

Abstract
Herein, we describe how cantilever-free scanning probes can be used to deposit precursor material and subsequently irradiate the precursor to initiate polymerization, resulting in a 3D lithographic method wherein the position, height and diameter of each feature can be tuned independently. Specifically, acrylate and methacrylate monomers were patterned onto thiol terminated glass and subsequently exposed to UV light produced brush polymers by a photoinduced radical acrylate polymerization reaction. Here, we report the first examples of glycan arrays, comprised of methacrylate brush polymers that are side-chain functionalized with α-glucose, by this new lithographic approach. Their binding with fluorophore labeled concanavalin A (ConA) was assayed by fluorescence microscopy. The fluorescence of these brush polymers was compared to glycan arrays composed of monolayers of α-mannosides and α-glucosides prepared by combining polymer pen lithography (PPL) with the thiol–ene photochemical reaction or the copper-catalyzed azide–alkyne cycloaddition. At high ConA concentration, the fluorescence signal of the brush polymer was nearly 20 times greater than that of the glycan monolayers, and the brush polymer arrays had a detection limit nearly two orders of magnitude better than their monolayer counterparts. Because of the ability of this method to control precisely the polymer length, the relationship between limit of detection and multivalency could be explored, and it was found that the longer polymers (136 nm) are an order of magnitude more sensitive towards ConA binding than the shorter polymers (8 nm) and that binding affinity decreased systematically with length. These glycan arrays are a new tool to study the role of multivalency on carbohydrate recognition, and the photopolymerization route towards forming multivalent glycan scaffolds described herein, is a promising route to create multiplexed glycan arrays with nanoscale feature dimensions.
Introduction
The emergence of massively parallel cantilever-free scanning probe molecular printing tools,1 such as polymer pen lithography (PPL)2 and beam pen lithography (BPL),3 points towards new capabilities in electronics, materials science, chemistry, and biology. To date, arrays with as many as 11 million pens have been produced, enabling printing over cm2 areas, while maintaining the nanometer-scale feature diameter and positional control that are characteristic of scanning probe lithography. By coating the tips with gold and etching nanoscopic apertures into the metal coatings, BPL combines the advantages of piezoactuated, massively parallel tip-based lithography with photolithography. In addition, individual pen actuation is a recent advance that endows each pen in an array the ability to create a distinct pattern.4 These tools are especially promising for biochemical applications because of their ability to print many types of biological structures and direct chemical reactions with dimensions on the scale of features found in biological and living systems. Furthermore, the direct deposition of the ink that occurs in scanning probe lithography prevents destruction or denaturation of delicate biologically active probes that would otherwise occur with conventional nanolithography methods.1a,5 Initial efforts in this direction have resulted in protein arrays that have been used to investigate mesenchymal stem cell differentiation.6
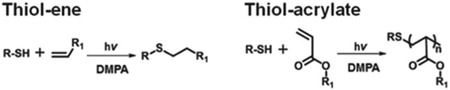
Glycan microarrays have become an invaluable tool for investigating the role of glycans and glycan binding proteins (GBPs) on differentiation and development, cancer metastasis, and viral and bacterial infection.7 Previous work with PPL has also shown promise for the preparation of glycan microarrays.8 Using PPL to induce the CuI catalyzed azide–alkyne cycloaddition (CuAAC) reaction,9 alkyne functionalized monosaccharides were patterned onto azide terminated surfaces, but these PPL-generated monolayer arrays possess only limited ability to detect GBPs. Because glycan recognition occurs on cell surfaces, the 3D structure of the carbohydrates plays a critical role in specificity and binding affinity as a result of the multivalent recognition that dominates the interactions between glycans and GBPs,7k,10 and the carbohydrate monolayers were unable to access these binding modes. Various surface chemistries for the patterning of multivalent scaffolds have been explored as alternative routes towards glycan array preparation,7,11 but the synthesis of the required complex carbohydrates is laborious,12 and arrays prepared with different immobilization strategies often provide markedly dissimilar and contradictory data.7f,i,13 Alternatively, synthetic glycopolymers have been explored as a route to create structurally complex architectures, while significantly reducing synthetic effort, but strategies for immobilizing these materials into arrays with micrometer-scale feature control and pattern flexibility are still required.11g,14 Ideally one would like to generate features that behave as multivalent scaffolds, that can be prepared from readily available starting materials, and that can be patterned using easy to implement surface chemistry. Herein, we describe a new approach that combines PPL or BPL with acrylate or methacrylate photopolymerization chemistry15 to generate arrays of microscale features of brush polymers that are side-chain functionalized with fluorophores or carbohydrates. These polymers exhibit excellent GBP binding ability by mimicking the multivalent glycan structures typically found on cell surfaces. Using the thousands of tips in a BPL array to localize light on a surface, the exposure time at each position on the surface during the photopolymerization could be varied precisely, providing in situ control over polymer length during patterning. Thus, in addition to achieving orders of magnitude improvement over binding sensitivity as a result of multivalency, the glycan arrays prepared by this 3D photolithography demonstrate that a relationship exists between polymer length and GBP limit of detection.
Results and discussion
Photochemical 3D lithography
Inspired by the thiol–ene reaction,15b,16 which has been used successfully in the context of carbohydrate immobilization,17 we turned to the thiol–acrylate photopolymerization as a potential route towards creating glycan-functionalized polymer brushes with pendant carbohydrate side groups because of (1) the compatibility of the radical reaction mechanism with the functional groups common to carbohydrates,18 and (2) the ability to induce the reaction with light made it an appropriate choice for high-throughput processing with BPL. In the presence of the photoinitiator 2,2-dimethoxy-2-phenyl-acetophenone (DMPA), UV light induces photopolymerization of acrylate or methacrylate monomers from a thiol initiator.15 By combining this polymerization with BPL arrays, control over feature diameter, feature height and feature spacing was possible. Moreover, the ease with which acrylate and methacrylate monomers are functionalized suggests that this approach could find broad use beyond glycan arrays. However, the photoinduced thiol-initiated acrylate polymerization has not been explored extensively as a tool to synthesize brush polymers, and there are no examples of glycan brush polymers prepared by the thiol-initiated acrylate radical photopolymerization. Therefore before glycan arrays could be synthesized, it was necessary to first develop methods to pattern brush polymers by combining the acrylate photopolymerization reaction with BPL.
The ability to combine PPL or BPL with acrylate photopolymerization to create micropatterns of brush polymers was investigated by depositing rhodamine-containing acrylate molecule 1 (ref. 17b) and irradiating with UV light. For comparison, a surface monolayer was formed by a UV initiated thiol–ene reaction of the thiol–rhodamine molecule 2 (ref. 17b) with an alkene terminated glass slide. In PPL patterning, an ink mixture containing the methacrylate or alkene molecules, DMPA and 2000 g mol−1 polyethylene glycol (PEG), was spin coated onto PPL pen arrays (8000 pens with 160 or 80 μm spacing between tips) that had been rendered hydrophilic by exposure to O2 plasma. The PEG was added to the ink mixture to assure uniform transport of the reactive components to the surface through the aqueous meniscus that forms between the tips and surface during patterning and subsequently prevent the spreading of ink following deposition.19 The PPL tip arrays were mounted onto an atomic force microscope (AFM) specially equipped with an XY tilting stage, an environmental chamber to control humidity, and lithography software to control the dwell time and z-piezo extension at each point in the pattern. The relative humidity was raised to 75–80%, and the ink was printed onto the surface by repeatedly bringing the tip arrays into contact with a thiol-20 or alkene-terminated21 glass slide, with dwell times varying from 50–100 000 ms. Following printing, the PPL-patterned surfaces were exposed to UV light (365 nm, 3 mW) for 3 h. After light exposure, the surfaces were washed with EtOH, H2O, and sonicated in EtOH, to remove unreacted molecules and the PEG matrix. Alternatively, to pattern by BPL (Fig. 1a), the same ink mixtures were spin coated onto BPL tip arrays (8000 tips with 100 μm spacing between tips), and the ink mixture was deposited under identical conditions as PPL printing. An important attribute of BPL is that the tips can be returned, with nanoscale fidelity, to any position in the pattern and locally irradiate the surface with precise dosages of light. To induce the photopolymerization by BPL, the tips were returned to the positions where ink was deposited, and the surface was irradiated through the apertures in the BPL array with exposure times of 2–20 min (365 nm, 90 mW). After patterning, the surfaces were washed with EtOH, H2O, and sonicated in EtOH, to remove unreacted molecules and the PEG matrix.
Fig. 1.

(a) Process for inducing the thiol–ene and (meth)acrylate polymerization reactions on a surface by beam pen lithography (BPL). (i) The tip-array, coated with an ink mixture (blue) containing the probe molecules and DMPA in a polyethylene glycol (PEG) matrix, deposits the ink mixture onto thiol-terminated glass slides. (ii) Light passing through the tips of the beam pens locally exposes the patterned surface to induce homolytic cleavage of the DMPA and initiate the polymerization of the monomers within the PEG matrix. (iii) Following rinsing of the surface to remove the PEG and excess molecules, only the covalently immobilized polymers remain. Methacrylate brush polymers are shown. (b) Molecules prepared for the BPL and polymer pen lithography (PPL)-induced thiol–ene and thiol–(meth)acrylate polymerization reactions.
The BPL induced radical photopolymerization is a new type of 3D scanning probe nanolithography22, where the feature diameter, feature height, feature position can all be controlled independently during printing. The photopolymerization is initiated by photochemical cleavage of the DMPA to form the thiol radical, which in turn initiates acrylate propagation. These thiol radicals, however, lack the stability of conventional radical initiators,23 and because the chain grows from the thiol, polymerization stops when the light is turned off. Because polymerization proceeds only during illumination, we reasoned that we could use this property of the thiol–acrylate photopolymerization to control brush polymer length by varying the dose of light delivered to each patterned feature. Previously, the thiol–ene reaction has been used to photopattern peptides, where the key advantage is that the bioorthogonal radical reactions between the methacrylate olefins and the thiols on the surface occur exclusively and do not damage the peptides.15b,24 In addition, the Hawker group has recently shown that photochemically induced atom-transfer radical addition chemistry can be used to create brush polymer gradients with control over height.23
In the current approach, feature position is controlled by the piezoactuator that moves the tip arrays, diameter is varied by the dwell-time between the pen arrays and the surface,2,25 and feature height is a function of the irradiation time. To demonstrate the control over feature diameter during thiol–ene and radical polymerization printing, fluorescent molecules 1 and 2 were patterned into a gradient by PPL with dwell times of 50–100 000 ms and subsequently irradiated for 3 h. Following washing, fluorescent patterns (λex = 532–587 nm, λem = 608–687 nm) were observed as a result of both the thiol–ene click photoreaction of 2 and the acrylate photopolymerization of 1, and both follow the expected linear relationship between the square root of dwell time and feature diameter.2,26 The normalized fluorescence intensity, defined as the peak fluorescence divided by the baseline fluorescence, was used to quantify fluorescence because of its relative independence of experimental conditions such as exposure time, light intensity, and optical path (Fig. 2a-c).27-29 Patterns of 2 had a normalized fluorescence intensity of 1.8 ± 0.1, and the intensity is independent of feature diameter, which is consistent with previous observations of fluorophore monolayers.8,28 In contrast, patterns of 1 (Fig. 2d-f) had a significantly higher normalized fluorescence intensity, 3.6 ± 0.2, which increases slightly with increasing dwell time. This increase in fluorescence intensity, compared to the monolayers prepared by the thiol–ene reaction, is attributed to the larger number of fluorophores that are immobilized in a feature that is composed of brush polymers that grow orthogonal to the surface. Using BPL, the ability to create patterns of brush polymers with varying heights was explored by printing 1 and varying the exposure time at the different positions in the array (Fig. 3). A 4 × 4 pattern of the ink mixture comprised of 1, PEG, and DMPA was printed with a dwell time of 1 s at each spot, resulting in features with a diameter of 2.41 ± 0.14 μm, and the spots were subsequently irradiated for 2, 5, 10 or 20 min. Following washing, the resulting patterns were analysed by fluorescence microscopy and AFM height imaging, which revealed linear increases in fluorescence intensity and height, respectively, with increasing UV exposure time (Fig. 3c). Control experiments further confirmed that patterns were the result of the thiol–ene and the acrylate reactions. When the inks were printed by either PPL or BPL, but not exposed to UV light, no patterns were visible by fluorescence microscopy after washing. Additionally, if the inks were printed onto bare glass surfaces, rather than alkene- or thiol-terminated surfaces and exposed to light, no patterns were observed.29 It should be noted that neither height nor fluorescence intensity are an accurate measure of polymer length, but the linear increases do suggest a chain-growth mechanism.15a,23,30 Chain growth, in combination with the inability of thiol-initiators to sustain polymerization in the absence of continuous irradiation, is essential for obtaining precise control over polymer chain length with exposure time. Although chain transfer is likely to occur, chain transfer does not affect the growth kinetics of brush polymers.15b,23,30a,31 Thus, 3D lithography capable of immobilizing polymers laden with functional side-chains – while directing feature diameter, feature height, and feature position with sub-micrometer resolution – is achieved by combining BPL with the photoinduced radical polymerization. Importantly, this is the first demonstration of patterning by first depositing material with a BPL array followed by exposure, and the first example of covalent photochemistry with BPL, showing that coordinated material deposition and photochemistry is possible through this technique.
Fig. 2.

(a) Fluorescence microscopy image (Nikon Eclipse Ti, λex = 532–587 nm, λem = 608–687 nm) of an array of 2 patterned by PPL on an alkene-terminated glass slide with dwell times of 50, 500, 5000, 50 000 and 100 000 ms. Inset shows the pattern prepared by a single tip. (b) Fluorescence intensity of the printed features. The x-axis corresponds to the white line from bottom to top in (a). (c) The relationship between the square root of dwell time and feature diameter during patterning of 2 by PPL. The cartoon represents the increasing feature size as a function of dwell time from left to right. Feature diameter has an average variation of 3%. (d) Fluorescence microscopy image (10×, λex = 532–587 nm, λem = 608–687 nm) of an array of 2 patterned by PPL on a thiol-terminated glass slide with dwell times of 50, 500, 5000, 50 000 and 100 000 ms. Inset shows the pattern prepared by a single tip. (e) Fluorescence intensity of features printed with a dwell time of 5 s. The x-axis corresponds to the white line from bottom to top in (d). (f) The relationship between the square root of dwell time and feature diameter during patterning of 1 by PPL. An average variation of 3% in diameter was observed. The cartoon represents the increasing feature size as a function of dwell time.
Fig. 3.

(a) Fluorescence image (Nikon Eclipse Ti, λex = 532–587 nm, λem = 608–687 nm) of an array of 1 printed by BPL with illumination times of 2, 5, 10 and 20 min. Inset shows an array prepared by a single tip. (b) Intensity profile of the features printed with different illumination times indicated by the white line in (a). Inset shows the cartoon image of the polymers formed on the surface. (c) The relationship between exposure time and fluorescence intensity or height. The variation in measured height was 5%, 1%, 7% and 43% from 2 min to 20 min, respectively.
Glycan arrays
Our primary aims in developing this new scanning probe lithography were to create a method to prepare glycan arrays with multivalent carbohydrates and to investigate how molecular architecture affects the binding affinity between patterned glycans and GBPs. The Bertozzi group has studied recently the binding of GBPs with microarrays composed of synthetic glycopolymers with varying glycan valency. They showed that arrays prepared with brush polymers of the highest saccharide valency had the strongest binding affinity with GBPs,7k consistent with previous studies of binding between synthetic polymers and GBPs in solution.10 So we expected, as an important validation of this new approach, that the high glycan valency of our brush polymers would increase significantly the binding towards ConA compared to arrays composed of glycan monolayers. One additional advantage of this new approach is that the pattern versatility enabled by combining BPL with in situ glycopolymer synthesis can be used to produce multiplexed arrays of polymers of systematically varied lengths on the same surface. Glucosides 3 and 4 and mannoside 5 were printed by either the thiol–ene or acrylate polymerization reactions onto thiol-terminated glass slides, and the resulting arrays were assayed against solutions containing different concentrations of the GBP concanavilin A (ConA). ConA was selected for these studies because it possesses many of the characteristic binding properties of GBPs that complicate the analysis of GBP–glycan interactions,7f including (1) multiple recognition sites, (2) cross specificity, and (3) low 1 : 1 binding affinity. Specifically ConA possesses two carbohydrate recognition domains separated by a distance of 6.8 nm and binds both α-mannosides (Ka = 103–104 M−1) and α-glucosides (Ka = 102 M−1), albeit with a preference for α-mannosides.32
Commercially available glucose methacrylate 3 was patterned into 4 × 4 dot patterns onto thiol-terminated glass surfaces using BPL tip arrays with a dwell time of 3 s at each spot, resulting in feature diameters of 3.97 ± 0.08 μm. The tips were returned to the positions where the inks were deposited, and the different spots were irradiated for exposure times of 2, 5, 10 or 20 min. The surfaces were washed and subsequently immersed in a solution of 1% BSA to reduce nonspecific protein adsorption on the surface21 and washed with PBS-Tween 20 (0.01 M PBS, 0.005% Tween 20, pH 7.4) and PBS (0.01 M PBS, pH 7.4) solutions. To assay binding against ConA, the substrates were immersed in a buffered solution of Cy3-labeled ConA (Cy3-ConA) (21.7 μM, 0.5 mg mL−1) for 4 h at 4 °C. The surfaces were then washed with PBS-Tween 20 and PBS solutions to remove unbound protein, and the binding of the fluorescent Cy3-ConA to the glycans in the arrays was determined by fluorescence microscopy (Fig. 4). Glycan arrays were also prepared with alkene–mannoside 5 (ref. 33) and alkene–glucoside 4 (ref. 34) on thiol-terminated glass slides by the PPL-induced thiol–ene reaction, which will form monolayers of glycans on the thiol-terminated surfaces, and their binding to Cy3-ConA was also measured. The binding data of the glycan polymers and glycan monolayers were also compared to our previously reported α-mannoside arrays that were prepared by using PPL to induce the CuAAC reaction on azide-terminated glass surfaces.8 As a control, the arrays were exposed to rhodamine-labeled peanut agglutinin (PNA), which is a galactose specific GBP that does not bind either α-glucosides or α-mannosides,35 to show that the observed patterns are not the result of nonspecific absorption.
Fig. 4.

(a) Fluorescence image (Nikon Eclipse Ti, λex = 532–587 nm, λem = 608–687 nm) of an array of 3 printed by BPL with illumination times of 2, 5, 10 and 20 min. Following washing, the surfaces were exposed to a 21.7 μM solution of Cy3-labeled ConA. Inset shows an array prepared by a single tip. (b) Binding of Cy3-ConA to glycan arrays prepared using different surface chemistries and technologies, including α-mannose immobilized by the CuAAC reaction by PPL (green), α-glucose by the acrylate polymerization of 3 by BPL (red), thiol–ene of α-mannose alkene by PPL (blue), thiol–ene reaction with 4 by PPL (purple), and binding of the acrylate surface of 3 with PNA (black) by PPL.
Upon exposing the arrays of 3 printed by the BPL-induced methacrylate photopolymerization to a 21.7 μM solution of Cy3-ConA (Fig. 4a), the brush polymers prepared with a 20 min exposure time displayed a normalized fluorescence intensity of 17.8 ± 1.0. Under the same conditions, monolayers of mannosides prepared by the thiol–ene or the CuAAC click reactions had normalized fluorescence intensities of 1.9 ± 0.1 and 1.50 ± 0.3, respectively, where a normalized fluorescence intensity of 1.0 indicates no binding. The monolayers of glucoside 4 prepared by the thiol–ene reaction at this concentration had a normalized fluorescence intensity of 1.3 ± 0.1, which is lower than the value obtained for arrays of the mannoside 5, and no binding was observed at lower concentrations of Cy3-ConA to monolayers of 4, which is consistent with the preference of ConA for α-mannosides over α-glucosides.32 Despite this preference, the signal for the patterns of the multivalent glucoside 3 that were prepared by the acrylate photopolymerization are approximately 20 times greater than those obtained with α-mannoside monolayers by the thiol–ene and the CuAAC reactions. To explore this effect further, the arrays of 3 and 4 were exposed to solutions of varying concentrations of Cy3-ConA (5.4, 3.0, 1.7 and 0.43 μM) (Fig. 4b). With the monolayer patterns of 4 prepared by the thiol–ene reaction, a normalized fluorescence of 1.7 ± 0.1 was observed at 5.4 μM−1, and no appreciable signal was detectable at lower concentrations. In contrast, arrays of 3 prepared by the BPL-induced thiol–acrylate photopolymerization decreased steadily with decreasing Cy3-ConA concentration, but still had appreciable signal at 0.43 μM, which is at least one order of magnitude more sensitive than the glycan monolayers prepared by the thiol–ene reaction, and nearly two orders of magnitude more sensitive than α-mannoside monolayers prepared by the CuAAC click reaction. In all cases, no binding to PNA was observed,29 confirming that the observed binding was the result of specific interactions between the proteins and the immobilized glycans.
The role of chain length on binding was explored by determining the detection threshold of the different brush polymers of 3, whose lengths were varied by altering the UV irradiation time. To first show that the chain lengths do indeed vary with irradiation time, the height of the glycan arrays were measured by AFM height imaging in non-contact mode before and after Cy3-ConA binding (Fig. 5a). The heights of the features printed with different exposures times could not be measured accurately prior to Cy3-ConA binding, presumably because they were adsorbed onto the surfaces. The feature heights were readily determined following Cy3-ConA binding (Fig. 5b) because the brush polymer–ConA aggregates form rods. We hypothesize this occurs following ConA binding because of the increased steric crowding that forces the chains to lengthen (Fig. 5c), although the degree to which they straighten could not be determined precisely. The feature heights of the ConA–brush polymer aggregates increase approximately linearly with exposure time, further supporting the chain growth polymerization mechanism, with the heights ranging from 8.0 ± 0.8 nm for a 2 min exposure time to 136 ± 3 nm for a 20 min exposure time. Moreover, the chain length significantly impacts the limit of detection of ConA, which is consistent with previous studies of binding to glycopolymers (Fig. 5d).11g While the tallest brush polymers were able to detect binding at concentrations as low as 0.43 μM, binding sensitivity decreased with decreasing height. The shortest polymers, by contrast, did not exhibit detectable binding towards Cy3-ConA below 3 μM. The two-to-threefold difference in fluorescence intensity between the long and short polymers when exposed to Cy3-ConA is likely a reflection of the higher number of glucose epitopes on the spots with longer polymers, and it remains to be determined whether the Cy3-ConA can penetrate the dense polymer brushes.36 In the context of the current study, we cannot determine whether the differences in fluorescence intensity arise from statistical or multivalent effects. Instead, we can only conclude that the limit of detection and signal intensity depend upon polymer chain length. The role of polymer valency, polymer spacing, and cooperativity on binding to these glycopolymer arrays are the subject of ongoing studies.
Fig. 5.

(a) AFM height image of a 4 × 4 pattern of 3 bound to Cy3-ConA. (b) The height profile of the features printed with different illumination times indicated by the white line in (a). (c) Cartoon image shows our hypothesis for why the height increased after Cy3-ConA was bound to the sugar. (d) Binding of Cy3-ConA to glycan arrays prepared using surface initiated acrylate polymerization by BPL with different UV exposure times, 20 min (black), 10 min (red), 5 min (blue) and 2 min (green).
Conclusion
We have combined thiol–acrylate photopolymerization chemistry with cantilever-free scanning probe lithography as a new approach towards creating brush-polymer microarrays, over large areas, with nanometer-scale control over feature position, diameter, and height. The novelty of this work does not lie in the discovery of new reactions or new lectin binding modes but rather in the combination of known polymer science with emerging nanolithographic capabilities afforded by BPL to synthesize glycopolymers, whose binding are consistent with previous reports. From the perspective of performing combinatorial chemistry at the tip of a scanning probe, this work validates three important concepts: (1) the same tips can be used to pattern materials and direct light to them to drive photochemistry, (2) combinatorial covalent photochemistry can be performed using BPL, and (3) arrays of biomaterials constructed through these approaches can be used to address challenging scientific problems, in this case understanding GBP binding. Multiplexing can potentially be accomplished by carrying out this same chemistry within a microfluidic cell capable of introducing different monomers. In addition, the chemical scheme for synthesizing brush polymers is broadly applicable as functionalized acrylate and methacrylate monomers are widely available or easily prepared, and the radical chain growth mechanism is compatible with a wide range of functional groups. While in these studies, every pen wrote a duplicate of the same arrangement of features, considering the recent advances in individual actuation in cantilever-free scanning probe lithography,4 one can imagine using these methods to generate centimeter-scale arrays consisting of nanoscale features in which the chemical conditions are exquisitely tuned. Therefore, this combination of localized photochemistry and functional-group tolerant chemistry could find applications in diverse problems in biology, materials chemistry, and organic electronics.
Supplementary Material
Acknowledgements
A.B.B. is grateful to the Air Force Office of Scientific Research (Young Investigator Award FA9550-13-1-0188) and the National Science Foundation (DBI-1340038). C.A.M is grateful for generous support from the following awards: AFOSR FA9550-12-1-0141, AFOSR FA9550-12-1-0280, NSF DBI-1152139, AOARD FA2386-10-1-4065, DARPA/MTO N66001-08-1-2044, and the Chicago Biomedical Consortium/Searle Funds at The Chicago Community Trust C2006-00997/L-003 for generous support. K.A.B. gratefully acknowledges support from Northwestern University's International Institute for Nanotechnology. The research was supported by the Center for Cancer Nanotechnology Excellence (CCNE) initiative of the National Institutes of Health (NIH) under Award No. U54 CA151880.
Footnotes
Electronic supplementary information (ESI) available: Organic synthesis, microfabrication and lithography, fluorescence binding studies, atomic force microscopy control experiments. See DOI: 10.1039/c3sc53315h
Notes and references
- 1.(a) Braunschweig AB, Huo FW and Mirkin CA, Nat. Chem, 2009, 1, 353; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Giam LR and Mirkin CA, Angew. Chem., Int. Ed, 2011, 50, 7482. [DOI] [PubMed] [Google Scholar]
- 2.Huo FW, Zheng ZJ, Zheng GF, Giam LR, Zhang H and Mirkin CA, Science, 2008, 321, 1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huo FW, Zheng GF, Liao X, Giam LR, Chai JA, Chen XD, Shim WY and Mirkin CA, Nat. Nanotechnol, 2010, 5, 637. [DOI] [PubMed] [Google Scholar]
- 4.(a) Brown KA, Eichelsdoerfer DJ and Mirkin CA, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom, 2013, 31, 06F201; [Google Scholar]; (b) Liao X, Brown KA, Schmucker AL, Liu G, He S, Shim W and Mirkin CA, Nat. Commun, 2013, 4, 2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Zheng ZJ, Daniel WL, Giam LR, Huo FW, Senesi AJ, Zheng GF and Mirkin CA, Angew. Chem., Int. Ed, 2009, 48, 7626; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brinkmann F, Hirtz M, Greiner AM, Weschenfelder M, Waterkotte B, Bastmeyer M and Fuchs H, Small, 2013, 9, 3266. [DOI] [PubMed] [Google Scholar]
- 6.Giam LR, Massich MD, Hao LL, Wong LS, Mader CC and Mirkin CA, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Essentials of Glycobiology, ed. Varki A, Cummings R, Esko J, Freeze H, Hart G and Marth J, Cold Spring Harbor Laboratory Press, Plainview, NY, 1999; [PubMed] [Google Scholar]; (b) Koeller KM and Wong CH, Nat. Biotechnol, 2000, 18, 835; [DOI] [PubMed] [Google Scholar]; (c) Bertozzi CR and Kiessling LL, Science, 2001, 291, 2357; [DOI] [PubMed] [Google Scholar]; (d) Blixt O, Head S, Mondala T, Scanlan C, Huflejt ME, Alvarez R, Bryan MC, Fazio F, Calarese D, Stevens J, Razi N, Stevens DJ, Skehel JJ, van Die I, Burton DR, Wilson IA, Cummings R, Bovin N, Wong CH and Paulson JC, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 17033; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Dube DH and Bertozzi CR, Nat. Rev. Drug Discovery, 2005, 4, 477; [DOI] [PubMed] [Google Scholar]; (f) Manimala JC, Roach TA, Li ZT and Gildersleeve JC, Angew. Chem., Int. Ed, 2006, 45, 3607; [DOI] [PubMed] [Google Scholar]; (g) Paulson JC, Blixt O and Collins BE, Nat. Chem. Biol, 2006, 2, 238; [DOI] [PubMed] [Google Scholar]; (h) Song EH and Pohl NLB, Curr. Opin. Chem. Biol, 2009, 13, 626; [DOI] [PubMed] [Google Scholar]; (i) Oyelaran O and Gildersleeve JC, Curr. Opin. Chem. Biol, 2009, 13, 406; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kiessling LL and Splain RA, Annu. Rev. Biochem, 2010, 79, 619; [DOI] [PubMed] [Google Scholar]; (k) Godula K and Bertozzi CR, J. Am. Chem. Soc, 2012, 134, 15732; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kennedy DC, Grunstein D, Lai CH and Seeberger PH, Chem.–Eur. J, 2013, 19, 3794. [DOI] [PubMed] [Google Scholar]
- 8.Bian S, He JJ, Schesing KB and Braunschweig AB, Small, 2012, 8, 2000. [DOI] [PubMed] [Google Scholar]
- 9.(a) Kolb HC, Finn MG and Sharpless KB, Angew. Chem., Int. Ed, 2001, 40, 2004; [DOI] [PubMed] [Google Scholar]; (b) Kohn M, Wacker R, Peters C, Schroder H, Soulere L, Breinbauer R, Niemeyer CM and Waldmann H, Angew. Chem., Int. Ed, 2003, 42, 5830. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kanai M, Mortell KH and Kiessling LL, J. Am. Chem. Soc, 1997, 119, 9931; [Google Scholar]; (b) Mann DA, Kanai M, Maly DJ and Kiessling LL, J. Am. Chem. Soc, 1998, 120, 10575; [Google Scholar]; (c) Mammen M, Choi SK and Whitesides GM, Angew. Chem., Int. Ed, 1998, 37, 2754; [DOI] [PubMed] [Google Scholar]; (d) Houseman BT and Mrksich M, Top. Curr. Chem, 2002, 218, 1; [Google Scholar]; (e) Gestwicki JE, Cairo CW, Strong LE, Oetjen KA and Kiessling LL, J. Am. Chem. Soc, 2002, 124, 14922; [DOI] [PubMed] [Google Scholar]; (f) Lundquist JJ and Toone EJ, Chem. Rev, 2002, 102, 555; [DOI] [PubMed] [Google Scholar]; (g) Zhang YL, Li QA, Rodriguez LG and Gildersleeve JC, J. Am. Chem. Soc, 2010, 132, 9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Fazio F, Bryan MC, Blixt O, Paulson JC and Wong CH, J. Am. Chem. Soc, 2002, 124, 14397; [DOI] [PubMed] [Google Scholar]; (b) Lee MR and Shin I, Org. Lett, 2005, 7, 4269; [DOI] [PubMed] [Google Scholar]; (c) Chevolot Y, Bouillon C, Vidal S, Morvan F, Meyer A, Cloarec JP, Jochum A, Praly JP, Vasseur JJ and Souteyrand E, Angew. Chem., Int. Ed, 2007, 46, 2398; [DOI] [PubMed] [Google Scholar]; (d) Pei Z, Yu H, Theurer M, Walden A, Nilsson P, Yan M and Ramstrom O, ChemBioChem, 2007, 8, 166; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang D, Carroll GT, Turro NJ, Koberstein JT, Kovac P, Saksena R, Adamo R, Herzenberg LA and Steinman L, Proteomics, 2007, 7, 180; [DOI] [PubMed] [Google Scholar]; (f) Michel O and Ravoo BJ, Langmuir, 2008, 24, 12116; [DOI] [PubMed] [Google Scholar]; (g) Godula K, Rabuka D, Nam KT and Bertozzi CR, Angew. Chem., Int. Ed, 2009, 48, 4973; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Weinrich D, Kohn M, Jonkheijm P, Westerlind U, Dehmelt L, Engelkamp H, Christianen PC, Kuhlmann J, Maan JC, Nusse D, Schroder H, Wacker R, Voges E, Breinbauer R, Kunz H, Niemeyer CM and Waldmann H, ChemBioChem, 2010, 11, 235. [DOI] [PubMed] [Google Scholar]
- 12.Godula K and Bertozzi CR, J. Am. Chem. Soc, 2010, 132, 9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roach TA, Li ZT and Gildersleeve JC, Glycobiology, 2008, 18, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vázquez-Dorbatt V, Lee J, Lin E-W and Maynard HD, ChemBioChem, 2012, 13, 2478. [DOI] [PubMed] [Google Scholar]
- 15.(a) Cramer NB and Bowman CN, J. Polym. Sci., Part A: Polym. Chem, 2001, 39, 3311; [Google Scholar]; (b) Hoyle CE and Bowman CN, Angew. Chem., Int. Ed, 2010, 49, 1540. [DOI] [PubMed] [Google Scholar]
- 16.(a) Hoyle CE, Lee TY and Roper TJ, J. Polym. Sci., Part A: Polym. Chem, 2004, 42, 5301; [Google Scholar]; (b) Dondoni A, Angew. Chem., Int. Ed, 2008, 47, 8995; [DOI] [PubMed] [Google Scholar]; (c) Kade MJ, Burke DJ and Hawker CJ, J. Polym. Sci., Part A: Polym. Chem, 2010, 48,743; [Google Scholar]; (d) Gupta N, Lin BF, Campos LM, Dimitriou MD, Hikita ST, Treat ND, Tirrell MV, Clegg DO, Kramer EJ and Hawker CJ, Nat. Chem, 2012, 4, 424. [DOI] [PubMed] [Google Scholar]
- 17.(a) Mehlich J and Ravoo BJ, Org. Biomol. Chem, 2011, 9, 4108; [DOI] [PubMed] [Google Scholar]; (b) Vico RV, Voskuhl J and Ravoo BJ, Langmuir, 2011, 27, 1391; [DOI] [PubMed] [Google Scholar]; (c) Wojcik F, O'Brien AG, Gotze S, Seeberger PH and Hartmann L, Chem.–Eur. J, 2013, 19, 3090. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hutchison JB, Stark PF, Hawker CJ and Anseth KS, Chem. Mater, 2005, 17, 4789; [Google Scholar]; (b) Pyun J and Matyjaszewski K, Chem. Mater, 2001, 13, 3436. [Google Scholar]
- 19.Huang L, Braunschweig AB, Shim W, Qin LD, Lim JK, Hurst SJ, Huo FW, Xue C, Jong JW and Mirkin CA, Small, 2009, 6, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halliwell CM and Cass AE, Anal. Chem, 2001, 73, 2476. [DOI] [PubMed] [Google Scholar]
- 21.Wendeln C, Heile A, Arlinghaus HF and Ravoo BJ, Langmuir, 2010, 26, 4933. [DOI] [PubMed] [Google Scholar]
- 22.(a) Zhou F, Zheng Z, Yu B, Liu W and Huck WT, J. Am. Chem. Soc, 2006, 128, 16253; [DOI] [PubMed] [Google Scholar]; (b) Benetti EM, Acikgoz C, Sui XF, Vratzov B, Hempenius MA, Huskens J and Vancso GJ, Adv. Funct. Mater, 2011, 21, 2088; [Google Scholar]; (c) Zhou X, Wang X, Shen Y, Xie Z and Zheng Z, Angew. Chem., Int. Ed, 2011, 50, 6506. [DOI] [PubMed] [Google Scholar]
- 23.(a) Poelma JE, Fors BP, Meyers GF, Kramer JW and Hawker CJ, Angew. Chem., Int. Ed, 2013, 52, 6844; [DOI] [PubMed] [Google Scholar]; (b) Fors BP, Poelma JE, Menyo MS, Robb MJ, Spokoyny DM, Kramer JW, Waite JH and Hawker CJ, J. Am. Chem. Soc, 2013, 135, 14106; [DOI] [PubMed] [Google Scholar]; (c) Khire VS, Benoit DSW, Anseth KS and Bowman CN, J. Polym. Sci., Part A: Polym. Chem, 2006, 44, 7027. [Google Scholar]
- 24.Jonkheijm P, Weinrich D, Koehn M, Engelkamp H, Christianen PCM, Kuhlmann J, Maan JC, Nuesse D, Schroeder H, Wacker R, Breinbauer R, Niemeyer CM and Waldmann H, Angew. Chem., Int. Ed, 2008, 47, 4421. [DOI] [PubMed] [Google Scholar]
- 25.Liao X, Braunschweig AB, Zheng ZJ and Mirkin CA, Small, 2009, 6, 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Piner RD, Zhu J, Xu F, Hong SH and Mirkin CA, Science, 1999, 283, 661; [DOI] [PubMed] [Google Scholar]; (b) Weeks BL, Noy A, Miller AE and De Yoreo JJ, Phys. Rev. Lett, 2002, 88, 255505; [DOI] [PubMed] [Google Scholar]; (c) Giam LR, Wang YH and Mirkin CA, J. Phys. Chem. A, 2009, 113, 3779. [DOI] [PubMed] [Google Scholar]
- 27.Cook NP, Torres V, Jain D and Marti AA, J. Am. Chem. Soc, 2011, 133, 11121. [DOI] [PubMed] [Google Scholar]
- 28.(a) Braunschweig AB, Senesi AJ and Mirkin CA, J. Am. Chem. Soc, 2009, 131, 922; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bian S, Schesing KB and Braunschweig AB, Chem. Commun, 2012, 48, 4995. [DOI] [PubMed] [Google Scholar]
- 29.See ESI†.
- 30.(a) Husseman M, Malmstrom EE, McNamara M, Mate M, Mecerreyes D, Benoit DG, Hedrick JL, Mansky P, Huang E, Russell TP and Hawker CJ, Macromolecules, 1999, 32, 1424; [Google Scholar]; (b) Matyjaszewski K, Miller PJ, Shukla N, Immaraporn B, Gelman A, Luokala BB, Siclovan TM, Kickelbick G, Vallant T, Hoffmann H and Pakula T, Macromolecules, 1999, 32, 8716. [Google Scholar]
- 31.(a) Rydholm AE, Held NL, Bowman CN and Anseth KS, Macromolecules, 2006, 39, 7882; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Polizzotti BD, Fairbanks BD and Anseth KS, Biomacromolecules, 2008, 9, 1084. [DOI] [PubMed] [Google Scholar]
- 32.(a) Ambrosi M, Cameron NR and Davis BG, Org. Biomol. Chem, 2005, 3, 1593; [DOI] [PubMed] [Google Scholar]; (b) Liang PH, Wang SK and Wong CH, J. Am. Chem. Soc, 2007, 129, 11177; [DOI] [PubMed] [Google Scholar]; (c) Bittiger HS and Schnebli HP, Concanavalin A as a Tool, John Wiley & Sons, London, New York, 1976. [Google Scholar]
- 33.Cumpstey I, Butters TD, Tennant-Eyles RJ, Fairbanks AJ, France RR and Wormald MR, Carbohydr. Res, 2003, 338, 1937. [DOI] [PubMed] [Google Scholar]
- 34.Ruiz JRJ, Osswald G, Petersen M and Fessner WD, J. Mol. Catal. B: Enzym, 2001, 11, 189. [Google Scholar]
- 35.Pendland JC and Boucias DG, Eur. J. Cell Biol, 1993, 60, 322. [PubMed] [Google Scholar]
- 36.Meng XL, Fang Y, Wan LS, Huang XJ and Xu ZK, Langmuir, 2012, 28, 13616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.