

Abstract
Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by impaired communication skills, ataxia, motor and balance deficits, intellectual disabilities, and seizures. The genetic cause of AS is the neuronal loss of UBE3A expression in the brain. A novel approach, described here, is a stem cell gene therapy which uses lentivector-transduced hematopoietic stem and progenitor cells to deliver functional UBE3A to affected cells. We have demonstrated both the prevention and reversal of AS phenotypes upon transplantation and engraftment of human CD34+ cells transduced with a Ube3a lentivector in a novel immunodeficient Ube3amat−/pat+ IL2rg−/y mouse model of AS. A significant improvement in motor and cognitive behavioral assays as well as normalized delta power measured by electroencephalogram was observed in neonates and adults transplanted with the gene modified cells. Human hematopoietic profiles observed in the lymphoid organs by detection of human immune cells were normal. Expression of UBE3A was detected in the brains of the adult treatment group following immunohistochemical staining illustrating engraftment of the gene-modified cells expressing UBE3A in the brain. As demonstrated with our data, this stem cell gene therapy approach offers a promising treatment strategy for AS, not requiring a critical treatment window.
Introduction
Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by developmental delay, impaired communication skills, ataxia, motor and balance deficits, poor attention, intellectual disabilities, microcephaly, and seizures (1–3). The genetic cause of AS is loss of expression in the brain of UBE3A (ubiquitin-protein ligase E6-AP), due typically to a 4-Mb de novo deletion of the maternal 15q11-q13 region (4–6). Due to brain-specific imprinting, the paternal allele is silenced, thus loss of the maternal allele causes UBE3A deficiency throughout the brain (7).
Novel strategies to treat AS are critical as there is no FDA approved cure or corrective therapy available to patients. Therapies in development for genetic precision medicine for AS have resulted in: (1) silencing or cleavage of the paternal antisense transcript that stops expression of the maternal allele, (2) molecular reversal of Ube3a expression levels and (3) some degree of functional phenotypic rescue in a few but not all affected domains. These include therapies by antisense oligonucleotides (ASO), viral vector delivery and artificial transcription factors (ATFs) (8–12). In fact, two ASO compounds are in Phase I clinical trials (GeneTx NCT04259281; Roche NCT04428281). These novel therapies are promising as treatment modalities for AS; however, a number of these approaches had limited functional phenotypic rescue preclinically, may have off-target genetic effects, and require repeated dosing. In addition, dietary and traditional pharmaceutical treatments have shown ‘alleviation’ of one or more symptom domains found in the mouse model, including but not limited to NNZ-2566, minocycline, ketone esters, dietary methylation, ErbB inhibitors and topoisomerase inhibitor drugs (13–19). However, despite various research attempts, clinical trials in AS have been limited.
A novel approach to corrective therapy being pursued by our laboratories is the use of hematopoietic stem and progenitor cells (HSPC), which offer a promising approach for life-long delivery of functional UBE3A to affected cells, i.e. neurons. This process would entail modifying autologous HSPC with a lentiviral vector expressing a modified form of UBE3A. The engrafted and genetically modified HSPC would produce cell progeny that systemically express the enzyme and provide functional UBE3A through a process called ‘cross-correction’ (20–22). Lentiviral vectors are highly effective in delivering transgenes to HSPC due to their ability to stably transduce both dividing and non-dividing cells (22–29). They provide long-term expression of the introduced genes due to their ability to integrate and are less prone to gene silencing or off-target effects in differentiated cells. Newly developed lentiviral vectors have undergone several refinements to increase safety and transgene expression. We chose to utilize human HSPC over mouse HSPC due to our desire to expedite into clinical translation and eliminate the need for bridging studies of mHSPC to hHSPC.
Transplantation of UBE3A-expressing HSPC has the potential to prevent, halt, or reverse symptoms associated with AS by providing UBE3A to the central nervous system from the engraftment of microglia derived from the vector transduced CD34+ cells (22,30–34). This was recently demonstrated for other metabolic diseases including metachromatic leukodystrophy, adrenoleukodystrophy and Tay-Sachs/Sandhoff disease, in which the hematopoietic system was used to deliver therapeutic levels of functional enzymes via engraftment of modified HSPC (22,30–32).
The experiments described herein took an innovative step to evaluate the concept of cross-correction for AS. First, we generated a lentiviral vector that expressed a modified form of the mouse Ube3a isoform 3 (mAS8) and confirmed its expression and functionality. In parallel, we generated a novel immunodeficient AS mouse model Ube3amat−/pat+ IL2rg−/y that was capable of receiving human CD34+ HSPC for transplantation and engraftment followed by multi-lineage hematopoiesis throughout the hematopoietic system and also confirmed that no AS-relevant behavioral phenotypes presented in the new model, given the interleukin 2 gamma chain loss. Finally, we administered our therapeutic via HSPC transplantation in neonates and adults.
Our data illustrate robust functional prevention and rescue of AS phenotypes in both neonate and adult AS mice following treatment with human CD34+ HSPC transduced with an Ube3a-expressing lentiviral vector. This strongly suggests that cross-correction is a beneficial strategy to pursue and that this stem cell gene-therapy approach offers a potential therapeutic intervention for AS patients.
Results
Expression and functionality of the Ube3a-expressing lentiviral vector
A lentiviral vector backbone, CCLc-MNDU3-x, was used to develop the various constructs used in this study (Fig. 1A). A control EGFP-alone vector was created by inserting a PGK-EGFP expression cassette downstream from the MNDU3 promoter (Fig. 1B). A modified mouse Ube3a isoform 3 or a modified human Ube3a isoform 1 were inserted upstream of the PGK-EGFP expression cassette under the control of an MNDU3 promoter (Fig. 1C and D). To evaluate whether UBE3A was expressed from the lentiviral vector and whether it was functional at ubiquitinating a target protein, western blots and an in vitro S5A ubiquitination assay were performed, respectively. Human CD34+ cells were left non-transduced (NT) or were transduced with the EGFP control or a lentiviral vector expressing a modified form of the human Ube3a isoform 1 (hAS8) and derived into macrophages in vitro. Cell extracts were evaluated for UBE3A expression via western blot. As displayed in Figure 1E, successful detection of UBE3A was observed in the cells transduced with the hAS8 lentiviral vector compared with the NT and EGFP control transduced cells. To determine whether the lentiviral vector expressed UBE3A was functional, an S5A ubiquitination assay was performed. As displayed in Figure 1F, ubiquitination of S5A, as determined by the banding pattern, was detected in the hAS8 lane compared with the control cell lanes which displayed no banding pattern. A similar banding pattern was observed in the positive (+) control lane.
Figure 1 .
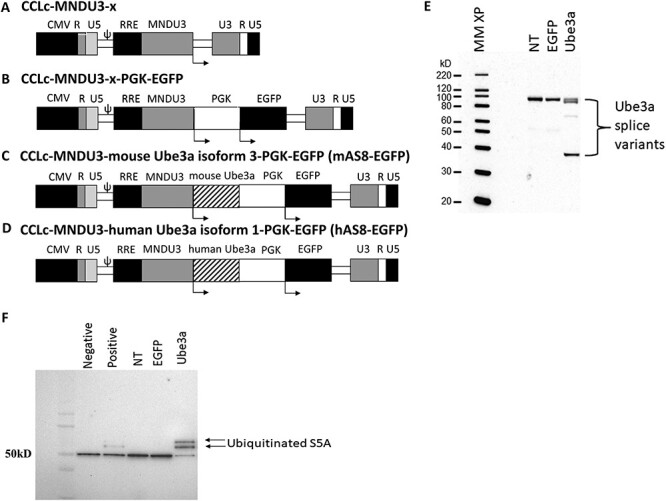
Schematic and functionality of the Ube3a-expressing lentiviral vector: (A) A self-inactivating lentiviral vector backbone, CCLc-MNDU3-X, was used to generate the Ube3a expressing lentiviral vectors. (B) EGFP vector used as an empty vector control. (C) The modified mouse Ube3a isoform 3 (mAS8) was cloned under the control of the MNDU3 promoter. EGFP was cloned downstream under the control of a PGK promoter. (D) The modified human UBE3A isoform 1 was cloned under the control of the MNDU3 promoter. EGFP was cloned downstream under the control of a PGK promoter. (E) Human CD34+ HSPC were transduced with the Ube3a lentivector and derived into mature macrophages. Cell extracts were then generated and evaluated for overexpression of UBE3A by western blot. (F) An S5A ubiquitination assay was performed on the macrophage cell extracts. Control NT, EGFP vector alone (EGFP) transduced cells, and negative and positive controls supplied by the manufacturer were used as controls.
Colony forming unit assay and the derivation of mature macrophages
To initially evaluate the safety of the Ube3a-expressing lentiviral vector after transduction of human CD34+ HSPC, a colony forming unit (CFU) assay was performed. This was performed to evaluate whether similar differentiation patterns, as demonstrated by the development of granulocyte/macrophage (GM), granulocyte/erythrocyte/megakaryocyte/macrophage (GEMM) and burst forming unit-erythroid colonies (BFU-E), were observed in all cultures. Transduced cells, either EGFP-control or hAS8 were first sorted based on EGFP expression and then cultured in methylcellulose media containing cytokines allowing for cell differentiation. As displayed in Figure 2A, similar GM, GEMM and BFU-E colony counts were observed in the hAS8 cultures as compared with the NT and EGFP control vector-transduced colonies. To further evaluate the differentiation potential of the hAS8 vector transduced CD34+ cells, the CFU colonies were cultured in macrophage specific media. After a 4-day incubation to allow for macrophage development, the cells were analyzed for macrophage cell-specific surface markers including CD14, CD4 and HLADR. hAS8 macrophages displayed, on average, 97.8, 97.9 and 99.2% expression of CD4, CD14 and HLADR, respectively. This compared with NT macrophages which displayed 97.6, 97.6 and 98.9% and EGFP control macrophages, which displayed 98.2, 97.8 and 97.9% of CD4, CD14 and HLADR, respectively. Representative flow cytometry histograms are displayed in Figure 2B.
Figure 2 .
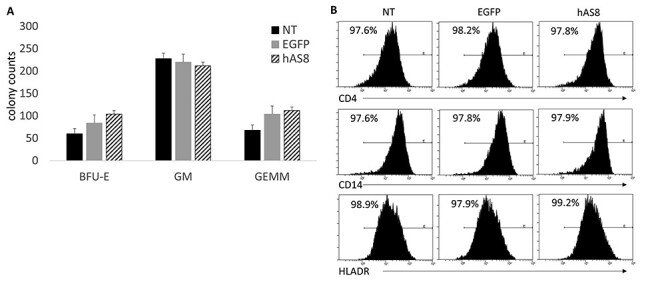
CFU assay and macrophage derivation of Ube3a vector-transduced human CD34+ HSPC: Human CD34+ HPSC were left NT or transduced with either the EGFP alone control vector (EGFP) or the Ube3a vector (hAS8). (A) Cells were cultured in methylcellulose media for 12 days when specific colonies (BFU-E, GM and GEMM) were counted. (B) CFUs were further differentiated into macrophages and analyzed by flow cytometry for the cell surface markers CD4, CD14 and HLADR.
Together, these data demonstrated an initial safety profile of the Ube3a lentiviral vector in primary CD34+ cells post-transduction in expanding and differentiating into the multiple colonies. Furthermore, phenotypically normal macrophages were able to be developed, in vitro. These data presented an initial safety profile of the Ube3a vector transduced cells. These experiments allowed for the further investigation of this therapeutic approach in vivo in a disease-specific mouse model of AS.
Generation and optimization of an immunodeficient Ube3a-deficient mouse model
For novel gene therapies to be translated into a clinical setting, in vivo studies in relevant disease-specific models evaluating the efficacy of genetically modified cells are essential. The Ube3a-deficient mouse model, which contains a deletion in exon 2 of Ube3a (AS mice), have been used to study AS and therapeutic interventions (35). Since the Ube3a gene is paternally imprinted, AS phenotypes occur only when the mutation is inherited on the maternal allele (35,36). This mouse model has pronounced behavioral phenotypes relevant to AS including robust motor deficits, cognitive impairments, seizure susceptibility, abnormal epileptiform discharges and spiking events in electroencephalogram (EEG), and a characteristic EEG signature of elevated delta spectral power (31,33–35). In order to evaluate our therapeutic candidate, human CD34+ cells transduced with a murine Ube3a lentiviral vector and an immunodeficient AS-specific model needed to be developed. We tested a suite of immunocompromised mice with various degrees of immunodeficiency for AS behavioral phenotypes and human cell engraftment. A partial degree of immunodeficiency was required to transplant human CD34+ HSPC without immune activation and rejection of these therapeutic cells to successfully reconstitute mice with human immune cells. To assess if immune deficiency, was causing deleterious behavioral deficits, we evaluated Rag2 (JAX #025730; B6.129S-Rag2tm1Fwa CD47tm1Fpl Il2rgtm1Wjl/J), Rag1 (JAX #002216; B6.129S7-Rag1tm1Mom/J) and IL2rg-null mice (JAX stock #003174; B6.129S4-IL2rgtm1Wjl/J) on a battery of AS-relevant outcomes and quantified human cell engraftment, Supplementary Material, Fig. S1A–F, 2. These studies revealed that the optimal immunodeficient strain to cross with Ube3a mat−/pat+ mice were the IL2rg null mice (IL2rg−/y), generating a Ube3amat−/pat+ IL2rg−/y immunodeficient model for studying the in vivo efficacy of the Ube3a-expressing lentiviral vector in human CD34+ cells.
Experimental cohorts evaluating in vivo efficacy of the Ube3a vector transduced human CD34+ cells
Four treatment groups were analyzed for prevention or functional rescue in behavioral assays relevant to AS and reproduced rigorously in our laboratory and others (31,36,37). The treatment groups were: (1) WT = control wildtype that had an IL2rg null mutation (IL2rg −/y) for appropriate control of the immune system influence and a WT (Ube3a+/+) genotype, Ube3amat+/pat+ IL2rg−/y, (2) HET = Ube3amat−/pat+ IL2rg−/y, the innovative AS model with a maternal deletion of Ube3a and capable of human cell engraftment attributed to the IL2rg −/y null mutation, (3) NT-HET = Ube3amat−/pat+ IL2rg−/y, the innovative AS model, transplanted with NT human CD34+ HSPC to control for the effect of HSPC alone and (4) Ube3a-HET = Ube3amat−/pat+ IL2rg−/y that are the novel immunodeficient AS model transplanted with Ube3a vector transduced human CD34+ HSPC.
Neonatal treatment with HSPC transduced with a Ube3a-expressing lentiviral vector prevented AS phenotypes in rigorous motor assessments
To evaluate whether AS phenotypes could be prevented prior to their initial onset, neonates were transplanted with the Ube3a vector transduced HSPC. Prevention of AS phenotypes in the Ube3a-HET treatment group was observed by these mice scoring indistinguishably from the WT group on multiple assays of motor deficits 8 weeks post-transplant. HET and NT-HET illustrated typical AS deficits of reduced motor activity and impaired motor coordination in four tests of motor abilities. This is the first report of significant gait impairments in an AS mouse model, although our group has previously reported this in the AS rat (37,38). Locomotion in a novel open field was collected by assessing the total distance traversed and the horizontal movements using beam breaks in a novel arena. Group differences in horizontal activity counts were observed using a multi-factor repeated measure ANOVA (Fig. 3A; F(3, 67) = 8.194, P < 0.0001). Holm-Sidak corrected posthoc analysis for multiple comparisons highlighted that both NT-HET (P < 0.0001) and HET (P < 0.0028) differed from WT while our treatment group, Ube3a-HET, did not differ from WT (P > 0.05). Holm-Sidak corrected posthoc analysis for multiple comparisons highlighted that both NT-HET (P < 0.0001) and HET (P < 0.0002) differed from WT while our treatment group, Ube3a-HET, did not differ from WT (P = 0.191). The rigor of this posthoc analysis is remarkable. The Ube3a-HET group did not differ from WT at any timepoint across the 30 min assay (0–5 min, P = 0.389, 6–10 min, P = 0.5027, 11–15 min, P = 0.132, 16–20 min, P = 0.5418, 21–25 min P = 0.995, 26–30 min P = 0.8991), while the HET and WT adjusted P’s from Holm Sidak provided (0–5 min, P < 0.0052, 6–10 min, P < 0.0076, 11–15 min, P < 0.004, 16–20 min, P < 0.024, 21–25 min, 26–30 min) differed in four of the six 5-min time bins and the NT-HET and WT differ on every 5-min bin of the 30-min task (0–5 min, P < 0.0002, 6–10 min, P < 0.000, 11–15 min, P < 0.0004, 16–20 min, P < 0.003, 21–25 min, P < 0.0287, 26–30 min, P < 0.0003). In corroboration, the total distance also illustrated group differences using a multi-factor repeated measure ANOVA (Fig. 3B; F(3, 67) = 9.487, P < 0.0001). Holm-Sidak corrected posthoc analysis for multiple comparisons highlighted that both NT-HET (P < 0.0001) and HET (P < 0.0002) differed from WT while our treatment group, Ube3a-HET, did not differ from WT (P = 0.191). The Ube3a-HET did not differ from WT at any timepoint across the 30-min assay, while the HET and WT adjusted P’s from Holm Sidak differed in five of the six 5-min time bins and the NT-HET and WT differ on every 5-min bin of the 30-min task. Sexes were combined as there was no sex difference in any group in the open field (Fig. 3A: WT F(1, 14) = 1.637, P > 0.05; HET F(1, 18) = 0.002, P > 0.05; NT-HET F(1, 18) = 3.062, P > 0.05; Ube3a-HET F(1, 13) = 0.048, P > 0.05).
Figure 3 .
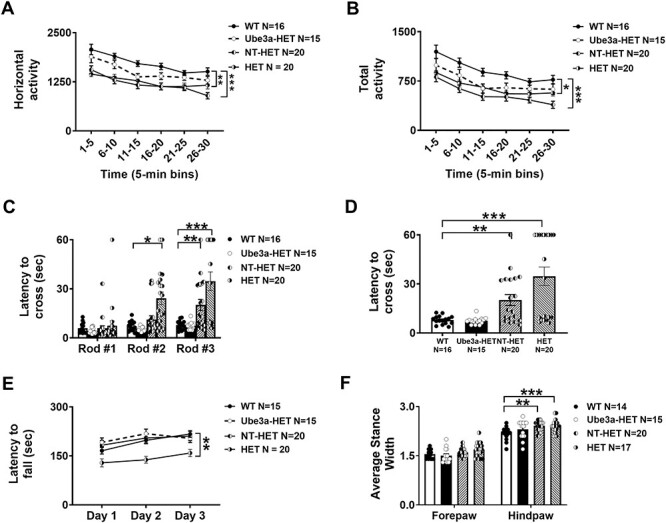
Locomotor ability, balance, motor coordination and gait from Ube3a lentivector transduced HSPC-transplanted neonates: Rigorous assessment of motor translational phenotypes using four standard motor behavioral tests in treated and untreated Ube3amat+/pat+ IL2rg−/y mice that were irradiated and transplanted as neonates with either NT (NT-HET (hatched bar) or the Ube3a lentivector transduced (Ube3a-HET (black bar)) human CD34+ HSPC. Eight weeks post-transplant, mice were subjected to (A, B) open field locomotion, (C, D) balance beam, (E) rotarod and (F) treadmill walking. In all tests, Ube3amat+/pat+ IL2rg−/y deficient mice transplanted with the Ube3a vector transduced human CD34+ HSPC (Ube3a-HET) exhibited wildtype values of performance. (A, B) Open field activity was increased by both the total distance and horizontal activity metrics. (C) Eight weeks post-transplant, mice were also tested on the beam walking assay. Beams decrease in width and are more difficult to cross going from Rod #1 to Rod #2 to Rod #3. (D) Highlight of Rod #3 of C, the most challenging coordination test with remarkable improvements. (E) Latency to fall from the rotarod was significantly improved in the Ube3amat+/pat+ IL2rg−/y mice transplanted with Ube3a vector-transduced human CD34+ HSPC (Ube3a-HET) compared with the NT (NT-HET) cell controls. (F) DigiGait™ analyses showed a narrowing of wide stances in the Ube3a-HET treated group. Data are expressed as ± standard error of mean (SEM). *P < 0.05, **P < 0.001, ***P < 0.0001 indicate when the HET and NT-HET groups differ from the control WT group.
A balance beam walking motor task was conducted as previously described (39). All groups displayed longer latencies to cross the rod-shaped beams as they became thinner and more difficult to traverse, as expected. Group differences were detected using a multi-factor repeated measure ANOVA (Fig. 3C and D; F(3, 67) = 17.02, P < 0.0001). Interestingly, Ube3a-HET mice transplanted with the Ube3a lentivector transduced human CD34+ HSPC exhibited values that were not significantly different from the values seen in WT on the beam, illustrated via Holm-Sidak posthoc analysis. The Ube3a-HET did not differ from WT on rod #3 (Fig. 3D: rod #3, P > 0.999) while the HET (P < 0.0001) and NT-HET (P < 0.227) had much slower latencies to cross rod #3, highlighting the improved motor coordination of our treated group. Sexes were combined as there was no sex difference in any group in the beam walking assay (Fig. 3C: WT F(1, 14) = 0.9276, P > 0.05; HET F(1, 18) = 0.563, P > 0.05; NT-HET F(1, 18) = 0.314, P > 0.05; Ube3a-HET F(1, 13) = 0.107, P > 0.05). Another corroborating assay of motor coordination is the rotarod. AS mice have deficits in this task, reported by other laboratories (36–40). As expected, HET differed from WT in their latencies to fall from the accelerating rod (Fig. 3E; F(3, 67) = 8.395, P < 0.0001) on days 2 (P < 0.0098) and 3 (P < 0.0133), illustrating poor motor coordination and a lack of motor learning. Strikingly, the Ube3a-HET group was similar to WT across all 3 days of testing using Holm-Sidak posthoc analyses (Day 1; P = 0.8356), (Day 2; P = 0.9572), and (Day 3; P = 9979). Group differences were detected using a multi-factor ANOVA (Fig. 3F; F(3, 65) = 5.782, P < 0.002). Sexes were combined as there was no sex difference in any group in the rotarod (Fig. 3E: WT F(1, 14) = 0.312, P > 0.05; HET F(1, 18) = 1.163, P > 0.05; NT-HET F(1, 18) = 1.196, P > 0.05; Ube3a-HET F(1, 13) = 0.0571, P > 0.05).
In multiple reports, AS patients exhibit wide stances on the Zenowalk Way (41). DigiGait analysis showed HET (P < 0.0026) and NT-HET (P < 0.002) differ from WT while the Ube3a-HET group showed a narrowing of these wide stances (P = 0.3486). Gait trajectories in the hindlimb and forelimb differ slightly, likely due to the differing functions between the fore and hind limbs in quadrupeds. Sexes were combined as there was no sex difference in any group in gait analysis (Fig. 3F: WT F(1, 14) = 5.486, P > 0.05; HET F(1, 18) = 2.301, P > 0.05; NT-HET F(1, 18) = 0.071, P > 0.05; Ube3a-HET F(1, 13) = 0.6627, P > 0.05).
Adult treatment with HSPC transduced with a Ube3a-secreting lentiviral vector rescued AS phenotypes in rigorous motor assessments
As described above, we have demonstrated prevention of AS phenotypes in Ube3amat−/pat+ IL2rg−/y neonates upon transplantation of human HSPC transduced with the Ube3a expressing lentivector. To evaluate whether AS phenotype progression could be halted or reversed, adult Ube3amat−/pat+ IL2rg−/y mice were transplanted with the Ube3a-expressing lentivector transduced cells after AS phenotypes were present. Rescue of AS phenotypes in the Ube3a-HET group was observed by these mice scoring indistinguishably from WT on multiple assays of motor deficits 6 weeks post-transplant. This was in contrast to the HET and NT-HET groups which illustrated typical AS deficits of reduced motor activity and impaired motor coordination in four tests of motor abilities. Locomotion in a novel open field was collected by assessing the total distance traversed and the horizontal movements using beam breaks in a novel arena. Group differences in horizontal activity counts were observed using a multi-factor repeated measure ANOVA (Fig. 4A; F(3, 63) = 9.650, P < 0.0001), followed by the Holm Sidak posthoc test (P = 0.3465). Similar to the treated neonates, in the adult treated groups, Holm-Sidak corrected for multiple comparisons still clearly highlighted that both NT-HET (P < 0.0397) and HET (P < 0.0001) differed from WT while our Ube3a-HET treatment group did not differ from WT (P = 0.3465). The Ube3a-HET do not differ from WT at any timepoint across the 30-min assay, while the HET, adjusted P’s from Holm Sidak differed in four of the six 5-min time bins. NT-HETs differed in three of the six 5-min time bins. In corroboration, the total distance also illustrated group differences using a multi-factor repeated measure ANOVA (Fig. 4B; F(3, 61) = 11.78, P < 0.0001). Holm-Sidak corrected posthoc analysis for multiple comparisons highlighted that both NT-HET (P < 0.0001) and HET (P < 0.0002) differed from WT while our treatment group, Ube3a-HET, did not differ from WT (P = 0.191). The Ube3a-HET group did not differ from WT at any timepoint across the 30-min assay, while the HET, adjusted P’s from Holm Sidak differed in five of the six 5-min time bins and the NT-HET differed on every 5-min bin of the 30-min task. Sexes were combined as there was no sex difference in any group in the open field (Fig. 4A: WT F(1, 18) = 0.332, P > 0.05; HET F(1, 15) = 0.189, P > 0.05; NT-HET F(1, 11) = 4.071, P > 0.05; Ube3a-HET F(1, 15) = 0.562, P > 0.05).
Figure 4 .
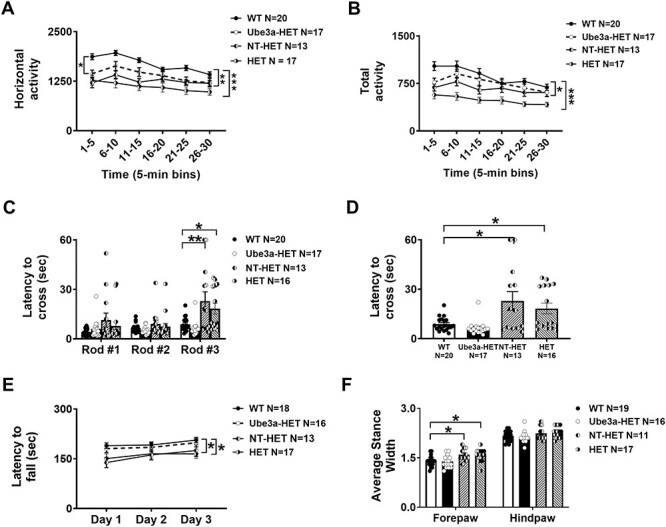
Locomotor ability, balance, motor coordination and gait from Ube3a lentivector transduced HSPC-transplanted adults: Rigorous assessment of motor translational phenotypes using four standard motor behavioral tests in treated and untreated Ube3amat−/pat+ IL2rg−/y mice that were treated with busulfan and transplanted as adults with either NT (NT-HET (hatched bar) or the Ube3a lentivector-transduced (Ube3a-HET (black bar)) human CD34+ HSPC. Four-five weeks old mice were conditioned with busulfan and transplanted via i.v. injection. Six weeks later mice were subjected to (A, B) open field locomotion, (C, D) balance beam, (E) rotarod and (F) treadmill walking. In all tests, Ube3a-deficient mice transplanted with the Ube3a vector-transduced human CD34+ HSPC (Ube3a-HET) exhibited wildtype values of performance. (A, B) Open field activity was increased by both the total distance and horizontal activity metrics. (C) Eight weeks post-transplant, mice were also tested on the beam walking assay. Beams decrease in width and are more difficult to cross going from Rod #1 to Rod #2 to Rod #3. (D) Highlight of Rod #3 of C. (E) Latency to fall from the rotarod was significantly improved in the Ube3a-deficient mice transplanted with Ube3a vector transduced human CD34+ HSPC (Ube3a-HET) compared with the NT (NT-HET) cells controls. (F) DigiGait™ analyses showed a narrowing of wide stances in the Ube3a-HET treated group. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.001, ***P < 0.0001 indicate when the HET and NT-HET groups differ from the control WT group.
As observed in Figure 4C, the WT and Ube3a-HET groups were the fastest to cross the rods and were indistinguishable from one another by time across the rods, displaying substantial improvement in motor coordination. A balance beam walking motor task was conducted in adults. All groups showed longer latencies to cross the rod shaped beams as they became thinner and more difficult to traverse (F(2, 124) = 11.73, P < 0.0001), as expected (39). Group differences using a multi-factor repeated measure ANOVA (Fig. 4C and D; F(3, 62) = 11.24, P < 0.0001). Amazingly, Ube3a-HET exhibited values that were not significantly different from the values seen in WT on the rods, illustrated via Holm-Sidak posthoc analysis. Ube3a-HET do not differ from WT on rod #3 (Fig. 4D: rod #3, P = 0.9931) while the HET (P < 0.0164) and NT-HET (P < 0.0002) had slower latencies to cross rod #3, highlighting improved motor coordination of our treated group. Sexes were combined as there was no sex difference in any group on the balance beam (Fig. 4C: WT F(1, 18) = 0.951, P > 0.05; HET F(1, 15) = 0.175, P > 0.05; NT-HET F(1, 11) = 0.132, P > 0.05; Ube3a-HET F(1, 15) = 0.066, P > 0.05). On the rotarod coordination assay, as expected, HET differed from WT in their latencies to fall from the accelerating rod (Fig. 4E, F(3, 59) = 3.30, P < 0.05) on day 3 (P < 0.05), illustrating poor motor coordination and a lack of motor learning. Strikingly, the Ube3a-HET group was similar to WT across all 3 days of testing using Holm-Sidak posthoc analyses (Day 1; P = 0.9321), (Day 2; P = 0.9691), and (Day 3; P = 0.9514). Sexes were combined as there was no sex difference in any group in the rotarod (Fig. 4E: WT F(1, 18) = 0.558, P > 0.05; HET F(1, 15) = 1.664, P > 0.05; NT-HET F(1, 11) = 2.82, P > 0.05; Ube3a-HET F(1, 15) = 3.825, P > 0.05). In evaluating gait analysis, group differences were detected using a multi-factor ANOVA (Fig. 4F; F(3, 65) = 5.782, P < 0.002). DigiGait analysis demonstrated that HET (P < 0.0026) and NT-HET (P < 0.002) groups differed from WT while the Ube3a-HET group demonstrated a narrowing of these wide stances (P = 0.3486). Gait trajectories in the hindlimb and forelimb differ slightly, likely due to the differing functions between the fore and hind limbs in quadrupeds. Sexes were combined as there was no sex difference in any group in gait metrics (Fig. 4F: WT F(1, 18) = 0.792, P > 0.05; HET F(1, 15) = 0.368, P > 0.05; NT-HET F(1, 11) = 0.588, P > 0.05; Ube3a-HET F(1, 15) = 0.584, P > 0.05).
Prevention and rescue of cognitive deficits in the novel AS model following transplantation with HSPC transduced with a Ube3a-expressing lentiviral vector
The AS rodent models exhibit learning and memory deficits as observed by us and others (36,37,42). As displayed in Figure 5, we demonstrated both the prevention and reversal of cognitive impairments, in neonates and adults, respectively, following 6–8 weeks of treatment with human CD34+ HSPC transduced with the Ube3a expressing lentiviral vector. All data were collected with automated Ethovision software and subsequently manually scored. For the Ube3amat−/pat+ IL2rg−/y neonate transplantations, as anticipated, the WT group spent more time investigating the novel object versus the familiar object. In contrast, the HET and NT-HET groups did not exhibit typical novel object preference (Fig. 5A: WT; t(15) = 3.833, P < 0.0006; HET; t(14) = 0.7699, P > 0.05; NT-HET; t(19) = 0.8703, P > 0.05). However, we observed cognitive rescue in the Ube3a-HET treated group (Fig. 5A: Ube3a-HET; t(14) = 3.302, P < 0.003). There was no object or side bias exhibited by any group (Fig. 5B: WT; t(15) = 0.1100, P > 0.05; HET; t(14) = 0.2968, P > 0.05; NT-HET; t(19) = 0.7160, P > 0.05; Ube3a-HET; t(14) = 0.5284, P > 0.05) since all groups explored the two identical objects similarly during the familiarization phase. Sexes were combined as there was no sex difference in any group in the open field (Fig. 5B: WT t(14) = 0.400, P > 0.05; HET t(18) = 0.265, P > 0.05; NT-HET t(18) = 1.029, P > 0.05; Ube3a-HET t(13) = 0.030, P > 0.05).
Figure 5 .
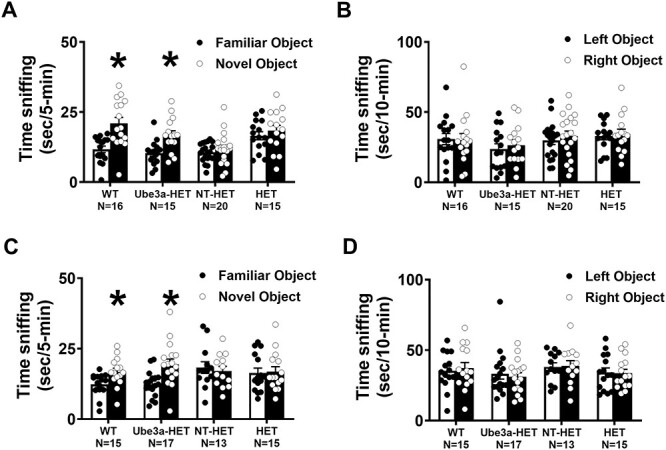
Novel object recognition with mice transplanted with Ube3a lentivector transduced HSPC: (A,B) Newborn Ube3a-deficient IL2rg-/y mice were transplanted with either nontransduced (NT-HET) or Ube3a vector-transduced (Ube3a-HET) human CD34+ HSPC. Wild type IL2rg-/y mice (WT) were used as a control. Eight weeks post-transplant, mice were assessed for learning and memory abilities using the novel object recognition test. (A) Ube3a-HET mice exhibited intact object recognition following a one-hour delay, similar to WT while, NT-HET mice did not spend more time with the novel object, exhibiting a lack of recognition memory. (B) All groups explored the two objects similarly during the familiarization phase. (C, D) Ube3a-deficient adult mice were transplanted with either nontransduced (NT-HET) or Ube3a vector-transduced (Ube3a-HET) human CD34+ HSPC. Wild type IL2rg-/y mice (WT) were used as a control. Six weeks post i.v. injection in adult mice, subjects were assessed for learning and memory abilities using the novel object recognition test. (C) Ube3a-HET mice exhibited intact object recognition following a one-hour delay, similar to WT while, NT-HET mice did not spend more time with the novel object, exhibiting a lack of recognition memory. (D) All groups explored the two objects similarly during the familiarization phase. *p < 0.05, novel versus familiar.
As displayed in Figure 5C and D, we demonstrated functional reversal of cognitive behavioral impairments in adult Ube3amat−/pat+ IL2rg−/y mice following treatment with human CD34+ HSPC transduced with a Ube3a expressing lentiviral vector. As anticipated, the WT group spent more time investigating the novel object versus the familiar object. In contrast, the HET and NT-HET groups did not exhibit typical novel object preference (Fig. 5C: WT; t(14) = 2.626, P < 0.0139; HET; t(16) = 0.1392, P > 0.05; NT-HET; t(12) = 0.1331, P > 0.05). Impressively, we observed cognitive rescue in the Ube3a-HET mice (Fig. 5C: Ube3a-HET; t(14) = 3.271, P < 0.0026). All groups explored the two objects similarly during the familiarization phase (Fig. 5D: WT; t(14) = 0.3698, P = 0.7193; HET; t(16) = 0.2968, P = 0.8903; NT-HET; t(12) = 0.1331, P = 0.8952; Ube3a-HET; t(14) = 0.1392, P = 0.6488). Sexes were combined as there was no sex difference in any group in the open field (Fig. 5D: WT t(13) = 0.036, P > 0.05; HET t(13) = 0.723, P > 0.05; NT-HET t(11) = 0.241, P > 0.05; Ube3a-HET t(15) = 0.539, P > 0.05).
Neonatal treatment with HSPC transduced with a Ube3a expressing lentiviral vector prevented the elevated delta power characteristic of AS
Several EEG abnormalities have been described in AS, including elevated delta power, which have been recapitulated in the Ube3a-deficent mouse models (43–45). To evaluate whether the CD34+ cells transduced with the Ube3a expressing lentivector rescued this deficit, the Ube3a-deficient mice were transplanted with these cells. As expected, we observed elevated delta power in the HET group when compared with WT littermate controls (Fig. 6: F(25, 362) = 4.312, P < 0.0001). However, strikingly, we observed a reduction of delta power in the Ube3a-HET (Fig. 6: F(25, 300) = 0.223, P > 0.999) group that was significantly lower than the NT-HET and HET mice (Fig. 6: F(25, 475) = 3.249, P < 0.0001). These results confirmed that delta power elevation could be corrected upon treatment with Ube3a-expressing HSPC.
Figure 6 .
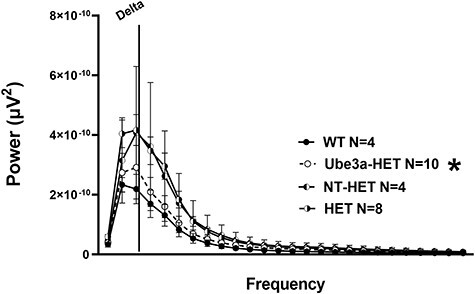
Delta power analysis in AS mice transplanted with Ube3a lentivector transduced HSPC: A PSD between using treated and untreated Ube3a-deficient IL2rg−/y mice that were irradiated and transplanted as neonates with either NT HET (semi-filled left) or the Ube3a lentivector transduced Ube3a-HET (black dotted line open circles) human CD34+ HSPC. AS mice on B6 background versus their WT background littermates illustrate observable differences in elevated delta power analogous to clinical phenotypes. Quantification of spectral power bands illustrates that treatment lowers/corrects the elevated delta spectral power toward the levels observed in WT mice.
Immunohistochemical detection of UBE3A expression in the brains of Ube3amat−/pat+ IL2rg−/y mice transplanted with the Ube3a-expressing vector transduced HSPC
To determine if UBE3A expression could be restored in the brains of mice engrafted with the Ube3a-epressing lentivector transduced human HSPC, transplanted mice were euthanized via transcardial perfusion and prepared for immunohistochemistry. Data suggest that the engrafted human cells were able to cross the blood brain barrier (BBB) when administered as adults, Figure 7A and B. Figure 7A and B highlights analysis of UBE3A expression in the cortex of the mouse brain 6-weeks post-HSPC transplant in adult mice. Figure 7A is an immunohistochemical image showing expression of UBE3A in the mouse cortex of non-transplanted WT mice, a Ube3a-HET (an AS mouse transplanted with Ube3a-epressing vector transduced human CD34+ HSPC), NT-HET (an AS mouse transplanted with NT human CD34+ HSPC) and a non-transplanted HET. Figure 7B shows the analysis that highlighted the significant increase in UBE3A positive cells, similar to the WT level, observed in transplanted Ube3a-HET mice compared with NT-HET and HET. Tukey’s corrected post hoc analysis for multiple comparisons revealed that our treatment group Ube3a-HET differed from HET (P = 0.0126) but did not differ from WT (P > 0.05). This data highlights the groundbreaking potential for adult rescue by intense positive staining for UBE3A with UBE3A-specific antibodies demonstrating restored expression in the brains of transplanted mice.
Figure 7 .
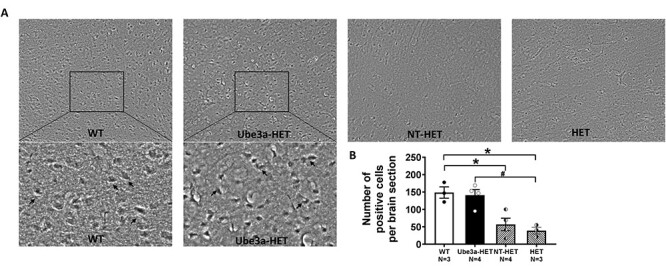
Immunohistochemical analysis of UBE3A expression in cortex of the mouse brain. Six weeks post-transplant in adult mice, subjects were assessed for UBE3A expression using DAB as a chromogen. (A) Immunohistochemical images showing expression of UBE3A in the mouse cortex of non-transplanted Ube3a wild type IL2rg−/y (WT) mice, those transplanted with Ube3a vector transduced (Ube3a-HET) human CD34+ HSPC, those transplanted with NT human CD34+ HSPC (NT-HET), and non-transplanted HET mice (HET). (B) A significant increase in the UBE3A positive cells, similar to the WT level, was observed in transplanted Ube3a-HET compared with NT-HET and HET when treated as adults. Data are expressed as mean ± SEM. *P < 0.05 indicates when the HET and NT-HET groups differ from the control WT group. #P < 0.05 indicates when the Ube3a-HET group differs from HET group.
Multi-lineage hematopoiesis of Ube3a vector transduced human CD34+ cells
To evaluate the in vivo engraftment and differentiation potential of Ube3a lentiviral vector transduced human CD34+ HSPC, cells were transplanted into immunodeficient NOD-Rag1−/-IL2rg−/y (NRG) mice. These mice contain deletions in their Rag1 and IL2 receptor gamma chain genes causing their immunodeficiency and allowing for the transplantation and engraftment of human CD34+ cells in the peripheral blood and various lymphoid organs. Human CD34+ HSPC were left either NT, transduced with the EGFP control vector, or transduced with the hAS8 Ube3a vector and transplanted into NRG mice. After 6-months post-transplant, mice were analyzed for engraftment in the peripheral blood, spleen, thymus and bone marrow.
Initial analyses determined that normal engraftment of hAS8 Ube3a lentiviral vector transduced CD34+ cells and development of human T cells was demonstrated in the peripheral blood of engrafted NRG mice. As displayed in Figure 8A, no significant difference (P > 0.05) in the development of CD3+/CD4+ T cells, CD3+/CD8+ T cells, or CD4+/CD8+ double positive T cells from hAS8 vector transduced human CD34+ cells was observed compared with EGFP-control vector transduced or NT human CD34+ cells. Similar levels of all T cells populations analyzed were observed in the peripheral blood of all mouse cohorts. On average, mice transplanted with the Ube3a vector-transduced cells displayed CD3+/CD4+ (76.1%), CD3+/CD8+ (38.2%), and CD3+/CD4+/CD8+ (24.4%) levels in the peripheral blood compared with mice transplanted with NT cells (CD3+/CD4+ (72.1%), CD3+/CD8+ (47.5%), and CD3+/CD4+/CD8+ (19.6%)) and EGFP alone vector transduced cells (CD3+/CD4+ (68.2%), CD3+/CD8+ (37.9%), and CD3+/CD4+/CD8+ (13.1%)). Similar to the peripheral blood, as displayed in Figure 8B, no significant difference (P > 0.05) in the development of CD3+/CD4+ T cells, CD3+/CD8+ T cells, or CD4+/CD8+ double positive T cells in the spleens of engrafted mice from hAS8 vector transduced human CD34+ cells was observed compared with EGFP control vector transduced or NT human CD34+ cells. On average, mice transplanted with the Ube3a vector transduced cells displayed CD3+/CD4+ (70.9%), CD3+/CD8+ (46.3%) and CD3+/CD4+/CD8+ (15.3%) levels in the spleen compared with mice transplanted with NT cells (CD3+/CD4+ (67.8%), CD3+/CD8+ (58.5%) and CD3+/CD4+/CD8+ (26.4%)) and EGFP control vector transduced cells (CD3+/CD4+ (57.9%), CD3+/CD8+ (52.0%) and CD3+/CD4+/CD8+ (19.1%)). We next analyzed the levels of T cells in the thymus of engrafted mice. As displayed in Figure 8C, no significant difference (P > 0.05) in the development of CD3+/CD4+ T cells, CD3+/CD8+ T cells or CD4+/CD8+ double positive T cells in the thymus of engrafted mice from hAS8 vector transduced human CD34+ cells was observed compared with EGFP control vector transduced or NT human CD34+ cells. On average, mice transplanted with the Ube3a vector transduced cells displayed CD3+/CD4+ (66.1%), CD3+/CD8+ (54.4%) and CD3+/CD4+/CD8+ (27.7%) levels in the thymus compared with mice transplanted with NT cells (CD3+/CD4+ (74.3%), CD3+/CD8+ (53.6%) and CD3+/CD4+/CD8+ (28.0%)) and EGFP control vector transduced cells (CD3+/CD4+ (65.2%), CD3+/CD8+ (57.1%) and CD3+/CD4+/CD8+ (22.3%)). These results demonstrate that human CD34+ HSC transduced with the hAS8 Ube3a-expressing lentiviral vector were capable of engraftment in the NRG mice and were able to differentiate into normal T cells in the peripheral blood, spleen and thymus of engrafted mice.
Figure 8 .
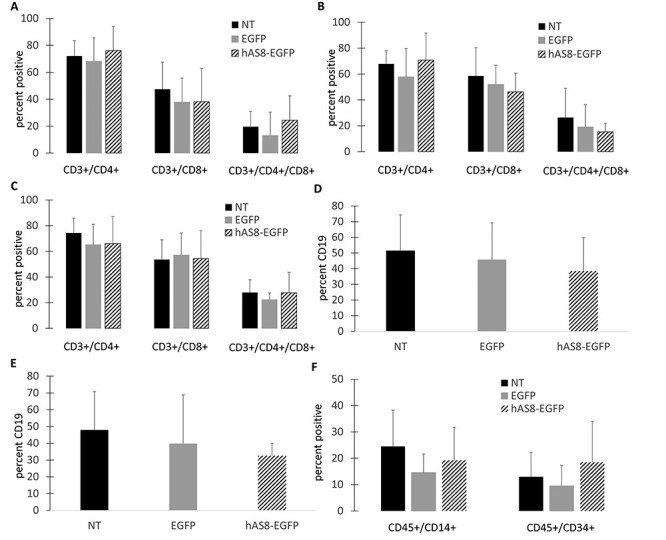
Engraftment and development of human immune cells in NRG mice: Human CD34+ HSPC were left NT or transduced with either the EGFP control (EGFP) or Ube3a-expressing (hAS8) lentiviral vector. Cells were transplanted into 2–5-day-old NRG mice. At 16 weeks post-transplant, mice were euthanized, and human T cells were analyzed for CD3, CD4, and CD8 expression in the (A) peripheral blood, (B) spleen, and (C) thymus. Human B cells were analyzed for CD45 and CD19 in the (D) spleen and (E) bone marrow. (F) Human macrophages (CD45/CD14) and human CD34+ HSPC (CD45/CD34) were analyzed in the bone marrow.
As a next step in evaluating the safety of the Ube3a vector transduced cells, human B cell analyses were performed on the spleen and bone marrow of engrafted NRG mice. As displayed in Figure 8D, no significant difference (P > 0.05) in the development of CD45+/CD19+ B cells in the spleen of engrafted mice from hAS8 vector transduced human CD34+ cells were observed compared with EGFP control vector transduced or NT human CD34+ cells. On average, mice transplanted with the Ube3a vector transduced cells displayed CD45+/CD19+ (38.5%) levels in the spleen compared with mice transplanted with NT cells (CD45+/CD19+ (51.6%) and EGFP control vector transduced cells (CD45+/CD19+ (45.8%). Similarly, as displayed in Figure 8E, no significant difference (P > 0.05) in the development of CD45+/CD19+ B cells in the bone marrow of engrafted mice from hAS8 vector transduced human CD34+ cells were observed compared with EGFP control vector transduced or NT human CD34+ cells. On average, mice transplanted with the Ube3a vector transduced cells displayed CD45+/CD19+ (32.6%) levels in the bone marrow compared with mice transplanted with NT cells CD45+/CD19+ (47.9%) and EGFP control vector transduced cells CD45+/CD19+ (39.8%). These results demonstrate that human CD34+ HSC transduced with the hAS8 Ube3a-expressing lentiviral vector were capable of engraftment in the NRG mice and were able to differentiate into normal B cells in the spleen and bone marrow of engrafted mice.
We next evaluated the levels of human macrophages and human CD34+ cells engrafted in the bone marrow of NRG mice transplanted with hAS8 Ube3a vector transduced cells. As displayed in Figure 8F, no significant difference (P > 0.05) in the development of CD45+/CD14+ macrophages or CD45+/CD34+ cells in the bone marrow of engrafted mice from hAS8 vector transduced human CD34+ cells was observed compared with EGFP control vector transduced or NT human CD34+ cells. On average, mice transplanted with the Ube3a vector-transduced cells displayed CD45+/CD14+ (19.2%) levels and CD45+/CD34+ (18.5%) levels in the bone marrow compared with mice transplanted with NT cells (CD45+/CD14+ (24.5%) and CD45+/CD34+ (13.0)) and EGFP control vector transduced cells (CD45+/CD14+ (14.6%) and CD45+/CD34+ (9.5)). These results demonstrate that human CD34+ HSC transduced with the hAS8 Ube3a-expressing lentiviral vector were capable of engraftment in the NRG mice and were able to differentiate into normal macrophages in the bone marrow of engrafted mice. These results also demonstrate that human CD34+ cells transduced with the Ube3a lentiviral vector were still present in the bone marrow of engrafted mice 6 months post-transplant.
Discussion
Currently, there is no FDA-approved therapy for AS; however, numerous therapeutic approaches are being evaluated both preclinically and clinically. While the initial preclinical study of Meng et al. using ASO did not observe improvements in motor abilities following ASO treatment, the therapeutic antisense used in that study did normalize body weight and improved performance on contextual fear conditioning of AS mice when given directly into the brain by intracerebroventricular injection in adult mice (8). Another technique that modified the imprint of the antisense transcript on the paternal allele included the artificial transcription actor (ATF), S1K (10). Viral vector delivery via adeno-associated virus (AAV) has also proven successful preclinically with efficacy in long-term potentiation and contextual fear conditioning but not in rotarod or other motor assays (9). Gene-replacement therapy for AS should be a reality in the coming years. In fact, this year, two clinical trials using ASO compounds began recruitment and treatment, highlighting the groundbreaking time in AS and gene therapy research (GeneTx NCT04259281; Roche NCT04428281). Another proof-of-concept for gene-replacement therapy utilized a sophisticated tamoxifen induced Cre-line. In this work, temporally controlled Cre-dependent induction of the maternal Ube3a allele found improvements or rescue in rotarod and marble burying yet only when restored embryonically and at juvenile ages, not as adolescents or adults (40,46).
Our report is the first to detect rescue in multiple behavioral assays across several impaired clinically relevant domains from treatment given to neonate and adult AS mice on a congenic C57BL/6 J background, which eliminated the large influence that background strain has on motor and seizure phenotypes (36,47,48). The work presented here highlights our effort to develop the use of HSPC transduced with a lentiviral vector expressing Ube3a, in an innovative preclinical model of AS, which offers a promising strategy for life-long delivery of functional UBE3A through the hematopoietic system to the brain. This cross-correction approach has been successfully demonstrated for other monogenic diseases including adrenoleukodystrophy, metachromatic leukodystrophy and Sandhoff disease (22,31,32,49). In previous studies, successful engraftment of gene-modified microglia in the brains of treated mice demonstrated that HSPC gene therapy can deliver therapeutic proteins to disease-affected cells (22,30,32–34,49). In our current study, we observed elevated levels of UBE3A in the brains of the Ube3a-HET treatment group, and rescue of motor, cognitive and EEG phenotypes following transplantation and engraftment of the genetically modified human CD34+ HSPC as neonates or adults.
The immunodeficient and Ube3a-deficient mice utilized in this study are capable of transplantation and engraftment of human cells following sub-lethal irradiation of neonates or busulfan treatment of adults. We chose to utilize human HSPC over mouse HSPC due to (a) early data that indicated the interleukin 2 common chain deficiency caused no behavioral outcomes in domains of interest to AS, (b) a fast forward approach to clinical translation not requiring bridging studies of mHSPC to hHSPC, (c) using human cells will allow us delineate the biological pathway that underlies cross correction, in the future, as the human cells should be distinguishable from mouse cells in brain and (d) feasibility, since an extraordinary number of mice would be required to collect donor mHSPC per subject treated mouse further in line with Animal Welfare’s RRR policy. The selection of mouse strains involved numerous iterations as to confirm that no behavioral phenotypes typically attributed to the loss of Ube3a, were not an unintended consequence of immune alterations when deriving the immunodeficient Ube3a-deficient model. This resulted in the use of an Ube3a-deficient and IL2 common gamma receptor chain-deficient model, Ube3amat−/pat+ IL2rg−/y allowing for the transplantation and engraftment of human cells. Next, we assessed the critical question of whether the gene-modified HSPC could prevent or rescue behavioral phenotypes in the mouse model. We performed functional experiments in cohorts treated as neonates or adults to evaluate whether providing treatment would only be useful if delivered before a critical therapeutic window early in life, or if later stage interventions could be similarly efficacious. Finally, elevated UBE3A expression was detected in the brains of adult treated mice. A limitation to the current work is the lack of co-localization staining of neurons; however, mechanistic studies beyond the scope of this current work are ongoing.
Functional reversal of AS symptoms in treated adult mice was remarkable in subjects displaying AS phenotypes of motor deficits, coordination and gait impairments, and cognitive deficits in novel objection recognition. This was an unexpected finding, as the prevailing dogma in the AS field has highlighted the importance of critical treatment periods, suggesting that improvements in functional/behavioral phenotypes beyond a critical period would be minimal or not possible (46). These conclusions are logical and well supported by behavioral outcomes using a novel inducible genetic mutant line and physiological outcomes from this same line (43–45). Yet, our robust adult rescue in multiple behavioral and physiological domains on multiple assays challenge this interpretation and highlight improvements in performance across multiple domains on the same scale as the neonatal treated mice. Differences in behavioral rescues may not be surprising given the highly disparate mouse models, as this earlier work was performed in a tamoxifen induced Cre-line. Recent studies have demonstrated imperfections with Cre tools for numerous reasons including undesirable expression patterns, unwanted germline expression of the Cre recombinase, mosaic expression of the Cre driver complicating genotyping, and recombination beings more or less efficient depending on the floxed locus (50,51). Another difference is that the behavioral experiments in the aforementioned studies were performed in an isogenic F1 hybrid background of 129/Sv and C57BL/6 mice while ours used a congenic C57BL/6J background throughout.
In earlier work, temporally controlled Cre-dependent induction of the maternal Ube3a allele found improvements or rescue in rotarod and marble burying were only observed embryonically and at juvenile ages, not as adolescents or adults (40,46). Our study also used rotarod, as it is a field gold standard assay, and we saw rotarod rescue in the neonate and adult cohorts, as well as in three additional motor tasks. While we did not perform marble burying, we performed other translational tasks such as gait mapping and EEG recordings. It is important to note, however, that not all behavioral phenotypes studied in laboratory animal models are robust, reliable, reproducible, nor optimal for enabling translational application to the clinic.
Our study, for the first time, illustrated rigorous reversal of many phenotypes in both neonate and adult treated mice using HSPC transduced with a Ube3a-expressing lentiviral vector. Gene modified lentiviral vectors have been applied in different clinical trials based on ex vivo correction of autologous HSPC and were demonstrated to be immunologically safe and therapeutically efficient in symptom correction in a wide range of single gene disorders (22,31,32). Yet still, for nearly all rare genetic disorders, including AS, there is a delay between developmental milestones not being met and an official diagnosis of a genetic syndrome. Thus, there needs to be testing of therapeutics beyond the prenatal/early neonatal state. While most AS research has highlighted this critical window, our HSPC therapeutic approach would address this oversight, as our observations of functional efficacy were nearly equivalent in neonates and adults (48).
A potential concern with the older in life approach is the busulfan conditioning cell ablation regimen used as adults (irradiation was used for the neonate transplants as analogous conditioning). We did not observe any overt side effects with the busulfan conditioning, and as it is being used clinically for other disorders, our data demonstrate strong evidence for the use of this therapy to treat AS (52).
Our data demonstrate strong evidence for the use of HSPC therapy to treat AS, which should provide long-term restoration of UBE3A in the brain. Additionally, this study challenges the current dogma in the AS field of therapeutic treatment windows, since treatment in adults was equally as efficacious as treatment in neonates, suggesting that therapeutic treatment beyond the critical period is possible for the AS community.
Materials and Methods
Lentiviral vector production
All lentiviral vectors used were generated from a self-inactivating third generation lentiviral vector backbone called CCLc-MNDU3-x (Fig. 1A). An EGFP-alone lentiviral vector, CCLc-MNDU3-PGK-EGFP, was constructed and used as an empty vector control (Fig. 1B). To construct the Ube3a expressing lentiviral vectors, modified mouse Ube3a isoform 3 (mAS8) or modified human Ube3a isoform 1 (hAS8) were manually synthesized and cloned into pcDNA 3.1 TOPO (ThermoFisher, Grand Island, NY). The sequences of the clones were confirmed (Laragen, Culver City, CA). The mAS8 and hAS8 TOPO clones were further subcloned individually under the control of the MNDU3 promoter in the ‘x’ site of the CCLc-MNDU3-x-PGK-EGFP backbone (Fig. 1C and D). Experiments involving recombinant DNA were performed following the NIH guidelines.
Lentiviral vectors were produced in human embryonic kidney (HEK)-293 cells by transient transfection with a 1:5:5 ratio of the envelope vesicular stomatitis virus glycoprotein (VSVG) plasmid, a packaging plasmid (Δ8.9) containing a capsid gene and a reverse transcriptase gene, and one of the transfer plasmids, either the control EGFP alone, the mouse Ube3a, or the human Ube3a plasmids. Forty-eight hours post-transfection, cell supernatants were concentrated by ultrafiltration. Titers for total vector transducing units were calculated by transduction of HEK-293 cells and analysis of EGFP expression by flow cytometry 48 h post-transduction. Flow cytometry was performed on a Beckman Coulter Cytomics FC500 and analyzed using CXP software.
Isolation and transduction of human CD34+ HSPC
Human CD34+ HSPC were purified from umbilical cord blood obtained from the UC Davis Umbilical Cord Blood Collection Program using Ficoll-Paque (GE Healthcare, Logan, UT) density gradient and CD34 specific magnetic bead separation (Miltenyi Biotec, Auburn, CA). The CD34+ cells were cultured for 48 h in XVIVO-10 media (Lonza, Anaheim, CA) containing 50 ng/ml Flt-3 ligand, thrombopoietin (TPO), and stem cell factor (SCF) (R&D Systems, Minneapolis, MN). After culturing for 48 h, the CD34+ cells were left either NT, transduced with the EGFP control vector, transduced with the mAS8 vector, or transduced with the hAS8 vector at an MOI of 20 and the addition of 8 mg/ml protamine sulfate for a minimum of 3 h at 37°C. The EGFP, mAS8, and the hAS8 CD34+ transduced cultures were either left unsorted or fluorescently active cell sorted (FACS) based on EGFP expression depending on the subsequent experiments in which they were utilized.
Colony-forming unit assays
The human CD34+ cells, either NT, FACS EGFP control vector, FACS mAS8 vector, or FACS hAS8 vector transduced (500 total cells) were cultured for 12 days in MethoCult medium (Stemcell Technologies, Vancouver, CA). Post-culture, total granulocyte/macrophage (GM) colonies, granulocyte/erythrocyte/megakaryocyte/macrophage (GEMM) colonies and burst forming unit-erythroid colonies (BFU-E), were counted by microscopy. Experiments were performed in triplicate.
In vitro derivation of macrophages and analysis of their phenotypic profiles
The colonies formed in the CFU assays were further differentiated into mature macrophages, in vitro, by culturing the cells in DMEM supplemented with 10% FBS, 10 ng/ml of granulocyte-macrophage colony stimulating factor (GM-CSF), 10 ng/ml macrophage colony stimulating factor (M-CSF) (R&D Systems, Minneapolis, MN) for 4 days. Macrophages were further analyzed by flow cytometry for surface expression of the macrophage cell surface markers, CD14, CD4, and HLA-DR. Macrophages were stained with a phycoerythrin (PE)-conjugated CD4, a PE-conjugated HLA-DR, or a PE-conjugated CD14 antibody (BD Biosciences, San Jose, CA). Flow cytometry was performed on a Beckman Coulter Cytomics FC500 and analyzed with CXP software. Experiments were performed in triplicate.
Western blot and ubiquitination analyses of Ube3a vector transduced cells
Human macrophages derived from the vector transduced human CD34+ cells were lysed in 1 × RIPA buffer supplemented with protease inhibitor cocktail (Millipore Sigma, St. Louis, MO) and protein concentrations were determined by Bradford assay (BioRad, Hercules, CA). For Western blots, 60 μg protein were separated on a Novex 10–20% Tris-Glycine gel (Invitrogen, Carlsbad, CA) with MOPS buffer. Protein was subsequently transferred onto a nitrocellulose membrane and protein loading was evaluated by Ponceau S stain. The membrane was rinsed with deionized water and incubated in blocking solution (5% non-fat dry milk in TBST; 50 mm Tris, 150 mm NaCl, 0.1% Tween-20) for 30 min at room temperature. Membranes were incubated with primary mouse UBE3A antibody (BD Biosciences, San Jose, CA #611416) at 1:500 dilution in blocking solution at 4°C overnight. After three 10-min washes with TBST the membrane was incubated with horseradish peroxidase conjugated rabbit anti mouse secondary antibody (1:2000 dilution in blocking solution) for 45 min at room temperature. After three more washes in TBST, proteins were visualized with Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, Logan, UT) using the ChemiDoc XRS Imaging System (BioRad, Hercules, CA). Cell lysates from the macrophages were further analyzed for ubiquitination of S5A using the Human E6AP/S5a Ubiquitination Kit (#K-230) (R&D Systems, Minneapolis, MN) according to the manufacturer’s protocol.
Generation of an immunodeficient mouse model of AS (Ube3amat−/pat+ IL2rg−/y)
Ube3a mutated immunodeficient mouse model was generated by crossing Ube3a deletion females (JAX stock #016590) with IL2rg null males (JAX stock #003174; B6.129S4-Il2rgtm1Wjl/J), Ube3amat−/pat+ IL2rg−/y. To identify mice, neonates were labelled by paw tattoo on postnatal day (PND) 2–3 using non-toxic animal tattoo ink (Ketchum Manufacturing Inc., Brockville, ON, Canada). At PND 5–7, tails of pups were clipped (0.5 cm) for genotyping, following the UC Davis IACUC policy regarding tissue collection. Genotyping was performed with RED Extract-N-Amp (Sigma Aldrich, St. Louis, MO) using primers R1965 GCTCAAGGTTGTATGCCTTGGTGCT, WTF1966 AGTTCTCAAGGTAAGCTGAGCTTGC, and ASF1967 TGCATCGCATTGTCTGAGTAGGTGTC for Ube3a, and IMR5330 GTGGGTAGCCAGCTCTTCAG, oIMR5331 CCTGGAGCTGGACAACAAAT, and oIMR7415 GCCAGAGGCCACTTGTGTAG for null for IL2rg. After weaning on PND 21, mice were socially housed in groups of 2–4 by sex. The resulting mice used in the experiments were identified genotypically as Ube3amat−/pat+ IL2rg−/y or Ube3amat+/pat+ IL2rg−/y.
Transplantation and generation of the humanized Ube3amat−/pat+ IL2rg−/y mice
Newborn Ube3amat−/pat+ IL2rg−/y mice (2–5 days old) were irradiated with 150 rads from a Cesium source and transplanted intrahepatically with 500 000 total human CD34+ HSPC/mouse. Eight weeks post-transplant, mice were bled via the tail vein and analyzed for engraftment by flow cytometry using a PE-CY7-conjugated anti-human CD45 antibody (BD Biosciences, San Jose, CA). Successfully engrafted mice were then evaluated for behavioral phenotypes. The order and age of testing were as follows: (1) Open field at 9 weeks of age, (2) Beam walking at 9 weeks of age, (3) Rotarod at 10 weeks of age, and (4) NOR at 11 weeks of age, (5) EEG at 15–17 weeks.
At 4–5 weeks of age, Ube3amat−/pat+ IL2rg−/y mice were treated with 20 mg/kg busulfan intraperitonially 48 and 24 h prior to transplanting with 500 000 total human CD34+ HSPC/mouse intravenously. Six weeks post-transplant, mice were bled via the tail vein and analyzed for engraftment by flow cytometry using a PE-CY7-conjugated anti-human CD45 antibody (BD Biosciences, San Jose, CA). Successfully engrafted mice were then evaluated for behavioral phenotypes. The order and age of testing were as follows: (1) Open field at 11–12 weeks of age, (2) Beam walking at 11–12 weeks of age, (3) Rotarod at 12–13 weeks of age, (4) NOR at 14–15 weeks of age.
Subjects for behavior and neurophysiology
All mice were housed in Techniplast cages (Techniplast, West Chester, PA, USA). Cages were housed in ventilated racks in a temperature (68–72°F) and humidity (~25%) controlled colony room on a 12:12 light/dark cycle. Standard rodent chow and tap water were available ad libitum. In addition to standard bedding, a Nestlet square, shredded brown paper, and a cardboard tube (Jonesville Corporation, Jonesville, MI) were provided in each cage. All experimental procedures were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees (IACUC) #21494 (PI, Silverman) of the University of California, Davis.
Behavioral assays
Open field
General exploratory locomotion in a novel open field arena was evaluated as previously described (53,54). Briefly, each subject was tested in a VersaMax Animal Activity Monitoring System (Accuscan, Columbus, OH, USA) for 30-min in a ~30 lux testing room. Total distance traversed, horizontal activity, vertical activity and time spent in the center were automatically measured to assess gross motor abilities in mice.
Beam walking
A beam-walking motor task was conducted as previously described in (39,55,56). Fifty-nine centimeter long round rods were suspended 68 cm above a cushioned landing pad. A goal box at the end of the beam consisted of a 12 cm diameter cylinder to provide motivation to cross the beam. Each mouse was placed at one end of the beam and the time to cross to the goal box on the other end was measured. Testing sequence moved from largest diameter to smallest diameter rods in order of increased difficulty. On the day prior to testing, all animals were given two practice trials on the largest diameter round beam in order to become accustom to the procedure. On the test day, each animal was sequentially tested on three round rods (35, 18 and 13 mm). Testing sequence was based on presentations of decreasing diameter to present increasing levels of difficulty. Each mouse was given two trials on each beam, separated by ~30 min. The time to transverse the beam was recorded and averaged across the two trials for each beam. A maximum time of 60 s was assigned to individuals that failed to cross the beam in that duration. In the small number of cases where mice fell from the beam, a score of 60-s was assigned.
Rotarod
Motor coordination, balance and motor learning were tested with an accelerating rotarod (Ugo Basile, Gemonio, Italy) as previously described in (39,57). Mice were placed on a rotating cylinder that slowly accelerated from 5 to 40 revolutions per min over 5 min. Mice were given three trials per day with a 60-min inter-trial rest interval and tested for three consecutive days for a total of nine trials. Performance was scored as latency to fall off the cylinder with a maximum latency of 5 min.
Gait analysis
Treadmill gait analysis was performed using the DigiGait™ system (Mouse Specifics Inc., USA) (39,58). Mouse paws were painted with non-toxic red food coloring to augment dark green paw tattoos that generated conflict in DigiGait™ analysis 1 min prior to introduction to the walking chamber to reliably capture the entire paw surface area. Before data collection, each subject was acclimated to the Perspex walking chamber for 1 min and the treadmill was slowly accelerated to the final speed of 20 cm/s to allow mice to adjust to walking on the belt. Digital images of paw placement were recorded through a clear treadmill from the ventral plane of the animal. Mice were tested in a single session at a 20 cm/s treadmill speed maintaining a normal pace walk for WT mice. Non-performers were defined as mice who were unable to sustain walking at 20 cm/s without colliding with the posterior bumper for at least 3 s. There is no practice effect or repeated exposure, and therefore, mice were allowed retrial and retest if they were unable to adjust to walking on the belt easily. The treadmill belt and the encasing Perspex chamber were cleaned with 70% (v/v) ethanol in between tests. For each mouse, videos of ∼5 s duration of all sessions were analyzed using the DigiGait™ Imaging and Analysis software v12.2 (Mouse Specifics Inc., USA). Contrast filters were determined on a mouse-by-mouse case to facilitate consistent recognition of all four paws. All analysis was conducted in a single session by experimenter blind to genotype. Stride length (distance a paw makes during a single stride) and frequency (number of strides per second to maintain pace) were automatically calculated. Data was averaged between left and right paws for fore and hind paws.
Novel object recognition
The novel object recognition test was conducted as previously described in opaque matte white (P95 White, Tap Plastics, Sacramento, CA, USA) arenas (41 cm l x 41 cm w x 30 cm h) (39,55). The assay consisted of four sessions: a 30-min habituation session, a second 10-min habituation phase, a 10-min familiarization session and a 5-min recognition test. On Day 1, each subject was habituated to a clean empty arena for 30-min. 24-h later, each subject was returned to the empty arena for an additional 10-min habituation session. The mouse was then removed from testing arena and was placed in a clean temporary holding cage while two identical objects were placed in the arena. Subjects were returned to the testing arena and given a 10-min of familiarization period in which they had time to investigate the two identical objects. After the familiarization, phase subjects were returned to their holding cages for a 1-h interval period. One familiar object and one novel object were placed in the arena, where the two identical objects had been located during the familiarization phase. After the 1-h interval, each subject was returned to the arena for a 5-min recognition test. The familiarization session and the recognition test were recorded using Ethovision XT video tracking software (version 9.0, Noldus Information Technologies, Leesburg, VA, USA). Sniffing was defined as head facing the object with the nose point within 2 cm or less from the object. Time spent sniffing each object was scored by an investigator blind to both genotype and treatment. Recognition memory was defined as spending significantly more time sniffing the novel object compared with the familiar object. Total time spent sniffing both objects was used as a measure of general exploration. Time spent sniffing two identical objects during the familiarization phase confirmed the lack of an innate side bias. Within genotype repeated-measures ANOVA was used to analyze novel object recognition using novel versus familiar objects as comparison. F, degrees of freedom, and P-values are reported.
EEG implantation and acquisition
These methods are described in detail by our laboratory in previous publications (58–60). Wireless EEG transmitters were implanted in anesthetized test animals using continuous isoflurane (2–4%). A 2–3 cm midline incision was made over the skull and trapezius muscles, then expanded to expose the subcutaneous space. Implants were placed in the subcutaneous pocket lateral to the spine to avoid discomfort of the animal and displacement due to movement. Attached to the implant were four biopotential leads made of a Nickel-Colbalt based alloy insulated in medical-grade silicone, making up two channels that included a signal and reference lead. These leads were threaded toward the cranial part of the incisions for EEG and EMG placement. The periosteum was cleaned from the skull using a sterile cotton-tip applicator and scalpel then two 1 mm diameter burr holes were drilled (1.0 mm anterior and 1.0 mm lateral; −3.0 mm posterior and 1.0 mm lateral) relative to bregma. Steel surgical screws were placed in the burr holes and the biopotential leads were attached by removing the end of the silicone covering and tying the lead to its respective screw. Once in place, the skull screws and lead connections were secured using dental cement. For EMG lead placement, the trapezius muscles of the animal were exposed, and each lead was looped through and sutured to prevent displacement. Finally, the incision was sutured using non-resorbable suture material and the animals were placed in a heated recovery cage where they received Carpofen (5 mg/kg; i.p.) directly after surgery and 24 h post-surgery as an analgesic. Subjects were individually caged with ad libitum access to food and water for 1-week before EEG acquisition and monitored daily to ensure proper incision healing and recovery. Each implantation surgery took <45-min and no fatalities were observed.
EEG data acquisition, processing and analysis
After a 1-week recovery from surgical implantation, individually housed mice were assigned to PhysioTel RPC receiver plates that transmitted data from the EEG implants to a computer via the data exchange matrix using Ponemah software (Data Sciences International, St. Paul, MN). EEG and EMG data were collected at a sampling rate of 500 Hz with a 0.1 Hz high-pass and 100 Hz low-pass bandpass filter. Activity, temperature and signal strength were collected at a sampling rate of 200 Hz. Data acquired in Ponemah were read into Python and further processed with a bandpass filter from 0 to 50 Hz to focus on our frequencies of interest, as previously described (64).
Spectral analysis was performed in Python using MEG and EEG analysis and visualization (MNE) open-source software, as previously described in (59,61). Frequency bands were defined as delta 0.5–4 Hz, theta 5–9 Hz, alpha 9–12 Hz, beta 13–30 Hz and gamma 30–50 Hz. Spectral power was analyzed using the Welch’s method, which windows over the signal and averages across spectral samples. For power spectral densities (PSD) investigated in Cohort 2, analysis started 3 h into recording and finished 3 h prior to the end of recording and PTZ administration, resulting in an 18-h sampling window. PSD analysis in Cohort 3 also began 3 h into recording but continued over the 3-day recording resulting in a 69-h sampling window. No statistical difference was detected in PSD within genotype between samples, therefore, both cohorts were combined. Total delta power was determined by adding the density data detected in the 0.5–4 Hz frequency range while total power summed all the PSD data in the 0.5–50 Hz frequency range. Relative delta frequencies were calculated by dividing total delta power by total power per animal and averaging across genotype.
Immunohistochemistry labeling and analysis
Upon completion of the behavioral, motor and cognitive studies, mice were euthanized, perfused and the brain tissues were obtained, snap frozen and labeled for UBE3A, as previously described in (61). Sagittal sections (40 μM) were cryosectioned and further labeled with ImmPACT DAB Peroxidase Substrate (Vector Labs, Burlingame, CA) utilizing the Vectastain ABC Kit (HP1–26), and in conformity with recommendations from the manufacturers included protocols. In brief, to begin the process tissues underwent peroxidase quenching using a 0.3% hydrogen peroxide solution in water for 30 min, followed by immersion in a 10% blocking solution in PBS (SEA BLOCK Blocking Buffer, ThermoFisher, Grand Island, NY) for 1 h, which was followed by immersion in primary antibody UBE3A (Monoclonal Anti-UBE3A antibody produced in mouse, SAB1404508) at a concentration ratio of 1:500 with incubation overnight at 4°C. On the second day, the tissues were immersed in a biotinylated secondary antibody solution at a concentration of 1:200 with incubation for 1 h (Goat Anti-Mouse IgG Antibody, BA-9200, Vector Labs, Burlingame, CA), followed by Vectastain ABC reagent incubation for 30 min (Vector Labs, Burlingame, CA) and concluded with immersion into ImmPACT DAB Peroxidase Substrate (Vector Labs, Burlingame, CA) at manufacturers recommended concentration for 8 min. In between each step, all tissues were washed using PBST (0.1% Triton) for ~15 min. Serial sections were mounted onto uncharged slides and cover slipped using Permount Mounting Medium (ThermoFisher, Grand Island, NY). Bright field immunohistochemistry stained slides were scanned using the 20× objective (0.8, M27) with bright field illumination on an Axio Scan (Zeiss).
Transplantation of NOD-RAG1−/− IL2rg−/y Mice
NOD-RAG1−/− IL2rg−/y (NRG) mice (stock number 007799) were obtained from The Jackson Laboratory (Sacramento, CA). Two to five-day old NRG mice (N = 15 per cohort) were sublethally irradiated with 100 rads from a Cesium source and transplanted intrahepatically with either the NT, EGFP control vector transduced, or the hAS8 vector transduced human CD34+ HSPC (300 000 total cells). To determine engraftment levels, at 12 weeks post-transplant, mice were bled via the tail vein and were analyzed by flow cytometry with a PE-CY7-conjugated anti-human CD45 antibody (BD Biosciences, San Jose, CA). Flow cytometry was performed on a Beckman Coulter FC-500 and analyzed by CXP software. Mice were used in accordance with institutional and IACUC guidelines.
Engraftment and multi-lineage hematopoiesis of transplanted human CD34+ cells
To evaluate the engraftment and multi-lineage differentiation of the human CD34+ cells transduced with the hAS8 lentiviral vector, cells from the peripheral blood, bone marrow, thymus and spleen were analyzed from the transplanted NRG mice. Total cells were obtained from the above-mentioned organs and were labeled with antibodies specific for human immune cells including T cells, B cells, macrophages and CD34+ cells. The following antibodies were used in these studies: T cell antibodies used were a Brilliant violet (BV) 421-conjugated CD3 antibody, an allophycocyanin (APC) H7-conjugated CD4 antibody and an Alexa fluor (AF) 700-conjugated CD8 antibody (BD Biosciences, San Jose, CA). B cell antibodies used were a PE-CY7-conjugated CD45 antibody and a PE-conjugated CD19 antibody (BD Biosciences, San Jose, CA). Macrophage antibodies used were a PE-CY7-conjugated CD45 antibody and a PE-conjugated CD14 antibody (BD Biosciences). CD34+ cell antibodies used were a PE-CY7-conjugated CD45 antibody and a PE-conjugated CD34 antibody (BD Biosciences, San Jose, CA). Flow cytometry was performed using a BD Fortessa and analyzed with CXP software.
Supplementary Material
Acknowledgements
We thank Ms Heather Boyle at the MIND Institute for maintaining the mouse colonies. We are grateful for the families of individuals affected with AS that inspired and accelerated these research discoveries, in particular Dr Allyson Berent and Ms Paula Evans. Funding was provided by the UC Davis Health MIND Institute, NIH R01NS097808 (J.L.S.; A.A.), the Foundation for Angelman Syndrome Therapeutics (FAST) (J.S.A., J.L.S. and D.J.S.) and the MIND Institute’s Intellectual and Developmental Disabilities Research Center (IDDRC) Grant P50HD103526 (PI Abbeduto).
Conflicts of Interest statement. A patent application has been filed for the Ube3a-expressing lentiviral vector. There are no additional competing interests.
Author Contributions
Conceived and designed the experiments: J.S.A., J.L.S. and D.J.S.; Performed the experiments: A.A.; N.A.C.; J.S.A.; J.B.; H.N.; H.O.; P.D.; D.L.C.; Analyzed and interpreted the data: A.A.; J.S.A.; K.D.F.; D.J.S. and J.L.S.; Provided regents, tools and funding: J.S.A., D.J.S. and J.L.S.; Drafted the article: A.A.; J.L.S. and J.S.A.; Revised the article: all authors.
References
- 1. Williams, C.A. (2005) Neurological aspects of the Angelman syndrome. Brain Dev., 27, 88–94. [DOI] [PubMed] [Google Scholar]
- 2. Buiting, K., Williams, C. and Horsthemke, B. (2016) Angelman syndrome-insights into a rare neurogenetic disorder. Nat. Rev. Neurol., 12, 584–593. [DOI] [PubMed] [Google Scholar]
- 3. Williams, C.A., Driscoll, D.J. and Dagli, A.I. (2010) Clinical and genetic aspects of Angelman syndrome. Genet. Med., 12, 385–395. [DOI] [PubMed] [Google Scholar]
- 4. Chamberlain, S.J. and Lalande, M. (2010) Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11-q13. Neurobiol. Dis., 39, 13–20. [DOI] [PubMed] [Google Scholar]
- 5. Matsuura, T., Sutcliffe, J.S., Fang, P., Galjaard, R.J., Jiang, Y.H., Benton, C.S., Rommens, J.M. and Beaudet, A.L. (1997) De novo truncating mutations in E6-AP ubiquiting-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet., 15, 74–77. [DOI] [PubMed] [Google Scholar]
- 6. Kishino, T., Lalande, M. and Wagstaff, J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet., 15, 70–73. [DOI] [PubMed] [Google Scholar]
- 7. Albrecht, U., Sutcliffe, J.S., Cattanach, B.M., Beechey, C.V., Armstrong, D., Eichele, G. and Beaudet, A.L. (1997) Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet., 17, 75–78. [DOI] [PubMed] [Google Scholar]
- 8. Meng, L., Person, R.E., Huang, W., Zhu, P.J., Costa-Mattioli, M. and Beaudet, A.L. (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet., 9, e1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Daily, J.L., Nash, K., Jinwal, U., Golde, T., Rogers, J., Peters, M.M., Burdine, R.D., Dickey, C., Banko, J.L. and Weeber, E.J. (2011) Adeno-associated virus-mediated rescue of the cognitive defects in a mouse model for Angelman syndrome. PLoS One, 6, e27221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bailus, B.J., Pyles, B., Mcalister, M.M., O'Geen, H., Lockwood, S.H., Adams, A.N., Nguyen, J.T., Yu, A., Berman, R.F. and Segal, D.J. (2016) Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an angelman syndrome mouse brain. Mol. Ther., 24, 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bailus, B.J. and Segal, D.J. (2014) The prospect of molecular therapy for Angelman syndrome and other monogenic neurologic disorders. BMC Neurosci., 15, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pyles, B., Bailus, B.J., O'Geen, H. and Segal, D.J. (2019) Purified protein delivery to activate an Epingenetically silenced allele in mouse brain. Methods Mol. Biol., 1767, 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ciarlone, S.L., Grieco, J.C., Agostino, D.P.D. and Weeber, E.J. (2016) Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis., 96, 38–46. [DOI] [PubMed] [Google Scholar]
- 14. Ciarlone, S.L., Wang, X., Rogawski, M.A. and Weeber, E.J. (2017) Effects of the synthetic neurosteroid ganaxolone on seizure activity and behavioral deficits in an Angelman syndrome mouse model. Neuropharmacology, 116, 142–150. [DOI] [PubMed] [Google Scholar]
- 15. Grieco, J.C., Ciarlone, S.L., Gieron-korthals, M., Schoenberg, M.R., Smith, A.G., Philpot, R.M., Heussler, H.S., Banko, J.L. and Weeber, E.J. (2014) An open-label pilot trial of minocycline in children as a treatment for Angelman syndrome. BMC Neurol., 14, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bird, L.M., Tan, W., Bacino, C.A., Peters, S.U., Skinner, S.A., Anselm, I., Barbieri-Welge, R., Bauer-Carlin, A., Gentile, J.K., Glaze, D.G. et al. (2012) A therapeutic trial of pro-methylation dietary supplements in Angelman syndrome. Am. J. Med. Genet. A, 155, 2956–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Woerden, G.M., Harris, K.D., Hojjati, M.R., Gustin, R.M., Qiu, S., de Avila Freire, R., Jiang, Y.H., Elgersma, Y. and Weeber, E.J. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat. Neurosci., 10, 280–282. [DOI] [PubMed] [Google Scholar]
- 18. Weeber, E.J., Jiang, Y., Elgersma, Y., Varga, A.W., Carrasquillo, Y., Brown, S.E., Christian, J.M., Mirnikjoo, B., Silva, A., Beaudet, A.L. et al. (2003) Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J. Neurosci., 23, 2634–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang, H., Allen, J.A., Mabb, A.M., Kind, I.F., Miriyala, J., Taylor-Blake, B., Sciaky, N., Dutton, J.W., Jr., Lee, H.M., Chen, X. et al. (2012) Topoisomerase inhibitors unsilence the dormant allele in Ube3a in in neurons. Nature, 481, 185–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wynn, R.F., Mercer, J., Page, J., Carr, T.F., Jones, S. and Wraith, J.E. (2009) Use of enzyme replacement therapy (Laronidase) before hematopoietic stem cell transplantation for Mucopolysaccharidosis I: experience in 18 patients. J. Pediatr., 154, 135–139. [DOI] [PubMed] [Google Scholar]
- 21. Wynn, R.F., Wraith, J.E., Mercer, J., O'Meara, A., Tylee, K., Thornley, M., Church, H.J. and Bigger, B.W. (2009) Improved metabolic correction in patients with lysosomal storage disease treated with hematopoietic stem cell transplant compared with enzyme replacement therapy. J. Pediatr., 154, 609–611. [DOI] [PubMed] [Google Scholar]
- 22. Biffi, A., Montini, E., Lorioli, L., Cesani, M., Fumagalli, F., Plati, T., Baldoli, C., Martino, S., Calabria, A., Canale, S. et al. (2013) Lentiviral hematopoietic stem cell gene therapy Benefi ts metachromatic Leukodystrophy. Science, 341, 853–855. [DOI] [PubMed] [Google Scholar]
- 23. Biffi, A. and Naldini, L. (2005) Gene therapy of storage disorders by retroviral and lentiviral vectors. Hum. Gene Ther., 16, 1133–1142. [DOI] [PubMed] [Google Scholar]
- 24. Schambach, A., Zychlinski, D., Ehrnstroem, B. and Baum, C. (2013) Biosafety features of lentiviral vectors. Hum. Gene Ther., 142, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young, L.S., Searle, P.F., Onion, D. and Mautner, V. (2006) Viral gene therapy strategies: from basic science to clinical application. J. Pathol., 208, 299–318. [DOI] [PubMed] [Google Scholar]
- 26. Zufferey, R., Nagy, D., Mandel, R.J., Naldini, L. and Trono, D. (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol., 15, 871–875. [DOI] [PubMed] [Google Scholar]
- 27. Anderson, J.S., Javien, J., Nolta, J.A. and Bauer, G. (2009) Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIM5α protein, a CCR5 shRNA, and a TAR decoy. Mol. Ther., 17, 2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barclay, S.L., Yang, Y., Zhang, S., Fong, R., Barraza, A., Nolta, J.A., Torbett, B.E., Abedi, M., Bauer, G. and Anderson, J.S. (2015) Safety and efficacy of a tCD25 Preselective combination anti-HIV lentiviral vector in human hematopoietic stem and progenitor cells. Stem Cells, 33, 870–879. [DOI] [PubMed] [Google Scholar]
- 29. Walker, J.E., Chen, R.X., McGee, J., Nacey, C., Pollard, R.B., Abedi, M., Bauer, G., Nolta, J.A. and Anderson, J.S. (2012) Generation of an HIV-1-resistant immune system with CD34(+) hematopoietic stem cells transduced with a triple-combination anti-HIV lentiviral vector. J. Virol., 86, 5719–5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Biffi, A. (2012) Genetically-modified hematopoietic stem cells and their progeny for widespread and efficient protein delivery to diseased sites: the case of lysosomal storage disorders. Curr. Gene Ther., 12, 381–388. [DOI] [PubMed] [Google Scholar]
- 31. Eichler, F., Duncan, C., Musolino, P.L., Orchard, P.J., De Oliveira, S., Thrasher, A.J., Armant, M., Dansereau, C., Lund, T.C., Miller, W.P. et al. (2017) Hematopoietic stem-cell gene therapy for cerebral Adrenoleukodystrophy. N. Engl. J. Med., 377, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beegle, J., Hendrix, K., Maciel, H., Nolta, J.A. and Anderson, J.S. (2020) Improvement of motor and behavioral activity in Sandhoff mice transplanted with human CD34+ cells transduced with a HexA/HexB expressing lentiviral vector. J. Gene Med., 22, e3205. [DOI] [PubMed] [Google Scholar]
- 33. Asheuer, M., Pflumio, F., Benhamida, S., Dubart-Kupperschmitt, A., Fouquet, F., Imai, Y., Aubourg, P. and Cartier, N. (2004) Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proc. Natl. Acad. Sci. USA., 101, 3557–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sergijenko, A., Langford-Smith, A., Liao, A.Y., Pickford, C.E., McDermott, J., Nowinski, G., Langford-Smith, K.J., Merry, C.L., Jones, S.A., Wraith, J.E. et al. (2013) Myeloid/microglial driven autologous hematopoietic stem cell gene therapy corrects a Neuronopathic lysosomal disease. Mol. Ther., 21, 1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jiang, Y., Armstrong, D., Albrecht, U., Atkins, C.M., Noebels, J.L., Eichele, G., Sweatt, J.D. and Beaudet, A.L. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron, 21, 799–811. [DOI] [PubMed] [Google Scholar]
- 36. Born, H.A., Dao, A.T., Levine, A.T., Lee, W.L., Mehta, N.M., Mehra, S., Weeber, E.J. and Anderson, A.E. (2017) Strain-dependence of the Angelman syndrome phenotypes in Ube3a maternal deficiency mice. Sci. Rep., 7, 8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berg, E.L., Pride, M.C., Petkova, S.P., Lee, R.D., Copping, N.A., Shen, Y., Adhikari, A., Fenton, T.A., Pedersen, L.R., Noakes, L.S. et al. (2020) Translational outcomes in a full gene deletion of ubiquitin protein ligase E3A rat model of Angelman syndrome. Transl. Psychiatry, 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dodge, A., Peters, M.M., Greene, H.E., Dietrick, C., Botelho, R., Chung, D., Willman, J., Nenninger, A.W., Ciarlone, S., Kamath, S.G. et al. (2020) Generation of a novel rat model of Angelman syndrome with a complete Ube3a gene deletion. Autism Res., 13, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adhikari, A., Copping, N.A., Onaga, B., Pride, M.C., Coulson, R.L., Yang, M., Yasui, D.H., LaSalle, J.M. and Silverman, J.L. (2018) Cognitive deficits in the Snord116 deletion mouse model for Prader-Willi syndrome. Neurobiol. Learn. Mem., 165, 106874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sonzogni, M., Hakonen, J., Bernabe Kleijn, M., Silva-Santos, S., Judson, M.C., Philpot, B.D., van Woerden, G.M. and Elgersma, Y. (2019) Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mo. Autism, 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grieco, J.C., Gouelle, A. and Weeber, E.J. (2018) Identification of spatiotemporal gait parameters and pressure-related characteristics in children with Angelman syndrome: a pilot study. J. Appl. Res. Intellect. Disabil., 31, 1219–1224. [DOI] [PubMed] [Google Scholar]
- 42. Huang, H.S., Burns, A.J., Nonneman, R.J., Baker, L.K., Riddick, N.V., Nikolova, V.D., Riday, T.T., Yashiro, K., Philpot, B.D. and Moy, S.S. (2013) Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav. Brain Res., 243, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sidorov, M.S., Deck, G.M., Dolatshahi, M., Thibert, R.L., Bird, L.M., Chu, C.J. and Philpot, B.D. (2017) Delta rhythmicity is a reliable EEG biomarker in Angelman syndrome: a parallel mouse and human analysis. J. Neurodev. Disord., 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Judson, M.C., Wallace, M.L., Sidorov, M.S., Burette, A.C., Gu, B., van Woerden, G.M., King, I.F., Han, J.E., Zylka, M.J., Elgersma, Y. et al. (2016) GABAergic neuron-specific loss of Ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron, 90, 56–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. den Bakker, H., Sidorov, M.S., Fan, Z., Lee, D.J., Bird, L.M., Chu, C.J. and Philpot, B.D. (2018) Abnormal coherence and sleep composition in children with Angelman syndrome: a retrospective EEG study. Mol. Autism., 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Silva-santos, S., van Woerden, G.M., Bruinsma, C.F., Mientjes, E., Jolfaei, M.A., Distel, B., Kushner, S.A. and Elgersma, Y. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest., 125, 2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Copping, N.A., Adhikari, A., Petkova, S.P. and Silverman, J.L. (2019) Genetic backgrounds have unique seizure response profiles and behavioral outcomes following convulsant administration. Epilepsy Behav., 101, 106547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sittig, L.J., Carbonetto, P., Engel, K.A., Krauss, K.S., Barrios-Camacho, C.M. and Palmer, A.A. (2016) Genetic background limits generalizability of genotype-phenotype relationships. Neuron, 91, 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Biffi, A., Capotondo, A., Fasano, S., del Carro, U., Marchesini, S., Azuma, H., Malaguti, M.C., Amadio, S., Brambilla, R., Grompe, M. et al. (2006) Gene therapy of metachromatic leukodystrophy reverses neurological damage and deficits in mice. J. Clin. Invest., 116, 3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo, L., Ambrozkiewicz, M.C., Benseler, F., Chen, C., Dumontier, E., Falkner, S., Furlanis, E., Gomez, A.M., Hoshina, N., Huang, W.H. et al. (2020) Optimizing nervous system-specific gene targeting with Cre driver lines: prevalence of germline recombination and influencing factors. Neuron, 106, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vogt, N. (2020) Beware the unexpected in Cre drivers. Nat. Methods, 17, 364. [DOI] [PubMed] [Google Scholar]
- 52. Squeri, G., Passerini, L., Ferro, F., Laudisa, C., Tomasoni, D., Deodato, F., Donati, M.A., Gasperini, S., Aiuti, A., Bernardo, M.E. et al. (2019) Targeting a pre-existing anti-transgene T cell response for effective gene therapy of MPS-I in the mouse model of the disease. Mol. Ther., 27, 1215–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Copping, N.A., Berg, E.L., Foley, G.M., Schaffler, M.D., Onaga, B.L., Buscher, N., Silverman, J.L. and Yang, M. (2017) Touchscreen learning deficits and normal social approach behavior in the Shank3B model of Phelan-McDermid syndrome and autism. Neuroscience, 345, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gompers, A.L., Su-Feher, L., Ellegood, J., Copping, N.A., Riyadh, M.A., Stradleigh, T.W., Pride, M.C., Schaffler, M.D., Wade, A.A., Catta-Preta, R. et al. (2017) Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci., 20, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vogel Ciernia, A., Pride, M.C., Durbin-Johnson, B., Noronha, A., Chang, A., Yasui, D.H., Crawley, J.N. and LaSalle, J.M. (2017) Early motor phenotype detection in a female mouse model of Rett syndrome is improved by cross-fostering. Hum. Mol. Genet., 26, 1839–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carter, R.J., Morton, J. and Dunnett, S.B. (2001) Motor coordination and balance in rodents. Curr. Protoc. Neurosci., Chapter 8, 15, 8.12.1--8.12.14. [DOI] [PubMed] [Google Scholar]
- 57. Flannery, B.M., Silverman, J.L., Bruun, D.A., Puhger, K.R., McCoy, M.R., Hammock, B.D., Crawley, J.N. and Lein, P.J. (2015) Behavioral assessment of NIH Swiss mice acutely intoxicated with tetramethylenedisulfotetramine. Neurotoxicol. Teratol., 47, 36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dhamne, S.C., Silverman, J.L., Super, C.E., Lammers, S.H.T., Hameed, M.Q., Modi, M.E., Copping, N.A., Pride, M.C., Smith, D.G., Rotenberg, A. et al. (2017) Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol. Autism., 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Haigh, J.L., Adhikari, A., Copping, N.A., Stradleigh, T., Wade, A.A., Catta-Preta, R., Su-Feher, L., Zdilar, I., Morse, S., Fenton, T.A. et al. (2021) Deletion of a non-canonical promoter regulatory element causes loss of Scn1a expression and epileptic phenotypes in mice. Genom. Med. [DOI] [PMC free article] [PubMed]
- 60. Copping, N.A. and Silverman, J.L. (2021) Abnormal electrophysiological phenotypes and sleep deficits in a mouse model of Angelman Syndrome. Mol. Autism., 12, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Judson, M.C., Sosa-Pagan, J.O., Del Cid, W.A., Han, J.E. and Philpot, B.D. (2014) Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J. Comp. Neurol., 522, 1874–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.