

Summary
Protein degradation technologies represent a powerful functional genomics tool, allowing fast and controllable target protein depletion. Establishing these systems requires a knock-in of the degradation tag into both endogenous target gene alleles. Here, we provide a step-by-step protocol for the efficient generation of biallelic degradation tag knock-ins in mouse and human cell lines using CRISPR-Cas9. We use knockin of an endogenous Kansl3 degradation tag in mouse embryonic stem (ES) cells as an example but provide modifications for application in other cell types.
For complete details on the use and execution of this protocol, please refer to Radzisheuskaya et al. (2021).
Subject areas: CRISPR, Genetics, Molecular Biology
Graphical Abstract
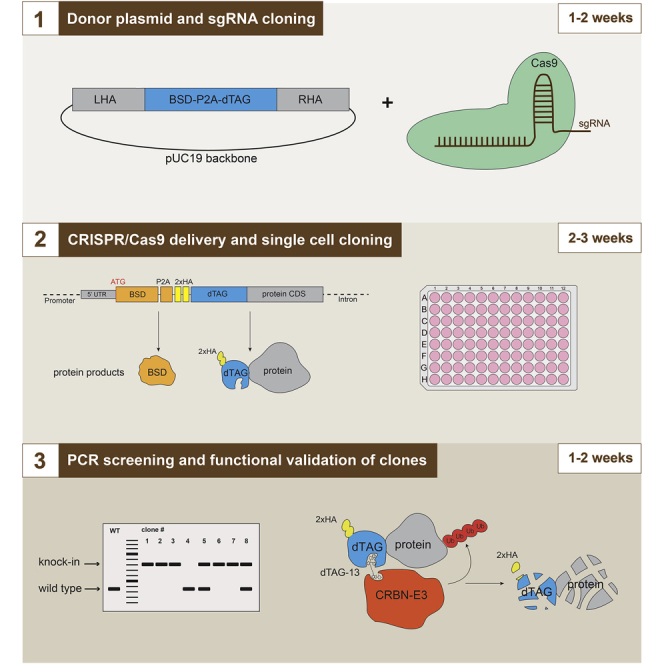
Highlights
-
•
Efficient generation of biallelic degradation tag knock-ins in mouse and human cells
-
•
Extensive advice on donor plasmid design and ligation-free vector construction
-
•
Recommendations for functional validation of the degron-fused protein
Protein degradation technologies represent a powerful functional genomics tool, allowing fast and controllable target protein depletion. Establishing these systems requires a knock-in of the degradation tag into both endogenous target gene alleles. Here, we provide a step-by-step protocol for the efficient generation of biallelic degradation tag knock-ins in mouse and human cell lines using CRISPR-Cas9. We use knock-in of an endogenous Kansl3 degradation tag in mouse ES cells as an example but provide modifications for application in other cell types.
Before you begin
Timing: 4–5 days
The protocol below describes generation of biallelic knock-in cell lines expressing a protein of interest fused to a FKBP12F36V degradation tag (dTAG). To study the immediate consequences of protein loss, we routinely utilize this degron system due to its superior selectivity and ease of use over other degradation tags, as it co-opts the endogenous E3 ubiquitin ligase system for target protein degradation and does not require overexpression of exogenous components (Nabet et al., 2018). In such a system, addition of a small molecule, heterobifunctional dTAG ligand (e.g., dTAG-13 or dTAGV-1), to the cell culture medium recruits E3 ligases, Cereblon (CRBN) or von Hippel-Lindau (VHL) respectively, leading to rapid polyubiquitination and proteasomal degradation of the tagged target protein (Figure 1). Both CRBN and VHL are widely expressed proteins. However, if not expressed in the cellular model of interest, one can consider an auxin-inducible degron (Nishimura et al., 2009) (Natsume et al., 2016) or a SMASh-tag (Chung et al., 2015).
Figure 1.

Schematic representation of the dTAG-KANSL3 degron system
To determine which target protein terminus to tag in order to best preserve protein function , one should survey previous experimental evidence with epitope tags on the protein of interest. If such data are lacking, avoid a terminus with a critical functional domain. In the protocol below, we tagged the N-terminus of mouse Kansl3 based on our previous observation that addition of an N-terminal triple FLAG-tag fully preserved KANSL3 protein functionality.
We have observed that adding a degron-tag to either protein terminus can lead to lower protein expression, which most likely is due to protein degradation in the absence of the dTAG-ligand. Such auto-degradation is target- and cell line-dependent and difficult to predict a priori. If the cDNA for the protein of interest is available, it is worthwhile to test beforehand if the C-terminal and N-terminal dTAG-fusion versions of the protein are stably expressed in the cell line of interest while still preserving their function. Gateway cloning-compatible lentiviral expression vectors to create such fusions are available at Addgene (catalog numbers 91797 and 91798).
The protocol below describes specific steps for creating degradation tag knock-ins using mouse ES cells with the option of either plasmid-based delivery of Cas9/sgRNA via lipofection or Cas9/gRNA ribonucleoprotein (RNP) complex delivery via nucleofection. We have also used this protocol in a variety of mouse and human primary and cancer cell lines and describe the necessary protocol modifications depending on the cell type of interest.
-
1.
Design and order the necessary primers.
All primers in this protocol were designed using NCBI Primer Designing Tool – NIH (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and are listed in the Key Resources Table. The primers can be ordered from any manufacturer without enhanced purification.
Note: This protocol is applicable to mouse ES cells cultured in either serum/LIF or 2i/LIF conditions. Here we describe serum/LIF culture as an example (see medium composition in the materials and equipment section). For detailed instructions on culturing mouse ES cells in 2i/LIF conditions refer to (Mulas et al., 2019).
-
2.
Establish a culture of mycoplasma-negative low passage mouse ES cells without signs of spontaneous cell differentiation. Grow cells on 0.1% gelatin and passage every 2–3 days using trypsin at a density of 1.5–3.0 X 104 cells/cm2.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-KANSL3 | Sigma | Cat# HPA035018, RRID: AB_10601763 |
| Anti-HA | Cell Signaling | Cat# 3724 S, RRID: AB_1549585 |
| Anti-GAPDH | Abcam | Cat# ab181602, RRID: AB_2630358 |
| IRDye® 800CW Goat anti-Rabbit IgG | LI-COR Biosciences | Cat# 925-32211, RRID: AB_2651127 |
| IRDye® 680RD Goat anti-Mouse IgG | LI-COR Biosciences | Cat# 926-68070, RRID: AB_10956588 |
| Bacterial and virus strains | ||
| DH5alpha chemically competent cells | Lab prepred or Thermo Fisher | 18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| dTAGV-1 | Tocris | 6914 |
| dTAG-13-NEG | Tocris | 6916 |
| dTAG-13 | Tocris | 6605 |
| Blasticidin S HCl, powder | Thermo Fisher | R21001 |
| GlutaMAX | Thermo Fisher | 35050061 |
| 2-Mercaptoethanol | Thermo Fisher | 21985023 |
| Sodium pyruvate (100 mM) | Thermo Fisher | 11360070 |
| MEM Non-Essential Amino Acids Solution (100×) | Thermo Fisher | 11140050 |
| 10,000 I.U./mL Penicillin - 10,000 μg/mL Streptomycin (100×) | MSK | N/A |
| LIF | Lab prepared | N/A |
| Critical commercial assays | ||
| KAPA HiFi HotStart ReadyMix (2×) | Roche | KK2602 |
| Eco53kI | NEB | R0116S |
| Calf Intestinal Alkaline Phosphatase (CIP) | NEB | M0290 |
| QIAquick PCR & Gel Cleanup Kit (100) | QIAGEN | 28506 |
| In-Fusion® HD Cloning Plus | Takara | 638911 |
| QIAprep Spin Miniprep Kit (250) | QIAGEN | 27106 |
| X-Gal and IPTG Ready To Use Dropper | BioWorld | 21530077-1 |
| Taq DNA Polymerase Kit (1000 U) | QIAGEN | 201205 |
| BbsI-HF | NEB | R3539S |
| Quick Ligation Kit | NEB | M2200S |
| Alt-R® CRISPR-Cas9 crRNA | IDT | custom sequence |
| Alt-R® CRISPR-Cas9 tracrRNA | IDT | 1072532 |
| Alt-R® S.p. Cas9 Nuclease V3 | IDT | 1081058 |
| SG Cell Line 4D-Nucleofector X Kit S | Lonza | V4XC-3032 |
| P3 Primary Cell 4D-Nucleofector X Kit S | Lonza | V4XP-3032 |
| FBS | MSK | N/A |
| Glasgow Minimum Essential Media | Sigma | G5154 |
| Proteinase K | Roche | 3115801001 |
| NP-40 Substitute | Fisher Scientific | 50-488-856 |
| Tween 20 | Fisher Scientific | BP337-500 |
| Lipofectamine 2000 | Thermo Fisher | 11668030 |
| Opti-MEM | Thermo Fisher | 31985088 |
| Experimental models: cell lines | ||
| E14 mouse embryonic stem cells | Joshua Brickman Lab | RRID: CVCL_C320 |
| Oligonucleotides | ||
| M13 RP: CAGGAAACAGCTATGAC | This study | N/A |
| M13 FP: TGTAAAACGACGGCCAGT | This study | N/A |
| blast-dTAG-amp FP: TGCTTGACTACTGACA TGGCCAAGCCTTTGTCTCA |
This study | N/A |
| blast-dTAG-amp RP: AGATCCGCCGCCACCCGACCC | This study | N/A |
| eGFP-dTAG-amp FP: TGCTTGACTACTGACAT GGTGTCTAAGGGCGAAGAG |
This study | N/A |
| eSpCas9 FP: GCATATACGATACAAGGCTG | This study | N/A |
| K3-ki gDNA FP: CGTCCTGTTTAGGGGACGA | This study | N/A |
| K3-ki gDNA RP: CATGCACTGAAAGCTGGAAGAG | This study | N/A |
| K3-ki-sgRNA FP: CACCGGAAGTCCCTCTCCCCACCC | This study | N/A |
| K3-ki-sgRNA RP: AAACGGGTGGGGAGAGGGACTTCC | This study | N/A |
| K3 left homology arm: tccccgggtaccgagACCTTTAGC TTTTGATGAATTCATGTTTGTGATCCCTCTGG TTTGCAAAAACCTTTACCACCCAGGAAGAAAA TTGCCCAGCAAATTTAGGTTTTTGTTTTTGAGATGGGA TCTCAATATATTGCTCTGGCTGTCCTGGAACCCACTC TGTAGACTAGGCTGCCCTTGAACTCCCGGAGATCCG TCTGCTTCCGCCTCCCCAGTGCTGGGATTAAAGGCG TGCGTCACCACGCCTTGGCAATTGCAAAGGTCATG TTTTTGTGAACTGTGGAGTCTTGTCAGTTTACAGTTA TAAAATTGTGTATGGCACCGTTCTGTTTGGTGGCGG TGTGGGAGGACACATAGATGTGCATAATCAAAGA TCGAATGCTGAGCATAAAGACATGTTTTGATAG TGAAAGTTGGGAGAAATGGAAGCTATTGAATTCA CAAGCCCCCTTTCACTGCAGGTGACTAGCA TGCAGATATTCACACCCTGACTTGGCTGCTTGACTACTGAC |
This study | N/A |
| K3 right homology arm: ggtggcggcggatctATGGCCCA TAGGGGTGGGGAGAGGGACTTCCAGACTTCAGC TCGGCGGATGGGTACCTCCCTGCTCTTCCAGC TTTCAGTGCATGAGCGGGAACTGGACCTCGTTTTCC TGGATCATAGCTATGCAAAACCGTGGAG TGCCCACCCAGATGCCAGTAGTGCCCGCCCC ACCCGCATGCTCTTTGTAACCCCCCG TCGGCAGCAGGAGAATACTATGTGAGTTTGAG AATACCATGTGAGTTTGGGTTTCCTTCTTAAGGAAGTGA TGCGTGGCGGGGCTTAGATGTTCTTAGGAGTGA TTGGGGCTGCAGGGTGAATGGAGAGCTTGGA TGAGCAGCATCAGAACGGCAGGGCAAAG TTGGACACCTACACAGACATTGCCAAACATGAG TTAGGAAAGGGAGATGCGCAAAGGAAAACAGCC TAGCTTGCTTAGTTCCCAGTAACTAGATAATTCCTTC TTCCCTTTACTGCAGCATCTAGTTTGTTTGGC AGTGGCTACAATctcgaattcactggc |
This study | N/A |
| Recombinant DNA | ||
| pUC19 | Addgene | 50005 |
| espCas9(1.1)-T2A-mCherry | A gift from Dr. Ian Chambers, Centre for Regenerative Medicine, University of Edinburgh | N/A |
| pCRIS-PITCH-BSD-P2A-dTAG | Addgene | 91792 |
| pLEX305-C-dTAG | Addgene | 91798 |
| pLEX305-N-dTAG | Addgene | 91797 |
| pUC19-eGFP-P2A-dTAG-Ing3 | Addgene | 170754 |
| pET28a-Cas9-His | Addgene | 98158 |
| pUC19_dTAG-KANSL3-targeting | Addgene | 170755 |
| Software and algorithms | ||
| SnapGene | SnapGene | https://www.snapgene.com/ |
| Image Studio Lite Software | LI-COR Biosciences | N/A |
| CHOPCHOP | http://chopchop.cbu.uib.no/ | N/A |
| Other | ||
| KANSL3 dTAG knock-in locus.gb | Mendeley Data: https://doi.org/10.17632/mjd8zdmr6h.1 | N/A |
Materials and equipment
Mouse ES cell culture medium composition: Glasgow Minimum Essential Media, 10% FBS, 1× Penicillin/Streptomycin, 2 mM GlutaMAX, 100 μM β-Mercaptoethanol, 1 mM sodium pyruvate, 0.1 mM MEM Non-Essential Amino Acids Solution and 1:500 homemade Leukemia Inhibitory Factor (LIF).
Lysis buffer for genomic DNA extraction: 25 mM KCl, 5 mM Tris pH 8.0, 1.25 mM MgCl2, 0.2% NP-40, 0.2% Tween 20, 0.4 μg/μL Proteinase K (freshly diluted from a 50× stock). Store lysis buffer without Proteinase K at 20°C–25°C.
Equipment: 4D-nucleofector X Unit (Lonza, AAF-1002X), 4D-nucleofector Core Unit (Lonza, AAF-1002B), Sony MA900 Cell sorter (Sony, MA900)
Suggested nucleofection programs (Amaxa 4D-nucleofector)
| Cell line | Nucleofection kit | Program |
|---|---|---|
| Mouse neural stem cells | SG Cell Line | DN-100 (2 pulses) |
| Human glioma stem cells | SG Cell Line | EN-138 |
| Mouse embryonic stem cells | P3 Primary Cell | CA-120 |
| Mouse hematopoietic stem cells | P3 Primary Cell | EO-100 |
| Mouse leukemic cells | P3 Primary Cell | EO-100 |
| THP-1 cells | SG Cell Line | FF-100 |
| OCI-AML3 cells | SG Cell Line | FF-100 |
Step-by-step method details
sgRNA design and cloning into eSpCas9(1.1)-T2A-mCherry expression vector
We design sgRNAs using the ChopChop tool (http://chopchop.cbu.uib.no/) by searching the intended knock-in site, including the upstream and downstream 100 bp nucleotide sequence, and selecting the appropriate genome for off-target scoring. Optimally, one should select a sgRNA that cuts the target sequence within 10–20 bp of the intended integration site and that has the lowest predicted off-target score. If performing a knock-in using nucleofection with preassembled Cas9/gRNA ribonucleoprotein (RNP) complex (see section 17), order a synthetic crRNA from a commercial vendor (e.g., IDT) and omit steps 1–8 and 16 in the protocol.
-
1.
sgRNA design
Design and order sgRNA oligos as follows:
Forward: 5’ CACC+”20 bp sgRNA sequence” 3’
Reverse: 5’ AAAC+”reverse complement of the 20 bp sgRNA sequence” 3’
Example for dTAG-KANSL3 knock-in generation:
Forward: CACC+GGAAGTCCCTCTCCCCACCC
Reverse: AAAC+GGGTGGGGAGAGGGACTTCC
The selected sgRNA induces cutting in the Kansl3 gene locus 8 bp after the START codon.
Note: In contrast to WT-SpCas9, eSpCas9(1.1) is sensitive to gRNA-DNA mismatches at the 5’end and it is, therefore, not advisable to add an extra G in front of the sgRNA sequence to ensure efficient transcription from the human U6 promoter (Zhang et al., 2017), as is common practice for WT-SpCas9 vectors. The 20 bp sgRNA sequence itself must start with G or A (A/GN19) (Wang et al., 2019; Zhang et al., 2017). Consider a delivery vector expressing WT-SpCas9 (such as PX330, Addgene catalog number 42230) or Cas9/gRNA RNP delivery, if the only available sgRNAs in close proximity to the intended knock-in site start with T or C.
-
2.
Digestion of eSpCas9(1.1)-T2A-mCherry vector for sgRNA cloning
Perform digestion at 37°C for at least 2 h, followed by purification of the digested vector backbone by gel electrophoresis on a 1% agarose gel using the QIAquick PCR & Gel Cleanup Kit. The expected backbone size is 9264 bp.
Note: Do not dephosphorylate the backbone as the phospho-groups on the vector backbone overhangs are required for successful cloning of the unphosphorylated sgRNA oligos.
| Component | Volume (μL) |
|---|---|
| 10× CutSmart buffer | 3 |
| BbsI-HF | 1 |
| 10 μg espCas9-mCherry vector (1 μg/ul) | 10 |
| ddH2O | 16 |
| Total | 30 |
-
3.
Oligo annealing
Prepare the following reaction in a PCR tube, heat in thermocycler for 5 min at 95°C and gradually ramp down to 20°C at 5°C/min.
| Component | Volume (μL) |
|---|---|
| NEB buffer 3 | 5 |
| sgRNA oligo top (100 μM) | 1 |
| sgRNA oligo bottom (100 μM) | 1 |
| ddH2O | 43 |
| Total | 50 |
Dilute annealed oligos 1:20 in ddH2O before ligation reaction.
-
4.
Ligation of the annealed sgRNA oligos into the eSpCas9-mCherry expression vector
Prepare the following reaction in a 0.2 mL PCR tube. Perform ligation at 20°C–25°C for 1 h using the Quick Ligation kit.
| Component | Volume (μL) |
|---|---|
| 2× Quick Ligation buffer | 5 |
| 50 ng BbsI-digested espCas9 vector (100 ng/μL) | 0.5 |
| Annealed sgRNA oligos (1:20 diluted) | 1 |
| Quick Ligase | 0.5 |
| ddH2O | 3 |
| Total | 10 |
Note: It is important to set up an empty ligation reaction with water instead of the annealed oligos. This serves as a control for incomplete backbone digestion.
-
5.
Bacterial transformation (day 1)
Transform 3 μL of the ligation reaction into a competent E. coli strain (e.g., DH5alpha) according to the protocol supplied by the manufacturer and plate the reaction on agar plates containing ampicillin for antibiotic selection. Competent DH5alpha bacteria can be purchased from many different vendors including ThermoFisher, NEB or Zymo Research.
-
6.
Pick colonies for plasmid preparations (day 2) (Troubleshooting 1)
Inoculate 1.5 mL of TB medium containing 100 μg/mL ampicillin with a single colony picked with a sterile pipette tip. Incubate and shake cultures at 37°C for 16–18 h.
Note: We use regular 2ml Eppendorf tubes for growing mini prep cultures. Make sure to make a hole in the lid of the tube with an 18G needle to allow sufficient air exchange during incubation. Incubation of the bacterial culture can be performed on a standard laboratory Thermomixer. The sgRNA cloning efficiency is generally high with much higher colony numbers in the cloning as opposed to the empty ligation plates. Picking two or three colonies is usually sufficient for identifying a plasmid with the correct sgRNA.
-
7.
Isolation of plasmid DNA (day 3)
Perform plasmid isolation using for instance a QIAprep spin miniprep kit according to the manufacturer’s instructions (https://www.qiagen.com/us/resources/download.aspx?id=22df6325-9579-4aa0-819c-788f73d81a09&lang=en).
-
8.
Validation of cloned sgRNAs
Verify the correctly cloned sgRNA sequence by Sanger sequencing with eSpCas9-forward primer 5' GCATATACGATACAAGGCTG 3'.
Donor plasmid design and construction
-
9.
Design donor plasmid following these guidelines:
A typical donor plasmid structure is presented in Figure 2.
Figure 2.

Generation of the donor plasmid by In-Fusion assembly
Any basic vector backbone can be used to construct the donor plasmid. We generally use pUC19 (Addgene catalog number 50005), as it allows blue-white screening of bacterial colonies for insertion of fragments after In-Fusion assembly. The pUC19 vector is linearized by digestion with restriction enzyme Eco53kI to prepare for the In-Fusion reaction, which assembles the donor plasmid consisting of the vector backbone, left and right homology arms (LHA and RHA) for homology directed repair (HDR), a selection marker and the dTAG degron tag. All fragments require a sequence homology of 15 bp for sequential In-Fusion assembly, which can be added by PCR amplification (Figure 2).
We generally use homology arms around 400–700 bp in length to generate knock-ins in mouse and human cell lines that are synthesized by IDT (gBlocks Gene Fragments service) or other manufacturers. However, some promoter regions have very high frequency of GC nucleotides, which precludes them from being synthesized. Furthermore, human cell lines from diverse origins can contain multiple single nucleotide polymorphisms (SNPs) in non-protein coding regions, which increases the risk of mismatches in the homology arms of the donor template causing reduced HDR efficiency. In these cases, we advise to amplify the homology arms from genomic DNA of the target cell line by PCR.
The efficiency of HDR differs between cell lines and having a good strategy to select for clones with correct integration can drastically decrease the time required for screening. We generally have good success generating endogenous dTAG knock-ins in diverse cell lines by using either an antibiotic selection marker (e.g., blasticidin/BSD, hygromycin, or neomycin) or a fluorescent protein (e.g., eGFP or tagBFP) allowing for fast and efficient selection of in-frame integrations (Figure 2). After successful knock-in, the selection marker and the dTAG-fused protein of interest are separated by a P2A protein self-cleavage site within one transcript, leading to production of two separate proteins: the selection marker as well as the dTAG-fusion protein (Figure 3). To allow for identification of biallelic integrations, two different selection cassettes can be used at the same time. In our experience, this works for most cell lines, except mouse ES cells, where we recover biallelic integrations with the same selection cassette in most clones.
Figure 3.

Targeting strategy chosen for the Kansl3 locus
A star indicates a silent mutation introduced into the Kansl3 open reading frame to prevent cutting of the modified locus and the targeting vector. CAC codon was changed to CAT (both encode histidine), CGG codon was changed to AGG (both encode arginine). In this case the sequence allowed to mutate two PAM nucleotides. This is often not possible without consequent amino acid change and introducing one mutation is usually sufficient.
It is important to ensure that Cas9 is unable to cut and edit the donor plasmid or the genomic locus once integration has occurred. Frequently, if the selected sgRNA overlaps with the start or stop codon, the binding sequence will be disrupted by the inserted DNA sequence. However, if the sgRNA sequence stays intact in the donor DNA after the integration, it is important to introduce silent mutations of the PAM site or the sgRNA seed region (8–10 bp upstream of the PAM site) to prevent Cas9 from cutting the donor plasmid and the final integrated cassette.
To simplify cell line validation and subsequent functional experiments, it is useful to include an epitope tag (e.g., HA or FLAG) as a part of the construct (Figure 3, sequence of edited Kansl3 dTAG knock-in locus available at Mendeley Data).
To generate the dTAG-KANSL3 knock-in donor vector, used as an example in this protocol, we selected the 500 bp genomic sequence before and after the start codon of Kansl3 and ordered both homology arms from IDT. The CGG PAM site of the sgRNA in the RHA was changed to CTA to prevent CRISPR editing of the donor construct. The DNA sequences of LHA and RHA are listed in Key Resources Table.
-
10.
Amplification of the selection cassette
PCR amplify either the BSD-P2A-dTAG cassette from pCRIS-PITCH-BSD-P2A-dTAG vector (Addgene catalog number 91792) or eGFP-P2A-dTAG from pUC19-eGFP-P2A-dTAG-Ing3 vector (Addgene catalog number 170754).-
a.Set up PCR reaction:
Component Volume (μL) 2× KAPA HiFi HotStart ReadyMix 25 10 μM forward primer (blast-dTAG-amp FP or eGFP-dTAG-amp FP) 1.5 10 μM reverse primer (blast-dTAG-amp RP) 1.5 10 ng template vector (10 ng/μL) 1 ddH2O 21 Total 50 -
b.Run PCR according to the following conditions:
PCR cycling conditions
Steps Temperature Time Cycles Initial Denaturation 95°C 3 min 1 Denaturation 98°C 20 s 25–30 Annealing 60°C 15 s Extension 72°C 30 s Final Extension 72°C 2 min 1 Hold 4°C forever
-
a.
After PCR amplification, load the reaction on a 0.8% agarose gel, excise the PCR product with the correct size (909 bp for BSD-P2A-dTAG, 1233 bp for eGFP-P2A-dTAG) and isolate DNA with a gel extraction kit according to manufacturer’s protocol (https://www.qiagen.com/cn/resources/download.aspx?id=a72e2c07-7816-436f-b920-98a0ede5159a&lang=en).
-
11.
Linearization of donor plasmid backbone with Eco53kI
Prepare reaction in a PCR tube and digest for 2 h at 37°C, followed by 20 min incubation at 65°C to heat-inactivate the restriction enzyme. Add 1 μL Calf Intestinal Alkaline Phosphatase (CIP) to the tube and incubate for one additional hour at 37°C. Proceed with loading the reaction on a 0.8% agarose gel, excise the linearized backbone and isolate backbone DNA from the gel (2686 bp) using the QIAquick PCR and gel cleanup kit. Freeze the linearized pUC19 vector backbone at −20°C and reuse for subsequent In-Fusion assembly reactions.
| Component | Volume (μL) |
|---|---|
| 10× CutSmart buffer | 3 |
| 10 μg pUC19 vector (1μg/μL) | 10 |
| Eco53kI | 1 |
| ddH2O | 16 |
| Total | 30 |
-
12.
In-Fusion HD cloning reaction for targeting vector assembly and transformation
The fragment amounts in the table below correspond to an insert to vector molar ratio of 2:1 and give very good results in In-Fusion assembly.-
a.Prepare the reaction below in a PCR tube, mix and incubate for 15 min at 50°C.
-
b.Transform 5 μL of the reaction into 1 vial of competent cells (e.g., DH5alpha, Stellar, or TOP10).
-
c.Add 2–3 drops of IPTG and X-gal solution onto LB-amp plate, spread equally and dry for 30 min.
-
d.Plate bacteria on LB-amp plates containing IPTG and X-gal for blue-white screening. pUC19 vectors that contain an insertion will appear white due to disruption of the lacZ gene.
Component Amount 5× In-Fusion HD master mix 2 μL pUC19 linearized vector backbone 50 ng Fragment 1 (LHA) 20 ng Fragment 2 (RHA) 20 ng Fragment 3 (BSD/eGFP-P2A-dTAG) 35 ng ddH2O to 10 μL Total 10 e.Optional: Perform diagnostic colony PCR
-
a.
Pick 5–10 white colonies and resuspend them in 10 μL of water. Incubate 5 μL of bacterial suspension at 98°C for 5 min and use as a template to set up a diagnostic PCR reaction. Save the other 5 μL of the bacterial suspension for inoculation of the positive clones.
One PCR primer should bind on the pUC19 backbone and the other on the inserted fragment. If the homology arms were prepared by PCR from genomic DNA, then this step can simultaneously select for colonies with correctly amplified homology arm sequence. We had good success with designing primers to yield different sized products for amplifying from the LHA, RHA and pUC19 backbone simultaneously.
-
i.
Set up diagnostic colony PCR reaction:
| Component | Volume (μL) |
|---|---|
| 10× Coral Load Buffer | 2 |
| 10 μM forward primer (e.g., M13 forward) | 0.4 |
| 10 μM reverse primer (e.g., RHA binding) | 0.4 |
| 10 mM dNTPs | 0.4 |
| Taq polymerase | 0.1 |
| Bacterial suspension | 5 |
| ddH2O | 11.7 |
| Total | 20 |
-
ii.
Run PCR according to the following conditions:
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 94°C | 5 min | 1 |
| Denaturation | 94°C | 30 s | 22 |
| Annealing | 55°C | 30 s | |
| Extension | 72°C | 30 s | |
| Final Extension | 72°C | 5 min | 1 |
| Hold | 4°C | Forever | |
-
13.
Inoculate at least 2 PCR-positive colonies into 1.5 mL LB or TB medium containing 100 μg/mL ampicillin. Incubate culture while shaking at 37°C for 16–18 h.
-
14.
Isolate plasmid DNA by using for instance a QIAprep spin miniprep kit according to the manufacturer’s instructions.
-
15.
Perform validation of donor plasmids by Sanger sequencing using M13 forward, M13 reverse and an internal primer. For BSD-P2A-dTAG-KANSL3 donor plasmid the following sequencing primers were utilized to span the whole insert: M13 forward 5' TGTAAAACGACGGCCAGT 3', M13 Reverse 5' CAGGAAACAGCTATGAC 3', and blast-dTAG-amp RP 5' AGATCCGCCGCCACCCGACCC 3'.
Delivery of sgRNA, Cas9, and the donor plasmid into the cells
For mouse ES cells we have achieved very good results expressing sgRNA/Cas9 from a plasmid and delivering all components via lipofection. However, other cell lines are difficult to transfect and furthermore display a lower propensity for integrating exogenous DNA via HDR compared to ES cells. In these cases, nucleofection with preassembled Cas9/gRNA ribonucleoprotein (RNP) complex produces superior knock-in results. We provide protocols for both options here.
-
16.Lipofection-based DNA delivery
-
a.Plate 5 X 105 mouse ES cells in 3 mL of media in a well of 6-well plate just before the lipofection.
-
b.For each transfection, prepare the following and mix each tube gently by flicking:
Tube A 2 μg donor plasmid 2 μg espCas9/sgRNA plasmid Opti-MEM to the final volume of 250 μL Tube B 240 μL Opti-MEM 10 μL Lipofectamine 2000 -
c.Incubate 5 min at RT (no more than 25 min).
-
d.Combine diluted DNA (Tube A) and Lipofectamine (Tube B) (total volume 500 μL). Mix gently by flicking the tube and incubate at RT for 20 min (solution may appear cloudy).
-
e.Add 500 μL of complexes to each well containing cells and medium. Rock the plate gently.
-
f.Change media the next day.Alternatives: If using Lipofectamine 3000 instead of Lipofectamine 2000, note that this is more toxic to mouse ES cells cultured in 2i/LIF conditions.
-
a.
-
17.Delivery of Cas9/gRNA ribonucleoproteins by nucleofection with Amaxa 4D-nucleofector (Troubleshooting 2)
-
a.Reconstitute synthetic Alt-R tracrRNA and crRNA in buffer (generally provided by manufacturer such as duplex buffer from IDT) at a concentration of 100 μM. Store reconstituted synthetic RNAs at −20°C.
-
b.To anneal tracrRNA/crRNA, mix an equal amount of 100 μM tracrRNA and crRNA in a PCR tube, heat to 95°C for 5 min and leave at 20°C–25°C for 30 min. Prepare 2 μL aliquots of annealed RNAs and place on ice if not immediately used for RNP complex formation. Store unused annealed tracr/crRNA at −20°C for later use.
-
c.For RNP complex formation, add 10 μg of recombinant SpCas9 protein to 2 μL of annealed tracr/crRNA, incubate for 10 min at 20°C–25°C and place on ice until ready to nucleofect. Prepare the RNP complex fresh before nucleofection.Note: Recombinant SpCas9 protein can be purchased from IDT, other vendors or prepared in-house. We have used both commercially available Cas9 as well as in-house prepared SpCas9 protein produced in BL21(DE3) E.coli using expression plasmid pET28a-Cas9-His (Addgene catalog number 98158) with similar results.
-
d.Nucleofect RNP complex and donor plasmid into recipient cells using Amaxa 4D-nucleofector (Lonza). The choice of nucleofection program and solution is cell line-dependent and requires optimization beforehand. For commonly used cell lines, optimized nucleofection conditions are found on the Lonza website. The nucleofection programs for cell types we have successfully used in the lab are provided in the materials and equipment section.
-
i.Mix 16.4 μL cell line specific nucleofection buffer (e.g., P3 Primary Cell) with 3.6 μL buffer supplement.
-
ii.Add 500 ng pUC19 donor plasmid (in the smallest volume possible) to 20 μL nucleofection buffer mix.
-
iii.Resuspend 2×105 cells in nucleofection buffer/plasmid mix.
-
iv.Transfer cell suspension to tube with assembled RNP complex and mix solutions by carefully pipetting up and down with a P200 pipette tip.
-
v.Transfer nucleofection mix into 16-well Nucleocuvette strip.
 CRITICAL: pipette slowly to avoid introducing air bubbles; tap the cuvette gently to remove any air bubbles.
CRITICAL: pipette slowly to avoid introducing air bubbles; tap the cuvette gently to remove any air bubbles. -
vi.Perform nucleofection with desired program.
-
vii.Transfer nucleofected cells to 6 well tissue culture plate with pre-warmed culture medium using a P200 pipette tip.
-
i.
-
e.Change media the next day to remove dead cells
 Pause point: 2–3 days after transfection or nucleofection the plate contains a mixed population of cells with different editing outcomes. The mixed cells can be cryopreserved for later selection and single-cell clone generation if desired.
Pause point: 2–3 days after transfection or nucleofection the plate contains a mixed population of cells with different editing outcomes. The mixed cells can be cryopreserved for later selection and single-cell clone generation if desired.
-
a.
Selection, single-cell clone generation, and screening
-
18.
Enrichment for cells with correct integrations and single-cell sorting
Antibiotic selection strategy
If the DNA was delivered using lipofection, start antibiotic selection for cassette integration 24 h after transfection. In the case of nucleofected cells, 3–5 days of recovery is required before starting the selection. It is important to set up a control well with untransfected cells and to continue selection until all the cells in this control well have died. Once antibiotic selection is complete, perform single cell sorting of cells with high mCherry (Cas9) expression into 96-well plates (only applicable if plasmid based delivery was used). In our experience, cells with high Cas9 protein levels display best knock-in efficiency. (Troubleshooting 3)
Note: Single cell sorting can be very stressful for cells and not all cell types are amendable to it. Minimizing the stress during sorting by choosing a big nozzle size, at least 100 μm for murine and 130 μm for human cell lines, can strongly enhance the number of clones recovered. (Troubleshooting 4)
Optional: If testing several different strategies for knock-in generation, use the pool of cells before single cell cloning to perform PCR-based screening to check for the presence of any successful integration (using protocol steps 19 d-f). We also have found that genomic DNA from the pooled population is useful to test different primers and optimize PCR conditions necessary for the screening step of single cell clones.
Fluorescent protein selection strategy
Once the cells have recovered after lipofection or nucleofection (3–5 days), perform single cell sorting of the GFP-positive cells into 96-well plates.
Note: We find it very convenient to use round bottom 96-well plates when generating clones of mouse ES cells, as this makes the emerging colonies easily identifiable under the microscope. As both antibiotic and fluorescent protein pre-selection provides a strong enrichment of cells with at least one correct integration event, and mouse ES cells grow easily as single cell clones, it is sufficient to sort two 96-well plates. Depending on how easily a cell line can grow from single cells, adjust the number of plates sorted accordingly. Furthermore, it is advisable to cryopreserve the pooled knock-in population in case the first round of single cell clones does not yield homozygous knock-in clones.
-
19.Screening of the generated clones
-
a.We generally pick mouse ES cell single cell clones 9–10 days after single cell sorting when cells cover approximately 60% of the well surface.
-
b.Identify expanding clones either by eye or with a microscope.CRITICAL: visually inspect clones with a microscope and exclude wells with more than one cell colony
-
c.Trypsinize the cells and resuspend in 100 μL of medium. Transfer 10 μL of the cell suspension into a fresh gelatin coated 96-well plate containing 130 μL of fresh media and place into the incubator. Transfer 100 μL of the suspension into a transparent 96-well PCR plate for screening.Note: If using a fluorescent reporter strategy, it is worthwhile to utilize the information from index sorting (record of fluorescence signal of the single sorted cells), when deciding which clones to screen/expand. Clones with higher fluorescent signal often indicate modification of both alleles.
-
d.Spin PCR plates to pellet the cells at 300 g for 5 min. Carefully remove the supernatant.
-
e.Add 20 μL of lysis buffer (see materials and equipment) per well. Use sticky film to seal the plates, vortex and quickly spin down to collect the liquid at the bottom of the plate.
-
f.Incubate in the PCR machine with 96-well compatibility using the following program:
Temperature Time 65°C 10 min 98°C 5 min 12°C forever -
g.Dilute the genomic DNA with 100 μL of TE (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA) and use 2 μL per PCR reaction. Run the PCR reaction below on a 1% agarose gel. Primer sequences can be found in Key Resources TableOptional: addition of Q-solution (provided in Qiagen PCR Taq polymerase kit) can improve amplification of some regions. (Troubleshooting 5)
-
i.Set up PCR reaction:
Component Volume (μL) 10× Coral Load Buffer 2 10 μM forward primer (K3-ki FP) 0.4 10 μM reverse primer 1 (K3-ki RP) 0.4 10 mM dNTPs 0.4 Taq polymerase 0.1 Genomic DNA 2 ddH2O 14.7 Total 20 -
ii.Run PCR according to the following conditions:
PCR cycling conditions
Steps Temperature Time Cycles Initial Denaturation 94°C 5 min 1 Denaturation 94°C 30 s 35 Annealing 60°C 30 s Extension 72°C 3 min Final Extension 72°C 5 min 1 Hold 4°C forever Note: We usually design one of the primers in the genomic region just outside one of the homology arms and the other one within the second homology arm. This way, a larger band is generated if successful integration has taken place. At the same time, absence of the smaller wild type band indicates biallelic integration (Figure 4A). An example of screening Kansl3 knock-in clones is represented in Figure 4B. To ensure correct integration in the selected clones, additional primers can be designed to completely cover the integration site.Alternatives: Screening of single cell clones with diluted genomic DNA from step 19f can also be performed using quantitative RT-PCR. For this strategy the amplicon should be 70–110 bp in length. We generally prefer regular PCR for screening, as it allows amplifications of several products in one reaction and can yield simultaneous information of presence of unmodified wild type locus as well as different selection marker knock-ins due to size differences in amplicon length.
-
i.
-
h.Purify PCR bands from the gel and analyze by Sanger sequencing to ensure correct integration without mutations. Primers used for PCR amplification can be used for sequencing of the PCR products.
-
a.
Figure 4.

Screening of the knock-in clones
(A) Overview of the typical screening strategy.
(B) Example screening result. Clones 1,2,3 and 6 demonstrate biallelic knock-in. Clone 5 demonstrates monoallelic knock-in. Clone 4 demonstrates a correctly integrated cassette in one allele and a potential cassette duplication event in the second allele.
Functional validation of the degron-fused protein
The best way to validate the successful dTAG knock-in cell line is to perform western blotting for an HA or any other epitope tag introduced during the knock-in procedure before and after dTAG ligand treatment. A concentration of 500 nM dTAG-ligand works for most targets and cell lines. In addition, antibody to the protein of interest should be used to ensure complete degradation and to confirm successful modification of both alleles. The degron-tag adds approximately 15 kDa to the protein size and a corresponding molecular shift should be expected. An example western blot validation for Kansl3 dTAG knock-in clones is shown in Figure 5A.
Figure 5.

Functional validation of the knock-in clones
(A) Western blot of HA tag, KANSL3 and GAPDH in three Kansl3 degron knock-in clones and wild type ES cells with and without dTAG-13 treatment for 24 h. An unspecific band is indicated with an asterisk.
(B) Western blot of HA-tagged KANSL3 and GAPDH in Kansl3 degron knock-in cell line at different times after dTAG-13 addition (Radzisheuskaya et al., 2021).
(C) Bar plots representing mean ± SD fold cell expansion and percent of Trypan blue positive cells after 4 days of dTAG-13 treatment in Kansl3 degron knock-in cell line (Radzisheuskaya et al., 2021).
(D) Western blot of HA tag and GAPDH in dTAG-TAK1 expressing glioma stem cells treated with 500 nM dTAG13-NEsG, dTAG-13 or dTAGV-1.
To establish the degradation kinetics of your protein of interest we recommend analyzing knock-in cells upon different durations of dTAG-ligand treatment (Figure 5B). In most cases the target protein should be degraded after 1–2 h.
If depletion of the target protein is known to confer a specific cellular phenotype, dTAG ligand treatment should recapitulate this effect in the case that both alleles are successfully targeted, and the protein is efficiently degraded by the ligand. Since Kansl3 is an essential gene, dTAG treatment of the knock-in cell lines induces decreased cell proliferation and concomitant loss of cell viability (Figure 5C).
However, not all protein targets can be efficiently degraded in a short time frame by recruiting CRBN E3 ligase complex using dTAG-13 (Nabet et al., 2020). Shown in Figure 5D is another dTAG-fusion protein where degradation via the VHL E3 ligase using the alternative dTAGV-1 ligand shows superior degradation results after short-term (2 h) and long-term (4 days) treatment compared to dTAG-13. (Troubleshooting 6).
Expected outcomes
After lipofection of mouse ES cells, we normally observe 40%–70% mCherry positivity and perform single cell sorting on the 5% highest expressing cells and/or the fluorescent protein marker positive cells present on the donor template (e.g., eGFP). The percentage of eGFP positive cells in the pool can vary dramatically from 0.1%–30% for different knock-ins, but very few cells are needed for single cell cloning and even pools with < 1% eGFP positive cells can ultimately yield several homozygous knock-in clones.
The yield of homozygous knock-ins in mouse ES cells is generally very high, if utilizing antibiotic or fluorescent protein-based selection (>80% of screened clones have biallelic targeting even when using a single selection marker). We generally screen 10–20 clones by PCR, cryo-preserve at least 6 homozygous clones and use 3 clones for further functional validation. Other cell types such as mouse neural stem cells or human glioma stem cells have efficiency rates of <10% biallelic integration and require the screening of more clones for these cell types.
The efficiency and success of generating homozygous clones is also target protein-, knock-in strategy- and cell line-dependent.
Limitations
Performing dTAG-degron knock-ins by enriching for successful integrations using an antibiotic or fluorescent selection marker only works if the gene to be tagged is expressed in the cell line of interest. Furthermore, for genes with low expression levels, such as transcription factors, antibiotic selection is more sensitive and preferable over the utilization of a fluorescent marker, as a sufficient amount of protein needs to accumulate for detection by flow cytometry.
Especially when attempting homozygous knock-ins in cancer cells, be aware of the copy number of your intended target locus. To improve the odds of tagging more than two alleles, use at least two different selection markers. We also observe unintended repair outcomes, including big deletions, more frequently in cancer cells.
In order to study complete loss of protein function, optimally all the alleles of a gene in a cell need to be tagged with dTAG which requires selection and expansion of clones from single cells. However, not all cell lines are amendable to single cell cloning. Utilizing multiple selection markers would allow to work with a pool of cells instead, however, generation of clones is still the preferred method.
Both N-terminal and C-terminal tagging sometimes leads to destabilization of the protein or loss of protein functionality. This could indicate that either this particular target is not suitable for dTAG-based degradation and another degron tag such as auxin-inducible degron (Nishimura et al., 2009) (Natsume et al., 2016) or SMASh-tag (Chung et al., 2015) could be considered instead. Alternatively, the protein target cannot be tagged in general.
Troubleshooting
Problem 1
During the sgRNA cloning, the number of colonies on the ligation plate is the same as the number of colonies on the control “empty” ligation plate.
Potential solution
This indicates incomplete digestion of the vector backbone or imprecise excision from the gel. Increase duration of the digestion reaction to 16–24 h and increase electrophoresis time.
Problem 2
No plasmid and/or RNP is delivered or high cell death after nucleofection.
Potential solution
Try to optimize the nucleofection conditions by using the cell line optimization kit from Lonza (Cat # V4XC-9064) to test different conditions for your cell line of interest.
Problem 3
No cells survive after antibiotic selection.
Potential solution
Wait 1–2 additional days before starting selection or lower the concentration of the antibiotic. As the selection marker is expressed from the same promoter as your gene of interest, especially in the case of lowly expressed genes, the concentration of the antibiotic selection needs to be adjusted accordingly. It is advisable to determine the sensitivity of the cell line of choice to the antibiotics to be used before starting to generate the targeted cell lines. Use the lowest concentration at which >99% of cells are killed within 3–4 days of selection for blasticidin or puromycin, and 4–7 days for neomycin.
Problem 4
No cells grow out after single cell sorting step.
Potential solution
To decrease the stress during single cell sorting, choose a bigger nozzle size (e.g., 130 μm) and perform the sort with a very low flow rate. For cells grown in serum-free conditions, increasing the concentration of growth factors or supplements in the medium can enhance recovery of clones. Sorting cells into semi-solid medium (e.g., Matrigel, soft agar, or MethoCult) can help some cell lines to grow in colonies.
Alternatively, either seed cells at very low density in big cell culture dishes and subsequently pick single colonies or manually seed cells in 96-well plates aiming for < 1 cell/well instead of single cell sorting to generate clonal lines if a cell sorter is not readily available.
Problem 5
No PCR product is obtained when amplifying homology arms from genomic DNA or when screening for knock-in clones.
Potential solution
Addition of 3% DMSO to the PCR reaction can help amplify regions with high GC content, which is often the case for promoters. Switching to an alternative High-Fidelity polymerase such as Phusion, Q5 or KOD can improve PCR results for certain amplicons. For PCR-based screening of knock-in clones, we have obtained good results using the Taq polymerase kit from Qiagen. Results can also be improved by changing the PCR program or designing alternative primers.
Problem 6
The protein shows very slow degradation dynamics upon treatment with dTAG-ligand with substantial residual protein left even after 8–24 h.
Potential solution
Expression levels of different E3 ligases can vary between cell lines and better degradation dynamics can be achieved for some targets by recruiting the VHL E3 ligase complex (by using dTAGV-1) instead of the CRBN E3 ligase system (by using dTAG-13 or dTAG-47) (Figure 5). We recommend to empirically test all three heterobifunctional dTAG molecules for degradation of your protein of interest. Alternatively, consider the use of the auxin-inducible degron system by creating a mini-AID fusion protein instead (Natsume et al., 2016).
Problem 7
No knock-in is achieved.
Potential solution
Check if your intended genomic locus is efficiently cut by your sgRNA by delivering Cas9/sgRNA with your optimized delivery method without providing the donor plasmid, wait for 3–4 days for optimal editing, isolate genomic DNA from your cells, amplify a 500–800 bp region surrounding the cut site, and send PCR product for Sanger sequencing to Genewiz or a similar company providing sequencing services.
Visually inspect the resulting chromatogram: there should be a clear disruption of the sequence at the predicted cut site. For a more quantitative method to assess cutting efficiency, we use the online tool TIDE (http://shinyapps.datacurators.nl/tide/) which allows estimation of the spectrum and frequency of insertions and deletions generated in a pool of cells from the sequence trace data. Of note: this method also required the chromatogram of a PCR product from unmodified genomic DNA as a reference.
Problem 8
No clones are homozygous knock-ins.
Potential solution
The simultaneous use of two donor vectors with different selection markers (antibiotic and/or fluorescent protein) can help with selection of clones with biallelic integration. In our experience, mouse ES cells are very efficient in performing HDR and generally one selection marker is sufficient to generate homozygous knock-in clones. However, if your protein of interest is essential for the maintenance of your cell line and the degron tag impedes protein function, homozygous knock-ins will be selected against. Try to tag the other terminus of the protein.
Problem 9
After successful endogenous tagging of the protein of interest, the function of the protein is compromised, or unintended degradation occurs before treatment with dTAG-ligand, as evidenced by reduced protein levels.
Potential solution
Place degron tag on the other terminus of the protein.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kristian Helin (helink@mskcc.org).
Materials availability
Plasmids and cell lines used in this protocol will be made available upon request. The sequence of the edited Kansl3 dTAG locus was deposited to Mendeley Data (https://doi.org/10.17632/mjd8zdmr6h.1).
Data and code availability
This study did not generate datasets and did not involve creating a code.
Acknowledgments
We thank members of the Helin Laboratory for discussions; Robin Armstrong for critical reading of the manuscript; and Sarah Teed for technical assistance. H.D. was in part funded by a Danish Cancer Society Fellowship (R146-A9194). A.R. was in part funded by the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement 659171. The work in the Helin Laboratory was supported by the Danish Cancer Society (R167-A10877), through a center grant from the NNF to the NNF Center for Stem Cell Biology (NNF17CC0027852) and through the Memorial Sloan Kettering Cancer Center Support Grant (NIH P30 CA008748).
Author contributions
H.D. and A.R. performed the experiments and analyzed the data. H.D., A.R., and K.H. wrote the manuscript.
Declaration of interests
K.H. is a consultant for Inthera Bioscience AG and a scientific advisor for Hannibal Health Innovation.
Contributor Information
Helene Damhofer, Email: damhofeh@mskcc.org.
Kristian Helin, Email: helink@mskcc.org.
References
- Chung H.K., Jacobs C.L., Huo Y., Yang J., Krumm S.A., Plemper R.K., Tsien R.Y., Lin M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015;11:713–720. doi: 10.1038/nchembio.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulas C., Kalkan T., von Meyenn F., Leitch H.G., Nichols J., Smith A. Defined conditions for propagation and manipulation of mouse embryonic stem cells. Development. 2019;146:dev173146. doi: 10.1242/dev.173146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Ferguson F.M., Seong B.K.A., Kuljanin M., Leggett A.L., Mohardt M.L., Robichaud A., Conway A.S., Buckley D.L., Mancias J.D. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat Commun. 2020;11:4687–4688. doi: 10.1038/s41467-020-18377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabet B., Roberts J.M., Buckley D.L., Paulk J., Dastjerdi S., Yang A., Leggett A.L., Erb M.A., Lawlor M.A., Souza A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018;14:431–441. doi: 10.1038/s41589-018-0021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume T., Kiyomitsu T., Saga Y., Kanemaki M.T. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016;15:210–218. doi: 10.1016/j.celrep.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Nishimura K., Fukagawa T., Takisawa H., Kakimoto T., Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Radzisheuskaya A., Shliaha P.V., Grinev V.V., Shlyueva D., Damhofer H., Koche R., Gorshkov V., Kovalchuk S., Zhan Y., Rodriguez K.L. Complex-dependent histone acetyltransferase activity of KAT8 determines its role in transcription and cellular homeostasis. Mol. Cell. 2021;81:1749–1765.e8. doi: 10.1016/j.molcel.2021.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Zhang C., Wang B., Li B., Wang Q., Liu D., Wang H., Zhou Y., Shi L., Lan F. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019;10:4284–4314. doi: 10.1038/s41467-019-12281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Zhang H., Li T., Chen K., Qiu J.-L., Gao C. Perfectly matched 20-nucleotide guide RNA sequences enable robust genome editing using high-fidelity SpCas9 nucleases. Genome Biol. 2017;18:191–197. doi: 10.1186/s13059-017-1325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets and did not involve creating a code.