyopathy teams.
Keywords: Cascade testing, Genetic Counseling, Genetic Testing, Genetics Services, Pediatrics, Variant Classification, Variant of Uncertain Significance (VUS), Workforce
INTRODUCTION
Genetic testing for rare, highly-penetrant variants has become important to diagnose and stratify pediatric cardiac disease in electrophysiology and cardiomyopathy (Gigli et al., 2019; Priori et al., 2013a). The presence of a clear etiology from a genetic test may confirm or clarify a diagnosis and may affect ongoing clinical care. Equally important, identification of a known genetic etiology allows for evaluation of at-risk family members to assess the possibility of developing the same condition.
The value of genetic counselors in cardiology practice has been demonstrated (Arscott et al., 2016; Ingles, Yeates, & Semsarian, 2011). They interpret genetic information, provide education and counseling, coordinate test logistics, and evaluate genetic test results. Cardiovascular genetic counselors are especially adept at evaluating variants of uncertain significance (Reuter, Grove, Orland, Spoonamore, & Caleshu, 2018) and for identification of eligible family members for additional family genetic screening (Helm et al., 2018).
Over the past decade, clinical labs have increased the number of genes available on panels for cardiomyopathies and arrhythmias. A number of authors have noted a corresponding increase in ‘background noise’, mainly due to reporting of Variants of Uncertain Significance (VUS)(Ackerman, 2015; Ghouse et al., 2018; Kapplinger et al., 2011; Ouellette et al., 2018). The complexity of variant interpretation was confirmed by the need for the American College of Medical Genetics (ACMG) standards and guidelines for variant classification issued in 2015.(Richards et al., 2015) Since then, the ACMG guidelines have been subject to ongoing evaluations assessing the efficacy and utility of the guidelines in clinical practice, as well as the ongoing logistical, social, and ethical considerations of variant reporting (Amendola et al., 2016; Bombard et al., 2019; Hellwig et al., 2018; Lin et al., 2017; Patel et al., 2017; Wong et al., 2019).
While the increase in tests ordered and VUS returned has been discussed in editorials (Ackerman, 2015) and is commonly “known” in clinical practice, few systematic reports are available to guide pediatric electrophysiology and cardiomyopathy providers with regard to variant interpretation. Pugh et al showed an increase in uncertain results in gene testing as more genes are included on panels for dilated cardiomyopathy; however, their work only addressed a single condition and included a broad range of ages.(Pugh et al., 2014) Lahrouchi et al reported a “highly unfavorable ratio of VUS to pathogenic and likely pathogenic variants” in a cohort of sudden death cases (Lahrouchi et al., 2017). Christiaans et al took a case-by-case look at complex genetic test results and the challenges of pre- and post-test counseling (Christiaans, Mook, Alders, Bikker, & Lekanne Dit Deprez, 2019). Our study is unique by virtue of analyzing a pediatric cohort with a clinically diverse range of diagnoses. We sought to quantify the contribution of each type of genetic test result, with particular interest in the increasing complexity of VUS and the increasing number of genes per test over the last 12 years. A quantitative understanding of variant reporting over time is an important factor for planning for future clinical support, including hiring genetic counselors.
METHODS
Study Design
We performed a retrospective medical record review encompassing the years 2006–2017 in a tertiary pediatric referral center. During the study period, there were 4–5 electrophysiologists and 2–4 heart failure specialists seeing these patients at any given time. Between 2006 and 2017, a certified genetic counselor was available on a case-by-case basis; a full time certified genetic counselor was hired in January 2017 to present.
Sample
We identified consecutive patients with diagnoses of cardiomyopathy or channelopathy by ICD-10 codes I42.0, I42.1, I42.2, I42.5, I42.8, I42.9, I45.81, I47.2, I49.01, I49.4, I49.8, I49.9, Q24.8, and Z86.74 as well as by review of previously collected disease-specific departmental records. Relatives mentioned in any patient’s medical record were identified and included in the retrospective review if medical records were available at our center. Pedigree information was maintained with Progeny Genetics software version 10.2 (Delray Beach, FL).
Data collected from the chart review
During chart review, we collected the following information regarding genetic test results: date of test, type of test, variants reported, and classification of variants reported. We also collected demographic information as described in Table I.
Table I:
Patient and Test Characteristics
| Number (%) or median [IQR] | ||
|---|---|---|
| Female | 277 (47) | |
| Age at time of test | 12.9 [5.0, 18.6] | |
| Race | Asian/Middle Eastern | 24 (4) |
| Black/African American | 58 (10) | |
| Hispanic | 114 (20) | |
| White | 378 (65) | |
| Mixed | 5 (1) | |
| Other | 5 (1) | |
| Proband Tests Ordered | ||
| Panel | Arrhythmia | 170 |
| Cardiomyopathy | 128 | |
| Combined | 22 | |
| Whole Exome, Whole Genome, or Mitochondrial |
28 | |
| Family Variant Tests | 273 | |
IQR: Interquartile range.
Data Analysis
Testing options and panel composition at the various individual commercial clinical genetic testing laboratories have changed over time. For main analyses, all arrhythmia panels were grouped. This included “Arrhythmia” panels defined by the testing laboratory as well as smaller panels for long QT syndrome (LQTS), Brugada syndrome, or catecholaminergic polymorphic ventricular tachycardia (CPVT). Similarly, all “Cardiomyopathy” panels and smaller panels for hypertrophic cardiomyopathy, dilated cardiomyopathy, and arrhythmogenic (right) ventricular cardiomyopathy were grouped for main analyses. Exome, genome, and mitochondrial testing were evaluated separately. Results from exome and genome sequencing were excluded from calculations requiring assessment of “number of genes tested” due to inability to effectively quantify genes evaluated on commercial exome or genome sequencing.
Variant Classification
For initial analysis, we used the variant classification assigned by the reference laboratory. Most laboratories classified variants as benign (B), likely benign (LB), variants of uncertain significance (VUS), likely pathogenic (LP), or pathogenic (P). Familion/Transgenomic laboratories (New Haven, CT) classified variants as Class I, II, and III, which were reassigned as LP, VUS, and LB, respectively.
Reclassification was performed using the ClinVar database, https://www.ncbi.nlm.nih.gov/clinvar (queried February 27, 2020) as previously described (Pottinger et al., 2020). Reclassification was based on a consensus from ≥ 70% of adjudications published to ClinVar after 2015 using the following reference laboratories: Ambry Genetics, Blueprint Genetics, GeneDx, Harvard/Laboratory for Molecular Medicine, Invitae, and Stanford. Variants that did not meet the 70% adjudication threshold were not reclassified.
Clinically Actionable Variants
We defined “Clinically Actionable Variants” as the combination of LP/P variant classifications. We defined “Variants of Interest” as the combination of VUS, LP, and P classifications. We included VUS within “Variants of Interest” because VUS require genetic counselors to provide additional information and interpretation to providers and families.
Statistical Analysis
Descriptive statistics summarized family, patient, genetic test, and gene level characteristics. Graphical visualizations were provided to assess trends in genes and variants over time. Spearman correlation coefficients estimated pairwise associations between number of genes tested, number of variants returned, number of variants of interest, and year at both the patient level and aggregate year level. Analyses focused on the first genetic test for each patient, unless otherwise specified.
RESULTS
Patient and Test Characteristics
The median age was 12.9 years at time of genetic testing and 277 (47%) were female (Table I). There was a weak positive correlation between year of testing and patient age (r=0.13, p=0.001). Results from genetic testing were returned to 583 patients drawn from 337 unique families over 12 years. Among the 583 patients, 621 genetic tests were ordered, resulting in 914 variants. All variants were discussed with patients and families by either a genetic counselor or a cardiologist.
About half of tests ordered were a gene panel (316/621, 52%). Among the patients with panel tests, only one panel was ordered for 91% of patients (276/303). Two panels were ordered for 25 patients (9%) and three panels were ordered for 2 patients (<1%). Family variant tests were the next most common test ordered (273/621, 44%). Only 16 evaluations (2%) were genome or exome analysis and another 16 were single-gene tests (all NKX2–5, 2%).
Increase in Total Variant Reporting
We first analyzed our results at the variant level because each variant result returned to a patient requires evaluation, adjudication, and counseling. The total number of variants returned each year increased dramatically during the study period (Figure I). As the Figure illustrates, the increase has accelerated over the past five years, tripling between the years 2013 (34 variants) and 2017 (100 variants). Across all years of our study, 114 (28.2%) of results were clinically actionable (LP/P) and 202 (29.0%) were VUS. Unsurprisingly, our data reflected a reduction in LB/B variant reporting to almost zero after the publication of the 2015 ACMG variant reporting guidelines.(Richards et al., 2015). Of the 914 unique variants reported, 584 variants (64%) were returned from panel tests and 308 (34%) were returned from family variant tests.
Figure I. Variants Reported Over Time.
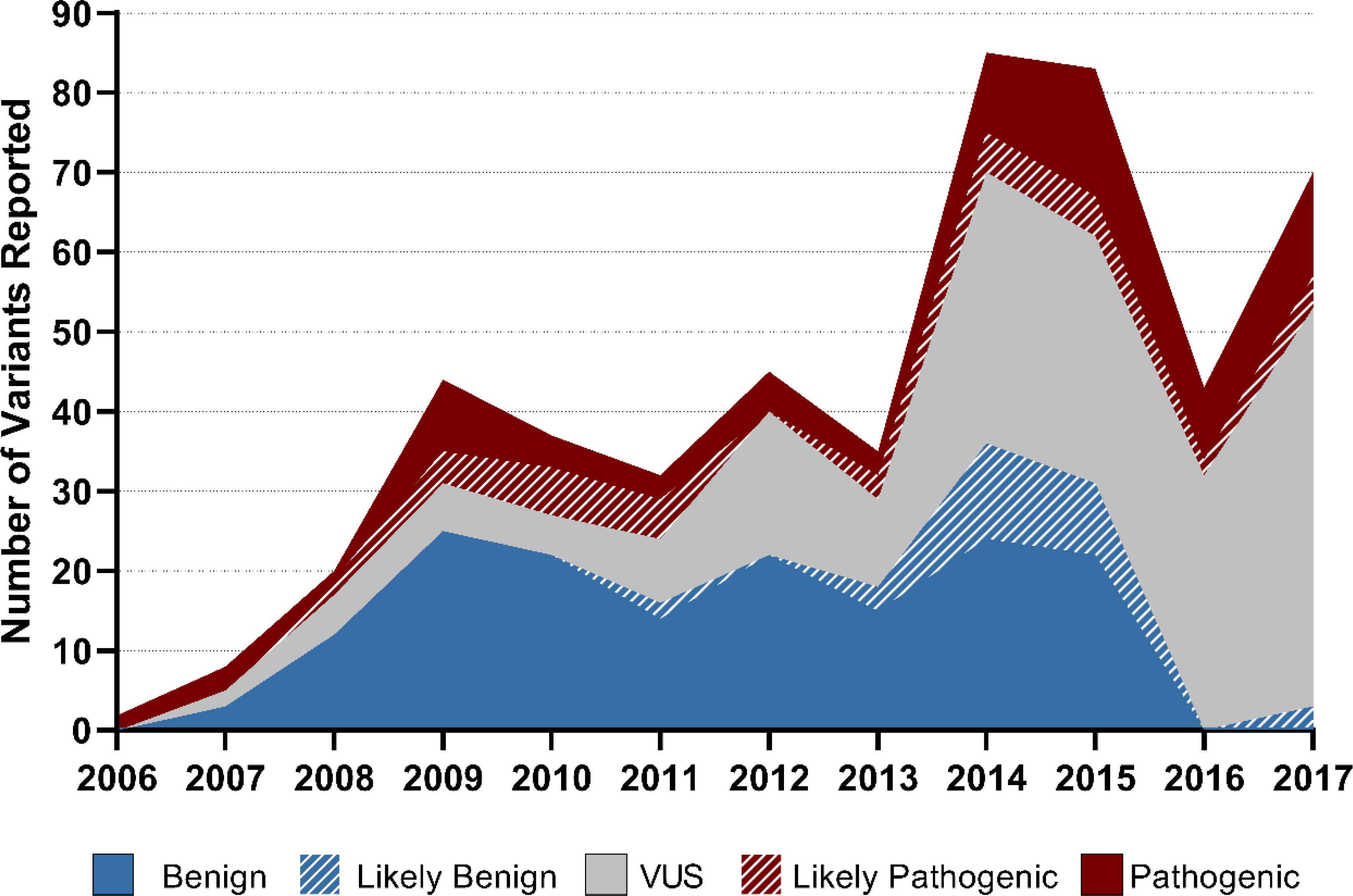
The total number of variants returned each year increased during the study period. The increase accelerated after 2012. Likely Benign/Benign variants were rarely reported after 2015. VUS: Variants of uncertain significance
The total number of patients who received a panel test also increased over time (1 patient in 2006 versus 47 patients in 2017, r=0.61, p=0.03). Similarly, there was an increase in the number of family variant tests ordered (5 patients in 2006 versus 65 patients in 2017, r=0.90, p=0.0001). Only 22 variants were derived from genome or exome sequencing (2%). In summary, the total number of results increased markedly between 2006 and 2017, whether that is measured by number of variants, number of probands tested with a panel, or number of family variant screening tests.
Decrease in Proportion of Clinically Actionable Variants
Across all years and test types, in 235 (40%) of patients, at least one LP/P variant was found. We did not see a substantially different fraction of clinically actionable results returned when we analyzed these data at the level of probands only (37%, n=125) as compared to families (39%, n=130). The proportion of total results classified as LP/P decreased slightly over time from 2006 to 2017 (r=–0.20, p=0.005, Figure II). In panel tests, the number of VUS began to outnumber the LP/P variants reported to families in 2011 and VUS remained greater than 50% of variants reported on panel tests thereafter.
Figure II. Proportion of Variant Classification Over Time.
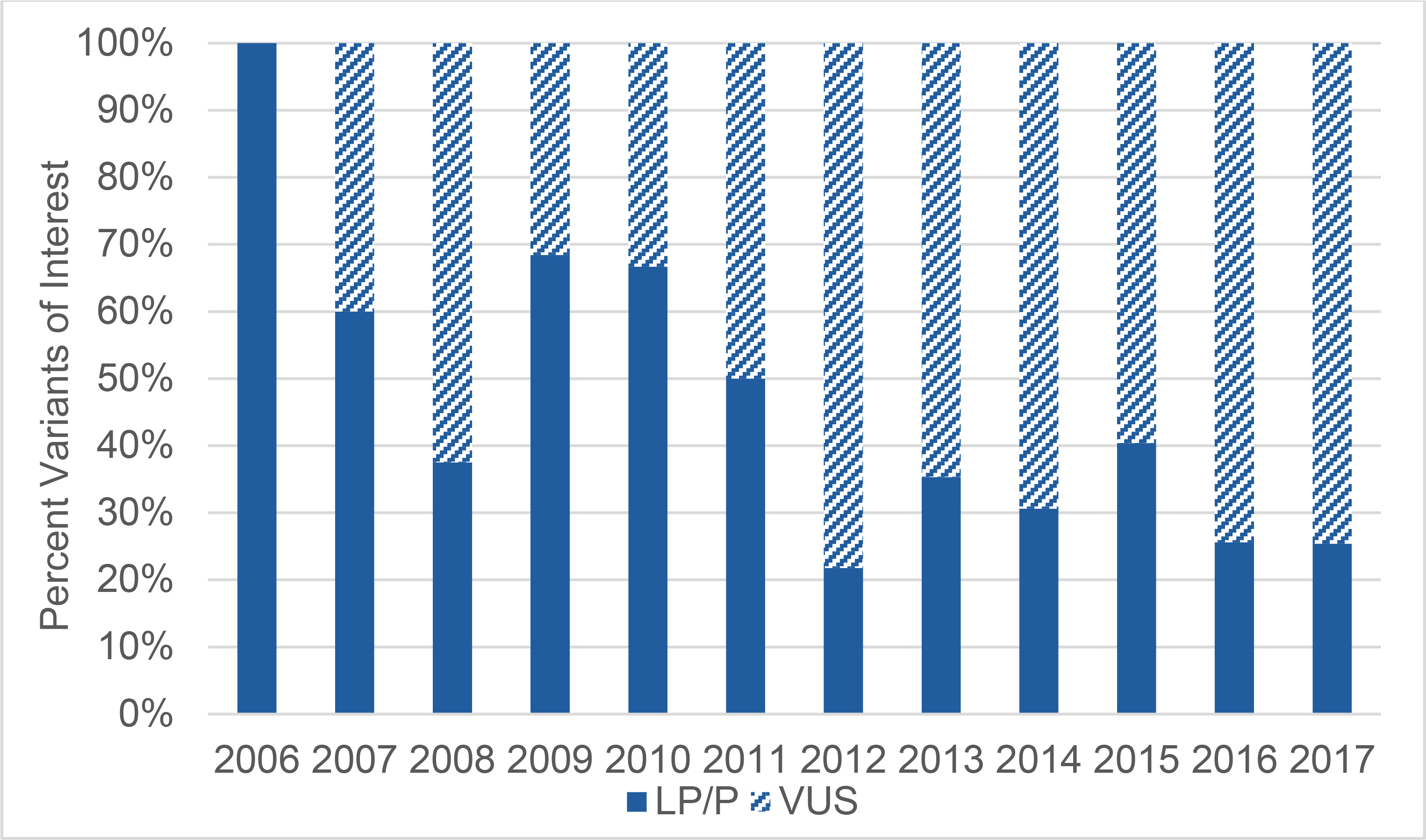
The total variants of interest in each year are normalized to 100%. The percentage of LP/P is shown in dark blue. Over time, there was a weak negative correlation between year of test and percentage of LP/P variants reported (Spearman correlation, r=−0.2, p=0.0045). LP/P: Likely pathogenic/pathogenic variants; VUS: Variants of uncertain significance
Increase in Number of Genes Tested Per Panel
There was a strong positive correlation between the average number of genes tested per panel and the year of testing (r=0.97, p <0.001). As illustrated in Figure III, there was a relatively slow increase in the number of genes per panel from 2006 to 2013. We noted a large change in 2014: the mean number of genes per panel increased, the variability in number of genes per panel increased, and the maximum number of genes per panel increased. There was a weak positive correlation between the number of genes tested in a panel and the number of variants of interest reported at the patient level (r= 0.35, p <0.001).
Figure III. Distribution of Genes Tested Per Panel, by Year.
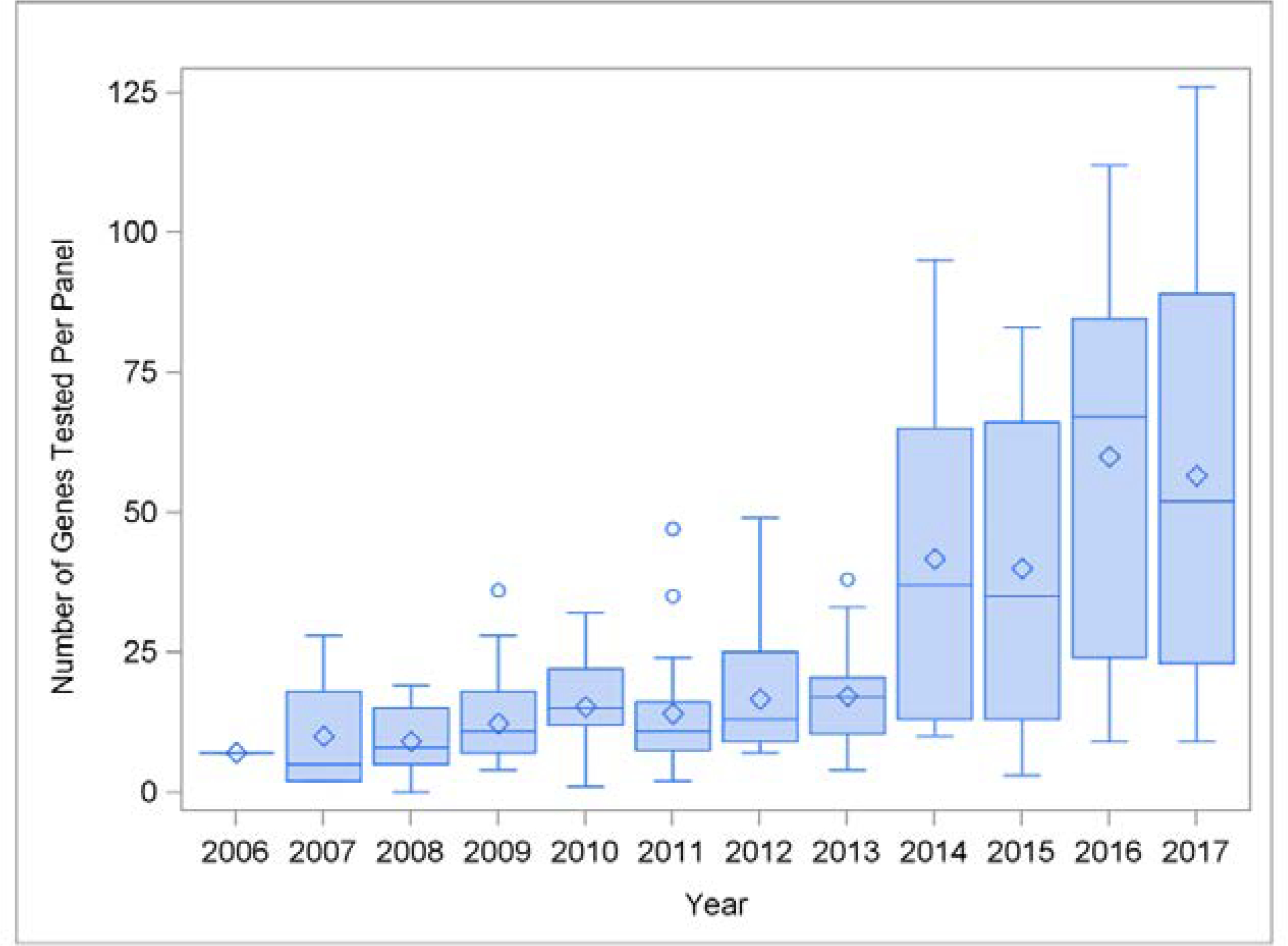
The plot shows the distribution of genes/panel over time. All cardiac panels were tabulated, including arrhythmia panels, cardiomyopathy panels, and combined cardiac panels, although each clinical laboratory had their own naming conventions and the names of panels changed over time. There was an increase in the number of genes tested per panel over time, as well as an increase in the diversity of panel sizes. Standard conventions for box and whiskers plots were used (median = center horizontal line; mean = diamond; outliers = circles).
Overall, 203 genes were tested via panel or family variant analysis. One or more variants were reported in 91/203 genes (45%). About half of those 91 genes (40/91) had clinically actionable variants detected, and another 41 genes had a VUS as the highest class of result. Furthermore, variants were not equally distributed among the 91 genes. There were only nine genes in which more than 10 variants of interest were reported (Figure IV). 46 genes had between 2 and 9 variants of interest reported and 27 genes had only 1 variant of interest reported. An additional 10 genes reported only LB/B variants. See the Online Supplement for a detailed tabulation.
Figure IV. Genes with More than Ten Variants of Interest Reported.
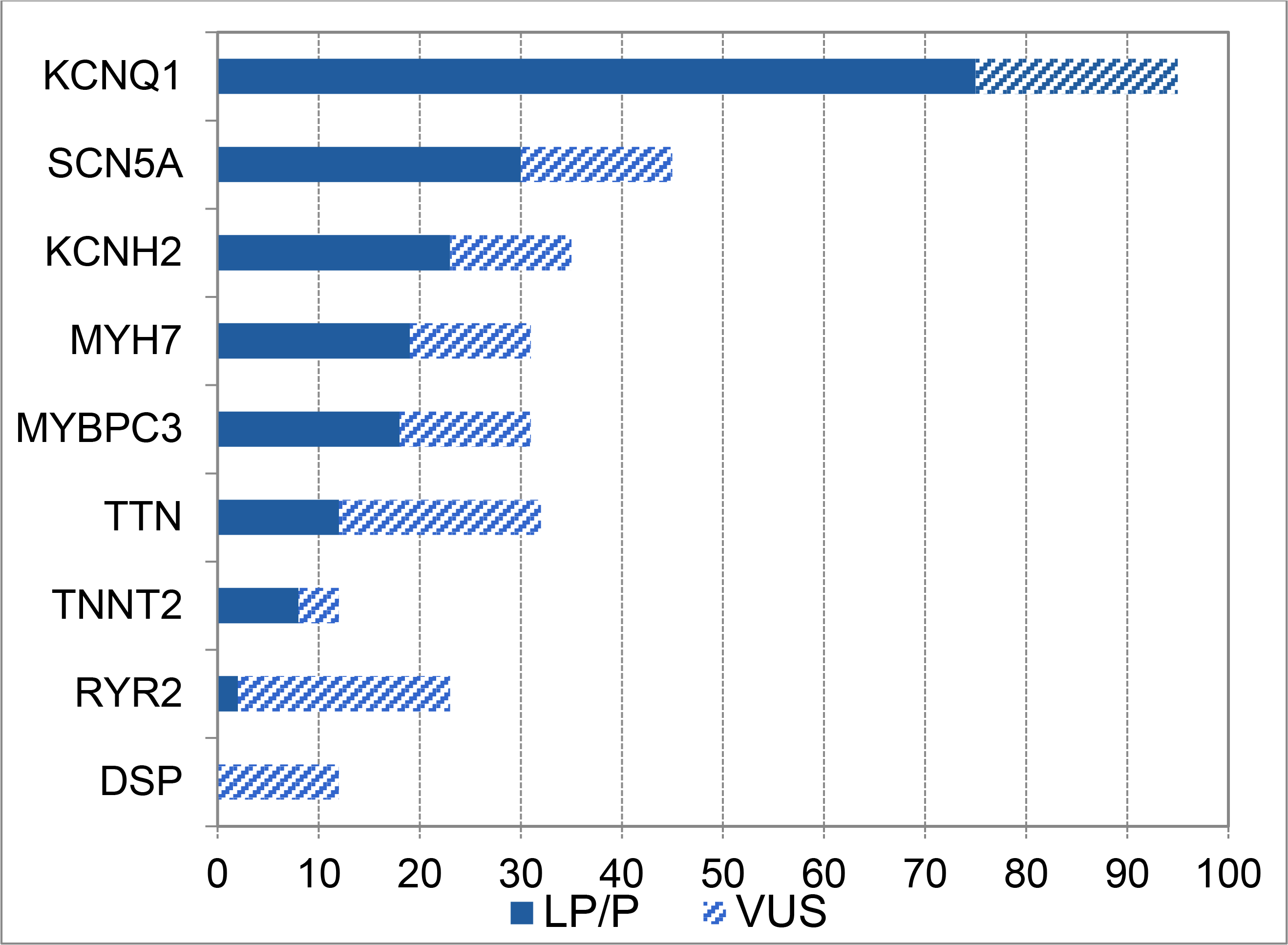
More than 10 variants of interest were reported in these nine genes, which represent the highest-yield educational targets for practitioners in pediatric electrophysiology and cardiomyopathy. A full tabulation of variant distribution by gene is in the Online Supplement. LP/P, Likely pathogenic or pathogenic variant; VUS, variant of uncertain significance.
We also considered the relative yield of various disease-specific panels (Figure V). The yield of clinically actionable variants in LQTS remains the highest, with CPVT being the least likely disease panel to yield a clinically actionable result in our population.
Figure V. Yield of Genetic Testing in Common Panels.
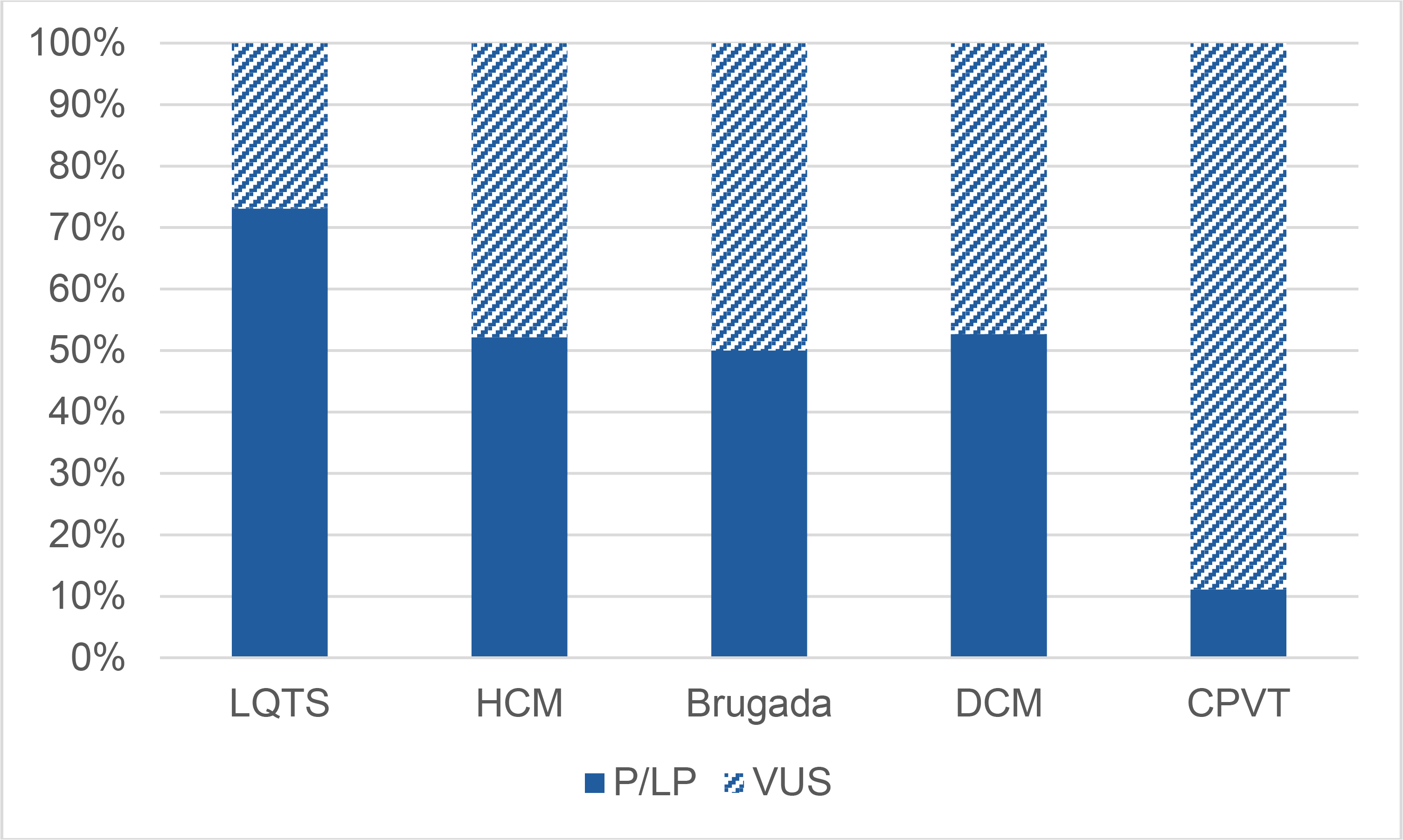
The total number of panels ordered for each phenotype indication were normalized to 100%. The percentage of panels returning LP/P variant(s) is shown in dark blue as a percentage of the total number of panels.
LQTS: Long QT Syndrome;HCM: Hypertrophic Cardiomyopathy; DCM: Dilated Cardiomyopathy; CPVT: catecholaminergic polymorphic ventricular tachycardia
Reclassification
We used the ClinVar database to update classifications (Table II). Of the 422 variants investigated, 330 (78.1%) had sufficient data in ClinVar for reclassification. Reclassification changed the clinical interpretation in 71/330 variants (21.5%). A change from VUS to LP/P occurred in 9/330 variants (2.7%), and from LP/P to VUS in 25 variants (7.6%). One variant was reclassified from LP/P to LB/B. The remaining reclassifications were from VUS to LB/B or LB/B to VUS.
Table II.
Reclassification of variants
| New Classification |
||||
|---|---|---|---|---|
| Original Classification | LB/B | VUS | LP/P | Total |
| LB/B | 56 | 1 | 0 | 57 |
| VUS | 35 | 122 | 9 | 166 |
| LP/P | 1 | 25 | 81 | 107 |
| Total | 92 | 148 | 90 | 330 |
LB/B: Likely benign/benign
VUS: Variant of uncertain significance
LP/P: Likely pathogenic/pathogenic (referred to as “clinically actionable variants” in the text)
Genetic Testing Within Families
Since cardiomyopathy and arrhythmia syndromes usually have an autosomal dominant inheritance pattern (with variable penetrance), we also analyzed our data by family. Of the 337 families, 301 had at least one panel or whole exome/genome test. There were 36 families with only family variant testing; these families presented to our practice with a variant previously detected in a family member. Of the 301 families with panel tests, 108 families had at least one clinically actionable variant (36%). Another 93/301 families had no clinically actionable variants but had at least one VUS (31%) and 100/301 families had no variants of interest reported (33%). The range of family members tested was 1 to 13. In 122 families, only one family member (the proband) was tested. An additional family member was tested in 81 families, and two or more additional family members were tested in 134 families. Unsurprisingly, family variant testing was more common in families with a LP/P variant, where cascade screening is now an accepted clinical strategy. However, even among families with LP/P variants, the median number of family members tested was 2 (IQR 1–4).
DISCUSSION
Increase in Genetic Data Returned to Families
The most important contribution here is to quantify the increase in complexity of genetic testing in pediatric cardiology, which has implications for the clinical work associated with genetic testing. We document an increase in the total volume of variants reported to patients, an increase in the number of genes/panel, and a decrease in the proportion of clinically actionable variants per panel (and a reciprocal increase in proportion of VUS).
Over this period, the most striking change in our population was the increase in the number of genes tested per panel and the range of genes tested per panel. Early in our experience, all panels were small and grew slowly. In cardiology, the marked increase in genes tested began in 2014. This increase in heterogeneity reflects the increased diversity of test options available from clinical testing laboratories.
Proportion of Clinically Actionable Variants Is Decreasing
The proportion of clinically actionable variants reported to patients decreased while the proportion of VUS increased. In our early testing, approximately 60% of variants were LP/P. By 2012, the percentage of LP/P was approximately 30%, with a corresponding increase in VUS. The “genetic purgatory” of VUS has been the subject of several publications (Ackerman, 2015; Ghouse et al., 2018; Kapplinger et al., 2011; Ouellette et al., 2018), and we provide supporting data that quantify the increase in VUS.
Reclassification of Variants Could Impact Care
Cardiac genetic counselors navigate result disclosure with families (Ingles et al., 2011) and reclassification adds complexity. In our study, 22% of variants changed classification and approximately 10% of variants had a reclassification that would change clinical interpretation. Upgrades from VUS to LP/P (9 variants, 2.7%) may cause providers to add new family cascade recommendations and to change the interpretation of disease for the proband. The downgrade of 25 variants (7.6%) from LP/P to VUS could also affect family cascade screening. Family members who tested negative for a variant classified LP/P may have been released from follow-up. A new VUS classification may require re-contact. Our study underscores the need for genetic counselors to spend time and effort on reclassification, often leading to subsequent education or clinical intervention for families.
Finally, the high rate of reclassifications (22%) highlights the era-sensitive nature of genetic test results. Classifications of individual variants may continue to shift in the future and this should be explained to families at the time of initial variant return.
Increase in Number of Genes to Understand
There were 203 unique genes tested within our patient population and 91 genes had variants reported. It is a significant endeavor for genetic counselors to learn about 203 (or 91) individual genes, and one would imagine that it would be even more daunting for physicians whose primary training is in clinical cardiology. Further, many of these genes have only preliminary evidence for their inclusion in clinical genetic testing.(Ingles et al., 2019) By definition, preliminary evidence genes have little published data to guide providers in counseling patients. The time required to understand so many genes is one of the main reasons that we recommend that a genetic counselor be involved in every return-of-results conversation.
Several recent articles have documented that the rise of genomics is associated with uncertainty among providers (Best et al., 2020; Stark et al., 2019). An initial step in genetic education is to learn the genes that have the highest prevalence of pathogenic variants in the provider’s specialty. In our study, a small number of genes hosted most of the clinically actionable variants. There were only 9 genes with over 10 variants of interest reported: KCNQ1, SCN5A, KCNH2, MYH7, MYBPC3, TTN, TNNT2, RYR2, and DSP (Figure IV). Unsurprisingly, given our study population, these genes are the leading genetic causes of LQTS, Brugada syndrome, dilated and/or hypertrophic cardiomyopathies, and CPVT (McNally & Puckelwartz, 2015; Priori et al., 2013b). These genes are a good focus for initial education in anyone in pediatrics or family-centered care who will care for families with electrophysiology or cardiomyopathy.
However, familiarity with these major genes is not synonymous with expertise, and the complexity of results in this field is increasing: a third of our patients had a clinically actionable variant, another third had a VUS, and the volume of genetic data is growing. Genetic counselors will continue to evaluate the changing nature of available genetic tests and stay abreast of variant re-classifications. Recent data show that reclassification conversations are complex and may be misunderstood by families (Wong et al., 2019). It is ideal for genetic counselors to be integrated into clinical practice in cardiology as they have been in multiple other subspecialties (Stopfer, 2000; Trepanier et al., 2004). They are a well-informed resource about the ever-evolving genetic contributors to cardiac disease.
LIMITATIONS
This was a descriptive, retrospective, single-center study and may represent idiosyncratic results from a center with a changing staff of physicians and genetic counselors over time. The ClinVar database is a public-access resource and it is not intended as a source to return data for clinical results to any individual patient.
CONCLUSION
In pediatric electrophysiology and cardiomyopathy, the work of reporting genetic results is increasing in clinical practice. More tests are being ordered and the number of genes in each panel is increasing, while the percentage of clinically actionable variants is decreasing. In addition, reclassification may occur in approximately 1 out of every 5 variants. We recommend the inclusion of a genetic counselor in pediatric electrophysiology and cardiomyopathy teams to provide service to patients and providers in genetic testing result interpretation, understanding, and integration in to clinical care.
Supplementary Material
ACKNOWLEDGEMENTS
Sources of Funding Research reported in this publication was supported, in part, by the National Institutes of Health, National Heart, Lung and Blood Institute, grant number K23HL130554, the American Heart Association Mentored Clinical and Population Research Award (Dallas, TX), and the Smeds Family Foundation (Chicago, IL).
Footnotes
CONFLICTS OF INTEREST
Authors Sara Cherny, Rachael Olson, Kathryn Chiodo, Lauren C. Balmert, and Gregory Webster declare that they have no conflicts of interest.
Human Subjects and Informed Consent
Approval for retrospective review was granted by the Institutional Review Board for the Ann and Robert H. Lurie Children’s Hospital of Chicago. No informed consent was required. This study conforms to recognized standards of the Federal Policy for the Protection of Human Subjects.
Animal Studies
No non-human animal studies were carried out by the authors for this article.
DATA AVAILABILITY
The data underlying this article will be shared on reasonable request to the corresponding author, subject to any restrictions from our Institutional Review Board at the time of request.
REFERENCES
- Ackerman MJ (2015). Genetic purgatory and the cardiac channelopathies: Exposing the variants of uncertain/unknown significance issue. Heart Rhythm, 12(11), 2325–2331. doi: 10.1016/j.hrthm.2015.07.002 [DOI] [PubMed] [Google Scholar]
- Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, … Rehm HL (2016). Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet, 99(1), 247. doi: 10.1016/j.ajhg.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arscott P, Caleshu C, Kotzer K, Kreykes S, Kruisselbrink T, Orland K, … Cherny S (2016). A Case for Inclusion of Genetic Counselors in Cardiac Care. Cardiol Rev, 24(2), 49–55. doi: 10.1097/crd.0000000000000081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best S, Stark Z, Brown H, Long JC, Hewage K, Gaff C, … Taylor N (2020). The leadership behaviors needed to implement clinical genomics at scale: a qualitative study. Genet Med. doi: 10.1038/s41436-020-0818-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombard Y, Brothers KB, Fitzgerald-Butt S, Garrison NA, Jamal L, James CA, … Levy HP (2019). The Responsibility to Recontact Research Participants after Reinterpretation of Genetic and Genomic Research Results. Am J Hum Genet, 104(4), 578–595. doi: 10.1016/j.ajhg.2019.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiaans I, Mook ORF, Alders M, Bikker H, & Lekanne Dit Deprez RH (2019). Large next-generation sequencing gene panels in genetic heart disease: challenges in clinical practice. Neth Heart J, 27(6), 299–303. doi: 10.1007/s12471-019-1251-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghouse J, Skov MW, Bigseth RS, Ahlberg G, Kanters JK, & Olesen MS (2018). Distinguishing pathogenic mutations from background genetic noise in cardiology: The use of large genome databases for genetic interpretation. Clin Genet, 93(3), 459–466. doi: 10.1111/cge.13066 [DOI] [PubMed] [Google Scholar]
- Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, … Mestroni L (2019). Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol, 74(11), 1480–1490. doi: 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwig LD, Biesecker BB, Lewis KL, Biesecker LG, James CA, & Klein WMP (2018). Ability of Patients to Distinguish Among Cardiac Genomic Variant Subclassifications. Circ Genom Precis Med, 11(6), e001975. doi: 10.1161/circgen.117.001975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm BM, Freeze SL, Spoonamore KG, Ware SM, Ayers MD, & Kean AC (2018). The Genetic Counselor in the Pediatric Arrhythmia Clinic: Review and Assessment of Services. J Genet Couns, 27(3), 558–564. doi: 10.1007/s10897-017-0169-5 [DOI] [PubMed] [Google Scholar]
- Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, … Funke B (2019). Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes. Circ Genom Precis Med, 12(2), e002460. doi: 10.1161/circgen.119.002460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingles J, Yeates L, & Semsarian C (2011). The emerging role of the cardiac genetic counselor. Heart Rhythm, 8(12), 1958–1962. doi: 10.1016/j.hrthm.2011.07.017 [DOI] [PubMed] [Google Scholar]
- Kapplinger JD, Landstrom AP, Salisbury BA, Callis TE, Pollevick GD, Tester DJ, … Ackerman MJ (2011). Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia-associated mutations from background genetic noise. J Am Coll Cardiol, 57(23), 2317–2327. doi: 10.1016/j.jacc.2010.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahrouchi N, Raju H, Lodder EM, Papatheodorou E, Ware JS, Papadakis M, … Behr ER (2017). Utility of Post-Mortem Genetic Testing in Cases of Sudden Arrhythmic Death Syndrome. J Am Coll Cardiol, 69(17), 2134–2145. doi: 10.1016/j.jacc.2017.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Williams N, Wang D, Coetzee W, Zhou B, Eng LS, … Tang Y (2017). Applying High-Resolution Variant Classification to Cardiac Arrhythmogenic Gene Testing in a Demographically Diverse Cohort of Sudden Unexplained Deaths. Circ Cardiovasc Genet, 10(6). doi: 10.1161/circgenetics.117.001839 [DOI] [PubMed] [Google Scholar]
- McNally EM, & Puckelwartz MJ (2015). Genetic Variation in Cardiomyopathy and Cardiovascular Disorders. Circ J, 79(7), 1409–1415. doi: 10.1253/circj.CJ-15-0536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette AC, Mathew J, Manickaraj AK, Manase G, Zahavich L, Wilson J, … Mital S (2018). Clinical genetic testing in pediatric cardiomyopathy: Is bigger better? Clin Genet, 93(1), 33–40. doi: 10.1111/cge.13024 [DOI] [PubMed] [Google Scholar]
- Patel RY, Shah N, Jackson AR, Ghosh R, Pawliczek P, Paithankar S, … Milosavljevic A (2017). ClinGen Pathogenicity Calculator: a configurable system for assessing pathogenicity of genetic variants. Genome Med, 9(1), 3. doi: 10.1186/s13073-016-0391-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottinger TD, Puckelwartz MJ, Pesce LL, Robinson A, Kearns S, Pacheco JA, … McNally EM (2020). Pathogenic and Uncertain Genetic Variants Have Clinical Cardiac Correlates in Diverse Biobank Participants. J Am Heart Assoc, 9(3), e013808. doi: 10.1161/jaha.119.013808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, … Tracy C (2013a). Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm, 10(12), e85–108. doi: 10.1016/j.hrthm.2013.07.021 [DOI] [PubMed] [Google Scholar]
- Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, … Tracy C (2013b). HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm, 10(12), 1932–1963. doi: 10.1016/j.hrthm.2013.05.014 [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, … Funke BH (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med, 16(8), 601–608. doi: 10.1038/gim.2013.204 [DOI] [PubMed] [Google Scholar]
- Reuter C, Grove ME, Orland K, Spoonamore K, & Caleshu C (2018). Clinical Cardiovascular Genetic Counselors Take a Leading Role in Team-based Variant Classification. J Genet Couns, 27(4), 751–760. doi: 10.1007/s10897-017-0175-7 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, Nisselle A, McClaren B, Lynch F, Best S, Long JC, … Gaff CL (2019). Attitudes of Australian health professionals towards rapid genomic testing in neonatal and paediatric intensive care. Eur J Hum Genet, 27(10), 1493–1501. doi: 10.1038/s41431-019-0429-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopfer JE (2000). Genetic counseling and clinical cancer genetics services. Semin Surg Oncol, 18(4), 347–357. [DOI] [PubMed] [Google Scholar]
- Trepanier A, Ahrens M, McKinnon W, Peters J, Stopfer J, Grumet SC, … Vockley CW (2004). Genetic cancer risk assessment and counseling: recommendations of the national society of genetic counselors. J Genet Couns, 13(2), 83–114. doi: 10.1023/b:Jogc.0000018821.48330.77 [DOI] [PubMed] [Google Scholar]
- Wong EK, Bartels K, Hathaway J, Burns C, Yeates L, Semsarian C, … Ingles J (2019). Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur J Hum Genet, 27(7), 1134–1142. doi: 10.1038/s41431-019-0377-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author, subject to any restrictions from our Institutional Review Board at the time of request.