

Abstract
At least 5% of children present unexpected difficulties in expressing and understanding spoken language. This condition is highly heritable and often co-occurs with other neurodevelopmental disorders such as dyslexia and ADHD. Through an exome sequencing analysis, we identified a rare missense variant (chr16:84405221, GRCh38.p12) in the ATP2C2 gene. ATP2C2 was implicated in language disorders by linkage and association studies, and exactly the same variant was reported previously in a different exome sequencing study for language impairment (LI). We followed up this finding by genotyping the mutation in cohorts selected for LI and comorbid disorders. We found that the variant had a higher frequency in LI cases (1.8%, N = 360) compared with cohorts selected for dyslexia (0.8%, N = 520) and ADHD (0.7%, N = 150), which presented frequencies comparable to reference databases (0.9%, N = 24 046 gnomAD controls). Additionally, we observed that carriers of the rare variant identified from a general population cohort (N = 42, ALSPAC cohort) presented, as a group, lower scores on a range of reading and language-related measures compared to controls (N = 1825; minimum P = 0.002 for non-word reading). ATP2C2 encodes for an ATPase (SPCA2) that transports calcium and manganese ions into the Golgi lumen. Our functional characterization suggested that the rare variant influences the ATPase activity of SPCA2. Thus, our results further support the role of ATP2C2 locus in language-related phenotypes and pinpoint the possible effects of a specific rare variant at molecular level.
Introduction
Speech and language disorders represent one of the most common childhood disabilities and account for a large proportion (23%) of paediatric referrals and statements of special educational need (School census, England, January 2019, https://www.gov.uk/government/statistics/special-educational-needs-in-england-january-2019).
Speech and language deficits are often the secondary manifestations of a pre-existent neurological or medical condition (e.g. Down syndrome, intellectual disability, autism spectrum disorder, hearing loss, traumatic brain injury and mental retardation). Severe and persistent language difficulties can be observed also despite normal intelligence and access to adequate educational opportunities. In this case, children were diagnosed with specific language impairment (SLI) (1). Recently, the use of this term has been replaced by the developmental language disorder (DLD) definition (2,3), which captures a wider group of children presenting co-occurring conditions such as intellectual disability, ADHD and developmental coordination disorder. Previous genetic research and the samples described here refer to participants recruited under the SLI criteria, and therefore, we will use the terms SLI and LI hereafter.
SLI affects up to 8% of pre-school children (4,5) and has a significant impact on social, academic and psychological outcomes (6–10). SLI often co-occurs with other neurodevelopmental disorders such as dyslexia (11,12) and attention deficit/hyperactivity disorder (ADHD) (13). The comorbidity observed between these conditions suggests that SLI, dyslexia and ADHD may be different manifestations of the same cognitive deficit or share common genetic factors (14). This hypothesis is supported by significant genetic overlap across studies as demonstrated by a recent genome-wide association study (GWAS) for dyslexia which showed significant shared contributions across disorders (15).
A strong genetic component for DLD, of up to 70%, was first proposed by twin and familial studies (16–20).
Initially, linkage studies conducted across multiplex families led to the identification of 5 susceptibility loci on chromosomes 2q36 (SLI5, OMIM #615432) (21), 7q35-q36 (SLI4—OMIM #612514) (22), 13q21 (SLI3—OMIM #607134) (23,24), 16q (SLI1—OMIM #606711) and 19q (SLI2—OMIM #606712) (25–28). Fine mapping within these regions reported associations of common variants at the ATP2C2, CMIP, CNTNAP2 genes with quantitative measures of language abilities (22,29), including the phonological memory non-word repetition task (NWR), which is considered as a strong endophenotype for SLI (30).
GWAS for language skills have been conducted in clinical and epidemiological samples but have been limited to a few studies of relatively small sample sizes (31). In the first case, the only locus reaching genome-wide significance in SLI individuals was found in the NOP9 gene, when including parent-of-origin effects (32,33). In the second case, the most significant result was the association between the locus 3p12.3, near ROBO2, and expressive vocabulary in the early phase of language acquisition (34). A GWAS conducted in two population cohorts reported an association close to statistical significance between ABCC13 gene and the NWR test (35). Other studies combined analyses for language and reading abilities using both a case–control (36) and quantitative design (37) reporting only marginally significant associations.
Chromosomal rearrangements and copy number variants (CNV) have also been implicated in the aetiology of LI. Children with SLI show a higher frequency of sex chromosome aneuploidies (38) and an increased CNV burden compared with population controls (39,40). A balanced translocation interrupting the gene SEMA6D (41) has been identified in a child with SLI. Rare deletions have been reported in the ZNF277 (42) and TM4SF20 (SLI5) genes (21). Larger de novo deletions have been reported across the ATP2C2 gene in a 10-year-old boy with receptive and expressive language delay (43) and at chromosome 15q13.1–13.3 in a child with LI (44). Single locus mutations have been implicated in severe forms of language disorders (31,45). De novo mutations in the FOXP2 gene cause childhood apraxia of speech (CAS), a severe oral-motor delay accompanied by deficits in expressive and receptive language (46).
Next-generation sequencing (NGS) technology is uncovering the role of rare variants in complex traits. NGS in individuals with CAS or SLI pinpointed to novel pathogenic variants in coding and non-coding regions (45,47,48). Whole exome sequencing (WES) analysis in 43 unrelated probands affected by severe SLI (49) reported several risk variants but suggested that a single mutation might not be sufficient to lead to a disorder unless in combination with other factors in line with previous studies (50–52).
Here, we followed up a specific finding from ongoing exome sequencing analyses on large pedigrees selected for having multiple members affected by SLI. We identified, in one family, a rare non-synonymous variant in the ATP2C2 gene (Chr16:84405221 GRCh38.p12, rs78887288, NM_001286527:exon3:c.G304A:p.V102M), previously implicated in SLI through association studies (29,43). Exactly the same variant was also reported in the WES study by Chen and collaborators (49) bolstering the idea that this rare variant has a role in susceptibility to language disorders. Nonetheless, given its rarity, larger screens and functional studies are required to support this role.
We screened the chr16:84405221 variant in larger cohorts of children with language difficulties. Due to the high rate of comorbidity between SLI, dyslexia and ADHD, the screening included cohorts selected for these conditions.
We found that this variant had a higher frequency in the SLI and LI cases compared with the control group and cohorts selected for dyslexia or ADHD. Additionally, this variant was associated with low performance in a range of reading and language measures. By means of a colorimetric ATPase assay, we further investigated the effects of the variant on the function of SPCA2, the calcium and manganese transporter encoded by ATP2C2. The mutation displayed a lower, albeit not significant, ATPase activity compared with SPCA2 WT.
Results
Identification of the missense variant Chr16:84405221
WES analysis in six families selected for SLI led to the identification of a rare variant (Chr16:84405221) in the ATP2C2 gene (Fig. 1). This variant was found in two children and their father, all diagnosed with SLI. The same variant was previously reported in an exome sequencing study performed on 43 unrelated probands affected by severe LI (49). ATP2C2 was previously associated with SLI (29). Sanger sequencing in the other family members showed imperfect segregation as the variant was not transmitted to the other two affected sons (Fig. 1). The variant was inherited from the father who also scored below expected (i.e. <15th %ile) on a standardized expressive language measure. The variant leads to a missense substitution from a valine to a methionine in exon three spanning the ATPase domain. Bioinformatics tools for pathogenicity suggest the change is likely to be benign (Table 1). The two algorithms that predicted a functional effect, albeit subtle, on protein activity and stability were the Mutation Assessor and the INPS-MD tools.
Figure 1 .
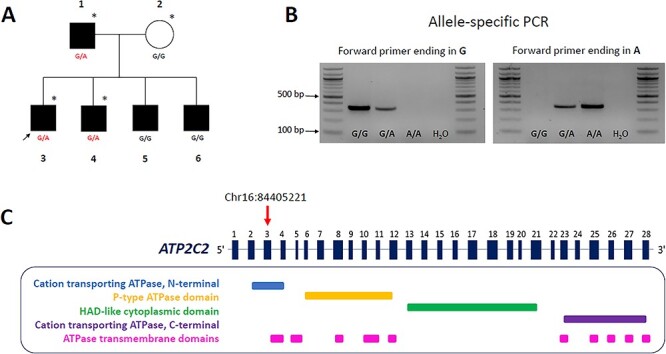
The rare ATP2C2 missense variant (Chr16:84405221). (A) Discovery pedigree. Father (1), mother (2), proband (3) and siblings (4–6). Subjects with SLI are shaded black and unaffected are white. Individuals included in the WES screening are indicated by an asterisk. (B) Allele-specific PCR. The presence of the missense variant was validated in follow up screening using PCR 3′-end mismatch primers. Depending on the allele present, the forward primer anneals perfectly to the template or forms a mismatch. The reaction gives an amplicon only in the first case. (C) On top, a schematic of the ATP2C2 gene. Exons are represented by numbered boxes. The rare variant is localized in the third exon (red arrow) and causes the substitution of the residue from valine to methionine. Exons and introns of ATP2C2 gene are not to scale. At the bottom, predicted domains of the protein (http://www.rcsb.org/pdb/protein/O75185) (118). The variant changes the amino acid sequence which codes for the cation transporting ATPase domain at the N-terminal of the protein.
Table 1.
Predictions of the possible effects of the ATP2C2 missense variant (chr16:84405221)
| Computational tool | Score (cut-off)a | Prediction |
|---|---|---|
| SIFT | 0.24 (0 < s < 0.05) | Tolerated |
| Mutation assessor | 1.535 (s > 3.5) | Low functional impact |
| PROVEAN | −0.399 (s ≤ −2.5) | Neutral |
| PolyPhen-2 | 0.002 (0.85 < s < 1.0) | Benign |
| MutPred | 0.114 (s > 0.50) | Benign |
| INPS-MD | −0.073 (s < −0.5) | Weak destabilising effect |
| MutationTaster | 21 (s ≥ 151) | Conservative |
aCut-off scores (s) for predicting functional effects.
Genotyping of Chr16:84405221 in the SLI, dyslexia and ADHD cohorts
The variant was genotyped in independent cohorts with LI, dyslexia or ADHD (Table 2). The frequency of the variant was 2.2% in the SLIC cohort selected for SLI and 1.8% across all the unrelated LI cases (combined LI samples, N = 360), including the discovery family and previously published data (49). The frequency was 0.8% in the combined dyslexia samples (N = 520) and 0.7% in the combined ADHD cases (N = 151).
Table 2.
Frequency of the rare allele A (Chr16:84405221) across different cohorts
| Cohort | Cohort type | N | Allele A count | Allele A frequency | Reference |
|---|---|---|---|---|---|
| SLIC | Clinical | 161 casesa | 7 | 2.2% | (26) |
| York | Longitudinal | 23 LI casesb | 0 | 0% | (53) |
| 18 dyslexia cases | 3* | 8.3% | |||
| 72 TD | 1 | 0.7% | |||
| Dyslexia | Clinical | 471 cases | 5 | 0.5% | (54) |
| ADHD | Clinical | 141 cases | 2 | 0.7% | (55) |
| ALSPAC | Longitudinal Epidemiological | 170 LI casesc | 5 | 1.5% | (56,57) |
| 31 dyslexia cases | 0 | 0% | |||
| 10 ADHD cases | 0 | 0% | |||
| Combined cases | |||||
| Combined SLI/LI samples | 360 casesd | 13 | 1.8% | ||
| Combined dyslexia samples | 520 cases | 8* | 0.8% | ||
| Combined ADHD samples | 151 cases | 2 | 0.7% | ||
| Controls | |||||
| ALSPAC | Longitudinal epidemiological | 681 controls | 14 | 1% | (56,57) |
| European population (gnomAD v2.1.1) | 24 046 controls | 426** | 0.9% | (58) | |
| gnomAD v2.1.1 | 59 718 controls | 584*** | 0.5% |
aIncluding the probands analyzed by Chen and colleagues through exome sequencing (Chen et al., 2017).
bIncludes N = 11 individuals with comorbid SLI and dyslexia.
cIncludes N = 9 individuals with comorbid LI and dyslexia.
dIncluding the six probands screened by whole exome sequencing.
* includes N = 1 homozygote; **includes N = 2 homozygotes; ***includes N = 3 homozygotes.
Abbreviations: ADHD: attention deficit hyperactivity disorder.
In the ALSPAC super-controls (N = 681), the variant occurred at a frequency of 1%. The frequency in the global gnomAD database is of 0.5% (N = 59 718) and of 0.9% in the European population (N = 24 046).
Although the frequency was higher in the LI cases (1.5%) than the controls (1%) derived from the ALSPAC cohort, the difference was not statistically significant when examined with a two-tailed Fisher’s exact test (P = 0.152).
Mean score of language and reading-related phenotypes
We evaluated the effects of the variant on a range of language-related quantitative measures available for the ALSPAC cohort (Supplementary Material, Table S4). The carriers (N = 42) had lower mean scores compared with the non-carriers (N = 1825) on all tested measures, reaching nominal statistical significance (t-test, two-tailed distribution) for six traits: READ (P = 0.016), SPELL (P = 0.006), PHONEME (P = 0.026), NW_READ (P = 0.002), VIQ (P = 0.006) and PIQ (P = 0.022) (Table 3). NW_READ survived a conservative Bonferroni correction for multiple comparison (10 tests, threshold for significance = 0.005; see also Supplementary Material, Table S5 for analysis through the Wald test and Supplementary Material, Fig. S2 for box plots). It is worth noting that the 10 tests are not completely independent given the correlation across these measures (59).
Table 3.
Mean score of different phenotypes across carriers and non-carriers (ALSPAC cohort)
| Genotype (total N) | Mean score (SD; N) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reading measures | Language measures | IQ | ||||||||
| READ | SPELL | PHONEME | NW_READ | WOLD | NWR | MEMSPAN | CCC_average7 | VIQ | PIQ | |
| GA or AA (42) | 26.6 | 6.4 | 17.8 | 4.3 | 7.6 | 7.2 | 3.3 | 31.1 | 104.4 | 97.4 |
| (9.8; 38) | (4.3; 37) | (9.5; 38) | (2.4; 39) | (1.8; 37) | (2.5; 37) | (0.8; 34) | (1.2; 37) | (13.6; 38) | (16.2; 39) | |
| GG (1825) | 30.1 | 8.3 | 21.2 | 5.5 | 7.8 | 7.5 | 3.5 | 31.3 | 112.1 | 103.7 |
| (8.7; 1707) | (4.3; 1696) | (9.4; 1710) | (2.4; 1763) | (1.9; 1711) | (2.4; 1711) | (0.8; 1622) | (1.1, 1722) | (16.6; 1703) | (16.8, 1701) | |
| P (t-test) | 0.016 | 0.006 | 0.026 | 0.002 | 0.533 | 0.503 | 0.131 | 0.311 | 0.006 | 0.022 |
Comparisons were evaluated with a Student’s t-test; P < 0.05 are indicated in bold. The threshold for Bonferroni correction based on 10 tests is 0.005.
Sample size varies depending on the availability of missing phenotypes. Details about each test can be found in Supplementary Material, Table S4.
READ, single-word reading accuracy; SPELL, single word spelling accuracy; PHONEME, phoneme awareness; NW_READ, nonword reading; WOLD, Listening and comprehension test; NWR, nonword repetition; MEMSPAN, working memory; CCC_average7, average of first seven scales from the Children’s Communication Checklist; VIQ, verbal IQ.
ATPase assay
An effect of allele A on the ATPase activity of SPCA2 was tested with a colorimetric assay to quantify the inorganic phosphate released during the catalytic reaction (Fig. 2A). Microsomes were isolated from HEK293 cells overexpressing the wildtype or mutated SPCA2-HA tagged protein. The activity of the overexpressed SPCA2 WT sample was higher compared with the negative controls (empty vector and untransfected cells) across all the conditions. SPCA2 p.V102M displayed on average a lower ATPase activity compared with that of the WT protein (Fig. 2A). WB confirmed the overexpression of SPCA2 for the WT and mutated constructs, while no protein was detected for the negative controls, suggesting a very minimal effect of baseline endogenous protein (Fig. 2B).
Figure 2 .
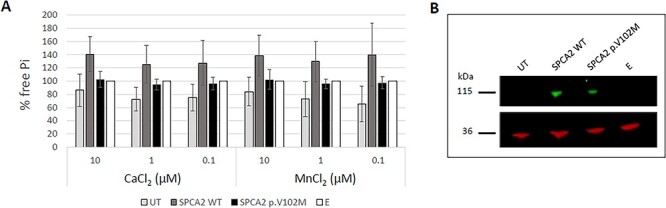
Effect of the mutation on ATPase activity. (A) Concentration of free inorganic phosphate (Pi) released during the ATPase catalytic reaction. The activity is expressed relative to the empty vector, normalized to 100%. The results are shown as mean percentages ± SD of two independent experiments. The samples are: untransfected HEK293 cells (UT), microsomes isolated from HEK293 cells transiently transfected to overexpress the recombinant SPCA2-HA tagged (SPCA2 WT) or mutated protein (SPCA2 p.V102M) and microsomes isolated from HEK293 cells transiently transfected with the empty vector (E). (B) Western blot. Each lane was loaded with 5 μg of microsomes. The membranes were probed with antibodies against SPCA2 (top, green) or GAPDH (36 kDa) (bottom, red). SPCA2 can be detected when overexpressed, while no signal is observed in the untransfected cells and the control samples. The band corresponds to the predicted SPCA2-HA tagged molecular weight (105.2 kDa).
Discussion
In this study, we investigated the effects of a rare ATP2C2 variant on LI and related disorders. Overall, we found that this variant had a higher frequency in LI samples (1.8%, N = 360) compared with controls (1%, N = 681, ALSPAC) and subjects with dyslexia (0.8%, N = 520) or ADHD (0.7%, N = 150). The frequency reached 2.2% in a clinical cohort ascertained for SLI (N = 161; Table 2). In a general population sample derived from the ALSPAC cohort, carriers of the rare variants presented lower scores compared with non-carries on a range of language-related measures, with the stronger effect observed for the non-word reading measure (P = 0.002; Table 3).
Taken together, our results suggest that the ATP2C2 missense variant is associated to LI and lower scores on reading and language measures. The presence of the ATP2C2 variant in controls shows that it is not sufficient on its own to cause a disorder and its association with a range of measures suggests a pleiotropic effect on cognitive traits. These observations are in line with the emerging findings in the field of complex traits genetics and, specifically, for neurodevelopmental and psychiatric disorders. For example, under the double-hit model, a risk factor might lead to a specific disorder or a range of conditions depending on the interactions with other genetic risk variants (51,52,60). Under this model, the rare ATP2C2 variant might increase the risk for LI or affect performance on reading- and language-related measures when combined with different susceptibility factors, as observed for other rare variants implicated in LI. These include a microdeletion in ZNF277 (42), a rare stop-gain variant in ERC1 (49) and nonsynonymous mutations in NFXL1 (61), GRIN2B and SRPX2 (Chen et al., 2017). Increasing evidence shows that the penetrance of rare risk factors is modulated by the cumulative effect of common risk alleles, even in severe neurodevelopmental disorders usually assumed to have a monogenic origin (62). Different manifestations determined by the interplay between common and rare variants have been reported for conditions such as schizophrenia (63,64), ADHD (65), autism spectrum disorder (66), intellectual disability (67) and outcomes such as educational attainment (68). Rare risk factors causing psychiatric disorders have also been reported to have an effect on cognitive abilities in the general population (69,70). This is in line with what we observe for the ATP2C2 variant which showed associations with poor performance on cognitive traits in the ALSPAC sample regardless of any disorder group assignment (Table 3). The variant is associated to lower mean scores on a range of cognitive measures, with the strongest effect observed for reading abilities rather than language skills. Our results support the emerging idea that genetic risk factors have generalized effects across cognitive and neurodevelopmental traits, rather than specific genotype–phenotype relationship. The same concept is illustrated also for common variants through polygenic risk scores (PRSs) analysis. For example, PRS for schizophrenia showed associations for a number of traits including poor cognitive outcomes (71) but also enhanced creativity (72), language performance (73) and hearing (74). Similarly, PRSs for general cognitive abilities show effects on reading and language measures (15,75–77).
The ATP2C2 gene encodes for an ATPase that transports Ca2+ and Mn2+ into the Golgi lumen (78). ATPases are emerging as a new class of proteins involved in neurodevelopmental disorders (79–82). Ca2+ is involved in the regulation of several neuronal processes such as neuronal migration, axon guidance, synaptic plasticity, memory, neuronal excitability, neurosecretion and aging (83–86). Excessive accumulation of Mn2+ inside the cells is toxic, induces apoptosis and causes manganism, a neurological disorder characterized by tremors and spasm (87,88). Additionally, dysregulation of these cations has been linked to Alzheimer’s, Parkinson’s and Huntington’s disease (89,90). The transport of Ca2+ and Mn2+ ions inside the Golgi is essential for the activity of enzymes involved in mechanisms such as neurotransmitter synthesis, membrane trafficking, metabolism, protein modification, sorting and folding (86). Previous studies have shown that ATP2C2 is expressed in postnatal rat hippocampal neurons (91) and in adult human brain (78,91,92), suggesting a possible role of ATP2C2 in maintaining ionic homeostasis in neurons.
The missense variant described in the current study is localized in the cation transporting N-terminal domain and causes the amino acid substitution from valine to methionine (Fig. 1). The latter residue is characterized by an unbranched side chain that provides increased flexibility. In addition to this, methionine has a sulphur group that can influence the binding preference for metal ions and be reversibly oxidized converting the hydrophobic amino acid into a highly polar one (93,94). Bioinformatic predictions do not attribute substantial detrimental effects to the substitution, and therefore, any potential effects are expected to be subtle (Table 1). We tested experimentally whether the rare allele A had an effect on the SPCA2 activity, through an ATPase assay. This approach allowed us to measure the hydrolysis of ATP in a quick and sensitive way. Independently of the concentrations tested, the rare variant displayed on average a lower ATPase activity compared with SPCA2 WT (Fig. 2). Although not statistically significant, the data suggested a possible impact of the variant on the velocity of the reaction and therefore the cation binding affinity. More specifically, the mutated transporter tended to present a lower ATP hydrolysis indicative of a lower turnover rate and consequently a higher ion affinity. In line with the bioinformatics prediction, the rare variant does not affect the functionality of the protein but might alter its performance which in turn, in combination with other factors, might contribute to neurodevelopmental phenotypes.
In summary, we identified a rare missense variant (chr16:84405221, GRCh38.p12) in ATP2C2, a previously reported candidate gene for LI. We showed that the rare allele A increases the risk for LI cohorts and is associated with more general cognitive abilities in a population-based cohort. Preliminary functional characterization of the rare allele suggested that the rare variant influences the ATPase activity of SPCA2 supporting follow up functional studies to fully understand the role of this variant and the ATP2C2 gene. WES studies for language disorders have been limited to samples, which makes the result interpretation particularly challenging. Our results demonstrate that combined analyses across disorders and phenotypes provide a valid route to validate WES findings. Taken together, our results further support a role of ATP2C2 in LI and pinpoint to a specific rare variant which might impact ATPasse activity of the SPCA2 protein.
Material and Methods
Discovery pedigree
An exome sequencing analysis was conducted in six pedigrees of SLI probands with multiple affected members per family, selected from an ongoing longitudinal study in the Midwestern USA (95,96). The full results of this screening will be reported in a separate manuscript. SLI probands were identified based on language performance 1 standard deviation (SD) or more below the mean on an age appropriate omnibus language test (95) and met the following entrance screening criteria: (i) a standard score above 85 on an initial age appropriate test of non-verbal IQ, (ii) normal hearing acuity, (iii) no history of neurological disorders or diagnosis of autism and (iv) intelligible speech sufficient for language transcription. Except for normal hearing acuity, the entrance screening criteria did not apply to siblings and parents of SLI probands, as their intellectual status was an outcome of the study (95).
Here, we report the analysis and follow up of a specific variant of interest (Chr16: 84405221) in one family (Fig. 1). A male SLI proband, his younger male sibling (Sib 1) and their parents were selected for WES. All family members including the other two siblings were genotyped for the ATP2C2 variant. Longitudinal phenotype data on the proband were assessed from age 6 to 19 years and the siblings were assessed also at multiple time points. Both mother and father were tested at a single time point. All siblings and the father met the criteria above for a clinical diagnosis of SLI. The sibling selected for WES was also delayed on the Test of Early Grammatical Impairment until age 7 years, 6 months (97). The children were also delayed in receptive vocabulary and oral reading across age, as well as in mean length of utterance when young. The study was approved by the institutional review boards at the University of Kansas and at the University of Nebraska Medical Center. Appropriate informed consent was obtained from the subjects and/or their caregivers.
Validation, genotyping and bioinformatics prediction for the variant
Sanger sequencing was used to validate the variant and test all available family members in the discovery pedigree.
The variant was then screened in larger cohorts with a TaqMan™ SNP Genotyping Assay run on a ViiA7 instrument (Life Technologies, Paisley, UK). The presence of the mutation was validated using PCR 3′-end mismatch primers (Fig. 1B). All primer sequences are reported in Supplementary Material, Table S1. For protocol details, see Supplementary Material, Tables S2 and S3, and Supplementary Material, Fig. S1. The functional impact of the rare ATP2C2 variant was estimated using the default settings of seven different in silico prediction algorithms (Table 1), SIFT (98), Mutation Assessor (99), PROVEAN (100), PolyPhen-2 (101), MutPred (102), INPS-MD (103) and MutationTaster (104).
Cohorts for follow-up analysis
The genotyping was conducted across different UK cohorts of children with LI, dyslexia or ADHD used in previous genetic studies (Table 2). We use the SLI term when participants were recruited under SLI criteria and the term LI to refer to individuals who did not receive a specific diagnosis but presented language difficulties on psychometric tests. Assignment to a case group was based either on existing clinical diagnoses (York and ADHD cohorts) or derived from poor performance on different psychometric tests as specified below. Exclusion criteria included non-European ethnicity, English as a second language, signs of other sensory or neurological conditions and missing information for the selected phenotypes.
The SLI Consortium (SLIC) cohort consisted of 263 families (263 probands, 258 siblings, 526 parents) (age range 5–19 years) recruited across different UK clinics (29). Participants were assigned to a case category when they scored ≥80 on performance IQ (PIQ) and had impaired expressive or receptive language skills (ELS or RLS score ≥ 1.5 SD below the normative mean of the general population) as assessed through the Clinical Evaluation of Language Fundamentals test (CELF-R, revised version) (105). If the same family comprised several cases, the most severe sibling was included into the analysis. Severity was determined by comparing the scores for ELS and RLS composite measure. If the cases performed at the same level on these measures, inclusion was determined by the most severe score on the NWR task. Overall, 161 unrelated cases met these criteria. A subset of this sample was used for the WES analysis conducted by Chen and colleagues where the rare variant was also reported (49). The ethical approval was given by local ethics committees of the hospitals involved in the consortium, and all subjects provided informed consent.
The York cohort consisted of 115 families (115 probands, 27 siblings, 184 parents), collected for a longitudinal study designed to investigate language and reading development (53). Families were recruited via advertisements and assessed for either having a family history of dyslexia or showing preschool language difficulties. Participants that did not meet these criteria and did not present other impairments were assigned to the typically developing (TD) group. At 8 years, the children were given a clinical diagnosis for SLI (N = 23, including 11 individuals with dyslexia) and dyslexia (N = 18). Ethical approval was obtained from the Yorkshire & The Humber—Humber Bridge NHS Research Ethics and the Ethics Committee at the University of York—Department of Psychology. Informed consent was given by all the participants and/or their parents.
Participants were assigned to a category of dyslexia from two other separate cohorts of families and individual cases. The criteria for the dyslexia, which is based on different cut-off points than the ones used for SLI, were PIQ ≥ 85 and a score of 1 SD below the population mean on the single-word reading (READ) test measured through the British Ability Scales (BAS) (106) or the Wide Range Achievement Test (WRAT-R, revised version) (107). The first cohort comprised 290 families (689 siblings, 580 parents) (age range 6–27 years) described before (108). All the subjects were recruited by the Dyslexia Clinic at the Royal Berkshire Hospital, Reading, UK. When multiple children from the same family met the above criteria, we referred to other psychometric measures and selected the individual with the most severe phenotype on the following tasks: BAS single-word spelling accuracy (SPELL) (106), phonological decoding ability (CCN) (109), phonemic awareness (SPOON) (110) and orthographic coding skill assessed through the irregular word (CCI) (109) or forced word choice test (OLSON) (111). The Dyslexia Cases cohort, N = 592 singletons (age range 6–18 years), was collected from either the Dyslexia Research Centre clinics in Oxford and Reading or the Aston Dyslexia and Development Clinic in Birmingham (54). In total, N = 471 participants met the above criteria for dyslexia. The study was approved by the Oxfordshire Psychiatric Research Ethics Committee and the Aston University Ethics Committee and informed consent was given by all the participants and/or their caregivers.
The ADHD cohort included 159 trios (age range 5–16 years) recruited from child psychiatry clinics in the UK and Ireland (55). Participants were interviewed employing the Child and Adolescent Psychiatric Assessment (112). All the probands fulfilled the DSM-IV (American Psychiatric Society, 1994) diagnostic criteria for ADHD. Ethical approvals were acquired from the Trinity College Dublin in Ireland and the University of Birmingham in UK. Informed consent was obtained from the parents of all the participants.
In addition to the clinical cohorts, we analyzed a subset of samples from the Avon Longitudinal Study of Parents and Children (ALSPAC, see Supplementary Material) (56,57) for which low-read-depth whole-genome sequence (WGS) data were generated as part of the UK10K project. Sequence data were provided by ALSPAC in the form of variant call files generated on the Illumina Hi-seq platform using paired-end 100 bp reads and a standard GATK workflow. Full details of sequence protocols have been reported before (113). For this manuscript, UK10K participants were excluded from the analysis if they reported a non-white European ethnicity, presented with overt hearing difficulties or were affected by neurological conditions, which may explain their language difficulties, such as autism, Asperger syndrome or pervasive developmental disorder. Individuals with missing information for the phenotypes of interest were also excluded.
The criteria for an assignment to a LI category were a PIQ ≥ 80 at age of 8.5 and a score either 1 SD below the population mean on the Wechsler Objective Language Dimensions (WOLD) comprehension test (114) at 8 years or 1 SD below the population mean on both tests of syntax, and intelligibility and verbal fluency, as assessed through the Children’s Communication Checklist (CCC) (115) at 7.5 years. Assignment to the dyslexia category was given based on a diagnosis reported by the mother through a questionnaire and a PIQ score higher or equal to 85 at the age of 8.5. Assignment to the ADHD category was given if the subject met the DAWBA DSM-IV diagnostic criteria and had a PIQ > 80 at the age of 8.5. Please note that the ALSPAC study website contains details of all the data that is available through a fully searchable data dictionary and variable search tool (https://www.bristol.ac.uk/alspac/researchers/our-data/).
Participants were assigned to a ‘super-control’ group (N = 681) when they performed above the mean on tests of receptive (WOLD comprehension) and expressive (CCC syntax and intelligibility and fluency scores) language and had both verbal and non-verbal IQ ≥ 80. From the controls, we also excluded any child who self-reported a need for special educational support or speech and language therapy and children who self-reported a diagnosis of neurodevelopmental disorder (ADHD, conduct disorder, anxiety disorder, depressive disorder, dyslexia, dyspraxia, dyscalculia, learning difficulties, speech and language difficulties or developmental delay).
Overall, in the ALSPAC sample, we identified: N = 170 LI cases, N = 31 dyslexia cases, N = 10 ADHD cases and N = 681 controls. Nine individuals in the LI group presented comorbidity with dyslexia.
Ethical approval for the study was obtained from the ALSPAC Law and Ethics Committee and the Local Research Ethics Committees. Consent for biological samples has been collected in accordance with the Human Tissue Act (2004). Informed consent was signed by the parents after having received a complete description of the study at the time of enrolment into the ALSPAC project, with the option for them or their children to withdraw at any time.
The gnomAD v2.1.1 database (https://gnomad.broadinstitute.org/) was used as a reference European population (58). Of note, this source includes the UK10K samples.
Quantitative analysis
While, in most cohorts, the number of carriers of the rare variant was too small to conduct any meaningful quantitative analysis, the ALSPAC dataset was suitable for this option. All the individuals with available genotype (N = 1867) were included into the analysis. Carriers (N = 42) and non-carriers (N = 1825) were compared for the mean score calculated for reading and language-related measures including: single-word reading accuracy (READ, target age: 7.5 year), single-word spelling accuracy (SPELL, 7.5 year), phoneme awareness (PHONEME, 7.5 year), working memory (MEMSPAN, 10.5 year), listening and comprehension test (WOLD, 8.5 year), phonological short-term memory test (NWR, 8.5 year), single-non-word reading accuracy (NW-READ, 9.5 year), average of the first seven scales from the Children’s Communication Checklist (CCC-average7, 7.5 year) and PIQ (8.5 year) (See Supplementary Material, Table S4 for more details). Statistical significance was calculated with the Student’s t-test (equal variances, two-tailed distribution).
ATPase assay
The human ATP2C2 coding sequence was cloned into the expression vector pcDNA™5/TO [Invitrogen, kind gift from Professor Galione (116)]. ATP2C2 cDNA was obtained from SK-BR-3 cells maintained under standard conditions and amplified by high fidelity PCR. The primers were designed to include a HindIII site prior the Kozak consensus sequence and a NotI site immediately before the stop codon (See Supplementary Material, Table S1 for the primer sequences). The ATP2C2 open reading frame was placed under the control of a constitutive CMV promoter and in frame with a downstream HA epitope tag. The SPCA2-HA tagged plasmid was modified to introduce the missense variant (allele A). The mutated construct (SPCA2 p.V102M) was built using the GeneArt™ Site-Directed Mutagenesis kit (Thermo Fisher) according to the manufacturer’s instructions. The sequences were verified by Sanger sequencing (See Supplementary Material, Table S1 for primer sequences).
Microsomes were prepared as described by Vandecaetsbeek (117) with minor modifications (See Supplementary Material for details). HEK293 cells were transfected with the ATP2C2 expressing constructs. The empty vector and untransfected cells were used as controls.
ATPase activity was evaluated using the PiColorLock™ Gold kit (Expedeon, Abcam) according to the manufacturer’s specifications. The assay was conducted in a 96-well plate and each sample run in duplicates. The amount of free inorganic phosphate (Pi) released from each reaction was calculated by plotting the absorbance against a series of Pi standards of known concentration.
The data generated were presented as mean percentages ± SD of two independent experiments. The activity of each sample was expressed relative to that detected in the empty vector, normalized to 100%. Pair-wise comparison between samples was performed by the Welch’s t-test (unequal variances, two-tailed distribution).
For western blot analysis, 5 μg of microsomes were mixed with Laemmli buffer and incubated at room temperature for 30 min. Samples (5 μg per well) were separated on a precast 4–12% NuPAGE™ polyacrylamide gradient gel (Invitrogen). The anti-SPCA2 (Abcam) and anti-GAPDH (Abcam) antibodies were diluted 1:4000 and 1:5000 in 0.1% TBS-Tween™20 (Fisher BioReagents™), respectively.
Supplementary Material
Acknowledgements
This work was, in part, completed while D.F.N. was at the Wellcome Trust Centre for Human Genetics, Oxford as an MRC Career Development Fellow (G1000569/1). We would like to thank the members of all teams who collected the data and the families who participated. We are extremely grateful to all the families who took part in the ALSPAC study, the midwives for their help in recruiting them and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. This publication is the work of the authors and DFN will serve as guarantors for the analysis of the ALSPAC data presented in this paper. A comprehensive list of grants funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). Whole-genome sequencing of the ALSPAC samples was performed as part of the UK10K Consortium (a full list of investigators who contributed to the generation of the data is available from www.UK10K.org.uk). ALSPAC GWAS data was generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. We thank Michael Gill for sharing data for the ADHD cohort and Arathi Jeyaratnam for the technical support.
Conflict of Interest statement.
The authors have declared that no competing interests existed at the time of publication.
Contributor Information
Angela Martinelli, School of Medicine, University of St Andrews, St Andrews, UK.
Mabel L Rice, Child Language Doctoral Program, University of Kansas, Lawrence, KS, USA.
Joel B Talcott, Aston Brain Centre, School of Life and Health Sciences, Aston University, Birmingham, UK.
Rebeca Diaz, School of Medicine, University of St Andrews, St Andrews, UK.
Shelley Smith, Department of Neurological Sciences, University of Nebraska Medical Center, Lincoln, NE, USA.
Muhammad Hashim Raza, Child Language Doctoral Program, University of Kansas, Lawrence, KS, USA.
Margaret J Snowling, Department of Experimental Psychology and St John's College, University of Oxford, Oxford, UK.
Charles Hulme, Department of Education, University of Oxford, Oxford, UK.
John Stein, Department of Physiology, University of Oxford, Oxford, UK.
Marianna E Hayiou-Thomas, Department of Psychology, University of York, York, UK.
Ziarih Hawi, Turner Institute for Brain and Mental Health, School of Psychological Sciences, Monash University, Clayton, VIC, Australia.
Lindsey Kent, School of Medicine, University of St Andrews, St Andrews, UK.
Samantha J Pitt, School of Medicine, University of St Andrews, St Andrews, UK.
Dianne F Newbury, Department of Biological and Medical Sciences, Oxford Brookes University, Oxford, UK.
Silvia Paracchini, School of Medicine, University of St Andrews, St Andrews, UK.
Funding
Royal Society to S.P.; Action Medical Research Action/The Chief Scientist Office (CSO), Scotland grant (GN2614); Cunningham Trust grant to S.P. and S.J.P.; St Andrews Bioinformatics Unit funded by the Wellcome Trust (grant 105621/Z/14/Z); The Waterloo Foundation to J.B.T. and S.P. [797–1720]; University of Kansas grant (NIH:NIDCD 5 R01 DC001803); Wellcome Trust Programme Grant (082036/B/07/Z); Oxford Brookes University funds, the Leverhulme Trust and the British Academy to D.F.N.; UK Medical Research Council and Wellcome (Grant ref: 217065/Z/19/Z); University of Bristol.
References
- 1. Bishop, D.V.M. (2014) Ten questions about terminology for children with unexplained language problems. Int. J. Lang. Commun. Disord., 49, 381–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bishop, D.V.M., Snowling, M.J., Thompson, P.A., Greenhalgh, T., Adams, C., Archibald, L., Baird, G., Bauer, A., Bellair, J., Boyle, C. et al. (2016) CATALISE: a multinational and multidisciplinary Delphi consensus study. Identifying language impairments in children. PLoS One, 11, e0158753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bishop, D.V.M. (2017) Why is it so hard to reach agreement on terminology? The case of developmental language disorder (DLD). Int. J. Lang. Commun. Disord., 52, 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Norbury, C.F., Gooch, D., Wray, C., Baird, G., Charman, T., Simonoff, E., Vamvakas, G. and Pickles, A. (2016) The impact of nonverbal ability on prevalence and clinical presentation of language disorder: evidence from a population study. J. Child Psychol. Psychiatry Allied Discip., 57, 1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tomblin, J.B., Records, N.L., Buckwalter, P., Zhang, X., Smith, E. and O’Brien, M. (1997) Prevalence of specific language impairment in kindergarten children. J. Speech. Lang. Hear. Res., 40, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conti-Ramsden, G., Durkin, K., Simkin, Z. and Knox, E. (2009) Specific language impairment and school outcomes. I: identifying and explaining variability at the end of compulsory education. Int. J. Lang. Commun. Disord., 44, 15–35. [DOI] [PubMed] [Google Scholar]
- 7. Conti-Ramsden, G., Mok, P.L.H., Pickles, A. and Durkin, K. (2013) Adolescents with a history of specific language impairment (SLI): strengths and difficulties in social, emotional and behavioral functioning. Res. Dev. Disabil., 34, 4161–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Snowling, M.J., Adams, J.W., Bishop, D.V.M. and Stothard, S.E. (2001) Educational attainments of school leavers with a preschool history of speech-language impairments. Int J Lang Commun Disord, 36, 173–183. [DOI] [PubMed] [Google Scholar]
- 9. Conti-Ramsden, G., Durkin, K., Toseeb, U., Botting, N. and Pickles, A. (2018) Education and employment outcomes of young adults with a history of developmental language disorder. Int. J. Lang. Commun. Disord., 53, 237–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Botting, N. (2020) Language, literacy and cognitive skills of young adults with developmental language disorder (DLD). Int. J. Lang. Commun. Disord., 55, 255–265. [DOI] [PubMed] [Google Scholar]
- 11. McArthur, G.M., Hogben, J.H., Edwards, V.T., Heath, S.M. and Mengler, E.D. (2000) On the “specifics” of specific reading disability and specific language impairment. J. Child Psychol. Psychiatry, 41, 869–874. [PubMed] [Google Scholar]
- 12. Snowling, M. and Stothard, S.E. (2000) Is preschool language impairment a risk factor for dyslexia in adolescence? J. Child Psychol. Psychiatry, 41, 587–600. [DOI] [PubMed] [Google Scholar]
- 13. Mueller, K.L. and Tomblin, J.B. (2012) Exmaining the comorbidity of language disorders and ADHD. Biol. Bull., 221, 18–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newbury, D.F., Paracchini, S., Scerri, T.S., Winchester, L., Addis, L., Richardson, A.J., Walter, J., Stein, J.F., Talcott, J.B. and Monaco, A.P. (2011) Investigation of dyslexia and SLI risk variants in reading- and language-impaired subjects. Behav. Genet., 41, 90–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gialluisi, A., Andlauer, T.F.M., Mirza-Schreiber, N., Moll, K., Becker, J., Hoffmann, P., Ludwig, K.U., Czamara, D., Pourcain, B.S., Honbolygó, F. et al. (2020) Genome-wide association study reveals new insights into the heritability and genetic correlates of developmental dyslexia. Mol. Psychiatry. 10.1038/s41380-020-00898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barry, J.G., Yasin, I. and Bishop, D.V. (2007) Heritable risk factors associated with language impairments. Genes Brain Behav., 6, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bishop, D.V.M., North, T. and Donlan, C. (1995) Genetic basis of specific language impairment: evidence from a twin study. Dev. Med. Child Neurol., 37, 56–71. [DOI] [PubMed] [Google Scholar]
- 18. Lewis, B.A. and Thompson, L.A. (1992) A study of developmental speech and language disorders in twins. J. Speech Hear. Res., 35, 1086–1094. [DOI] [PubMed] [Google Scholar]
- 19. Choudhury, N. and Benasich, A.A. (2003) A family aggregation study: the influence of family history and other risk factors on language development. J Speech Lang Hear Res., 46, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tomblin, J.B. and Buckwalter, P.R. (1998) Heritability of poor language achievement among twins. J. Speech, Lang. Hear. Res., 41, 188–199. [DOI] [PubMed] [Google Scholar]
- 21. Wiszniewski, W., Hunter, J.V., Hanchard, N.A., Willer, J.R., Shaw, C., Tian, Q., Illner, A., Wang, X., Cheung, S.W., Patel, A. et al. (2013) TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet., 93, 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vernes, S.C., Newbury, D.F., Abrahams, B.S., Winchester, L., Nicod, J., Groszer, M., Alarcón, M., Oliver, P.L., Davies, K.E., Geschwind, D.H. et al. (2008) A functional genetic link between distinct developmental language disorders. N. Engl. J. Med., 359, 2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartlett, C.W., Flax, J.F., Logue, M.W., Vieland, V.J., Bassett, A.S., Tallal, P. and Brzustowicz, L.M. (2002) A major susceptibility locus for specific language impairment is located on 13q21. Am. J. Hum. Genet., 71, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bartlett, C.W., Flax, J.F., Logue, M.W., Smith, B.J., Vieland, V.J., Tallal, P. and Brzustowicz, L.M. (2004) Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum. Hered., 57, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Newbury, D.F., Cleak, J.D., Ishikawa-Brush, Y., Marlow, A.J., Fisher, S.E., Monaco, A.P., Stott, C.M., Merricks, M.J., Goodyer, I.M., Bolton, P.F. et al. (2002) A genomewide scan identifies two novel loci involved in specific language impairment. Am. J. Hum. Genet., 70, 384–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. SLI Consortium (2004) Highly significant linkage to the SLI1 locus in an expanded sample of individuals affected by specific language impairment. Am. J. Hum. Genet., 74, 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Monaco, A.P. (2007) Multivariate linkage analysis of specific language impairment (SLI). Ann. Hum. Genet., 71, 660–673. [DOI] [PubMed] [Google Scholar]
- 28. Falcaro, M., Pickles, A., Newbury, D.F., Addis, L., Banfield, E., Fisher, S.E., Monaco, A.P., Simkin, Z., Conti-Ramsden, G., Newbury, D.F. et al. (2008) Genetic and phenotypic effects of phonological short-term memory and grammatical morphology in specific language impairment. Genes Brain Behav., 7, 393–402. [DOI] [PubMed] [Google Scholar]
- 29. Newbury, D.F., Winchester, L., Addis, L., Paracchini, S., Buckingham, L.L., Clark, A., Cohen, W., Cowie, H., Dworzynski, K., Everitt, A. et al. (2009) CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am. J. Hum. Genet., 85, 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bishop, D.V.M., North, T. and Donlan, C. (1996) Nonword repetition as a behavioural marker for inherited language impairment: evidence from a twin study. J. Child Psychol. Psychiatry Allied Discip., 37, 391–403. [DOI] [PubMed] [Google Scholar]
- 31. Graham, S.A. and Fisher, S.E. (2015) Understanding language from a genomic perspective. Annu. Rev. Genet., 49, 131–160. [DOI] [PubMed] [Google Scholar]
- 32. Nudel, R., Simpson, N.H., Baird, G., O’Hare, A., Conti-Ramsden, G., Bolton, P.F., Hennessy, E.R., Ring, S.M., Davey Smith, G., Francks, C. et al. (2014) Genome-wide association analyses of child genotype effects and parent-of-origin effects in specific language impairment. Genes. Brain Behav., 13, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pettigrew, K.A., Frinton, E., Nudel, R., Chan, M.T.M., Thompson, P., Hayiou-Thomas, M.E., Talcott, J.B., Stein, J., Monaco, A.P., Hulme, C. et al. (2016) Further evidence for a parent-of-origin effect at the NOP9 locus on language-related phenotypes. J. Neurodev. Disord., 8. 10.1186/s11689-016-9157-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. St Pourcain, B., Cents, R.A.M., Whitehouse, A.J.O., Haworth, C.M.A., Davis, O.S.P., O’reilly, P.F., Roulstone, S., Wren, Y., Ang, Q.W., Velders, F.P. et al. (2014) Common variation near ROBO2 is associated with expressive vocabulary in infancy. Nat. Commun., 5. 10.1038/ncomms5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luciano, M., Evans, D.M., Hansell, N.K., Medland, S.E., Montgomery, G.W., Martin, N.G., Wright, M.J. and Bates, T.C. (2013) A genome-wide association study for reading and language abilities in two population cohorts. Genes, Brain Behav., 12, 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eicher, J.D., Powers, N.R., Miller, L.L., Akshoomoff, N., Amaral, D.G., Bloss, C.S., Libiger, O., Schork, N.J., Darst, B.F., Casey, B.J. et al. (2013) Genome-wide association study of shared components of reading disability and language impairment. Genes. Brain Behav., 12, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gialluisi, A., Newbury, D.F., Wilcutt, E.G., Olson, R. K, DeFries, J.C., Brandler, W.M., Pennington, B.F., Smith, S.D., Scerri, T.S., Simpson N.H. et al. (2014) Genome-wide screening for DNA variants associated with reading and language traits. Genes, Brain Behav., 13, 686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Simpson, N.H., Addis, L., Brandler, W.M., Slonims, V., Clark, A., Watson, J., Scerri, T.S., Hennessy, E.R., Bolton, P.F., Conti-Ramsden, G. et al. (2014) Increased prevalence of sex chromosome aneuploidies in specific language impairment and dyslexia. Dev. Med. Child Neurol., 56, 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simpson, N.H., Ceroni, F., Reader, R.H., Covill, L.E., Knight, J.C., Hennessy, E.R., Bolton, P.F., Conti-Ramsden, G., O’Hare, A., Baird, G. et al. (2015) Genome-wide analysis identifies a role for common copy number variants in specific language impairment. Eur. J. Hum. Genet., 23, 1370–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalnak, N., Stamouli, S., Peyrard-Janvid, M., Rabkina, I., Becker, M., Klingberg, T., Kere, J., Forssberg, H. and Tammimies, K. (2018) Enrichment of rare copy number variation in children with developmental language disorder. Clin. Genet., 94, 313–320. [DOI] [PubMed] [Google Scholar]
- 41. Gulhan Ercan-Sencicek, A., Davis Wright, N.R., Sanders, S.J., Oakman, N., Valdes, L., Bakkaloglu, B., Doyle, N., Yrigollen, C.M., Morgan, T.M. and Grigorenko, E.L. (2011) A balanced t(10;15) translocation in a male patient with developmental language disorder. Eur. J. Med. Genet. 55, 128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ceroni, F., Simpson, N.H., Francks, C., Baird, G., Conti-Ramsden, G., Clark, A., Bolton, P.F., Hennessy, E.R., Donnelly, P., Bentley, D.R. et al. (2014) Homozygous microdeletion of exon 5 in ZNF277 in a girl with specific language impairment. Eur. J. Hum. Genet., 22, 1165–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith, A.W., Holden, K.R., Dwivedi, A., Dupont, B.R. and Lyons, M.J. (2014) Deletion of 16q24.1 supports a role for the ATP2C2 gene in specific language impairment. J. Child Neurol., 30, 517–521. [DOI] [PubMed] [Google Scholar]
- 44. Pettigrew, K.A., Reeves, E., Leavett, R., Hayiou-Thomas, M.E., Sharma, A., Simpson, N.H., Martinelli, A., Thompson, P., Hulme, C., Snowling, M.J. et al. (2015) Copy number variation screen identifies a rare de novo deletion at chromosome 15q13.1-13.3 in a child with language impairment. PLoS One, 10: e0134997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hildebrand, M.S., Jackson, V.E., Scerri, T.S., Van Reyk, O., Coleman, M., Braden, R.O., Turner, S., Rigbye, K.A., Boys, A., Barton, S. et al. (2020) Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology, 94, e2148–e2167. [DOI] [PubMed] [Google Scholar]
- 46. Lai, C.S.L., Fisher, S.E., Hurst, J.A., Vargha-Khadem, F. and Monaco, A.P. (2001) A forkhead-domain gene is mutated in a severe speech and language disorder. Nature, 413, 519–523. [DOI] [PubMed] [Google Scholar]
- 47. Devanna, P., Chen, X.S., Ho, J., Gajewski, D., Smith, S.D., Gialluisi, A., Francks, C., Fisher, S.E., Newbury, D.F. and Vernes, S.C. (2018) Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders. Mol. Psychiatry, 23, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eising, E., Carrion-Castillo, A., Vino, A., Strand, E.A., Jakielski, K.J., Scerri, T.S., Hildebrand, M.S., Webster, R., Ma, A., Mazoyer, B. et al. (2019) A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry, 24, 1065–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen, X.S., Reader, R.H., Hoischen, A., Veltman, J.A., Simpson, N.H., Francks, C., Newbury, D.F. and Fisher, S.E. (2017) Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci. Rep., 7, 46105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Girirajan, S., Brkanac, Z., Coe, B.P., Baker, C., Vives, L., Vu, T.H., Shafer, N., Bernier, R., Ferrero, G.B., Silengo, M. et al. (2011) Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet., 7, e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Girirajan, S., Rosenfeld, J.A., Cooper, G.M., Antonacci, F., Siswara, P., Itsara, A., Vives, L., Walsh, T., McCarthy, S.E., Baker, C. et al. (2010) A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat. Genet., 42, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. O’ Roak, B.J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B.P., Levy, R., Ko, A., Lee, C., Smith, J.D. et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nash, H.M., Hulme, C., Gooch, D. and Snowling, M.J. (2013) Preschool language profiles of children at family risk of dyslexia: continuities with specific language impairment. J. Child Psychol. Psychiatry Allied Discip., 54, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scerri, T.S., Macpherson, E., Martinelli, A., Wa, W.C., Monaco, A.P., Stein, J., Zheng, M., Suk-Han Ho, C., McBride, C., Snowling, M. et al. (2017) The DCDC2 deletion is not a risk factor for dyslexia. Transl. Psychiatry, 7, e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kent, L., Doerry, U., Hardy, E., Parmar, R., Gingell, K., Hawi, Z., Kirley, A., Lowe, N., Fitzgerald, M., Gill, M. and Craddock, N. (2002) Evidence that variation at the serotonin transporter gene influences susceptibility to attention deficit hyperactivity disorder (ADHD): analysis and pooled analysis. Mol. Psychiatry, 7, 908–912. [DOI] [PubMed] [Google Scholar]
- 56. Boyd, A., Golding, J., Macleod, J., Lawlor, D.A., Fraser, A., Henderson, J., Molloy, L., Ness, A., Ring, S. and Smith, G.D. (2013) Cohort profile: The’Children of the 90s’-the index offspring of the Avon longitudinal study of parents and children. Int. J. Epidemiol., 42, 111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fraser, A., Macdonald-wallis, C., Tilling, K., Boyd, A., Golding, J., Davey smith, G., Henderson, J., Macleod, J., Molloy, L., Ness, A. et al. (2013) Cohort profile: the Avon longitudinal study of parents and children: ALSPAC mothers cohort. Int. J. Epidemiol., 42, 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Scerri, T.S., Morris, A.P., Buckingham, L.L., Newbury, D.F., Miller, L.L., Monaco, A.P., Bishop, D.V.M. and Paracchini, S. (2011) DCDC2, KIAA0319 and CMIP are associated with reading-related traits. Biol. Psychiatry, 70, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Parenti, I., Rabaneda, L.G., Schoen, H. and Novarino, G. (2020) Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci, 43, 608–621. [DOI] [PubMed] [Google Scholar]
- 61. Villanueva, P., Nudel, R., Hoischen, A., Fernández, M.A., Simpson, N.H., Gilissen, C., Reader, R.H., Jara, L., Echeverry, M.M., Francks, C. et al. (2015) Exome sequencing in an admixed isolated population indicates NFXL1 variants confer a risk for specific language impairment. PLoS Genet., 11, e1004925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Niemi, M.E.K., Martin, H.C., Rice, D.L., Gallone, G., Gordon, S., Kelemen, M., McAloney, K., McRae, J., Radford, E.J., Yu, S. et al. (2018) Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature, 562, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tansey, K.E., Rees, E., Linden, D.E., Ripke, S., Chambert, K.D., Moran, J.L., McCarroll, S.A., Holmans, P., Kirov, G., Walters, J. et al. (2016) Common alleles contribute to schizophrenia in CNV carriers. Mol. Psychiatry, 21, 1085–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bergen, S.E., Ploner, A., Howrigan, D., O’Donovan, M.C., Smoller, J.W., Sullivan, P.F., Sebat, J., Neale, B. and Kendler, K.S. (2019) Joint contributions of rare copy number variants and common SNPs to risk for schizophrenia. Am. J. Psychiatry, 176, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martin, J., O’Donovan, M.C., Thapar, A., Langley, K. and Williams, N. (2015) The relative contribution of common and rare genetic variants to ADHD. Transl. Psychiatry, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weiner, D.J., Wigdor, E.M., Ripke, S., Walters, R.K., Kosmicki, J.A., Grove, J., Samocha, K.E., Goldstein, J.I., Okbay, A., Bybjerg-Grauholm, J. et al. (2017) Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat. Genet., 49, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kurki, M.I., Saarentaus, E., Pietiläinen, O., Gormley, P., Lal, D., Kerminen, S., Torniainen-Holm, M., Hämäläinen, E., Rahikkala, E., Keski-Filppula, R. et al. (2019) Contribution of rare and common variants to intellectual disability in a sub-isolate of northern Finland. Nat. Commun., 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ganna, A., Genovese, G., Howrigan, D.P., Byrnes, A., Kurki, M., Zekavat, S.M., Whelan, C.W., Kals, M., Nivard, M.G., Bloemendal, A. et al. (2016) Ultra-rare disruptive and damaging mutations influence educational attainment in the general population HHS public access. Nat. Neurosci., 19, 1563–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kendall, K.M., Bracher-Smith, M., Fitzpatrick, H., Lynham, A., Rees, E., Escott-Price, V., Owen, M.J., O’Donovan, M.C., Walters, J.T.R. and Kirov, G. (2019) Cognitive performance and functional outcomes of carriers of pathogenic copy number variants: analysis of the UK biobank. Br. J. Psychiatry, 214, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stefansson, H., Meyer-Lindenberg, A., Steinberg, S., Magnusdottir, B., Morgen, K., Arnarsdottir, S., Bjornsdottir, G., Walters, G.B., Jonsdottir, G.A., Doyle, O.M. et al. (2014) CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature, 505, 361–366. [DOI] [PubMed] [Google Scholar]
- 71. Hubbard, L., Tansey, K.E., Rai, D., Jones, P., Ripke, S., Chambert, K.D., Moran, J.L., Mccarroll, S.A., Linden, D.E.J., Owen, M.J. et al. (2016) Evidence of common genetic overlap between schizophrenia and cognition. Schizophr. Bull., 42, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Power, R.A., Steinberg, S., Bjornsdottir, G., Rietveld, C.A., Abdellaoui, A., Nivard, M.M., Johannesson, M., Galesloot, T.E., Hottenga, J.J., Willemsen, G. et al. (2015) Polygenic risk scores for schizophrenia and bipolar disorder predict creativity. Nat. Neurosci., 18, 953–955. [DOI] [PubMed] [Google Scholar]
- 73. Rajagopal, V.M., Ganna, A., Coleman, J.R.I., Allegrini, A.G., Voloudakis, G., Grove, J., Als, T.D., Horsda, H.T., Petersen, L., Appadurai, V. et al. (2020) Genome-wide association study of school grades identifies a genetic overlap between language ability, psychopathology and creativity. BioRxiv, doi 10.1101/2020.05.09.075226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schmitz, J., Abbondanza, F. and Paracchini, S. (2020) Genome-wide association study and polygenic risk score analysis for hearing measures in children. bioRxiv, 2020.07.22.215376. [DOI] [PubMed] [Google Scholar]
- 75. Gialluisi, A., Andlauer, T.F.M., Mirza-Schreiber, N., Moll, K., Becker, J., Hoffmann, P., Ludwig, K.U., Czamara, D., St Pourcain, B., Brandler, W. et al. (2019) Genome-wide association scan identifies new variants associated with a cognitive predictor of dyslexia. Transl. Psychiatry, 9, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Newbury, D.F., Gibson, J.L., Conti-Ramsden, G., Pickles, A., Durkin, K. and Toseeb, U. (2019) Using polygenic profiles to predict variation in language and psychosocial outcomes in early and middle childhood. J. Speech, Lang. Hear. Res., 62, 3381–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Verhoef, E., Demontis, D., Burgess, S., Shapland, C.Y., Dale, P.S., Okbay, A., Neale, B.M., Faraone, S.V., Stergiakouli, E., Davey Smith, G. et al. (2019) Disentangling polygenic associations between attention-deficit/hyperactivity disorder, educational attainment, literacy and language. Transl. Psychiatry, 9, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vanoevelen, J., Dode, L., Van Baelen, K., Fairclough, R.J., Missiaen, L., Raeymaekers, L. and Wuytack, F. (2005) The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J. Biol. Chem., 280, 22800–22808. [DOI] [PubMed] [Google Scholar]
- 79. Guillen Sacoto, M.J., Tchasovnikarova, I.A., Torti, E., Forster, C., Andrew, E.H., Anselm, I., Baranano, K.W., Briere, L.C., Cohen, J.S., Craigen, W.J. et al. (2020) De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial dysmorphism. Am. J. Hum. Genet., 107, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kaplanis, J., Samocha, K., Wiel, L., Zhang, Z., Arvai, K., Eberhardt, R., Gallone, G., Lelieveld, S., Martin, H., McRae, J. et al. (2020) Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature, 586, 757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Reuter, M.S., Tawamie, H., Buchert, R., Gebril, O.H., Froukh, T., Thiel, C., Uebe, S., Ekici, A.B., Krumbiegel, M., Zweier, C. et al. (2017) Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry, 74, 293–299. [DOI] [PubMed] [Google Scholar]
- 82. Snijders Blok, L., Rousseau, J., Twist, J., Ehresmann, S., Takaku, M., Venselaar, H., Rodan, L.H., Nowak, C.B., Douglas, J., Swoboda, K.J. et al. (2018) CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun., 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zheng, J.Q. and Poo, M.-M. (2007) Calcium Signaling in neuronal motility. Annu. Rev. Cell Dev. Biol., 23, 375–404. [DOI] [PubMed] [Google Scholar]
- 84. Dash, P.K., Moore, A.N., Kobori, N. and Runyan, J.D. (2007) Molecular activity underlying working memory. Molecular activity underlying working memory. Learn. Mem., 14, 554–563. [DOI] [PubMed] [Google Scholar]
- 85. Nikoletopoulou, V. and Tavernarakis, N. (2012) Calcium homeostasis in aging neurons. Calcium homeostasis in aging neurons. Front. Genet., 3, 200. 10.3389/fgene.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vangheluwe, P., Sepúlveda, M.R., Missiaen, L., Raeymaekers, L., Wuytack, F. and Vanoevelen, J. (2009) Intracellular ca 2+−and Mn 2+−transport ATPases. Chem. Rev., 109, 4733–4759. [DOI] [PubMed] [Google Scholar]
- 87. He, W. and Hu, Z. (2012) The role of the golgi-resident SPCA ca 2+/Mn 2+ pump in ionic homeostasis and neural function. The role of the golgi-resident SPCA ca 2+/Mn 2+ pump in ionic homeostasis and neural function. Neurochem. Res., 37, 455–468. [DOI] [PubMed] [Google Scholar]
- 88. Normandin, L. and Hazell, A.S. (2002) Manganese neurotoxicity: an update of pathophysiologic mechanisms. Metab. Brain Dis., 17, 375–387. [DOI] [PubMed] [Google Scholar]
- 89. Harischandra, D.S., Ghaisas, S., Zenitsky, G., Jin, H., Kanthasamy, A., Anantharam, V. and Kanthasamy, A.G. (2019) Manganese-induced neurotoxicity: new insights into the triad of protein misfolding, mitochondrial impairment, and neuroinflammation. Front. Neurosci., 13, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wojda, U., Salinska, E. and Kuznicki, J. (2008) Calcium ions in neuronal degeneration. IUBMB Life, 60, 575–590. [DOI] [PubMed] [Google Scholar]
- 91. Xiang, M., Mohamalawari, D. and Rao, R. (2005) A novel isoform of the secretory pathway Ca2+, Mn(2+)-ATPase, hSPCA2, has unusual properties and is expressed in the brain. J. Biol. Chem., 280, 11608–11614. [DOI] [PubMed] [Google Scholar]
- 92. Ishikawa, K.-I., Nagase, T., Suyama, M., Miyajima, N., Tanaka, A., Kotani, H., Nomura, N. and Ohara, O. (1998) Prediction of the coding sequences of unidentified human genes. X. the complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res., 5, 169–176. [DOI] [PubMed] [Google Scholar]
- 93. Aledo, J.C. (2019) Methionine in proteins: the Cinderella of the proteinogenic amino acids. Protein Sci., 28, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bozzi, A.T., Bane, L.B., Weihofen, W.A., McCabe, A.L., Singharoy, A., Chipot, C.J., Schulten, K. and Gaudet, R. (2016) Conserved methionine dictates substrate preference in Nramp-family divalent metal transporters. Proc. Natl. Acad. Sci. U. S. A., 113, 10310–10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rice, M.L., Smith, S.D. and Gayán, J. (2009) Convergent genetic linkage and associations to language, speech and reading measures in families of probands with specific language impairment. J. Neurodev. Disord., 1, 264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Andres, E.M., Earnest, K.K., Smith, S.D., Rice, M.L. and Raza, M.H. (2020) Pedigree-based gene mapping supports previous loci and reveals novel suggestive loci in specific language impairment. J. Speech, Lang. Hear. Res., 63, 4046–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Rice, M.L.; Wexler, K. (2001) Rice/Wexler Test of Early Grammatical Impairment. Psychological Corporation San Antoni. [Google Scholar]
- 98. Sim, N.L., Kumar, P., Hu, J., Henikoff, S., Schneider, G. and Ng, P.C. (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res., 40, W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Reva, B., Antipin, Y. and Sander, C. (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res., 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Choi, Y. and Chan, A.P. (2015) PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Adzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Kondrashov, A.S. and Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pejaver, V., Urresti, J., Lugo-Martinez, J., Pagel, K.A., Lin, G.N., Nam, H.-J., Mort, M., Cooper, D.N., Sebat, J., Iakoucheva, L.M. et al. (2020) Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 11, 5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Savojardo, C., Fariselli, P., Martelli, P.L. and Casadio, R. (2016) INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics, 32, 2542–2544. [DOI] [PubMed] [Google Scholar]
- 104. Schwarz, J.M., Cooper, D.N., Schuelke, M. and Seelow, D. (2014) Mutationtaster 2: mutation prediction for the deep-sequencing age. Nat. Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- 105. Semel, E.M., Wiig, E.H. and Secord, W. (1992) Clinical Evaluation of Language Fundamental, Revised. Psychological Corporation, San Antonio. [Google Scholar]
- 106. Elliot, C.D., Murray, D.J. and Pearson, L.S. (1983) British Abilities Scales. NFER-Nelson, Wind. UK. [Google Scholar]
- 107. Jastak, J. and Wilkinson, G.S. (1984) Wide Range Achievement Test–Revised (WRAT-R). Psychological Corporation, San Antonio, TX. [Google Scholar]
- 108. Scerri, T.S., Paracchini, S., Morris, A., Mac Phie, I.L., Talcott, J., Stein, J., Smith, S.D., Pennington, B.F., Olson, R.K., de Fries, J.C. and Monaco, A.P. (2010) Identification of candidate genes for dyslexia susceptibility on chromosome 18. PLoS One, 5, e13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Castles, A. and Coltheart, M. (1993) Varieties of developmental dyslexia. Cognition, 47, 149–180. [DOI] [PubMed] [Google Scholar]
- 110. Frederickson, N., Frith, U. and Reason, R. (1997) Phonological assessment battery (PhAB). Wind, NFER-Nelson. [Google Scholar]
- 111. Olson, R., Forsberg, H., Wise, B. and Rack, J. (1994) Measurement of word recognition, orthographic, and phonological skills. In Lyon, G.R. (ed), Frames of Reference for the Assessment of Learning Disabilities: New Views on Measurement Issues. Paul H. Brookes Publishing Co., Baltimore. [Google Scholar]
- 112. Angold, A., Prendergast, M., Cox, A., Harrington, R., Simonoff, E. and Rutter, M. (1995) The child and adolescent psychiatric assessment (CAPA). Psychol. Med., 25, 739–753. [DOI] [PubMed] [Google Scholar]
- 113. Walter, K., Min, J.L., Huang, J., Crooks, L., Memari, Y., McCarthy, S., Perry, J.R.B., Xu, C., Futema, M., Lawson, D. et al. (2015) The UK10K project identifies rare variants in health and disease. Nature, 526, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rust, J. (1996) WOLD Wechsler Objective Language Dimensions Manual. In WOLD Wechsler Objective Language Dimensions Manual. The Psychological Corporation, London, UK. [Google Scholar]
- 115. Bishop, D.V.M. (1998) Development of the children’s communication checklist (CCC): a method for assessing qualitative aspects of communicative impairment in children. J. Child Psychol. Psychiatry, 39, 879–891. [PubMed] [Google Scholar]
- 116. Rietdorf, K., Funnell, T.M., Ruas, M., Heinemann, J., Parrington, J. and Galione, A. (2011) Two-pore channels form homo- and heterodimers. J. Biol. Chem., 286, 37058–37062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vandecaetsbeek, I., Holemans, T., Wuytack, F. and Vangheluwe, P. (2014) High-throughput measurement of the Ca2+−dependent ATPase activity in COS microsomes. Cold Spring Harb. Protoc., 2014, 865–875. [DOI] [PubMed] [Google Scholar]
- 118. Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm, W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain, S. et al. (2002) The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr., 58, 899–907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.