

Summary
Understanding T-cell responses requires identifying viral peptides presented by human leukocyte antigens (HLAs). X-ray crystallography can be used to visualize their presentation. This protocol describes the expression, purification, and crystallization of HLA-A∗02:01, one of the most frequent HLA in the global population in complex with peptides derived from the SARS-CoV-2 nucleocapsid protein. This protocol can be applied to different HLA class I molecules bound to other peptides.
For complete details on the use and execution of this protocol, please refer to Szeto et al. (2021).
Subject areas: Immunology, Protein expression and purification, X-ray Crystallography
Graphical abstract
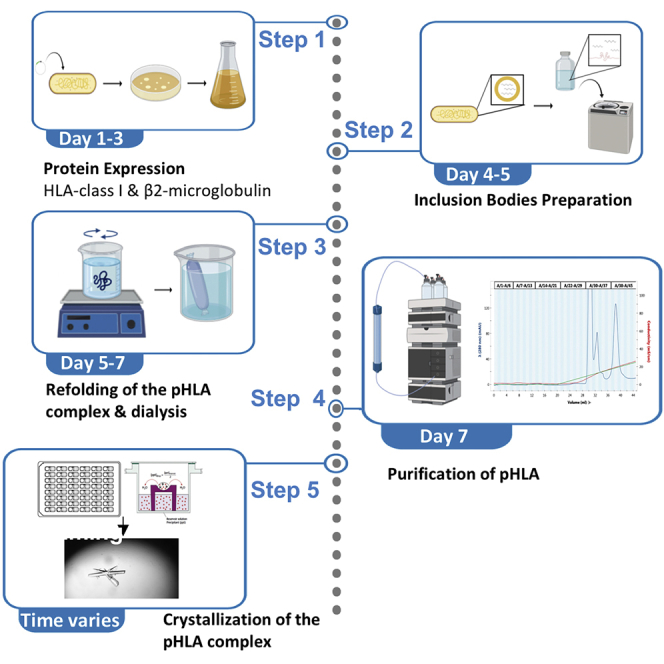
Highlights
-
•
Bacterial expression of HLA-A∗02:01 the most common HLA in the global population
-
•
Refolding of HLA-A∗02:01 with peptides derived from the SARS-CoV-2 nucleocapsid protein
-
•
Purification of the peptide-HLA complex
-
•
Crystallization of the peptide-HLA complex for subsequent X-ray data collection
Understanding T-cell responses requires identifying viral peptides presented by human leukocyte antigens (HLAs). X-ray crystallography can be used to visualize their presentation. This protocol describes the expression, purification, and crystallization of HLA-A∗02:01, one of the most frequent HLA in the global population in complex with peptides derived from the SARS-CoV-2 nucleocapsid protein. This protocol can be applied to different HLA class I molecules bound to other peptides.
Before you begin
Timing: 6 h
This protocol can be applied with a variety of different peptides. It has been optimized for HLA-A∗02:01 molecule but it has been used for different HLA-class I molecules by optimizing the refolding time (see below) depending on the HLA protein of interest. Here, we describe the complex formation with a SARS-CoV-2 Nucleocapsid protein-derived peptide N222-230 LLLDRLNQL. Other peptides can be substituted but the conditions may vary as described in Szeto C, et al., 2021 (iScience, https://doi.org/10.1016/j.isci.2021.102096) where five different SARS-CoV-2 peptides have been used.
Note: A DNA plasmid (here, pET-30a(+) vector, but any vector for protein expression in bacterial system can be used) encoding the heavy chain of the HLA-class I molecule of interest, here HLA-A∗02:01, and a plasmid encoding human β2-microglobulin need to be available and be kept at −20°C.
Note: The peptide of interest, here peptides derived from SARS-CoV-2 Nucleocapsid protein, needs to be at least 95% pure, usually as powder kept at −20°C.
Note: When working with bacterial cultures make sure everything is sterile. Clean everything (bench, pipettes, etc.) with 70% EtOH, and work in aseptic conditions (under flame) to avoid any contamination.
Note: For the protein expression, different bacterial transformation methods can be also used, such as electroporation, with the use of electrocompetent E. coli protein expression strains instead.
-
1.
Prepare all the buffers needed and place the ones indicated at 4°C in the fridge to cool down.
-
2.
Prepare LB/agar petri dishes and LB broth media and autoclave at least one day prior to the experiment.
-
3.Prepare the Diethylaminoethyl cellulose (DEAE-C) chromatography resin for the Step 5: Purification of the HLA-A∗02:01- SARS-CoV-2 peptide complex.
-
a.Make a 50% w/v of DEAE-C slurry in distilled water and add 5 mL or the mix into an econo-column and allow settling for 30–45 min. Measure the settled volume of the resin. This is the Column Volume (CV) to be used for measuring the washing solutions.
-
b.Wash DEAE resin with 5 CV of 10 mM Tris-HCl pH 8.0 and 1 M NaCl.
-
c.Equilibrate DEAE with 5 CV of 10 mM Tris-HCl pH 8.0.
-
d.Keep the column at ∼25°C
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| BL21 (DE3) E. coli chemically competent cells | Sigma-Aldrich | Cat# CMC0014 |
| Chemicals, peptides, and recombinant proteins | ||
| Additive Screen HT | Hampton Research | Cat# HR2-428 |
| Agar | Astral Scientific | Cat# BIOFB0010 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906 |
| Bromophenol blue | Sigma-Aldrich | Cat# B0126 |
| Deoxyribonuclease I from bovine pancreas (DNase I) | Sigma-Aldrich | Cat# DN25 |
| Di-sodium ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | Cat# E5134 |
| Dithiothreitol (DTT) | GoldBio | Cat# DTT10 |
| Glutathione, Reduced | GoldBio | Cat# G-155-500 |
| Glutathione, Oxidized | GoldBio | Cat# G-060-25 |
| Glycerol | Sigma-Aldrich | Cat# G5516 |
| Guanidine hydrochloride | Sigma-Aldrich | Cat# G3272 |
| Isopropyl β-d-1-thiogalactopyranoside (IPTG) | GoldBio | Cat# 12481C100 |
| Kanamycin monosulfate | GoldBio | Cat# K-120-50 |
| L-Arginine monohydrochloride | Sigma-Aldrich | Cat# A5131 |
| Lysozyme | GoldBio | Cat# L-040-25 |
| Magnesium Chloride (MgCl2) | Merck | Cat# 1058330250 |
| PEG/Ion HT Crystallization reagent kit | Hampton Research | Cat# HR2-139 |
| Phenylmethylsulfonyl fluoride (PMSF) | GoldBio | Cat# P470 |
| Polyethylene glycol 3350 (PEG3350) | Sigma-Aldrich | Cat# 88276 |
| Potassium formate | Merck | Cat# 294454 |
| SARS-CoV-2 Nucleocapsid protein-derived peptide N222-230 LLLDRLNQL | GenScript | N/A |
| Sodium chloride (NaCl) | Merck | Cat# 7647-14-5 |
| Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) prestained protein marker (10–180 kDa) | Bio-Rad | Cat# 1610394 |
| Tris | Thermo Fisher | Cat# FSBBP152-5 |
| Triton X-100 | Sigma-Aldrich | Cat# X100-1L |
| Tryptone | Thermo Fisher | Cat# LP0042R |
| Urea | Thermo Fisher | Cat# AJA817-5KG |
| Yeast Extract | Sigma-Aldrich | Cat# 09182-5KG-F |
| Recombinant DNA | ||
| pET-30a(+) vector encoding HLA-A∗02:01 heavy chain | GenScript | N/A |
| pET-30a(+) vector encoding β2-microglobulin | GenScript | N/A |
| Other | ||
| Acrodisk syringe filters, 0.22 um | Pall | Cat#4187 |
| Autoclave | N/A | N/A |
| Cell spreaders | N/A | N/A |
| Cellulose acetate filter papers | Sartorius | Cat#11104—47----N |
| Centrifuge concentration filters 10 kDa cut-off | Merck | Cat# 900000072879 |
| Crystal Clear Sealing Tape | Hampton Research | HR4-511 |
| Crystal crusher | Hampton Research | HR4-216 |
| Crystal plate fridge 20°C | N/A | N/A |
| Dialysis buckets, 15L | N/A | N/A |
| Dialysis membranes | Livingstone | Cat# DIALYSIS |
| Diethylaminoethyl (DEAE) Sepharose Fast Flow anion exchange chromatography resin | Cytiva | Cat# 17070910 |
| Econo-Column Chromatography Columns | Bio-Rad | Cat# 7374251 |
| Erlenmeyer Flasks, 2 Liters | Duran | Cat# 2121663 |
| FPLC | Bio-Rad | NGC Chromatography System Quest 10 |
| Fraction collection tubes for FPLC | N/A | N/A |
| Glass microfiber filters | MC Scientific | Cat# FF0483-047 |
| High speed centrifuge | Beckman Coulter | Avanti J-E |
| HiTrap Q HP anion exchange chromatography column | Cytiva | Cat# 29-0513-25 |
| Homogenizer | Hanchen Lab Homogenizer | AD200L-H |
| LAQUA-Twin pH 22 Meter Kit | Westlab | Cat# 3999960123 |
| Magnetic flea | N/A | N/A |
| Microscope | Leica | N/A |
| MRC Maxi 48-well sitting drop crystallization plates | Hampton Research | HR3-179 |
| NanoDrop 2000 | Thermo Scientific | N/A |
| Petri dishes, 90 mm | Techno Plas | Cat# S9014UV20 |
| Protein Electrophoresis Equipment | Bio-Rad | PowerPac Basic |
| Schott bottles, plasticware (measuring cylinders, beakers) | N/A | N/A |
| Shaker incubators | New Brunswick | Innova 43 |
| Solvac Filtering device | Pall | Cat#4020 |
| Stirrer | N/A | N/A |
| Tube rotator | Thermo Scientific | 88881004 |
| Water bath | N/A | N/A |
Materials and equipment
Alternatives: This protocol uses a BIO-RAD Fast Protein Liquid Chromatography (FPLC) system. There are other FPLC systems that can be used such as the AKTA FLPC from GE Healthcare.
Alternatives: This protocol uses sitting drop crystallization methodology but hanging drop can be used as well. Both methodologies are easy to perform, require a small amount of sample, and allow flexibility during screening and optimization. The advantages of the hanging drop technique include: the ability to view the drop through glass without the optical interference from plastic, flexibility, reduced chance of crystals sticking to the hardware, and easy access to the drop. The disadvantage is that extra time is required for set up. The advantages of the sitting drop technique include speed and simplicity. The disadvantages are that crystals can sometimes adhere to the sitting drop surface making manipulation difficult. This disadvantage can turn into an advantage where occasionally the surface of the sitting drop can assist in nucleation. Sitting drop crystallization may be performed using many different plates (Micro-Bridges or Glass Sitting Drop Rods with VDX or Linbro plates). All plates can be sealed with clear sealing tape for easy viewing and access (Hou et al., 2019).
Alternatives: This protocol uses a NanoDrop 2000 spectrophotometer for protein quantification. Regular spectrophotometers or Qubit Fluorometer can be alternatively used. The main advantage of the NanoDrop 2000 over regular spectrophotometers is that it uses microvolumes of the sample (0.5 μL), and over the Qubit is the ability to measure the purity of the sample and that it doesn't require any reagents so sample measurements do not come at a cost.
CRITICAL: When you prepare buffers, make sure you have read the Material Safety Data Sheet (MSDS), understand the safety procedure and take caution in handling any hazardous chemicals.
LB Agar
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast Extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| Agar | 1.5% (w/v) | 15 g |
| ddH2O | n/a | Up to 1L |
| Total | n/a | 1 L |
Note: LB/agar needs to be autoclaved before pouring it into petri dishes. The sterile filtered antibiotic needs to be added after the LB/agar cools down below 55°C for Kanamycin. Set a water bath to 50°C to help maintain temperature. The final concentration of Kanamycin used in the LB agar is 34 μg/mL. LB/agar/Kanamycin plates can be stored at 4°C and away from direct light for no longer than two weeks.
LB Broth
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast Extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| ddH2O | n/a | Up to 1L |
| Total | n/a | 1 L |
Note: LB broth needs to be autoclaved then cooled down below 55°C prior to the addition of 34 μg/mL Kanamycin.
LB broth/Kanamycin can be stored at ∼25°C for a few days, but it is recommended to be made fresh each time.
Tris-Buffered Saline (TBS)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH 8.0 (2 M) | 10 mM | 5 mL |
| NaCl (5 M) | 150 mM | 30 mL |
| ddH2O | n/a | Up to 1L |
| Total | n/a | 1 L |
Note: TBS can be stored at ∼25°C.
Inclusion Bodies – Enzymatic Lysis Buffer (for 6× 800 mL bacterial culture pellet)
| Reagent | Final concentration | Amount |
|---|---|---|
| Lysozyme (10 mg/mL) | 0.1 mg/mL | 2 mL |
| DNase I (5 mg/mL) | 50 μg/ mL | 2 mL |
| Triton X-100 (20%) | 1% | 10 mL |
| MgCl2 (2M) | 4 mM | 0.4 mL |
| ddH2O | n/a | 185.6 mL |
| Total | n/a | 200 mL |
Note: Make fresh each time.
Inclusion Bodies Wash Buffer 1 (600 mL, 4 washes)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH 8.0 (2 M) | 50 mM | 15 mL |
| NaCl (5 M) | 100 mM | 12 mL |
| Triton X-100 (20%) | 0.5% | 15 mL |
| ddH2O | n/a | 558 mL |
| Total | n/a | 600 mL |
Note: Chill the buffer solution at 4°C prior to use.
Inclusion Bodies Wash Buffer 2 (150 mL, 1 wash)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH 8.0 (2 M) | 50 mM | 15 mL |
| Na-EDTA (0.5 M) | 1 mM | 0.3 mL |
| ddH2O | n/a | 134.7 mL |
| Total | n/a | 150 mL |
Note: Chill the buffer solution at 4°C prior to use.
Guanidine HCI Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris-HCl pH 8.0 (2 M) | 20 mM | 0.2 mL |
| Guanidine-HCl (6 M) | 6 M | 11.46 g |
| NaEDTA (0.5 M) | 0.5 mM | 0.02 mL |
| DTT (1 M) | 1 mM | 0.02 mL |
| PMSF (0.2 M) | 0.2 mM | 0.02 mL |
| ddH2O | n/a | Up to 20 mL |
| Total | n/a | 20 mL |
Note: Make fresh each time.
Alternatives: This protocol uses a Guanidine-HCl buffer in the last step of Inclusion Bodies preparation, but 8 M Urea can be used instead. However, the protocol has been optimized for 6M Guanidine-HCl as the yield were consistently higher based on our experience.
Refolding buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Urea | 3 M | 90.07 g |
| L-Arginine HCl | 0.4 M | 42 g |
| Tris-HCl pH 8.0 (2 M) | 0.1 M | 25 mL |
| Na-EDTA (0.5 M) | 2 mM | 2 mL |
| Glutathione reduced | 0.16% (w/v) | 0.8 g |
| Glutathione oxidized | 0.03% (w/v) | 0.15 g |
| ddH2O | n/a | Up to 500 mL |
| Total | n/a | 500 mL |
Note: Make fresh each time.
Step-by-step method details
Expression of HLA-A∗02:01 and human β2-microglobulin and extraction of insoluble proteins (inclusion bodies)
This step describes the expression of HLA-A∗02:01 heavy chain and human β2-microglobulin proteins as well as their recovery from inclusion bodies from Escherichia coli expression using a mild solubilization process. This protocol can be applied to a variety of different HLA-class I molecules.
-
1.
HLA-A∗02:01 heavy chain and β2-microglobulin expression. (The protocol can be applied for both proteins)
Day 1Timing: 2 h
-
a.Chemical transformation of competent BL21 (DE3) E. coli cells with the pET-30a (+) vector encoding HLA-A∗02:01 heavy chain, or β2-microglobulin.Note: The gene sequence of β2-microglobulin (sequence below) was inserted in pET-30a (+) that was cut with NdeI/HindIII restriction enzymesIQRTPKIQVYSRHPAENGKSNFLNCYVSGFHPSDIEVDLLKNGERIEKVEHSDLSFSKDWSFYLLYYTEFTPTEKDEYACRVNHVTLSQPKIVKWDRDMThe gene sequence of HLA-A∗02:01 heavy chain (sequence below) was inserted in the pET-30a (+) vector that was cut with NdeI/HindIII restriction enzymes.GSHSMRYFFTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQRMEPRAPWIEQEGPEYWDGETRKVKAHSQTHRVDLGTLRGYYNQSEAGSHTVQRMYGCDVGSDWRFLRGYHQYAYDGKDYIALKEDLRSWTAADMAAQTTKHKWEAAHVAEQLRAYLEGTCVEWLRRYLENGKETLQRTDAPKTHMTHHAVSDHEATLRCWALSFYPAEITLTWQRDGEDQTQDTELVETRPAGDGTFQKWAAVVVPSGQEQRYTCHVQHEGLPKPLTLRWEPSSBoth sequences and successful sub-cloning into pET30a vector have been confirmed by sequencing by Genscript.
-
i.Thaw competent cells on ice.Note: competent cells are very “fragile”, do not warm up the cells by holding the bottom of the tube in your fingers.Note: work under a flame, or in a fume hood, to maintain aseptic conditions. Alternatively, if not applicable, make sure that everything is sterile by cleaning bench, pipettes, etc. with 70% EtOH to avoid any contamination.
-
ii.Add 0.5–1 μL (<100 ng) of plasmid to the competent cells.Note: Use 50–100 ng of plasmid. The volume of plasmid added to the competent cells shouldn’t exceed 10% of the final volume as the H2O may cause lysis of the cells.
-
iii.Incubate cells-DNA mixture on ice for 20 min.
-
iv.Heat shock competent cells at 42°C in a water bath (or heat block) for 45 s.
-
v.Add 1 mL of sterile LB broth to the tube and incubate at 37°C in a shaker incubator at 180 rpm for 1 h.
-
vi.Plate 50 and 100 μL of cells onto LB/agar plates with 34 μg/mL Kanamycin and place the plates in a stationary incubator for 14–17 h at 37°C.
Day 2Timing: 30 min
-
i.
-
b.Inoculate two liquid starter cultures of 150 mL LB broth each, with 34 μg/mL Kanamycin using a single colony from each LB agar plate and incubate for 14–17 h in a shaker incubator at 37°C at 180 rpm.Day 3Timing: 8 h
-
c.Large scale liquid cultures
-
i.Inoculate 6× 800 mL of LB broth containing 34 μg/mL Kanamycin with 8 mL of starter culture (100-fold dilution) and grow at 37°C until the OD 600 nm reaches 0.6 (approximately 3 h).Note: The volume of bacterial cultures determines the final protein yield and can be adjusted to your needsNote: When growing bacterial cultures, it is important not to let the OD get any higher than 0.6. The OD should be carefully monitored and checked often, especially when it gets above 0.2, as the cell growth is exponential.
-
ii.Add 800 μL of 0.5 M isopropyl-β-D-thiogalactopyranoside (IPTG) to each 800 mL culture to induce protein expression (HLA-A∗02:01, or β2-microglobulin) and incubate for 4 h.
-
iii.Harvest cells by centrifugation at 5000 × g for 15 min at 4°C. Resuspend cell pellets in MilliQ H2O (20 mL in total for the entire 6× 800 mL culture). We recommend using 50 mL falcon tubes for easy storage and manipulation.
-
iv.Store cell pellet in the −80°C (long term storage) or −20°C (short term storage) freezer.
-
i.
-
a.
Pause point: You can either proceed to the preparation of insoluble proteins (Inclusion bodies) or store the cell pellets as described above.
-
2.
Preparation and extraction of insoluble proteins (Inclusion Bodies)
Day 4Timing: 3 h
-
a.Thaw bacterial pellet in a 37°C water bath.
-
b.Use Enzymatic Lysis Buffer on a rotating wheel for 20 min at ∼25°C for enzymatic cell lysis and DNA degradation.Note: Use 200 mL of Enzymatic Lysis Buffer per 6× 800 mL bacterial culture pellet.Alternatives: Other methods can also be used for the lysis of bacterial cells (e.g., French press, sonication).
-
c.Centrifuge at 15,000 g at 4°C for 15 min.
-
d.Wash Inclusion Bodies.
-
i.Discard supernatant and resuspend pellet (from 6× 800 mL culture) in 150 mL of Wash Buffer 1.
-
ii.Add 1 mM DTT (150 μL of 1 M stock) and 0.2 mM PMSF (150 μL of 200 mM stock)
-
iii.Homogenize and centrifuge at 10,000 g at 4°C for 10 min.
-
iv.Repeat i-iii until pellet is clean of dark debris (minimum of 4 washes).
-
v.Discard supernatant and resuspend pellet in 150 mL of Wash Buffer 2.
-
vi.Add 1 mM DTT (150 μL of 1 M stock) and 0.2 mM PMSF (150 μL of 200 mM stock).
-
vii.Homogenize and centrifuge at 10,000 g at 4°C for 10 min.
-
viii.Discard supernatant and resuspend pellet in 5 mL Guanidine·HCl Buffer for 6× 800 mL culture (1 mL/L of culture), on a rotating wheel for 14–17 h at 4°C.
Note: If the mixture turns into a jelly-like consistency, add in 10 mM DTT.Note: For problems that may arise during this procedure please refer to troubleshooting problems 1 and 2 below.Day 5Timing: 3 h
-
i.
-
e.Centrifuge at 30,000 g at 4°C for 30 min and harvest the supernatant
-
f.Quantification by SDS-PAGE
-
i.Use 1 mg/mL Bovine Serum Albumin (BSA) as standard, with reduced loading dye (10% SDS, 500 mM DTT, 50% Glycerol, 250 mM Tris-HCl and 0.5% bromophenol blue dye, pH 6.8).
-
i.
-
a.
Load wells to look like this as an example:
-
1.
Protein marker
-
2.
2 μL of BSA
-
3.
4 μL of BSA
-
4.
8 μL of BSA
-
5.
1 μL of sample diluted 10 times in 8 M Urea
-
6.
1 μL of sample diluted 5 times in 8 M Urea
-
7.sample not diluted (if the sample looks yellow clear)
-
i.Look for the size and thickness of the band for the protein of interest and estimate the concentration of your sample by eye compared to the BSA control of known concentration. This is a rough estimation of the concentration, as IBs are not 100% pure it is difficult to get an accurate concentration using nanodrop.
-
i.
Note: The molecular mass of HLA α-chain is ∼32 kDa and human β2-microglobulin is ∼10 kDa.
Aliquot them out to:
-
•
10 mg for human β2-microglobulin
-
•30 mg for HLA-A∗02:01 heavy chain
-
ii.Store in −80°C or proceed to the next step (Refolding).
-
ii.
Refolding of the HLA class I molecule with human β2-microglobulin and the peptide of interest (SARS-CoV-2 N222-230 LLLDRLNQL) followed by dialysis
The refolding step produces soluble peptide-HLA complexes. During this step, the pHLA complex will be formed, which is composed of the HLA-A∗02:01 heavy chain, human β2-microglobulin, and the peptide of interest, in this case the SARS-CoV-2 N222-230 LLLDRLNQL peptide.
Day 5 continue
-
3.Refolding of the HLA-class I molecule with human β2-microglobulin and the peptide of interest
-
a.Add the following to 0.5 L of Refolding Buffer:
-
i.30 mg of HLA-A∗02:01 heavy chain
-
ii.10 mg of β2-microglobulin
-
iii.5 mg of the SARS-CoV-2 peptide dissolved in 0.4 mL 100% DMSO
-
i.
-
b.Stir at ∼25°C for 3 h
-
a.
Note: Refolding conditions (temperature and time) may vary according to the protein. For example, we have found that protein yields were higher for certain HLA molecules (HLA-A∗11:01 and HLA-A∗68:01) when allowed a refold time of 3 days at 4°C. If the yield is poor the quantity of the peptide can be increased (10 or 20 mg).
Day 5 continue – Day 7
-
4.Dialysis
-
a.Place refolding buffer (0.5 L) into dialysis membranes (10 kDa cut-off), and dialyze over 10 L of 10 mM Tris-HCl pH 8.0 for 12 h (3× times) at 4°C.
-
a.
Purification of the HLA-A∗02:01- SARS-CoV-2 N222-230 LLLDRLNQL complex
Day 7
-
5.Ion exchange chromatography - Diethylaminoethyl cellulose (DEAE-C)
-
a.Prepare, wash and equilibrate the DEAE-C resin as described in the “before you begin”.
-
b.Filter dialyzed protein sample using a 0.22 μm syringe filter and load onto the column at 4°C.
-
c.Elute bound proteins with 5 CV (column volume) of 10 mM Tris-HCl pH 8.0, 150 mM NaCl.
-
d.Visualize your protein purity on SDS-PAGE.
-
e.DEAE resin can be washed with 1 M NaCl followed by 10 mM Tris-HCl pH 8.0 and stored in 20% EtOH to be reused at 4°C.
-
a.
Day 7
-
6.Ion exchange chromatography - HiTrap Q HP, 1 mL
-
a.Concentrate the eluted protein using a centrifugal concentrator with a 10 kDa cut-off and buffer exchange over 10 mM Tris-HCl pH 8 four times.CRITICAL: This step is crucial as NaCl needs to be removed from the sample before loading it onto the HTQ column.Note: Protein of interest must be filtered with 0.22 μm filter before loading onto the column to remove particulate material. The starting volume of the sample should be less than 1 mL for the HTQ column and diluted in equilibration buffer (see below) to less than 5 mL (5 fold dilution). 5 mL loop should be used on FPLC machine for injection onto the HTQ column. Check the pressure limit for the column and set this parameter on the FPLC. If sample volume is higher than the volume of the loop multiple injections can be done.
-
b.Connect the column on the FPLC with desired buffer (Pump A: 10 mM Tris-HCl pH 8.0).
-
c.Wash the column with five column volumes of 10 mM Tris-HCl pH 8.0, 1 M NaCl (Pump B).
-
d.Equilibrate the column with five column volumes of 10 mM Tris-HCl pH 8.0 (Pump A).
-
e.Start fractionation before protein injection. Ensure the deep well or tubes are at the right position. Collect 1 mL fractions.
-
f.Inject protein sample and run at a flow rate of 1 mL/min.
-
g.After two loop volumes (10 mL) set a gradient from 0 to 20% of 10 mM Tris-HCl pH 8.0, 1 M NaCl (Pump B) over 20 min.
-
h.Usually, peptide-HLA complex elute at ∼15% of 10 mM Tris-HCl pH 8.0, 1 M NaCl although this can vary between different proteins.
-
i.Run SDS-PAGE gel of the fractions of interest.
-
j.Pool the fractions needed based on purification profile and gel analysis.
-
k.Concentrate the protein sample using a 10 kDa cut-off concentrator and measure the concentration of protein using a UV spectrophotometer at 280 nm absorbance. The protein sample needs to be concentrated to the desired concentration for crystallization. The HLA-A∗02:01- SARS-CoV-2 N222-230 LLLDRLNQL complex was concentrated at 5 mg/mL to set up crystal trays.
-
a.
Note: Beer-Lambert law is used to calculate concentration based on absorbance at 280 nm: A = εcl, where A = Absorbance; ε = Molar absorption coefficient, M-1cm-1; c = Molar concentration, M; l= optical path length, cm.
Note: The protein sample can be stored at 4°C short term or at −20°C long term. However, it is highly recommended to be used straight away to set up trays.
Note: For problems that may arise during this procedure please refer to troubleshooting problems 3 and 4 below.
Crystallization of the HLA-A∗02:01-SARS-CoV-2 N222-230 LLLDRLNQL complex
This step describes the crystallization process of the HLA-A∗02:01-N222-230 LLLDRLNQL (pHLA) complex.
The condition used is: 20% PEG3350 w/v, 0.2 M Potassium Formate, 1 mM CaCl2, via sitting-drop, vapor diffusion at 20°C with a protein: reservoir drop ratio of 1:1, at a protein concentration of 5 mg/mL in 10 mM Tris-HCl pH 8.0, 150 mM NaCl (Szeto et al., 2021).
This process can be applied for a variety of different proteins/ peptides/ protein complexes, however, many factors such as the crystallization condition, protein concentration, use of seeds and additives, can vary significantly.
Note: The purity of the protein sample should be evaluated prior to crystallization. For the initial crystallization screening, the protein sample should be at least 95% pure on SDS-PAGE. It is recommended, if possible, to also evaluate the homogeneity and monodispersity of the protein sample as both factors can significantly influence the crystallization outcome.
-
7.Set up sitting drop 48-well-plates according to or using reagent formulations from Hampton Research commercial screening kits. The kit used here is the PEG/Ion HT Crystallization reagent kit.
-
a.Prepare approximately 50 μL (per plate to be set up) of pHLA-A∗02:01 peptide complex with concentration at approximately 5 mg/mL in the buffer used for protein purification (10 mM Tris-HCl pH 8.0, 150 mM NaCl).Note: The concentration of the protein needed to form crystals can vary.
-
b.Pipette 100 μL of crystallization solution into the deep well
-
c.Pipette 1 μL of crystallization solution from each deep well onto the well, followed by 1 μL of the protein sample
-
d.Seal the plate with Crystal Clear plate Sealing Tape and store the plate at 20°CNote: Alternatively, hanging drop plates can be used with silicon glass cover slides.
-
e.Wait until crystals appear
-
a.
Note: Crystallization trial is a multi-factor process, and not all factors are under your control. Please see below troubleshooting problem 5 for some alternative tips that may facilitate the process.
-
8.
Optimization
Depending on the crystal quality such as size, shape, and diffraction quality, optimization steps may be needed. Here we optimized the initial conditions that crystals were grown in by using the Additive Screen HT and micro seeds from the drop that gave crystals.-
a.Set up sitting drop 48-well-plates using the condition that produced crystals.
-
b.Pipette 1 μL of the crystallization solution from each deep well onto the well, followed by 1 μL of the protein.Note: The concentration of the protein may need to be adjusted depending on the initial crystals. For example, if there are multiple clusters of crystals in the drop, or high precipitation, the protein concentration may need to be decreased.
-
c.Add 0.2 μL of additive included in the Additive Screen HT in your drop (protein and crystallization solution), in this case 1 mM CaCl2 was used.
-
d.Add crystal micro seeds if initial crystals have been obtained. Crush up the seeds finely and add 0.2 μL of crushed seeds into each drop.
-
e.Seal the plate with Crystal Clear plate Sealing Tape and store the plate at 20°C.
-
f.Wait until crystals appear.
-
a.
Expected outcomes
The yield of Inclusion Bodies for HLA-A∗02:01 heavy chain is typically 125 mg/ L of bacterial culture, and for human β2-microglobulin the typical yield is 48 mg/ L of bacterial culture. The purity of the Inclusion Bodies can be increased by increasing the number of washes with Inclusion Bodies Wash Buffer 1 (Figure 1).
Figure 1.

HLA-A∗02:01 SDS-PAGE gel
(A) Pure protein sample (1) after 4 washes.
(B) Impurities in protein sample (2) after 3 washes. First lane of each gel is the protein molecular weight marker in kDa (MW).
The final yield of pHLA is expected to be 0.5–5 mg, depending on the peptide and the HLA molecule. After the first purification step, using the DEAE-C resin, the purity of the pHLA complex is expected to be > 80% as shown in Figure 2. During the second purification step, the pHLA complex elutes in approximately 15 mS/cm in the HiTrap Q HP column (Figure 3). The purity of the complex is expected to be > 90% as shown in Figure 4.
Figure 2.
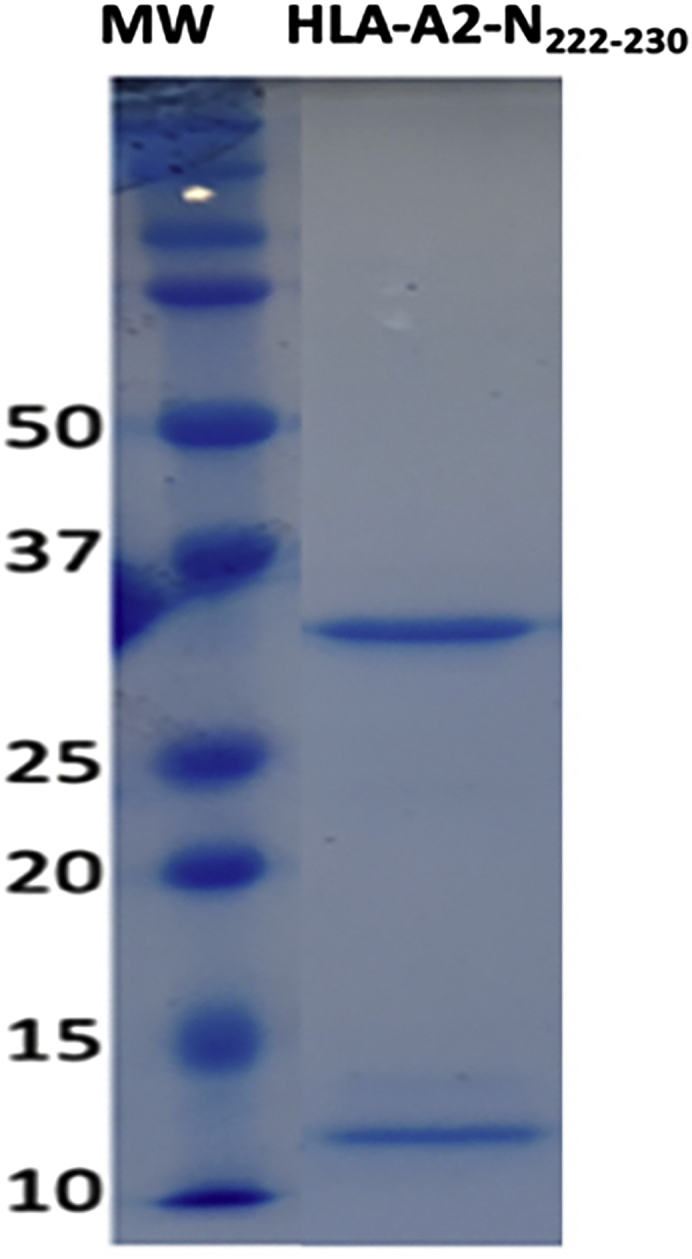
SDS-PAGE performed for DEAE elution for the HLA-A2 in complex with the SARS-CoV-2 N222-230 peptide
Elution peak was pooled together and an 8 μL aliquot was taken to run the SDS-PAGE. As the pHLA protein complex is non-covalently linked, on an SDS-PAGE gel, it is shown as two chains: a heavy α chain (∼32 kDa) and a β-2-microgolubin chain (∼10 kDa). First lane of the gel is the protein molecular weight marker in kDa (MW).
Figure 3.

Hi Trap Q HP purification chromatograph of pHLA complex
The blue trace represents the protein absorbance at 280 nm (mAU). Changes in conductivity occur with changing salt levels and the conductivity is shown with the red trace. The percentage of buffer B is given by the green trace. Excess of human β2-microglobulin elutes at around 8 mS/cm while pHLA complex elutes at around 12–18 mS/cm. Protein that hasn’t refolded is eluted at 100% buffer B (aggregation peaks). The top bar of the graph shows the numbers of the collected fractions (i.e., A/66-A/68).
Figure 4.
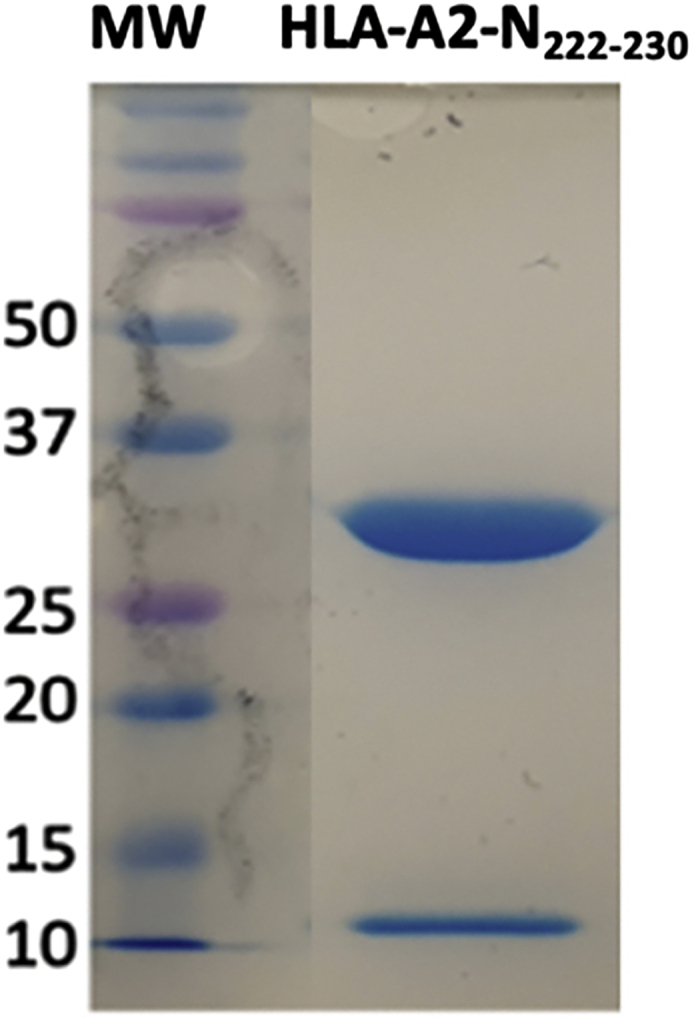
SDS-PAGE performed for Hi Trap Q elution for the HLA-A2 in complex with the SARS-CoV-2 N222-230 peptide
Elution peak was pooled together and an 8 μL aliquot was taken to run the SDS-PAGE. As pHLA protein complexes are non-covalently linked, on an SDS-PAGE gel, it is shown as two chains: a heavy α chain (~32 kDa) and a β-2-microgolubin chain (~10 kDa). First lane of the gel is the protein molecular weight marker in kDa (MW).
Crystals of the pHLA complex were grown in 20% PEG3350 w/v, 0.2 M Potassium Formate, 1 mM CaCl2 (Figure 5A) via sitting-drop, vapor diffusion at 20°C with a protein: reservoir drop ratio of 1:1, at a protein concentration of 5 mg/mL in 10 mM Tris-HCl pH 8.0, 150 mM NaCl (Szeto et al., 2021). Crystals appeared in 48 h. Optimization trials using micro-seeds from the drop shown in Figure 5A led to bigger crystals, as shown in Figure 5B, that appeared in 24 h.
Figure 5.

Crystals of the pHLA complex grown in 20% PEG3350 w/v, 0.2 M Potassium Formate, 1 mM CaCl2 via sitting-drop, vapor diffusion at 20°C with a protein: reservoir drop ratio of 1:1, at a protein concentration of 5 mg/mL
(A) Crystals appeared in 48 h.
(B) Optimization of crystals using micro-seeds from drop (A). Crystals started forming in 24 h.
Limitations
Limitations of the protocol are mentioned in the troubleshooting section below.
Troubleshooting
Problem 1
Inclusion bodies and cell debris are not pelleting at the base of the centrifuge tubes and are a “jelly-like” consistency (Step 2 - Preparation and extraction of insoluble proteins).
Potential solution
Firstly, check that the speed and time for centrifugation is adequate. We recommend using 250 mL centrifuge bottles to hold the 150 mL solutions of wash buffer. Secondly, the highly viscous consistency can be caused by excess nucleic acids so ensure that you have added the appropriate amount of DNase. Thirdly, check that the E. coli cells are resuspended in solution. If cells are not completely lysed, the wash buffer solutions can permeabilise cell membranes leading to spillage and contamination of inclusion bodies with nucleic acids and cellular debris. If enzymatic lysis is insufficient, resuspend the cell pellet in lysis buffer, homogenize the cell pellet and allow additional time (30 min–1 h) for enzymatic lysis to occur. If this is still insufficient, consider using a French Press or sonication to fully lyse cells before proceeding to inclusion body washing.
Problem 2
Inclusion bodies resuspended in 6 M Guanidium HCl turned into a jelly-like consistency after the 14–17 h incubation (Step 2 - Preparation and extraction of insoluble proteins).
Potential solution
This viscosity is caused by reformation of disulphide bonds from denatured proteins. Adding in small amounts of DTT (20 mM DTT) and allowing the mixture to continue rotating at ∼25°C will re-solubilize the protein.
Problem 3
The protein elution profile from HTQ (anion exchange chromatography) has a “shoulder” peak (Step 6 - Ion exchange chromatography - HiTrap Q HP).
Potential solution
A number of HLAs including HLA-A∗02:01 and the mouse MHC H-2Db elute with shoulder peaks (before or after the main peak). This could be due to oligomerisation at high protein concentrations that reflect multiple isoelectric points and therefore elution of proteins at different salt concentrations. It is always important to check both peaks on an SDS-PAGE to confirm purity of the refolded pHLA complex. If the gel shows contamination with other proteins, size-exclusion chromatography should be performed to further purify the protein sample. Remember that if the pHLA sample is used for crystallization trial, the purity of the protein is critical to enable crystal formation.
Problem 4
No protein has been eluted from HTQ during the NaCl elution gradient (Step 6 - Ion exchange chromatography - HiTrap Q HP).
Potential solution
Check the elution profile for the protein on the HTQ column for the flowthrough (0% Buffer B), a 280 nm peak at 8% Buffer B and then a 280 nm peak at 15%–20% Buffer B. The flowthrough (0% Buffer B) should show a large amount of aggregate or misfolded protein that does not bind to the column. If this is missing, please check the setup of the FPLC machine and check that the protein has been correctly loaded. The 8% Buffer B elution contains refolded β2-microglobulin that is not non-covalently linked to the α-chain. If the 0% and 8% peaks are present but the 15%–20% Buffer B peak is not, then the α-chain is not binding to the peptide as the HLA complex cannot be stabilized without peptide binding. If the 0% peak is present but both 8% and 15%–20% Buffer B peaks are missing, then refolding of inclusion bodies needs to be optimized, varying the time, temperature, amount of IBs, and multiple IB injections.
Problem 5
Crystals of the protein of interest are not obtained (Step 6 – Crystallization of the HLA-A∗02:01-SARS-CoV-2 N222-230 LLLDRLNQL complex).
Potential solution
Multiple factors affect the crystallization procedure including the solubility of the protein in certain buffers, the choice of the right precipitant, the pH of the solution, and the temperature (20°C, 16°C, or 4°C). Instead of setting up trays manually, fast screening of crystallization conditions using sitting drop techniques in 96-well plates with sealing film can be used instead by using commercially available sparse matrix or grid screen solution kits (Jancarik and Kim, 1991; Cudney et al., 1994; Tran et al., 2004) (plates and kits available at: Emerald Biostructures Inc.; Hampton Research; Molecular Dimensions; Qiagen-Nextal Biotechnology, Canada; Jena Bioscience, Germany; Axygen Biosciences), following the order: (i) sparse matrix, (ii) matrix screen PEG/ions, (iii) grid screen ammonium sulfate, (iv) grid screen PEGs, (v) grid screen PEG/LiCl, (vi) grid screen alcohols, (vii) grid screen salts. Shake crystallization solutions (i.e., sparse matrix or grid screen kit solutions) thoroughly before use. Dispense 100 μL of crystallization solution in the deep well and drops consisting of 0.5–1 μL of protein solution + 0.5–1 μL of crystallization solution (equal amounts).
Note: Pipette the crystallization solution into the protein solution and let the two diffuse together without mixing. Once taken from the refrigerator, the screening solutions should be kept approximately 30 min at ∼25°C to allow equilibration.
This procedure facilitates the finding of the condition where the protein forms crystals. Using this condition, optimization steps may be needed in order to obtain good quality crystals, such as the use of seeds and/ or additives, as well as a range of pH, protein and precipitant concentrations.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Professor Stephanie Gras, s.gras@latrobe.edu.au
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze datasets/code.
Acknowledgments
This work was funded by the Australian National Health and Medical Research Council (NHMRC) and an MRFF grant; D.S.M.C. and C.S. are supported with an AINSE Early Career Research Grant, and S.G. is supported by an NHMRC Senior Research Fellowship (#1159272).
Author contributions
Conceptualization, S.G. and D.S.M.C.; methodology, D.S.M.C., C.S., and D.J.; investigation, S.G. and D.S.M.C; writing – original draft, D.S.M.C.; writing – review & editing, D.S.M.C., C.S., D.J., and S.G.; funding acquisition, S.G.; resources, D.S.M.C., C.S., and S.G.; supervision, S.G.
Declaration of interests
The authors have no competing interests.
Contributor Information
Demetra S.M. Chatzileontiadou, Email: d.chatzileontiadou@latrobe.edu.au.
Stephanie Gras, Email: s.gras@latrobe.edu.au.
References
- Cudney R., Patel S., Weisgraber K., Newhouse Y., McPherson A. Screening and optimization strategies for macromolecular crystal growth. Acta Crystallogr. D. Biol. Crystallogr. 1994;50:414–423. doi: 10.1107/S0907444994002660. [DOI] [PubMed] [Google Scholar]
- Hou H. A systematic comparison of sitting and hanging-drop crystallization using traditional and cross-diffusion microbatch crystallization plates. J. Crystal. Growth. 2019;521:1–8. [Google Scholar]
- Jancarik J., Kim S.H. Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl. Crystallogr. 1991;24:409–411. [Google Scholar]
- Szeto C., Chatzileontiadou D.S.M., Nguyen A.T., Sloane H., Lobos C.A., Jayasinghe D., Halim H., Smith C., Riboldi-Tunnicliffe A., Grant E.J. The presentation of SARS-CoV-2 peptides by the common HLA-A∗02:01 molecule. iScience. 2021;24:102096. doi: 10.1016/j.isci.2021.102096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T.T., Sorel I., Lewit-Bentley A. Statistical experimental design of protein crystallization screening revisited. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1562–1568. doi: 10.1107/S0907444904015732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.