

Abstract
Objective:
To gain insight into the role of germline genetics in the development of chordoma, we evaluated data from two sets of familial chordoma patients, those with and without a germline duplication of the T gene (T-dup+ versus T-dup-), which was previously identified as a susceptibility mechanism in some families. We then compared the familial patients to chordoma patients in the United States general population reported to the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) program through 2015.
Methods:
Evaluation of family members included review of personal and family medical history, physical and neurologic examination, and pre- and post-contrast magnetic resonance imaging (MRI) of the skull-base and spine. Sixteen patients from six white chordoma families had a chordoma diagnosis at family referral. Screening MRIs of 35 relatives revealed clival lesions in six, four of which were excised and confirmed to be chordoma. Thus, data were available for 20 histologically confirmed familial chordoma patients. There were 1759 histologically confirmed chordoma patients with known race in SEER.
Results:
Median age at chordoma diagnosis differed across the groups: lowest in T-dup+ familial patients (26.8 years, range: 5.3–68.4), intermediate in T-dup- patients (46.2 years, range: 11.8–60.1) and highest in SEER patients (57 years, range: 0–98). There was a marked preponderance of skull-base tumors in familial chordoma patients (93% in T-dup+ and 83% in T-dup-) versus 38% in SEER (37% in whites, 53% in blacks, and 48.5% in Asian/Pacific Islander/American Indian/Alaska Native). Further, 29% of white and 16–17% of non-white SEER patients had mobile spine chordoma versus no familial patients. Several T-dup+ familial chordoma patients had putative second/multiple primary chordomas.
Conclusions:
The occurrence of young age at diagnosis, skull-base presentation, or multiple primary chordomas should encourage careful review of family history for patients diagnosed with chordoma as well as screening at-risk family members by MRI for early detection of chordoma. Further, given genetic predisposition in some familial chordoma patients, identification of a specific mutation in a family will permit surveillance to be limited to mutation carriers and consideration should be given for imaging the entire neuroaxis in any chordoma patient presenting at an early age or with a blood relative with chordoma. Finally, future studies should explore racial differences in age at diagnosis and presenting site in chordoma.
Keywords: chordoma, familial, T (brachyury), skull-base
Introduction
Chordoma is a rare bone cancer (<1 per 100,000) that is believed to arise from remnants of the notochord that persist along the axial skeleton into adulthood. Chordoma occurs more frequently in males than females and in Caucasians than African Americans.1,2 Chordoma incidence increases with age and is rare in young patients, especially in the first decade of life. The usual sites of origin are the skull-base, mobile spine and sacrum/coccyx. Although most chordomas are sporadic, ten multiple-case families with at least two blood relatives with histopathologically-confirmed chordoma have been reported.3–8 We previously identified autosomal dominant inheritance of a duplication of the T gene (brachyury) using a whole-genome human array-comparative genomic hybridization (array-CGH) chip followed by a quantitative polymerase chain reaction (PCR) assay for confirmation in four of seven families investigated.8
To gain insight into the role of germline genetics in the development of chordoma and to evaluate whether any clinical features might impact surveillance or screening, we evaluated clinical data from two sets of familial chordoma patients, those with a germline duplication of the T gene (T-dup+) and those without a T gene duplication (T-dup-). We then compared the familial chordoma patients to chordoma patients in the United States (US) general population reported to the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) program through 2015.
Methods
Chordoma families
We ascertained seven families, all Caucasian, with ≥2 blood relatives with histopathologically-confirmed chordoma through physician referral (Families 1, 3, 4 and 7), self-referral (Families 6 and 8), and evaluation of patients reported to four SEER registries between 1988 and 1998 (Family 2). We excluded Family 3 because of limited clinical information on the chordoma patients. Therefore, this report focuses on six families (Figure 1).
Figure 1.
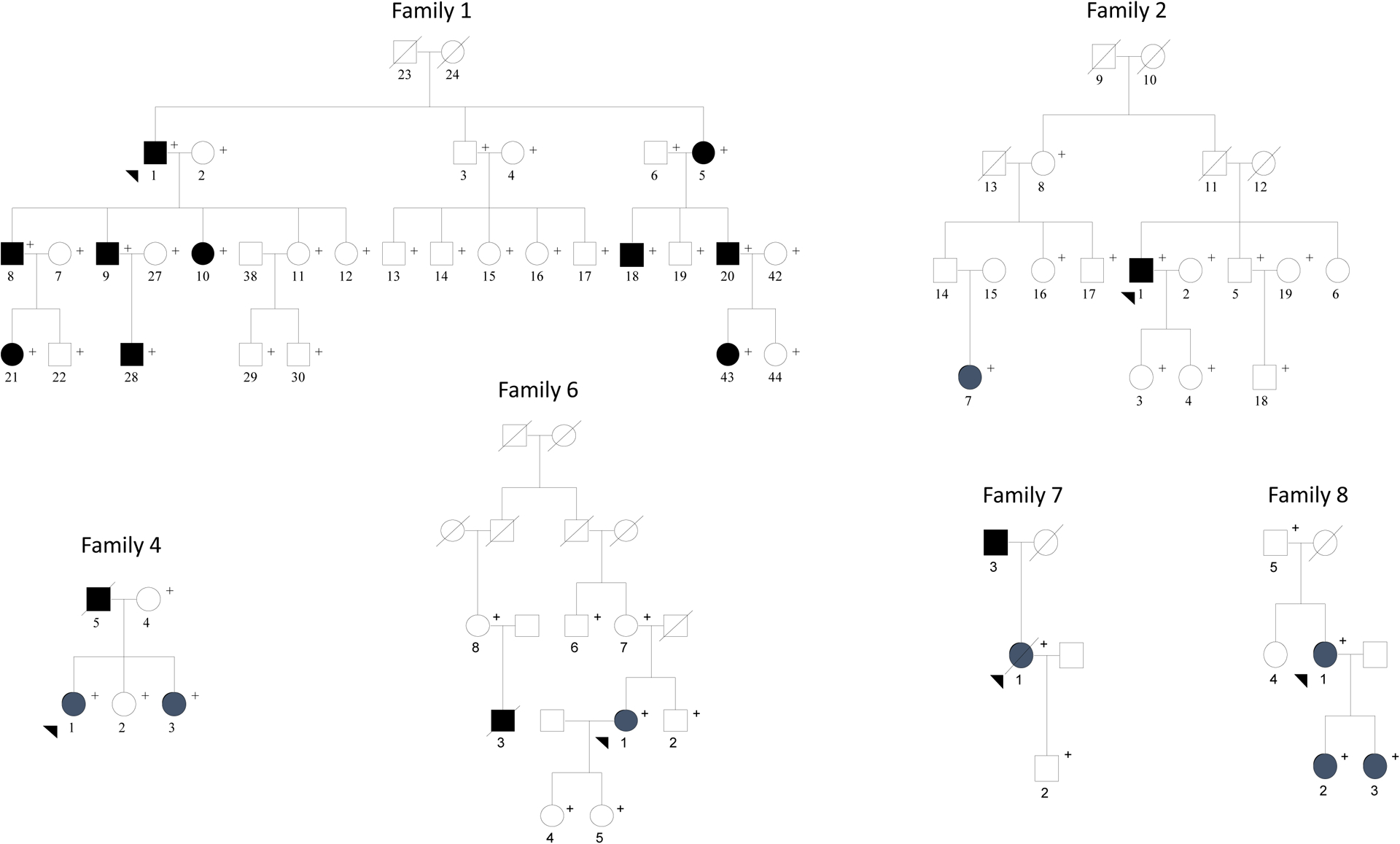
Chordoma Families
Pedigrees for the six families (Family 1, 2, 4, 6, 7, and 8) in the current study. Filled squares and circles indicate patient diagnosed with chordoma. Plus sign (+) shows the family members that were participants in the clinical study.
Evaluated individuals included living family members with chordoma and selected first and second-degree relatives. This study was approved by the institutional review board of the National Cancer Institute, National Institutes of Health (NIH). Patients gave written informed consent or assented before undergoing evaluation. Families were seen at the NIH Clinical Center. Some affected members of Families 2, 4, 6 and 7 were also seen at Massachusetts General Hospital (by NL). Molecular genetic studies previously revealed duplication of the T gene in Families 1, 4, and 8.6,8
At the NIH Clinical Center, evaluation, regardless of chordoma status, included review of personal and family medical history, physical and neurologic examination, and pre- and post-contrast magnetic resonance imaging (MRI) of the skull-base and spine including sacrum and coccyx. All chordoma diagnoses were confirmed by review of pathology materials/reports and medical records. Follow-up data were obtained during return clinic visits, review of outside medical records and pathology reports, and/or telephone conversations with patients or relative(s).
SEER
We used SEER to obtain population-based data for all histologically confirmed cases of chordoma among patients reported to 18 cancer registries during the period 1973–20159 for comparison with familial chordoma patients. We identified cases using the WHO’s International Classification of Diseases for Oncology, Third Edition (ICD-O-3) morphology code for chordoma (9370/3) and the ICDO-2 and ICDO-3 topography codes for sites of presentation.1 Sixteen cases with unknown race were excluded from the analyses. Because the scope of SEER data collection and reporting has varied over time, we limited analysis of each variable to the number of patients with known status. Data were analyzed using SEER*Stat v.8.3.5.10
Statistical Analysis
For categorical variable comparisons (e.g. presenting site of chordoma), Pearson chi-square or Fisher exact test was used depending on sample sizes. Since the incidence rates, distribution of chordomas at presenting sites, and ages at diagnosis of chordoma differed by race, and almost 90% of patients with this rare tumor and all members of the studied chordoma families were white, analyses of familial versus SEER chordoma patients were restricted to white chordoma patients. Because of the relatively small number of patients available for analysis, we assumed independence of patients with chordoma within families.
Results
Chordoma families
Fifty-one individuals from six chordoma families were study participants (Table 1, Figure 1). Sixteen patients had a diagnosis of chordoma at referral. Of these, 11 chordoma patients underwent MRI evaluation: skull-base and spine (n=8 skull-base chordomas and 1 sacral chordoma), and spine (n=2 skull-base chordomas). Thirty-five blood relatives who enrolled in the study without a diagnosis of chordoma were screened by MRI: skull-base and spine (n=34) and skull-base only (n=1).
Table 1.
Chordoma patients and relatives included in the Familial Chordoma Study
| Sixteen patients diagnosed with chordoma before study entry | Clinical findings in thirty-five relatives screened at NIH by MRI | ||||||
|---|---|---|---|---|---|---|---|
| Family ID | T dup+ Present | Imaged at NIH | Not imaged at NIH | Chordoma | Another abnormal finding | No abnormal findings | |
| Alive | Deceased | ||||||
| 1 | Yes | 4 | 0 | 0 | 4 | 21 | 13 |
| 2 | No | 1 | 1 | 0 | 0 | 12 | 6 |
| 4 | Yes | 2 | 0 | 1 | 0 | 0 | 13 |
| 6 | No | 1 | 0 | 1 | 0 | 0 | 5 |
| 7 | No | 0 | 2 | 0 | 0 | 0 | 1 |
| 8 | Yes | 3 | 0 | 0 | 0 | 0 | 2 |
| 11 | 3 | 2 | 4 | 3 | 28 | ||
Both relatives had MRI findings suggestive of clival chordoma but neither lesion was biopsied.
This relative had a cystic lesion within her cervical spinal cord at age 16 which was detected on the screening MRIs. It was resected and the histopathologic diagnosis was juvenile pilocytic astrocytoma.
This relative had been diagnosed elsewhere with a cerebellar astrocytoma at age 11 years which was resected. When she was seen at NIH, no abnormal MRI findings were detected.
Screening MRIs of relatives
Screening MRIs of relatives revealed clival lesions in six, all from Family 1. Four lesions were excised and confirmed to be chordoma (Table 1). The two other clival lesions were not biopsied; therefore, although their location and imaging characteristics were compatible with chordoma, no diagnosis could be confirmed. Thus, data were available for 20 histologically confirmed chordoma patients. For ease of presentation, T-dup+ (3 families, 14 patients) and T-dup- families (3 families, 6 patients) are presented separately (Tables 1–3, Supplemental table 1).
Table 3.
Location, extension, size and initial treatment of chordoma in 20 patients from six chordoma families
| Family | No. of patients | Tumor location and extension | Size (cm) | Major symptom(s) | Initial treatment1 |
|---|---|---|---|---|---|
| Cranial Tumors | |||||
| T dup+ Families | |||||
| Family 1 | 22 | clivus, nasopharynx | 2.5, 3.0 | snoring, nasal obstruction | GTR |
| 23 | clivus, nasopharynx | 3.0, 4.9 | nasal speech and obstruction, snoring2 | RR | |
| 24 | clivus, nasopharynx | 2.0, 2.4 | none | RR | |
| 12 | clivus, sphenoid sinus, cavernous sinus, Meckel’s cave, petrous apex | 5.4 | headaches, nausea, vomiting | PR + RT | |
| Family 4 | 12 | clivus | NA | nasal congestion and obstruction | BX + RT |
| 12 | clivus | 4 | nasal congestion and obstruction | STR + RT | |
| 12 | clivus, sphenoid sinus, nasopharynx | 4.5 | ear pain | PR | |
| Family 8 | 12 | clivus, nasopharynx | 3.9 | epistaxis, nasal congestion, obstruction | RR |
| 15 | clivus | NA | nasal congestion, obstructed breathing | RR | |
| 16 | clivus | 1.5 | none | RR | |
| Total | 13 | ||||
| T dup- Families | |||||
| Family 2 | 12 | clivus, nasal cavity; paranasal, ethmoid sphenoid and maxillary sinuses, nasopharynx | >6.0 | nasal congestion and obstruction | PR+ RT |
| 12 | clivus, pons | 3 | diplopia, headaches, R pupil > L, R ptosis | GTR + RT | |
| Family 6 | 12 | clivus, foramen magnum to C2 vertebrae | 4.2 | headaches from occipital to frontal region | PR + RT |
| 12 | clivus with sinu-nasal extension | >6.0 | aural fullness, ptosis AU, vertical diplopia, nasal obstruction, epistaxis | PR + RT | |
| Family 7 | 12 | clivus, cranio-cervical junction | ~5.0 | atrophy R tongue, pain R neck, shoulder, arm; numbness and tingling, R hand | STR + RT |
| Total: | 5 | ||||
| Sacrococcygeal Tumors | |||||
| T dup+ Family | |||||
| Family 1 | 12 | coccyx to S3 vertebrae, presacral region | ~9.5 | buttock pain on sitting | GTR + RT |
| T dup- Family | |||||
| Family 7 | 12 | S3-S5 vertebra, presacral region, paraspinal soft tissue, piriformis muscle | ~10.0 | pain in L buttock, sacrum, lower back | BX + palliative RT |
Initial treatment: treatment within 4 months of diagnosis. Types of surgery: GTR, gross total resection; RR, radical resection; PR, partial resection, BX, biopsy; STR, subtotal resection; RT, radiation therapy.
These patients were diagnosed because they presented with symptoms.
These two patients reported long standing symptoms but did not pursue a cause for them because their development had been so gradual. Subsequently, they were found to have skull base chordomas based on the NIH screening MRIs.
These patients were asymptomatic and diagnosed based on the NIH screening MRIs.
This patient was presumed to have enlarged tonsils and adenoids prior to initial surgery, so no pre-operative images are available.
This patient was asymptomatic but her local MD had her screened by neuroaxis MRIs because her mother and younger sister had been diagnosed with skull base chordomas.
Clinical findings, treatment and outcome in T-dup+ families
Among the 14 chordoma patients from T-dup+ families, 13 presented with skull-base tumors; one presented with sacrococcygeal chordoma (Table 2). Among the 13 skull-base chordomas, 8 occurred in females (61%) and 5 (39%) in males, with a median age at diagnosis of 25.5 years (range 5.3–68.4 years). All 13 skull-base chordomas arose from the clivus. In most, there was a soft tissue component that included only the nasopharynx. Two tumors were more extensive (Table 3, Supplemental table 1).
Table 2.
Characteristics of histologically confirmed chordoma in white patients from 6 chordoma families and white patients reported to SEER
| Families with a T gene duplication | Families with no T gene duplication | SEER | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Presenting site1 | Presenting site1 | Presenting site | |||||||||
| Feature | Skullbase | Sacrum/coccyx | Total | Skull base | Sacrum/coccyx | Total | Skull base | Mobile spine | Sacrum/coccyx | Other2 | Total |
| Gender No. (%) | |||||||||||
| Male | 5 (62.5) | 1 (33.3) | 6 (100.0) | 3 (100.0) | 0 (0.0) | 3 (100.0) | 315 (34.4) | 277 (30.3) | 294 (32.1) | 29 (3.2) | 915 (100.0) |
| Female | 8 (100.0) | 0 (0.0) | 8 (100.0) | 2 (66.2) | 1 (33.3) | 3 (100.0) | 251 (40.2) | 167 (26.8) | 181 (29.0) | 25 (4.0) | 624 (100.0) |
| Total | 13 (92.9) | 1 (7.1) | 14 (100.0) | 5 (83.3) | 1 (16.7) | 6 (100.0) | 566 (36.8) | 444 (28.8) | 475 (30.9) | 54 (3.5) | 1539 (100.0) |
| Age at diagnosis, years | |||||||||||
| Median | 25.5 | 28.1 | 26.8 | 46.4 | 37.4 | 46.2 | 49 | 60 | 65 | 64.5 | 58 |
| Range | 5.3–68.4 | 5.3–68.4 | 11.8–60.1 | 11.8–60.1 | 0–92 | 0–92 | 11–98 | 13–95 | 0–98 | ||
| Tumor size, cm (%) | |||||||||||
| < 2.0 | 1 (9.0) | 0 (0.0) | 1 (8.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 47 (14.5) | 12 (7.6) | 18 (5.2) | 3 (12.0) | 80 (9.5) |
| 2.0–3.9 | 5 (45.5) | 0 (0.0) | 6 (50.0) | 1 (20.0) | 0 (0.0) | 1 (16.6) | 161 (49.7) | 48 (30.4) | 23 (6.7) | 5 (20.0) | 237 (27.8) |
| 4.0–5.9 | 5 (45.5) | 0 (0.0 | 4 (33.3) | 2 (40.0) | 0 (0.0) | 2 (33.3) | 83 (25.6) | 53 (33.5) | 57 (16.5) | 5 (20.0) | 198 (23.2) |
| > 6.0 | 0 (0.0) | 1 (100.0) | 1 (8.3) | 2 (40.0) | 1 (100.0) | 3 (50.0) | 33 (10.2) | 45 (28.2) | 247 (71.6) | 12 (48.0) | 337 (39.6) |
| Total | 11 (100.0) | 1 (100.0) | 12 (100.0) | 5 (100.0) | 1 (100.0) | 6 (100.0) | 324 (100.0) | 158 (100.0) | 345 (100.0) | 25 (100.0) | 852 (100.0) |
| Treatment No. (%) | |||||||||||
| No treatment | 0 | 0 | 0 | 0 | 0 | 0 | |||||
| Surgery only | 10 (76.9) | 0 (0.0) | 10 (71.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||||
| Surgery and radiotherapy | 2 (15.4) | 1 (100.0) | 3 (21.4) | 5 (100.0) | 0 (0.0) | 5 (83.3) | |||||
| Radiotherapy only | 1 (7.7) | 0 (0.0) | 1 (7.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | |||||
| Other | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (100.0) | 1 (16.7) | |||||
| Total | 13 (100.0) | 1 (100.0) | 14 (100.0) | 5 (100.0) | 1 (100.0) | 6 (100.0) | |||||
| Length of follow-up, months | |||||||||||
| Median | 203.7 | 144.8 | 200.8 | 159.8 | 19.6 | 138.7 | 63 | 61 | 53 | 47 | 58 |
| Range | 98.5–449.5 | 98.5–449.5 | 19.6–289 | 19.6–289 | 0–455 | 0–469 | 0–461 | 0–368 | 0–469 | ||
| Overall Mortality | |||||||||||
| Alive | 10 (76.9) | 0 (0.0) | 10 (71.4) | 4 (80.0) | 0 (0.0) | 4 (66.7) | 341 (63.6) | 202 (50.1) | 215 (50.4) | 24 (50.0) | 782 (55.3) |
| Chordoma death | 1 (7.7) | 1 (7.1) | 2 (14.3) | 1 (20.0) | 1 (100.0) | 2 (33.3) | 140 (26.1) | 133 (33.0) | 126 (29.5) | 16 (33.3) | 415 (29.3) |
| Non-chordoma death | 2 (15.4) | 0 (0.0) | 2 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 55 (10.3) | 68 (16.9) | 86 (20.1) | 8 (16.7) | 217 (15.3) |
| Total | 13 (100.0) | 1 (100.0) | 14 (100.0) | 5 (100.0) | 1 (100.0) | 6 (100.0) | 536 (100.0) | 403 (100.0) | 427 (100.0) | 48 (100.0) | 1414 (99.9) |
No patient with familial chordoma presented with a chordoma of the mobile spine or “other” sites.
”Other” sites include extra-axial and ill-defined sites
Of the 11 measured skull-based chordomas, six were diagnosed because of presenting symptoms (median diameter 4.2 cm, range: 2.5–5.4 cm) and these tumors were larger than the five chordomas identified through MRI screening (median diameter 2.4 cm, range: 1.5–4.9 cm) (see Table 3 and Supplemental Table 1 for symptom details). Only one chordoma measured <2.0 cm; it was diagnosed in an asymptomatic child (Patient 8–3) screened because of her chordoma family history. For patients diagnosed because of symptoms, median time between symptom onset and diagnosis was 24 months (range, 3 weeks-48 months).
The T-dup+ sacrococcygeal chordoma patient was a male diagnosed at 28 years. He presented with buttock pain and had an 18-month delay between symptom onset and diagnosis (Table 3, Supplemental Table 1). His tumor originated in the mid-coccyx, was ~9.5 cm in maximal diameter and extended into the retrorectal and presacral space.
Neuroaxis MRIs conducted on 13 patients revealed two with lesions suspected of being additional primary chordomas. Five (38.5%) also had ≥1 lesion with imaging findings consistent with benign notochordal cell tumor (BNCT). Details of the other relevant clinical findings are reported in Supplemental Table 1 and Supplemental clinical note.
All T-dup+ patients with primary skull-base chordomas were treated initially with surgery; 10 (76.9%) had no additional therapy and three (23.1%) received adjuvant external beam radiotherapy (Table 3, Supplemental Table 1). The sacrococcygeal chordoma patient underwent gross total surgical resection of extensive but localized tumor. Because microscopic tumor was present at surgical margins and in rectal tissue, the region was subsequently treated with adjuvant fractionated radiation.
After a median follow-up of 203.7 months, ten (76.9%) skull-base chordoma patients were alive at last contact (Table 2, Supplemental table 1). Among these patients, seven had no evidence of disease, two had stable tumor, and one developed pulmonary metastases without local recurrence 26.6 years after initial diagnosis. Three T-dup+ skull-base chordoma patients have died, with one death directly attributable to chordoma. Patient 4–5 developed a recurrence in the paranasal sinuses 31 years after initial diagnosis and treatment at age 8. The tumor progressed despite serial resections, and the patient died at age 47 from recurrent disease.
The sacrococcygeal chordoma patient had widespread metastases when seen at NIH. His disease progressed despite sequential treatment with multiple courses of different single chemotherapeutic agents, and he died at age 40, 144 months after diagnosis.
Table 4 shows clinical findings from three other screened T-dup+ relatives. Initial screening MRI identified asymptomatic clival lesions in two patients (1–8 and 1–43) at ages 44 and 6 years, respectively. Individual 1–44, the younger sister of 1–43, had no abnormalities suggestive of chordoma on screening MRI at 3 years. At 13, a neck CT scan to evaluate a symptomatic thyroglossal duct cyst unexpectedly revealed a 1.6 cm nasopharyngeal mass associated with bony erosion of the clivus. None of these clival lesions has been biopsied or excised.
Table 4.
Clinical findings in 3 screened relatives (Family 1) with T-dup+ but no biopsy-confirmed chordoma
| Patient ID | Sex | Skull base findings, size of skull base lesion (APxTxV1 in cm); age (yrs) First exam |
Skull base findings, size of skull base lesion (APxTxV in cm); age (yrs) Last exam |
Symptoms | Biopsy or surgery of lesion | Findings on spinal MRIs; age (yrs) | Other clinical findings; age (yrs) | Length of follow-up (months); outcome; age (yrs) |
|---|---|---|---|---|---|---|---|---|
| 1–8 | M | cyst in nasopharynx associated with a bone defect in clivus; 1.6 × 0.8 × 1.9; 44.6 | no change in size; 49 | no | no | sacral perineural meningeal cyst; 44.6 and 49 | diabetes, 49 | 188; AW2; 60.3 |
| 1–43 | F | cyst in nasopharynx with dorsal extension indenting intact clivus; 1.4 × 1.2 × 1.5; 6.3 | enhancing cyst in nasopharynx that erodes ventral clivus; 2.3 × 1.3 × 1.7; 15 | NA3 | no | ND4 | none, 15 | 149; AW; 18.7 |
| 1–44 | F | none; 3 | enhancing cyst in nasopharynx eroding clivus; 1.6 × 1.2 × 1.5; 13.8 | NA | no | none at age 3 | ranula, 7; thyroglossal duct cyst, 13.8 | 176; AW; 17 |
APxTxY: anterior-posterior × transverse × vertical dimensions
AW: alive and well
NA: not available
ND: not done
Clinical findings, treatment, and outcome in T-dup- families
We evaluated six chordoma patients from three T-dup- families (Families 2, 6 and 7, Figure 1, Table 1) in which no other chordoma-related mutation(s) have been identified. Chordoma was diagnosed in two blood relatives in each family; males and females were equally affected.
The skull-base was the presenting site for five patients; the sixth patient had a primary sacrococcygeal chordoma (Table 3; Supplemental Table 1). The median age at diagnosis of the skull-base chordoma patients was 46.4 years. All five skull-base chordomas originated in the clivus but extended beyond it in anterior, superior, and/or posterior directions. All patients sought medical attention because of symptoms, which in three patients affected cranial nerves. The median time from initial symptoms to diagnosis was 7 months (range, 1–36 months).
The five T-dup- skull-base chordomas had a median diameter of 5.0 cm (range 3.5 – 12.8 cm). All were locally advanced and treated with radiotherapy following surgery. After a median of 159.8 months, four patients were alive at last follow-up. Two had no evidence of disease, one had stable tumor and one had local tumor progression to his optic nerve 24 years after initial diagnosis. The death of patient 6–3 was directly attributable to his tumor (Supplemental Table 1).
The T-dup- sacrococcygeal chordoma patient was diagnosed at 37 years, 98 months following her initial presentation with buttock pain (Table 3). Pulmonary metastases were found shortly after diagnosis. The tumor was treated with palliative high dose proton-photon radiotherapy. She died at age 39 from widely disseminated disease 19 months after diagnosis.
Spinal MRIs were available on 2/6 T-dup- patients. Neither had a lesion suspected of being a second primary chordoma or BNCT.
Ten families with at least two blood relatives with histopathologically confirmed chordoma have been described worldwide (Table 5).3–8,11–18 The six families described in this report (numbered in Table 5) include three of the four published T-dup+ chordoma families. The fourth T-dup+ chordoma family (Family 3) has branches in both South Africa and England; we present the two branches separately but treat it as a single family. Family 3 displayed characteristics like those for the other T-dup+ families: ≥3 affected first-degree relatives, an autosomal dominant pattern, preponderance of skull-base tumors, early ages at diagnosis (e.g., ages 3 and 8 years) and a member with putative multifocal chordoma diagnosed in the skull-base and sacrum.3 Among the three additional reported chordoma families in Table 5, one is notable for the occurrence of sacral chordomas in two adult siblings, another features skull-base chordomas in two very young brothers and the third describes skull-base chordomas in two adult siblings and two of their children. Geographically, the chordoma families were from the US, Italy, South Africa/England, Russia and China. Overall, among the 34 confirmed cases of familial chordoma in the 10 families the M:F ratio was 15:19 (1:1.3) with 30 skull-base tumors versus 4 sacral chordomas. The Chinese family showed a clinical pattern of chordoma similar to the four T-dup+ families, but mutation analysis results have not been reported.7
Table 5.
Ten families with two or more relatives with histopathologically-confirmed chordoma reported worldwide
| Family No. | Family in this paper? | Total relatives with Chordoma1 | Patients’ relationships | Gender | Tumor Locations | Median age and range at diagnosis (yrs)2 | Year first reported | References (see list of references) |
|---|---|---|---|---|---|---|---|---|
| Family 1 | Yes | 8 (plus 3)3 |
See figure 1 | 5M, 3F | Clivus (6), nasopharynx (1), sacrum (1) | 33 17–68 |
1998 | 6, 8, 14, 17, 18 |
| Family 2 | Yes | 2 | Distant cousins – see figure 1 | 1M, 1F | Clivus (2) | 28.9 11.8–46.1 |
2001 | 6, 8, 14, 18 |
| Family 3a (South African branch) | No | 3 (plus 2)4 |
Father, his mother, his daughter | 1M, 2F | Nasopharynx (1), clivus (1), upper cervical spine (1) | 22 3.0–51 |
1975 | 3, 11, 15, 18 |
| Family 3b (English branch) | No | 3 | Sisters, maternal first cousin | 0M, 3F | Clivus (3) | 21 8.0–40 |
2001 | 3, 8, 14, 18 |
| Family 4 | Yes | 3 | Father, two daughters | 1M, 2F | Clivus (3) | 8 7.7–25.5 |
1999 | 6, 8, 13, 16, 18 |
| Family 6 | Yes | 2 | Distant cousins – see figure 1 | 1M, 1F | Clivus (2) | 47.3 46.4–48.3 |
2009 | 6, 18 |
| Family 7 | Yes | 2 | Father, daughter | 1M, 1F | Clivus & upper cervical spine (1), sacrum (1) | 48.8 37.4–60.1 |
2009 | 6, 18 |
| Family 8 | Yes | 3 | Mother, daughters | 0M, 3F | Clivus (3) | 8.4 5.3–32.0 |
2009 | 6, 12, 18 |
| na | No | 2 | Sister, brother | 1M, 1F | Sacrum (2) | 52 52–52 |
1958 | 5 |
| na | No | 2 | Brothers | 2M | Nasal cavity (2) | 2.4 1.8–4 |
1964 | 4 |
| na | No | 4 | Daughter, father, paternal aunt, paternal first cousin | 2M, 2F | Clivus (3), nasal region (1) | 31 15–47 |
2015 | 7 |
| Total confirmed cases: | 34 | 15M:19F | ||||||
Histopathologically-confirmed chordoma
Based on patients with histopathologically-confirmed chordoma
The brother of the index case and two female paternal 1st cousins once removed have clival lesions on MRI or CT scans that are consistent with chordoma but which have not been biopsied. (see Table 4 for details).
Two additional distant relatives were reported to have had chordoma by Bhadra and Casey, 2006 (reference #4).
Population-based chordomas reported to SEER
Supplemental Table 2 shows the clinical characteristics of 1,759 histologically confirmed chordomas in patients with known race. Briefly, the overall incidence rate was 0.10 per 100,000. Males comprised 59.1% of cases resulting in a M:F ratio of 1.45:1. Most patients (87%) were white. There were significant differences in the distribution of chordoma presenting site by race. Blacks and Asian/Pacific Islander/American Indian/Alaska Native (A/PI/AI/AN) had higher frequencies of skull-base (p=0.015 and p=0.003, respectively) and lower frequencies of mobile spine (p=0.032 and p<0.0001, respectively) chordomas compared to white patients. Overall, median age at chordoma diagnosis was 57 years (range, 0–98 years) but non-white patients had lower median ages at chordoma diagnosis versus white patients (Supplemental Table 2). Chordoma was uncommon at younger ages; only 287 (16.3%) cases were diagnosed before age 35 years. However, a higher proportion of blacks (29.8%) and A/PI/AI/AN (23.9%) were diagnosed before 35 years compared to whites (15.0%) (p=0.002, blacks vs whites and p=0.003, A/PI/AI/AN vs whites).
Comparison of familial chordomas with population-based chordomas reported to SEER
Table 2 shows the 1539 SEER-reported white patients with chordoma for comparison with the familial chordoma patients. Familial chordoma patients were much more likely than chordoma patients in the US general population to present with skull-base tumors: 18/20 (90%) versus 566/1539 (36.8%), respectively (p<0.0001). In contrast, 29% of SEER chordoma patients, but no familial chordoma patients, presented with chordoma in the mobile spine (p<0.0001). T-dup+ familial chordoma patients were diagnosed at much younger ages (median 26.8 years) than patients in either T-dup- families (median 46.2 years) or SEER chordoma patients (median 58 years) (Table 2). Moreover, 77% of the T-dup+ skull-base tumors were diagnosed by 35 years versus only 20% of T-dup- (p=0.048, versus T-dup+) and 23.5% of SEER (p<0.0001, versus T-dup+) skull-base tumors. Several T-dup+ familial chordoma patients had putative second or multiple primary chordomas and/or BNCT(s) in the spine. No such clinical findings were observed in the small number of T-dup- familial chordoma patients available for examination. It was not possible to evaluate SEER chordoma patients for these features.
Discussion
To gain insight into the role of germline genetics in the development of chordoma, we evaluated data from two sets of familial chordoma patients, those with a germline duplication of the T gene (T-dup+) and those without a T gene duplication (T-dup-). We then compared the clinical data on familial chordoma patients with data from population-based chordoma patients diagnosed in the US between 1973 and 2015. The results demonstrate that T-dup+ familial chordoma patients differ from sporadic chordoma patients (SEER) in at least two aspects: the marked preponderance among the former of skull-base tumors and the young age at which these tumors were diagnosed. Skull-base tumors were also more frequent among T-dup- familial chordoma patients; their age at diagnosis, however, did not differ markedly from that in sporadic chordoma patients. Among the chordoma patients reported to SEER, non-white SEER patients had a higher frequency of skull-base chordomas, a lower frequency of mobile spine chordomas, and younger age at diagnosis compared with white SEER chordoma patients.
In the general population, skull-base chordoma was rare (3.7%) in young patients (<10 years).1 In contrast, four (31%) T-dup+ patients were diagnosed with skull-base chordomas before 10 years, three of which were symptomatic, suggesting that these tumors have very early onset and/or unusually rapid growth. In fact, most skull-base chordoma in T-dup+ patients occurred early with almost 77% of the T-dup+ skull-base tumors diagnosed in patients by 35 years. In contrast, only about 26% of skull-base tumors in SEER (23% in whites, 40% in blacks, and 39% in A/PI/AI/AN) and 20% of T-dup- skull-base tumors were diagnosed by age 35 years.
The site of chordoma presentation varied with age. In both the familial and sporadic (SEER) settings, the earlier presentation of skull-base versus sacrococcygeal chordomas may reflect their anatomical location: a slow growing tumor might be expected to produce symptoms earlier in the closed intracranial space compared with a tumor developing from the sacrum. Alternatively, there may be an intrinsic biological basis for the earlier initiation of chordomas in the skull-base versus the sacrum/coccyx, and it seems plausible that those with a genetic predisposition to chordoma would have the earliest onset.
Early age at diagnosis is a hallmark of cancers associated with rare deleterious germline mutations in cancer-predisposition genes, e.g., BRCA1/2, TP53, CDKN2A.19–22 Another feature of such cancers is the presence of disease in both of paired susceptible organs or at multiple sites. Among the patients reported here, two unrelated T-dup+ individuals had at least one neuroaxial lesion in addition to their presenting chordoma that was considered a second primary chordoma based on anatomic location and imaging characteristics. Because these lesions were asymptomatic, they were not biopsied. A third T-dup+ patient with multicentric chordomas has been reported.3 In Family 3, the young girl who was diagnosed and treated for skull-base chordoma at age 3 developed a sacral chordoma at age 6 that was incompletely resected and treated with radio- and chemotherapy. The tumor recurred when she was 19 and again at 25 years; both times she underwent palliative debulking and radiotherapy. She died at age 28 but her cause of death was unknown.3
BNCTs are believed to be chordoma precursors. Like chordoma they are thought to derive from notochord remnants and have the same immunohistopathologic and molecular profiles. In a few instances chordomas have been reported to develop from a BNCT.23 With a reported incidence of 20%, BNCTs rarely develop into chordomas. However, in the presence of soft tissue extension, care must be taken to distinguish benign from malignant lesions. The presence of at least one BNCT in five (38.5%) T-dup+ patients should be considered another manifestation associated with T-dup+.
Presenting signs, symptoms and size of the familial skull-base chordomas reported here differed by T-dup status. Based on very small numbers, the T-dup+ skull-base chordomas diagnosed because of symptoms were smaller (median of 4.0 cm versus 5.0 cm in maximum diameter) and diagnosed later after symptom onset (median of 24 months versus 7 months) than symptomatic T-dup- tumors. These differences may reflect the direction of tumor growth from the clivus; most T-dup+ chordomas grew towards the nasopharynx which resulted in more non-specific symptoms than those from T-dup- chordomas that grew in other directions and impinged cranial nerves. The differences in size and growth patterns between the two groups of familial chordomas might explain treatment differences. Surgery alone (either radical or gross total resection) was the main treatment for most (10/13, 80%) T-dup+ skull-base chordomas whereas all T-dup- skull-base chordomas underwent adjuvant fractionated radiation therapy after surgery. Current treatment standards for sporadic chordoma patients recommend post-surgical adjuvant radiation therapy if en-bloc resection or negative tumor margins have not been achieved.24,25 Because optimal surgical outcomes depend in part on tumor location and extent of local invasion, complete pathological resection is often difficult to achieve for skull-base (location) and large sacral (local invasion) chordomas.
This study was limited by the small number of chordoma families available for evaluation and the relatively small numbers of non-white chordoma patients reported to SEER. In addition, clinical and follow-up details were not available for all family members. Further, given the scope of data collection in SEER, it was not possible to conduct comparisons for treatment, multiple primaries, or BNCTs. However, although limited numbers of families have been published to date, many more families likely exist, and patients would therefore benefit from collection of family history information at chordoma diagnosis to allow for screening and surveillance recommendations. In addition, given the differences observed between white and non-white SEER chordoma patients, future studies should explore racial differences in age at diagnosis and presenting site in chordoma.
Conclusions
In summary, the findings from the current study indicate that the occurrence of young age at diagnosis, skull-base presentation, or the presence of multiple primary chordomas should encourage careful review of family history for patients diagnosed with chordoma. Identification of a positive family history presents the opportunity for long-term screening of at-risk family members by MRI for early detection of chordoma. In addition, given the relevance of genetic predisposition in some familial chordoma patients,6,8 identification of a specific mutation in a family will permit surveillance to be limited to mutation carriers. Finally, consideration should be given for regularly imaging the entire neuroaxis in any chordoma patient presenting at an early age or with a blood relative with chordoma.
Supplementary Material
Acknowledgements:
We are indebted to the participating families, whose generosity and cooperation have made this study possible. We acknowledge the clinical contributions to this work that were made by Dr. Gladys Glenn.
Funding:
This work was supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health
References
- 1.McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12(1):1–11. [DOI] [PubMed] [Google Scholar]
- 2.Smoll NR, Gautschi OP, Radovanovic I, et al. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119(11):2029–2037. [DOI] [PubMed] [Google Scholar]
- 3.Bhadra AK, Casey AT. Familial chordoma. A report of two cases. J Bone Joint Surg Br. 2006;88(5):634–636. [DOI] [PubMed] [Google Scholar]
- 4.Enin IP. [Chordoma of the Nasopharynx in 2 Members of a Family]. Vestn Otorinolaringol. 1964;26:88–90. [PubMed] [Google Scholar]
- 5.Foote RF, Ablin G, Hall WW. Chordoma in siblings. Calif Med. 1958;88(5):383–386. [PMC free article] [PubMed] [Google Scholar]
- 6.Kelley MJ, Shi J, Ballew B, et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Human genetics. 2014;133(10):1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang KE, Wu Z, Tian K, et al. Familial chordoma: A case report and review of the literature. Oncol Lett. 2015;10(5):2937–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang XR, Ng D, Alcorta DA, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41(11):1176–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surveillance, Epidemiology and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence -- SEER 18 Regs Research Data_Hurricane Katrina Impacted Louisiana Cases, Nov 2017 Sub (1973–2015 varying) - Linked to County Attributes -- Total U.S. 1969–2016 Counties National Cancer Institute, DCCPS, Surveillance Research Program. [Google Scholar]
- 10.SEER*Stat. National CAncer Institute SEER*Stat Software. seer.cancer.gov/seerstat.
- 11.Chetty R, Levin CV, Kalan MR. Chordoma: a 20-year clinicopathologic review of the experience at Groote Schuur Hospital, Cape Town. J Surg Oncol. 1991;46(4):261–264. [DOI] [PubMed] [Google Scholar]
- 12.Coppens JR, Ric Harnsberger H, Finn MA, et al. Oronasopharyngeal chordomas. Acta Neurochir (Wien). 2009;151(8):901–907. [DOI] [PubMed] [Google Scholar]
- 13.Dalpra L, Malgara R, Miozzo M, et al. First cytogenetic study of a recurrent familial chordoma of the clivus. Int J Cancer. 1999;81(1):24–30. [DOI] [PubMed] [Google Scholar]
- 14.Kelley MJ, Korczak JF, Sheridan E, et al. Familial chordoma, a tumor of notochordal remnants, is linked to chromosome 7q33. Am J Hum Genet. 2001;69(2):454–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr WA, Allen KL, Haynes DR, et al. Letter: Familial nasopharyngeal chordoma. S Afr Med J. 1975;49(39):1584. [PubMed] [Google Scholar]
- 16.Miozzo M, Dalpra L, Riva P, et al. A tumor suppressor locus in familial and sporadic chordoma maps to 1p36. Int J Cancer. 2000;87(1):68–72. [PubMed] [Google Scholar]
- 17.Stepanek J, Cataldo SA, Ebersold MJ, et al. Familial chordoma with probable autosomal dominant inheritance. Am J Med Genet. 1998;75(3):335–336. [DOI] [PubMed] [Google Scholar]
- 18.Yang XR, Beerman M, Bergen AW, et al. Corroboration of a familial chordoma locus on chromosome 7q and evidence of genetic heterogeneity using single nucleotide polymorphisms (SNPs). Int J Cancer. 2005;116(3):487–491. [DOI] [PubMed] [Google Scholar]
- 19.Aoude LG, Wadt KA, Pritchard AL, et al. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res. 2015;28(2):148–160. [DOI] [PubMed] [Google Scholar]
- 20.Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122(23):3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Read J, Wadt KA, Hayward NK. Melanoma genetics. J Med Genet. 2016;53(1):1–14. [DOI] [PubMed] [Google Scholar]
- 22.Turnbull C, Hodgson S. Genetic predisposition to cancer. Clin Med (Lond). 2005;5(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamaguchi T, Yamato M, Saotome K. First histologically confirmed case of a classic chordoma arising in a precursor benign notochordal lesion: differential diagnosis of benign and malignant notochordal lesions. Skeletal Radiol. 2002;31(7):413–418. [DOI] [PubMed] [Google Scholar]
- 24.Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012;13(2):e69–76. [DOI] [PubMed] [Google Scholar]
- 25.Yolcu Y, Wahood W, Alvi MA, et al. Evaluating the Role of Adjuvant Radiotherapy in the Management of Sacral and Vertebral Chordoma: Results from a National Database. World Neurosurg. 2019;127:e1137–e1144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.