

Abstract
Planar chiral carbon nanorings and nanobelts (CNRs and CNBs), the sidewall segment molecules of chiral-type carbon nanotubes (CNTs), have attracted attention owing to their characteristic chiroptical properties. From the appropriate CNTs, axially or planar chiral CNRs and CNBs have been designed and synthesized, but multiply helical sidewall segments were almost unexplored due to the difficulty in simultaneous control of multiple chiralities. In this article, we have succeeded in the perfectly diastereo- and enantiocontrolled catalytic synthesis of a cycloparaphenylene with four helical and two planar chiralities showing good chiroptical responses as chiral organic molecules. The perfectly stereocontrolled multiply helical structure was confirmed by a single-crystal X-ray diffraction analysis. The experimental and theoretical studies established the importance of the highly symmetric multiply helical structure in the cylindrical axis in obtaining good chiroptical responses.
The perfectly diastereo- and enantiocontrolled catalytic synthesis of a cycloparaphenylene with four helical and two planar chiralities showing good chiroptical responses was achieved by the rhodium-catalyzed alkyne cyclotrimerization.
Introduction
Carbon nanorings1 and nanobelts2 (CNRs and CNBs), the sidewall segment molecules of carbon nanotubes (CNTs), have attracted attention for their unique characters, derived from their curved and strained structures.3 Recently, chiral CNRs and CNBs have also attracted attention owing to their characteristic chiroptical properties, such as enhanced responses in electronic circular dichroism (ECD) and circularly polarized luminescence (CPL).4 So far, the following chiral CNRs and CNBs embedding configurationally stable non-centro chirality have been designed by cutting out the appropriate sidewall segments from the CNTs, and their bottom-up syntheses were accomplished. For example, Isobe designed planar chiral CNRs, as represented by CNR a5 (Fig. 1A), by cutting out the configurationally stable arene units,4b,5,6 and Miao/Chi designed CNBs possessing doubly stranded planar chiral arene units.2c Itami,7 Swager,8 Isobe,9 Müllen,10 and Mazaki11 designed axially chiral CNRs, as represented by CNR b,7a by cutting out the configurationally stable biaryl units (Fig. 1B). Our research group designed planar chiral cyclophenylenes (CPs), as represented by CP c,12 by the introduction of asymmetric substituents into the achiral CPs (Fig. 1C). Although some complicated chiral CNRs9d and CNBs2c are accompanied by [4]helicene units on their peripheries as subsidiary components, CNT segments with configurationally stable [n]helicene units (n ≥ 5) have not been reported. Recently, Segawa and Itami disclosed the design and the strain energy of helicene-containing CNBs, but the synthesis of these theoretically predicted molecules has not been achieved.13 Furthermore, no studies have been reported on the chiroptical properties caused by the helical chirality of the chiral CNRs and CNBs. Concerning the synthesis, chiral CNRs were often obtained as mixtures of stereoisomers because non-enantioselective aryl–aryl couplings were used.1 Although the use of chiral cholesterol stearate could afford CNR a with 17% ee,5 and the kinetically controlled Scholl reaction and thermal epimerization could afford the planar chiral CNB2c and CNR b,7a respectively, as a single diastereomer, their highly enantioselective syntheses have not been achieved. On the other hand, our research group synthesized CP c12 with 98% ee by the rhodium-catalyzed intramolecular alkyne cyclotrimerization,14 which demonstrated the high utility of alkyne cyclotrimerization for the enantioselective synthesis of chiral CNRs and CNBs.15,16
Fig. 1. Chiral sidewall segment molecules of CNTs. (A) Planar chiral CNR a.5 (B) Axially chiral CNR b.7 (C) Planar chiral CP c.12 (D) Multiply-helical CPP 1a (this work).

In this article, we report the perfectly diastereo- and enantiocontrolled catalytic synthesis (>99% ee) and good chiroptical responses of multiply helical cycloparaphenylene (CPP) (Sp,Sp)-(M,M,M,M)-1a with four helical and two planar chiralities, which is designed by cutting out configurationally stable helical units from the armchair-type CNT (Fig. 1D). Experimental and theoretical studies revealed the importance of the highly symmetric multiply helical structure in the cylindrical axis to realize the observed good chiroptical responses as chiral organic molecules.
Results and discussion
Diastereo- and enantioselective synthesis
The synthetic route to [12]CPP 1b as well as [8]CPP 1a is shown in Fig. 2. The palladium-catalyzed Sonogashira-coupling of known diiodide 2 and terminal alkyne 3 afforded tetrayne 4, and the subsequent acid hydrolysis afforded bisphenol 5 in 82% yield from 2. Bispropargylation of 5 with dibromide 6 generated dodecayne 7a and octadecayne 7b in 12% and 6% yields, respectively. Pleasingly, the intramolecular four sequential cyclotrimerizations of 7a proceeded at 40 °C by using a cationic rhodium(I)/(S)-H8-BINAP catalyst to give [8]CPP (Sp,Sp)-(M,M,M,M)-(−)-1a with four helical and two planar chiralities as a single diastereomer with 96% ee and 36% yield, although the room temperature reaction was sluggish. Screening of axially chiral biaryl bisphosphine ligands revealed that decreasing the dihedral angle linearly increases the ee value [dihedral angle: H8-BINAP (75°) > BINAP (74°) > Segphos (66°),17 ee value: H8-BINAP < BINAP < Segphos] and the use of (S)-Segphos generates 1a with >99% ee and 40% yield, which corresponds to 80% yield in each cyclotrimerization. As with 1a, the intramolecular six sequential cyclotrimerizations of 7b also proceeded at 40 °C and the use of (S)-Segphos generated (Sp,Sp,Rp)-(M,M,M,M,P,P)-(−)-1b with the highest ee value of 62%. Although a trace amount (ca. 5%) of another diastereomer [presumably (Sp,Sp,Sp)-(M,M,M,M,M,M)-1b] was generated, this diastereomer could not be isolated in a pure form. This ee value was lower than that of 1a, but the yield was improved to 59%, which corresponds to 92% yield in each cyclotrimerization. In these reactions, complex mixtures of by-products were generated presumably as a result of the competitive intermolecular side reactions.
Fig. 2. Rhodium-catalyzed diastereo- and enantioselective synthesis of CPPs with helical and planar chiralities. THP = 2-tetrahydropyranyl. p-TsOH = p-toluenesulfonic acid. DMF = N,N-dimethylformamide. cod = 1,5-cyclooctadiene. a Calculated dihedral angles of Ru/ligand complexes shown in ref. 17.

Structural analyses
(Sp,Sp)-(M,M,M,M)-1a gave a relatively simple 1H NMR spectrum due to its highly symmetrical structure (D2 symmetry in solution) (Fig. 3A). Three singlet peaks are observed in the aromatic region, one of which (pink) corresponds to the S-shaped helicene-core and the other two (red and blue) to the p-phenylene unit. If the p-phenylene rings rotate smoothly on the NMR time scale, red and blue hydrogens should be equivalent and afford one singlet peak. The obtained 1H NMR spectrum indicates that the rotation of p-phenylene units is suppressed, which results in the inner proton (blue) and the outer proton (red) placed in different magnetic environments affording two singlet peaks.
Fig. 3. 1H NMR spectra of (Sp,Sp)-(M,M,M,M)-1a (A) and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b (B).

On the other hand, compound 1b gave a more complicated 1H NMR spectrum shown in Fig. 3B. There are two possible diastereomers, (Sp,Sp,Sp)-(M,M,M,M,M,M)-1b and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b, of which (Sp,Sp,Sp)-(M,M,M,M,M,M)-isomer (D3 symmetry) should have the same number of 1H NMR peaks as those of (Sp,Sp)-(M,M,M,M)-1a. However, the observed 1H NMR has more peaks, suggesting that 1b has non-uniform multiple chiralities. Six singlet peaks in the aromatic region (6.6–8.0 ppm) and 12 doublet peaks in the methylene region (4.5–5.2 ppm) were observed, which match the number of 1H NMR peaks of the C2-symmetrical (Sp,Sp,Rp)-(M,M,M,M,P,P)-isomer. In the 13C NMR spectrum, the six alkyne peaks (75–90 ppm) of precursor 1b disappeared, and the same number of peaks appeared as expected in the (Sp,Sp,Rp)-(M,M,M,M,P,P)-isomer. (51 peaks in the aromatic region, 6 peaks in the methylene region, and 12 peaks in the n-Bu region). 2D NMR (NOESY, COSY, HMBC, and HSQC) were also recorded to increase the reliability of the structure, and all the above NMR analysis data confirm the formation of (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b.
A single crystal of (±)-1a, in which one molecule of CH2Cl2 is subsumed inside presumably due to the strong CH⋯π interaction (2.79 Å), was obtained by slow diffusion of n-hexane into a CH2Cl2 solution of (±)-1a, which was synthesized by the cyclotrimerization of 7a using (±)-BINAP. Its crystal structure was determined by a single-crystal X-ray diffraction analysis (Fig. 4). All four helical units have the same chirality, and this CPP has an ellipse shape (major diameter = 11.7 Å, minor diameter = 9.1 Å) with two greatly curved S-shaped helical units, which is in sharp contrast to the circle shape of unsubstituted [8]CPP.3a The conformation of the two p-phenylene units e and f, sandwiched between the S-shaped helical units, is not parallel but a V-shaped conformation (Fig. 4A). For the packing structure, each of the P-isomer and the M-isomer forms an independent columnar structure, and they are arranged alternately with C–H⋯O interactions between adjacent columns (Fig. 4B). Although we failed to obtain chiral single crystals of 1a and 1b, the absolute configurations of (Sp,Sp)-(M,M,M,M)-(−)-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-(−)-1b could be confirmed by comparison of the experimental ECD spectra of (−)-1a and (−)-1b, and theoretical ECD spectra of (Sp,Sp)-(M,M,M,M)-Me4-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-Me6-1b (Fig. S10–S13†).
Fig. 4. Single-crystal X-ray diffraction analysis of (±)-1a. (A) ORTEP drawings (top: top view, bottom: side view) showing thermal ellipsoids at 50% probability. (B) Packing structures (left) and bimolecular interactions (right).

The stability of chiralities in 1a and 1b was examined by heating the solution of (Sp,Sp)-(M,M,M,M)-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b. No epimerization and racemization was observed for (Sp,Sp)-(M,M,M,M)-1a even at 110 °C in toluene for 16 hours due to its stable planar and helical chiralities. In contrast, the racemization of (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b gradually proceeded even at 60 °C in (CH2Cl)2 (Fig. 5). The formation of a trace amount of an epimerization product [presumably (Sp,Sp,Sp)-(M,M,M,M,M,M)-1b, diastereomeric ratio = ca. 30 : 1] was detected at 80 °C in (CH2Cl)2 for 19 hours. The difference in the stability of chirality between 1a and 1b depends on the ring strain. The strain energy of 1b (ΔH = −43.9 kcal mol−1) is smaller than that of 1a (ΔH = −62.9 kcal mol−1) (Fig. S7 and S8†), which results in the lower chiral stability of 1b than 1a. Concerning the thermodynamic stabilities of two possible diastereomers (Sp,Sp,Sp)-(M,M,M,M,M,M)-1b and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b, DFT calculation revealed that (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b was slightly more stable than (Sp,Sp,Sp)-(M,M,M,M,M,M)-1b (ΔG = 0.55 kcal mol−1). (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b was generated at a higher ratio (>12 : 1) than its thermodynamic stability (calculated ratio = 2.4 : 1) because no epimerization proceeds at the reaction temperature (40 °C), and the cyclotrimerization reaction proceeds with kinetic control. The fact that the diastereomer of 1b, in which three planar chiralities are not identical, was kinetically predominant is consistent with the previously reported triply twisted Möbius molecules.18 On the other hand, (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b was obtained from 7b with significantly lower ee value (6 2% ee) than (Sp,Sp)-(M,M,M,M)-1a (>99% ee) derived from 7a. Since 1b was configurationally stable at the reaction temperature of 40 °C and perfect enantioselectivity (>99% ee) was observed in our previously reported enantioselective synthesis of a double [6]helicene-like molecule by the rhodium-catalyzed intramolecular cyclotrimerization of a triyne,19 the racemization process might be included during the sequential cyclotrimerization of 7b.
Fig. 5. Stability of the chiralities of (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b in (CH2Cl)2.

Photophysical and chiroptical properties
Absorption and fluorescence spectra of (Sp,Sp)-(M,M,M,M)-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b are shown in Fig. 6A, and the data are summarized in Table 1. 1a and 1b showed absorption up to the nearly visible region with absorption maxima at 420 nm and 386 nm, respectively. These wavelengths are much longer than the maximum absorption wavelengths of unsubstituted CPPs, which have absorption maxima at around 340 nm regardless of the ring-size. 1a and 1b showed yellow green and blue green fluorescence and their fluorescence maxima are 514 nm and 486 nm, respectively, and thus fluorescence peak-shifts in comparison with unsubstituted CPPs are moderate ([8]CPP: 533 nm [12]CPP: 450 nm).3a Fluorescence quantum yields of 1a and 1b are 40% and 48%, respectively, and the yield of 1a is notably higher than that of [8]CPP (ΦF = 10%).3a These optical properties may be derived from the S-shaped helical structures because those are close to the values of not the CPPs but our previously reported S-shaped double [6]helicene-like molecule (λabs = 361 nm, λlum = 466 nm, ΦF = 48%).20
Fig. 6. Photophysical and chiroptical properties. (A) Absorption (solid line) and fluorescence (broken line) spectra of [(Sp,Sp)-(M,M,M,M)-1a (red) and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b (blue) in CH2Cl2 at 25 °C. ECD (B) and CPL (C) spectra of 1a (solid line) [(Sp,Sp)-(M,M,M,M)-1a: red, (Rp,Rp)-(P,P,P,P)-1a: blue] and 1b (broken line) [(Sp,Sp,Rp)-(M,M,M,M,P,P)-1b: red, (Rp,Rp,Sp)-(P,P,P,P,M,M)-1b: blue] in CH2Cl2 at 25 °C. (D) Calculating the energy diagrams of (M,M)-8, (Sp,Sp)-(M,M,M,M)-Me4-1a, (Sp,Sp,Rp)-(M,M,M,M,P,P)-Me6-1b, and [8]CPP. (E) Calculating the transition dipole moments of (M,M)-8, (Sp,Sp)-(M,M,M,M)-Me4-1a, (Sp,Sp,Rp)-(M,M,M,M,P,P)-Me6-1b, and (R,R,R,R)-9.

Photophysical data of 1a and 1b.
| Compound | λ abs,max b (nm) | ε max c (M−1 cm−1) | λ em,max d (nm) | Φ F e |
|---|---|---|---|---|
| (Sp,Sp)-(M,M,M,M)-1aa | 420 | 31 300 | 514 | 0.40 |
| (Sp,Sp,Rp)-(M,M,M,M,P,P)-1ba | 386 | 62 500 | 486 | 0.48 |
In CH2Cl2 (1.0 × 10−5 M) at 25 °C.
λ abs, max: longest wavelength absorption maximum.
ε max: molar extinction coefficient.
λ em, max: fluorescence maximum.
Φ F: fluorescence quantum yield. The excitation wavelengths of 1a and 1b are 420 nm and 390 nm, respectively.
To understand the above optical properties, DFT and TDDFT calculations of (Sp,Sp)-(M,M,M,M)-Me4-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-Me6-1b were performed using Gaussian 16 at the B3LYP/6-31G(d) level of theory (Fig. 6D). HOMO/HOMO–1 and LUMO/LUMO+1 of Me4-1a pseudodegenerate, and HOMO–2/HOMO–1/HOMO and LUMO/LUMO+1/LUMO+2 of Me6-1b also pseudodegenerate. These pseudodegenerate molecular orbitals show good agreement with energy levels and orbital shapes of the partial structure, S-shaped double helicene-like molecule (M,M)-8. The p-phenylene spacer hardly contributes to the electronic structure of HOMO and LUMO (Fig. 6D, S3, and S4†), which diminishes the CPP character. The HOMO → LUMO transitions in Me4-1a and Me6-1b are allowable, which are different from the symmetry-forbidden HOMO → LUMO transitions of the CPPs. These transitions have large oscillator strengths like 8 and correspond to the longest wavelength absorption bands. However, as with the CPPs, the HOMO–LUMO energy gap decreases in this order, 8 (3.67 eV) > Me6-1b (3.50 eV) > Me4-1a (3.33 eV), as the bend increases as shown in Fig. S7 and S8,† which is consistent with the experimental fact that 1a has an absorption maximum in the longer wavelength region than 1b.
Next, chiroptical properties of (Sp,Sp)-(M,M,M,M)-1a and (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b were also measured (Fig. 6B and C), and the data are summarized in Table 2. The molar optical rotation, [M]25D, is extremely large for both 1a and 1b, especially 1a showed a value exceeding 45 000 degree, which is as large as that of [11]helicene.21,22 The existence of multiple identical helicities in the molecule may induce this large optical rotation value. Their ECD and CPL spectra exhibited mirror images corresponding to the enantiomers. The dissymmetry factor of ECD (gabs) and CPL (glum) of 1a are 0.0095 and 0.0049, respectively, which are twice as large as those of our previously reported planar chiral CP c,12 although these values are smaller than those of cyclochrysenylene4a and cycloanthracene.4b However, the gabs and glum values of 1b, possessing mixed P/M chirality, are 0.0027 and 0.0017, respectively, which are smaller than those of 1a.
Chiroptical data of 1a and 1b.
| Compound | [α]25Dd | [M]25De | g abs f/10−3 (wavelength) | g lum g/10−3 (wavelength) |
|---|---|---|---|---|
| (Sp,Sp)-(M,M,M,M)-1a (>99% ee)a | −3357° | −45 700° | −9.48 (461) | −4.92 (512) |
| (Rp,Rp)-(P,P,P,P)-1a (>99% ee)b | — | — | +9.58 (462) | +4.95 (512) |
| (Sp,Sp,Rp)-(M,M,M,M,P,P)-1b (62% ee)a | −466°c | −9520°c | −2.74 (422)c | −1.67 (490)c |
| (Rp,Rp,Sp)-(P,P,P,P,M,M)-1b (62% ee)b | — | — | +2.90 (424)c | +1.97 (488)c |
In CH2Cl2 (1.0 × 10−5 M) at 25 °C.
In CH2Cl2 (1.1 × 10−5 M) at 25 °C.
Values are calculated as 100% ee.
[α]25D: optical rotation.
[M]25D: molar optical rotation (= M[α]25D/100).
g abs: CD dissymmetry factor.
g lum: CPL dissymmetry factor.
The structure–chiroptical property relationship was also examined by TDDFT calculations (Fig. 6E). The gabs value is expressed by the following equation, in which μ, m, and θ are the electric transition dipole moment, magnetic transition dipole moment, and the angle between μ and m, respectively.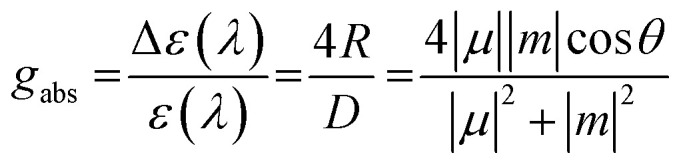
From the above equation, it is necessary to reach θ = 0° or 180° (cos θ = ± 1) and |m|/|μ| = 1 to realize a high gabs value. Thus, it is important to align μ and m in parallel and enhance |m|, while conventional chiral organic molecules have negligible |m|. To date, ideal topologies have been found in planar chiral cylindrical carbon nanohoops4,5 and highly symmetric multihelicenes.23,24 The TDDFT calculation of partial structure (M,M)-8 revealed that undesirable large μ is generated in the direction perpendicular to the C2 axis in the S0 → S1 transition, and θ is also undesirable (69°). On the other hand, in the S0 → S1 transition of (Sp,Sp)-(M,M,M,M)-Me4-1a, ideal small |μ| and large |m| as well as ideal θ (180°) are generated. Thus, the symmetrical arrangement of S-shaped helical units improves the gabs value.
According to the decomposition analysis by Isobe,6dμ of (Sp,Sp)-(M,M,M,M)-Me4-1a can be divided into two μlocal of two S-shaped helical units, and μlocal can be expressed schematically as the μ of (M,M)-8. The symmetrical arrangement of two μlocal in the z-axis results in the ideal θ value (180°). However, in (Sp,Sp,Rp)-(M,M,M,M,P,P)-Me6-1b, θ becomes 117° because three μlocal lose symmetry in the z-axis. From the above discussion, the hypothetical molecule (R,R,R,R)-9, corresponding to the twisted [8]CPP unit of (Sp,Sp)-(M,M,M,M)-Me4-1a, would have a small μ value while maintaining the ideal θ value. Indeed, the TDDFT calculation of 9 revealed that the parallel arrangement of μ and m is maintained, and |m|/|μ| is significantly larger than that of (Sp,Sp)-(M,M,M,M)-Me4-1a. Thus, it is expected that a molecule with excellent chiroptical responses can be created if a method for inducing and stabilizing the multiply helical CPP structure with appropriate dihedral angles like (R,R,R,R)-9 can be developed.
Conclusions
In conclusion, we have achieved the perfectly diastereo- and enantiocontrolled catalytic synthesis of cycloparaphenylene (CPP) 1a containing two double [5]helicene-like molecules, the structure of which was confirmed by a single-crystal X-ray diffraction analysis. A CPP containing three double [5]helicene-like molecules has also been synthesized, although the ee value was moderate. The stability of chiralities in 1a and 1b were examined by heating the solution of 1a and 1b, which revealed that no epimerization and racemization were observed for 1a even at 110 °C, whereas the racemization of 1b gradually proceeded even at 60 °C. CPP 1a showed good chiroptical responses, such as the large molar optical rotation ([M]25D = 45 700°), and the dissymmetry factors of ECD (gabs = 0.0095) and CPL (glum = 0.0049). The experimental and theoretical studies established the importance of the highly symmetric multiply helical structure in the cylindrical axis in obtaining good chiroptical responses. Finally, we proposed that the hypothetical multiply helical molecule (R,R,R,R)-9 would show excellent chiroptical responses hence the method for inducing and stabilizing this multiply helical CPP structure with appropriate dihedral angles can be developed.
Author contributions
J. N. designed the project and carried out the experimental work. J. N. also carried out computational studies and wrote a draft under the guidance of Y. N. K. M. performed the X-ray crystal structure analysis and A.M. performed CPL measurements. Y. N., A. M., and M. U. analyzed and discussed the experimental work and computational studies. K. T. designed and directed the project, and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This research was supported partly by Grants-in-Aid for Scientific Research (No. JP19H00893 to K. T. and No. 17H06173 to M. U.) from JSPS (Japan) and CREST (No. JPMJCR19R2 to M. U.) from JST (Japan). We thank Takasago International Corporation for the gift of Segphos and H8-BINAP, and Umicore for their generous support in supplying the palladium and rhodium complexes. A generous allotment of computational resources from TSUBAME (Tokyo Institute of Technology) is gratefully acknowledged.
Electronic supplementary information (ESI) available. CCDC 2050855. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00861g
Notes and references
- For reviews, see: ; (a) Majewski M. A. Stępień M. Angew. Chem., Int. Ed. 2019;58:86. doi: 10.1002/anie.201807004. [DOI] [PubMed] [Google Scholar]; (b) Segawa Y. Yagi A. Matsui K. Itami K. Angew. Chem., Int. Ed. 2016;55:5136. doi: 10.1002/anie.201508384. [DOI] [PubMed] [Google Scholar]; (c1) Eisenberg D. Shenhar R. Rabinovitz M. Chem. Soc. Rev. 2010;39:2879. doi: 10.1039/b904087k. [DOI] [PubMed] [Google Scholar]; . For pioneering works, see: ; (d) Jasti R. Bhattacharjee J. Neaton J. B. Bertozzi C. R. J. Am. Chem. Soc. 2008;130:17646. doi: 10.1021/ja807126u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Takaba H. Omachi H. Yamamoto Y. Bouffard J. Itami K. Angew. Chem., Int. Ed. 2009;48:6112. doi: 10.1002/anie.200902617. [DOI] [PubMed] [Google Scholar]; (f) Yamago S. Watanabe Y. Iwamoto T. Angew. Chem., Int. Ed. 2010;49:757. doi: 10.1002/anie.200905659. [DOI] [PubMed] [Google Scholar]
- (a) Povie G. Segawa Y. Nishihara T. Miyauchi Y. Itami K. Science. 2017;356:172. doi: 10.1126/science.aam8158. [DOI] [PubMed] [Google Scholar]; (b) Povie G. Segawa Y. Nishihara T. Miyauchi Y. Itami K. J. Am. Chem. Soc. 2018;140:10054. doi: 10.1021/jacs.8b06842. [DOI] [PubMed] [Google Scholar]; (c) Cheung K. Y. Gui S. Deng C. Liang H. Xia Z. Liu Z. Chi L. Miao Q. Chem. 2019;5:838. [Google Scholar]; (d) Cheung K. Y. Watanabe K. Segawa Y. Itami K. Nat. Chem. 2021;13:255. doi: 10.1038/s41557-020-00627-5. [DOI] [PubMed] [Google Scholar]; (e) Chi C. Han Y. Dong S. Shao J. Fan W. Angew. Chem., Int. Ed. 2020;60:2658. doi: 10.1002/anie.202012651. [DOI] [PubMed] [Google Scholar]; (f) Xia Z. Pun S. H. Chen H. Miao Q. Angew. Chem., Int. Ed. 2021;60:10311. doi: 10.1002/anie.202100343. [DOI] [PubMed] [Google Scholar]
- For reviews, see: ; (a) Cheung K. Y. Segawa Y. Itami K. Chem.–Eur. J. 2020;26:14791. doi: 10.1002/chem.202002316. [DOI] [PubMed] [Google Scholar]; (b) Chen H. Gui S. Zhang Y. Liu Z. Miao Q. CCS Chem. 2020;2:613. [Google Scholar]; (c) Chen H. Miao Q. J. Phys. Org. Chem. 2020;33:1. [Google Scholar]; (d) Xu Y. von Delius M. Angew. Chem., Int. Ed. 2020;59:559. doi: 10.1002/anie.201906069. [DOI] [PubMed] [Google Scholar]; (e) Darzi E. R. Jasti R. Chem. Soc. Rev. 2015;44:6401. doi: 10.1039/c5cs00143a. [DOI] [PubMed] [Google Scholar]
- (a) Sato S. Yoshii A. Takahashi S. Furumi S. Takeuchi M. Isobe H. Proc. Natl. Acad. Sci. U. S. A. 2017;114:13097. doi: 10.1073/pnas.1717524114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang J. Zhuang G. Chen M. Lu D. Li Z. Huang Q. Jia H. Cui S. Shao X. Yang S. Du P. Angew. Chem., Int. Ed. 2020;59:1619. doi: 10.1002/anie.201909401. [DOI] [PubMed] [Google Scholar]
- Hitosugi S. Nakanishi W. Yamasaki T. Isobe H. Nat. Commun. 2011;2:492. [Google Scholar]
- (a) Hitosugi S. Yamasaki T. Isobe H. J. Am. Chem. Soc. 2012;134:12442. doi: 10.1021/ja305723j. [DOI] [PubMed] [Google Scholar]; (b) Matsuno T. Kamata S. Hitosugi S. Isobe H. Chem. Sci. 2013;4:3179. [Google Scholar]; (c) Sun Z. Sarkar P. Suenaga T. Sato S. Isobe H. Angew. Chem., Int. Ed. 2015;54:12800. doi: 10.1002/anie.201506424. [DOI] [PubMed] [Google Scholar]; (d) Kogashi K. Matsuno T. Sato S. Isobe H. Angew. Chem., Int. Ed. 2019;58:7385. doi: 10.1002/anie.201902893. [DOI] [PubMed] [Google Scholar]
- (a) Yagi A. Segawa Y. Itami K. J. Am. Chem. Soc. 2012;134:2962. doi: 10.1021/ja300001g. [DOI] [PubMed] [Google Scholar]; (b) Okada K. Yagi A. Segawa Y. Itami K. Chem. Sci. 2016;8:661. doi: 10.1039/c6sc04048a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batson J. M. Swager T. M. Synlett. 2013;24:2545. [Google Scholar]
- (a) Ikemoto K. Sato S. Isobe H. Chem. Lett. 2016;45:217. [Google Scholar]; (b) Sarkar P. Sun Z. Tokuhira T. Kotani M. Sato S. Isobe H. ACS Cent. Sci. 2016;2:740. doi: 10.1021/acscentsci.6b00240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang Y. Nanjo Y. Isobe H. Sato S. Org. Biomol. Chem. 2020;18:4949. doi: 10.1039/d0ob01064b. [DOI] [PubMed] [Google Scholar]; (d) Matsuno T. Yang Y. Nanjo Y. Isobe H. Sato S. Matsuno T. Yang Y. Nanjo Y. Isobe H. Sato S. Chem. Lett. 2021;50:110. [Google Scholar]
- Lorbach D. Keerthi A. Figueira-Duarte T. M. Baumgarten M. Wagner M. Müllen K. Angew. Chem., Int. Ed. 2016;55:418. doi: 10.1002/anie.201508180. [DOI] [PubMed] [Google Scholar]
- Sato K. Hasegawa M. Nojima Y. Hara N. Nishiuchi T. Imai Y. Mazaki Y. Chem.–Eur. J. 2020;26:1. [Google Scholar]
- Nogami J. Tanaka Y. Sugiyama H. Uekusa H. Muranaka A. Uchiyama M. Tanaka K. J. Am. Chem. Soc. 2020;142:9834. doi: 10.1021/jacs.0c03684. [DOI] [PubMed] [Google Scholar]
- Watanabe K. Segawa Y. Itami K. Chem. Commun. 2020;56:15044. doi: 10.1039/d0cc06373h. [DOI] [PubMed] [Google Scholar]
- For selected recent reviews, see: ; (a) Shibata Y. and Tanaka K., Rhodium(I)-Catalyzed [2+2+2] and [4+2] Cycloadditions, in Rhodium Catalysis in Organic Synthesis: Methods and Reactions, ed. K. Tanaka, Wiley-VCH, Weinheim, 2019, p. 183 [Google Scholar]; (b) Tanaka K. TCI Mail. 2018;179:3. [Google Scholar]; (c) Shibata Y. Tanaka K. Synthesis. 2012;44:32. [Google Scholar]; (d) Tanaka K. Heterocycles. 2012;85:1017. [Google Scholar]; (e) Tanaka K. Chem.–Asian J. 2009;4:508. doi: 10.1002/asia.200800378. [DOI] [PubMed] [Google Scholar]; (f) Tanaka K. Synlett. 2007;18:1977. [Google Scholar]
- For the enantioselective synthesis of a Möbius-shaped cycloparaphenylene, see: ; Nishigaki S. Shibata Y. Nakajima A. Okajima H. Masumoto Y. Osawa T. Muranaka A. Sugiyama H. Horikawa A. Uekusa H. Koshino H. Uchiyama M. Sakamoto A. Tanaka K. J. Am. Chem. Soc. 2019;141:14955. doi: 10.1021/jacs.9b06197. [DOI] [PubMed] [Google Scholar]
- Very recently, the stereoselective synthesis of two enantiomers of chiral conjugated nanohoops was reported. See:; Hermann M. Wassy D. Kohn J. Seitz P. Betschart M. U. Grimme S. Esser B. Angew. Chem., Int. Ed. 2021;60:10680. doi: 10.1002/anie.202016968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu H. Nagasaki I. Saito T. Tetrahedron. 2005;61:5405. [Google Scholar]
- (a) Naulet G. Sturm L. Robert A. Dechambenoit P. Röhricht F. Herges R. Bock H. Durola F. Chem. Sci. 2018;9:8930. doi: 10.1039/c8sc02877j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang X. Laffoon S. D. Chen D. Pérez-Estrada S. Danis A. S. Rodríguez-López J. Garcia-Garibay M. A. Zhu J. Moore J. S. J. Am. Chem. Soc. 2020;142:6493. doi: 10.1021/jacs.0c01430. [DOI] [PubMed] [Google Scholar]
- Kinoshita S. Yamano R. Shibata Y. Tanaka Y. Hanada K. Matsumoto T. Miyamoto K. Muranaka A. Uchiyama M. Tanaka K. Angew. Chem., Int. Ed. 2020;59:11020. doi: 10.1002/anie.202001794. [DOI] [PubMed] [Google Scholar]
- Kimura Y. Shibata Y. Noguchi K. Tanaka K. Eur. J. Org. Chem. 2019;2019:1390. [Google Scholar]
- Gingras M. Félix G. Peresutti R. Chem. Soc. Rev. 2013;42:1007. doi: 10.1039/c2cs35111k. [DOI] [PubMed] [Google Scholar]
- Sehnal P. Stará I. G. Šaman D. Tichý M. Míšek J. Cvačka J. Rulíšek L. Chocholoušová J. Vacek J. Goryl G. Szymonski M. Císařová I. Starý I. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13169. doi: 10.1073/pnas.0902612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima T. Kikkawa S. Azumaya I. Watanabe S. ChemistryOpen. 2018;7:278. doi: 10.1002/open.201800006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Tanaka H. Inoue Y. Mori T. ChemPhotoChem. 2018;2:386. [Google Scholar]; (b) Tanaka H. Ikenosako M. Kato Y. Fujiki M. Inoue Y. Mori T. Commun. Chem. 2018;1:38. [Google Scholar]; (c) Kubo H. Shimizu D. Hirose T. Matsuda K. Org. Lett. 2020;22:9276. doi: 10.1021/acs.orglett.0c03506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.