

Abstract
Direct reprogramming of fibroblasts to alternative cell fates by forced expression of transcription factors offers a platform to explore fundamental molecular events governing cell fate identity. The discovery and study of induced cardiomyocytes (iCMs) not only provides alternative therapeutic strategies for heart disease but also sheds lights on basic biology underlying CM fate determination. The iCM field has primarily focused on early transcriptome and epigenome repatterning, whereas little is known about how reprogramming iCMs remodel, erase, and exit the initial fibroblast lineage to acquire final cell identity. Here, we show that autophagy-related 5 (Atg5)–dependent autophagy, an evolutionarily conserved self-digestion process, was induced and required for iCM reprogramming. Unexpectedly, the autophagic factor Beclin1 (Becn1) was found to suppress iCM induction in an autophagy-independent manner. Depletion of Becn1 resulted in improved iCM induction from both murine and human fibroblasts. In a mouse genetic model, Becn1 haploinsufficiency further enhanced reprogramming factor–mediated heart function recovery and scar size reduction after myocardial infarction. Mechanistically, loss of Becn1 up-regulated Lef1 and down-regulated Wnt inhibitors, leading to activation of the canonical Wnt/β-catenin signaling pathway. In addition, Becn1 physically interacts with other classical class III phosphatidylinositol 3-kinase (PI3K III) complex components, the knockdown of which phenocopied Becn1 depletion in cardiac reprogramming. Collectively, our study revealed an inductive role of Atg5-dependent autophagy as well as a previously unrecognized autophagy-independent inhibitory function of Becn1 in iCM reprogramming.
INTRODUCTION
Research on organ development and cellular reprogramming has informed our understanding of cell identity determination. Forced expression of combinations of transcription factors transforms terminally differentiated cells into induced pluripotent cells (iPSCs) (1, 2) or induces conversion between somatic cell lineages without going through the pluripotent state (3–8). Direct cardiac reprogramming that converts fibroblasts into contractile induced cardiomyocytes (iCMs) has been achieved using three transcription factors, Gata4, Mef2c, and Tbx5 (G, M, and T, respectively) (9–16). The iCMs display CM-like features including molecular, structural, and functional characteristics (11, 13, 17–19). Delivery of GMT into injured murine myocardium led to decreased fibrosis and improved heart function after myocardial infarction (MI) (12, 15, 20), and cardiac reprogramming has emerged as a promising approach for cardiac regenerative therapy. Despite a growing interest in the basic biology of iCM reprogramming (19, 21–26), the molecular mechanism governing iCM fate induction remains to be fully understood. An incomplete understanding of this process will inevitably hinder future clinical applications of iCMs.
Autophagy is an evolutionally conserved process to maintain cellular homeostasis (27, 28). By engulfing cytosolic proteins and organelles into autophagosomes followed by lysosomal digestion, autophagy allows cells to recycle or spare damaged cytoplasmic contents. As such, autophagy has been implicated in the differentiation of multiple cell types and iPSC reprogramming through cytoplasmic remodeling to promote structural changes (29–31). During direct cardiac reprogramming, the reprogramming fibroblasts need to remove cellular contents related to fibroblast function and establish CM-like structures. However, whether and how autophagy regulates the conversion of fibroblasts into iCMs remains to be determined. Beclin1 (Becn1), the mammalian ortholog of yeast autophagy-related 6 (Atg6) (32), is a rate-limiting component of autophagy that interacts with class III phosphatidylinositol 3-kinase (PI3K III, also known as Vps34) complex (33–35). Upon phosphorylation by the upstream kinase ULK1 (Unc-51 like autophagy activating kinase 1), Becn1 binds with core components including Vps34, Vps15, and Atg14L or UVRAG (ultraviolet radiation resistance-associated protein), to facilitate autophagosome formation and maturation. Homozygous deletion of Becn1 leads to early lethality during embryonic development or disrupted cavitation of embryonic bodies derived from mouse embryonic stem cells (ESCs) (36, 37). However, knockdown of Becn1 in mouse embryonic fibroblasts increases iPSC generation efficiency, likely through its autophagic function (30).
In this study, we found that autophagy plays a critical role in iCM reprogramming. Genetic inhibition of autophagy blocked iCM conversion, whereas pharmacological activation of autophagy boosted iCM induction. Unexpectedly, we found that Becn1 interacts with the canonical PI3K III complex to negatively regulate iCM induction independent of autophagy. Loss of Becn1 led to Wnt/β-catenin activation and promoted iCM induction and maturation. Our work thus identifies a previously uncharacterized role of and interplay between Becn1 and Wnt signaling pathways in cellular reprogramming and provides mechanistic insights into cell fate decision involving autophagy-dependent and -independent regulatory networks.
RESULTS
Autophagy is induced early during cardiac reprogramming
We first validated iCM reprogramming by Western blot to evaluate the expression of green fluorescent protein (GFP) under the mouse alpha myosin heavy chain (αMHC) promoter (αMHC-GFP; Fig. 1A). To determine whether autophagy is activated during reprogramming, we measured the expression of LC3-II (microtubule-associated proteins 1A/1B light chain 3B-II) and p62 and also evaluated the formation of LC3 puncta (autophagosomes) (38, 39). LC3-II was increased from reprogramming day 3, whereas p62 was significantly reduced (P < 0.05; Fig. 1A and fig. S1, A to D). GFP+ LC3 puncta were detectable in iCMs as early as day 3 and became more pronounced around days 5 to 7 (Fig. 1B). In contrast, control fibroblasts infected with LacZ retrovirus showed minimal punctuated GFP-LC3 signal (Fig. 1B). Similarly, transmission electron microscopy analysis indicated an increase in the number of autophagosomes, double-membraned structures with wrapped mitochondria, and cell membranes, in iCMs compared to control fibroblasts (Fig. 1, C and D).
Fig. 1. Autophagy was activated during iCM reprogramming.
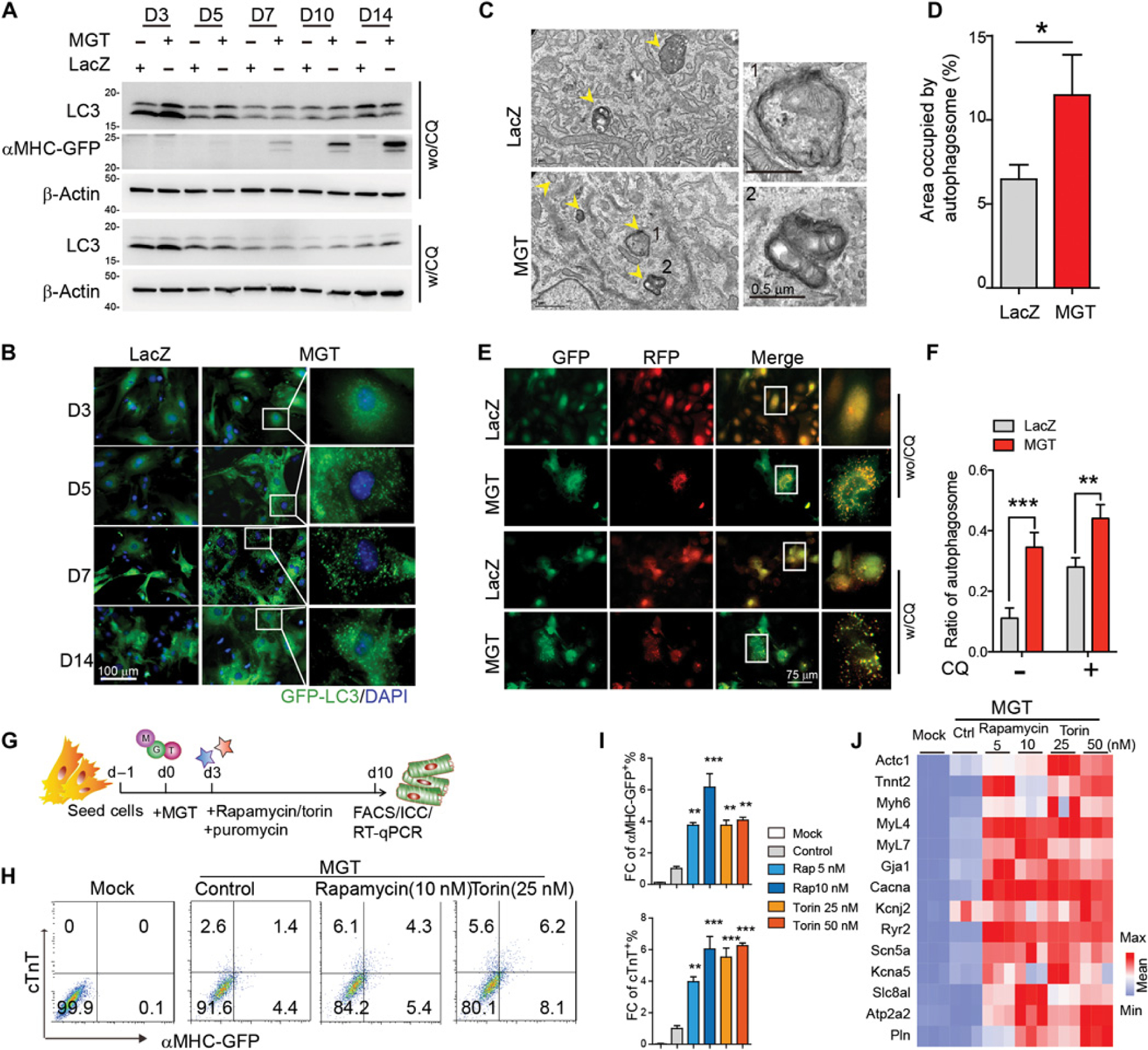
(A) Western blot of autophagy markers in murine CFs transduced with MGT or LacZ. Chloroquine (CQ) was added to treat cells for 4 hours at the concentration of 20 µm. w/CQ, with CQ treatment; wo/CQ, without CQ treatment. (B) Representative ICC images of GFP–LC3 puncta formation in explanted CFs transduced with either MGT or LacZ control virus at different time courses of reprogramming. (C and D) Representative TEM images (C) and quantification data (D) showing autophagic vacuoles (highlighted with yellow arrowheads). High magnification of labeled areas is shown in the right panels. (E and F) Representative images (E) and quantification (F) of mFRP-GFP–LC3 puncta formation in CFs transduced with either MGT or LacZ control virus at reprogramming day 5. CQ was added to culture media at the final concentration of 20 µm for 4 hours before sample collection. (G) Schematic depiction of experimental design on pharmacological treatment of iCMs. (H and I) Representative flow cytometry plots (H) and quantification data (I) for αMHC-GFP+ and cTnT+ cells 10 days after transduction of MGT followed by rapamycin and torin treatment. (J) Heatmap of cardiac gene expression in uninfected (mock) and MGT-infected iCMs treated with rapamycin, torin, or vehicle control. All experiments were repeated at least three times. Mean values from technical triplicates [except n = 15 to 20 for (D) and (F)] were used for statistics. Groups were compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Error bars indicate means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001.
We next assessed autophagic flux in the reprogramming cells. We first determined LC3 turnover in the presence of the lysosomal inhibitor chloroquine (CQ). MGT (polycistronic construct expressing Mef2c, Gata4, and Tbx5 sequentially)–infected fibroblasts had significantly higher LC3-II expression than control cells (P < 0.05), indicating higher autophagy flux in iCMs (Fig. 1A and fig. S1, A and B). We then took advantage of the tandem monomeric red fluorescent protein (mRFP)–GFP-LC3 reporter as an indicator of autophagy flux. Upon exposure to the acidic environment of autolysosomes, mRFP is stable, whereas GFP fluorescence is quenched: GFP+RFP+ puncta (yellow) represent autophagosomes, whereas RFP+ puncta (red) indicate autolysosomes. We analyzed the extent of green and red puncta colocalization and found enhanced autophagosome formation in reprogramming cells compared to control cells (Fig. 1, E and F). CQ treatment resulted in further enhanced accumulation of autophagosome in iCMs than control cardiac fibroblasts (CFs; Fig. 1, E and F). We also assessed mitophagy during reprogramming, observing no statistical difference in mitochondrial protein HSP60 (60 kDa heat shock protein) expression (fig. S1, E and F) or the colocalization ratio of lysosome and mitochondria markers (fig. S1, G and H) between control and reprogramming fibroblasts, suggesting minimal involvement of mitophagy in cardiac reprogramming. These results altogether indicate that autophagy is activated during early stage of iCM reprogramming.
To test whether autophagy was required for iCM reprogramming, we treated MGT-infected fibroblasts with rapamycin and torin, which activate autophagy through inhibition of mammalian target of rapamycin (mTOR) signaling (40, 41) (Fig. 1G). The drug treatments resulted in four- to eightfold increases in the percentage of αMHC-GFP+ and cardiac troponin T+ (cTnT+) iCMs (Fig. 1, H and I, and fig. S1, I and J). Evaluation of a panel of cardiac genes revealed higher expression in the rapamycin- or torin-treated iCMs than in control cells (Fig. 1J).
ATG5-dependent autophagy is required for iCM induction
The evolutionary conserved Atgs are essential regulators of autophagy induction and maturation (42). Among them, Atg5 is a key player of the Atg12-Atg5-Atg16 complex that functions to promote the elongation of the phagophoric membranes in autophagic vesicles (43, 44). To explore whether impaired autophagy would affect iCM reprogramming, we delivered lentivirus encoding a panel of short hairpin RNA (shRNA) targeting Atg5 together with MGT to fibroblasts (Fig. 2A). Atg5 endogenous expression was not affected by MGT induction (fig. S2, A and B). Knocking down Atg5 significantly reduced the percentage of cTnT+ iCMs compared to shNT (non-targeting shRNA) controls (P < 0.001; Fig. 2, B to F). Of note, individual shAtg5 oligonucleotides showed similar repressive effects on reprogramming as pooled oligonucleotides; thus, for the rest of the study, we used shAtg5 pool (referred as shAtg5), unless otherwise indicated (fig. S2C). Atg5 deficiency also led to a decrease in sarcomere gene expression (Fig. 2G) and a reduced number of cells positive for gap junction connenxin43 (Cx43+; fig. S2, D and E). Atg7 plays a similar role to Atg5 during autophagy, and silencing Atg7 phenocopied Atg5 depletion (Fig. 2, E and F, and fig. S2, D and E). In contrast to the effect of silencing Bmi1 on reprogramming, which was evident as early as day 3 (21), we observed a reduced percentage of cTnT+ cells starting at day 5 upon Atg5 knockdown (fig. S2, F to H). To assess the critical time window during which Atg5 was required, we introduced shAtg5 to reprogramming cultures at 0, 1, 4, 6, and 8 days after MGT retroviral infection. Depletion of Atg5 before reprogramming day 8 decreased the percentage of cTnT+ iCMs (Fig. 2H). These data suggest that Atg5 likely functions after epigenetic repatterning and early fate switch during iCM fate determination.
Fig. 2. Activation of autophagy promoted iCM reprogramming.

(A) Schematic depiction of experimental design using MGT and shRNAs. (B and C) Representative flow cytometry plots (B) and quantification data (C) for cTnT+ and αMHC-GFP+ cTnT+ cells 10 days after transduction of MGT and shRNAs targeting Atg5 or nontargeting control (NT). Cells that were not transduced with retrovirus were used as mock. (D) qPCR analysis of knockdown efficiency of shRNAs targeting Atg5 and Atg7, respectively, on reprogramming day 10. RQ, relative quantification of gene expression. (E and F) Representative images (E) and quantification (F) of cells transduced with MGT followed by shRNAs targeting Atg5 and Atg7 on reprogramming day 14. (G) qPCR analysis of cardiac gene expression in iCMs transduced with MGT and shAtg5 on reprogramming day 10. Values were normalized to control cells without MGT delivery (mock). (H) Quantification of flow cytometric analysis of cTnT+ iCMs transduced with lentiviral shNT or shAtg5 at different time points. Cells were collected on reprogramming day 10. FC, fold change. (I and J) Representative images (I) and quantification (J) showing the formation of autophagosomes in iCMs treated with shNT or shAtg5 on reprogramming day 5. w/CQ, with CQ treatment for 4 hours; wo/CQ, without CQ treatment. (K) Representative Western blot showing the autophagy flux in iCMs treated with shNT or shAtg5 on reprogramming day 5 and day 10. CQ was added to cells for 4 hours before sample collection. β-Actin serves as loading control. wo/CQ, without CQ treatment; w/CQ, with CQ treatment (20 µm) for 4 hours. (L) Quantification of autophagy flux detected by Western blot in the presence of shAtg5 from day 3 to day 14. (M) Expression of Atg5 after overexpression. (N) Representative flow cytometry plots showing gating cTnT+ cells out of Atg5-EGFP+ or EGFP+ cells. (O) Quantification of cTnT% cells in (N). All experiments were repeated at least three times. Mean values from technical triplicates [except n = 10 to 20 for (E) and (J)] were used for statistics. Groups were compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Error bars indicate means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001.
We next asked if and how autophagy was affected by depleting Atg5 during reprogramming. Knockdown Atg5 reduced GFP-LC3 puncta formation in MGT-infected fibroblasts (fig. S3, A and B). Consistently, autophagy flux analysis using tandem mRFP-GFP-LC3 reporter also indicated a decreased autophagosome ratio with or without CQ treatment (Fig. 2, I and J) upon Atg5 knockdown (Fig. 2K). As expected, we also observed diminished LC3II in the presence and absence of CQ treatment when Atg5 was knocked down (Fig. 2, K to L), further supporting that Atg5 positively regulates iCM induction and its involved autophagy.
Complementary to loss-of-function experiments, we simultaneously delivered retroviruses encoding enhanced GFP (EGFP)–tagged Atg5 in MGT-infected fibroblasts. Atg5 overexpression was apparent at both mRNA (Fig. 2M) and protein level (fig. S3C) and led to increased autophagy flux in iCMs compared to control cells (fig. S3, D to G). We quantified cTnT+ iCMs in GFP+ (Atg5 expressing) cells and found a higher percentage of iCMs upon Atg5 overexpression (Fig. 2, N and O). Together, our results suggest that autophagy is required for iCM reprogramming, and enhancing autophagy via Atg5 overexpression can boost iCM induction.
Loss of Becn1 facilitated the establishment of cardiomyocyte features in iCMs
Next, we targeted additional nodal points of autophagy. Unlike silencing Atgs, knockdown of Becn1, a well-described key regulator for autophagy initiation (42), resulted in an unexpected three- to fourfold increase in reprogramming efficiency (Fig. 3, A to C, and fig. S4, A and B). Transduction of shBecn1 (pooled and used afterward unless otherwise specified) together with MGT generated a remarkable number of uniformly shaped αMHC-GFP+ cells from reprogramming day 5 (Fig. 3D). Immunocytochemistry (ICC) results demonstrated that these uniformly shaped cells also expressed cTnT and αActinin (Fig. 3, E and F). Quantification of ICC revealed a marked increase in the number of αMHC-GFP+, cTnT+, and αActinin+ cells in shBecn1 culture compared to the shNT group (Fig. 3G). These observations were further confirmed by the higher expression of a panel of CM (cardiomyocyte) marker genes and reduced expression of fibroblast genes in shBecn1-treated CFs than controls (Fig. 3, H and I). We further validated our findings in several other types of fibroblasts (Fig. 3, J to L, and fig. S4C).
Fig. 3. Depletion of Beclin1 increased reprogramming efficiency and enhanced iCM maturation.

(A and B) Representative flow cytometry plots (A) and quantification data (B) for αMHC-GFP+ and cTnT+ cells 10 days after transduction of MGT together with shRNAs against Becn1 or nontargeting (NT) control on freshly isolated CFs. (C) Knockdown efficiency of shBecn1 evaluated by qPCR. (D) Morphology of iCMs treated with shNT or shBecn1 lentivirus on reprogramming day 7. (E to G) Representative ICC images (E and F) and quantification data (G) for αMHC-GFP (green), cTnT (magenta), and αActinin (red) on MGT-transduced CFs treated with indicated shRNAs. (H) Heatmap of cardiac gene expression in uninfected (CF) and MGT-infected iCMs treated with shNT or shBecn1 (biological duplicates in each group). (I) Quantification of fibroblast gene expression in MGT-transduced cells treated with shNT or shBecn1 by qPCR. FC, fold change. (J to L) Quantification of FACS data showing reprogramming efficiency of neonatal explanted cardiac fibroblast (ExCFs) (J), adult ExCFs (K), and MEFs (L). (M) Representative ICC images showing αMHC-GFP, cTnT, and connectin43 (CX43) in iCMs. Enlarged images showing the well-aligned CX43 molecules at two adjacent GFP+ cells and well-formed sarcomere structure in iCMs deficient for Becn1. (N) Quantification of cells showing sarcomere structure in shNT- or shBecn1-treated iCMs on reprogramming day 14. (O) Quantification of CX43+ iCMs on reprogramming day 14. (P and Q) iCM calcium transients (P) and quantification (Q) measured by Rhod3 dye labeling. Each trace corresponds to the spot numbered in the top panel. (R and S) Representative images (R) and quantification (S) of spontaneously contracting iCMs (yellow lines) 6 weeks after MGT and indicated shRNA transduction. (T) Distribution of iCMs aligned on MEA culture plate. Each white dotted cycle indicates individual microelectrode. (U) Representative simultaneous recordings of the iCMs 3 weeks after retroviral MGT plus shNT or shBecn1 transduction on the MEA culture plates. The number of individual electrodes corresponding to the plot is shown. (V and W) Representative flow plots (V) and quantification data (W) for percentage of cTnT 12 days after human MGT (hMGT) and indicated shRNA transduction into human H9F fibroblasts. hMGT, human MGT; hBecn1, human Becn1. (X) Knockdown efficiency of shRNA targeting human Becn1 accessed by qPCR. (Y) Representative ICC images and quantification data of human iCMs at 2 weeks after hMGT and indicated shRNA transduction. (Z) Heatmap of the relative expression of a set of cardiac genes determined by qPCR 2 weeks after hMGT and indicated shRNA transduction. All experiments were repeated at least three times. Mean values from technical triplicates [except n = 10 to 20 for (G), (N), (O), (Q) and (Y)] were used for statistics. Groups were compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Error bars indicate means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001.
We then evaluated structural and functional maturation of the Becn1-depleted iCMs. We performed ICC 2 weeks after viral infection and found that Becn1 knockdown resulted in enhanced expression of gap junction protein Cx43 and enhanced Cx43 localization to cell-cell borders (Fig. 3, M to O), suggesting the development of cell-cell junctions and electrical coupling. The number of iCMs displaying well-organized sarcomere structures markedly increased after Becn1 knockdown (Fig. 3N). We observed a threefold increase in the number of spontaneously beating cells showing periodic calcium oscillation in shBecn1 culture compared to the control group (Fig. 3, P to S). To characterize the detailed physiological properties of Becn1-depleted iCMs, we recorded the field potential of iCMs by microelectrode array (MEA) (45). Consistent with earlier results, we observed increased number of αMHC-GFP+ and cTnT+ iCMs on the MEA plate compared to the control (Fig. 3T). Although the field potential property varied among different electrodes, we detected an increased number of electrodes showing field potential and elevated frequency of field potential spikes in Becn1-depleted culture (Fig. 3U and fig. S4D). Because maturation of mitochondria is essential to meet the functional and metabolic demands of iCMs, we also characterized the dynamics of mitochondria by quantifying mitochondrial copy numbers in shBecn1 or shNT reprogramming fibroblasts at 1 to 3 weeks after viral transduction. Reverse transcription polymerase chain reaction (RT-PCR) amplifying mitochondria gene ND2 (NADH dehydrogenase 2) and Cox1 against genomic gene β−2-microglobulin demonstrated that average mitochondrial DNA (mtDNA) copy number was gradually increased over time (fig. S4E). Depletion of Becn1 increased mtDNA copy numbers around two- to threefold more, suggesting that knockdown of Becn1 enhanced mitochondria biogenesis in iCMs. Together, our results demonstrated that Becn1 down-regulation promotes MGT-mediated iCM reprogramming efficiency and improves iCM quality in vitro.
To test whether human BECN1 could suppress iCM conversion from human CFs, we introduced retrovirus encoding human MGT (hMGT) into H9Fs (H9 embryonic cell derived fibroblasts) as we recently reported (46). Flow cytometry and ICC analyses indicated an increased reprogramming efficiency when hBECN1 is knocked down (Fig. 3, V to Y). Consistently, we also observed activation of human cardiac genes upon loss of hBECN1 (Fig. 3Z).
Enhanced reprogramming outcomes in a murine genetic model of Becn1 haploinsufficiency
To translate our in vitro findings to an in vivo setting where heart disease can be modeled, we used a murine genetic model of Becn1 loss of function whereby exons 1 and 2 were replaced by a neomycin resistance (neo) cassette (Fig. 4, A and B) (33, 43). The homozygous knockout mice (Becn1−/−) show early embryonic lethality. In contrast, heterozygous mutant mice (Becn1+/−) are viable and manifest attenuated autophagy in multiple tissues (36, 47). Overall heart morphology, size, organization, and function were indistinguishable between wild-type (Becn1+/+) and Becn1+/− mice (fig. S5, A to D). On the basis of aforementioned findings that dosage of Becn1 influences the efficiency of iCM induction, we reasoned that fibroblasts derived from Becn1+/− mice might manifest enhanced cardiac reprogramming. To test this idea, we crossed αMHC-GFP reporter mice with Becn1+/− mice and obtained αMHC-GFP+ Becn1+/+ (control) and αMHC-GFP+ Becn1+/− mice (Fig. 4C). Decreased Becn1 mRNA expression was confirmed in CFs from the het mice (Fig. 4D). We found that Becn1+/− CFs were more amenable to be converted into iCMs than the littermate control cells (Fig. 4, E and F, and fig. S5, E and F), further illustrating the effect of Becn1 dosage on iCM induction.
Fig. 4. Heterozygous loss of function of Becn1 enhanced reprogramming and improved heart function after MI.
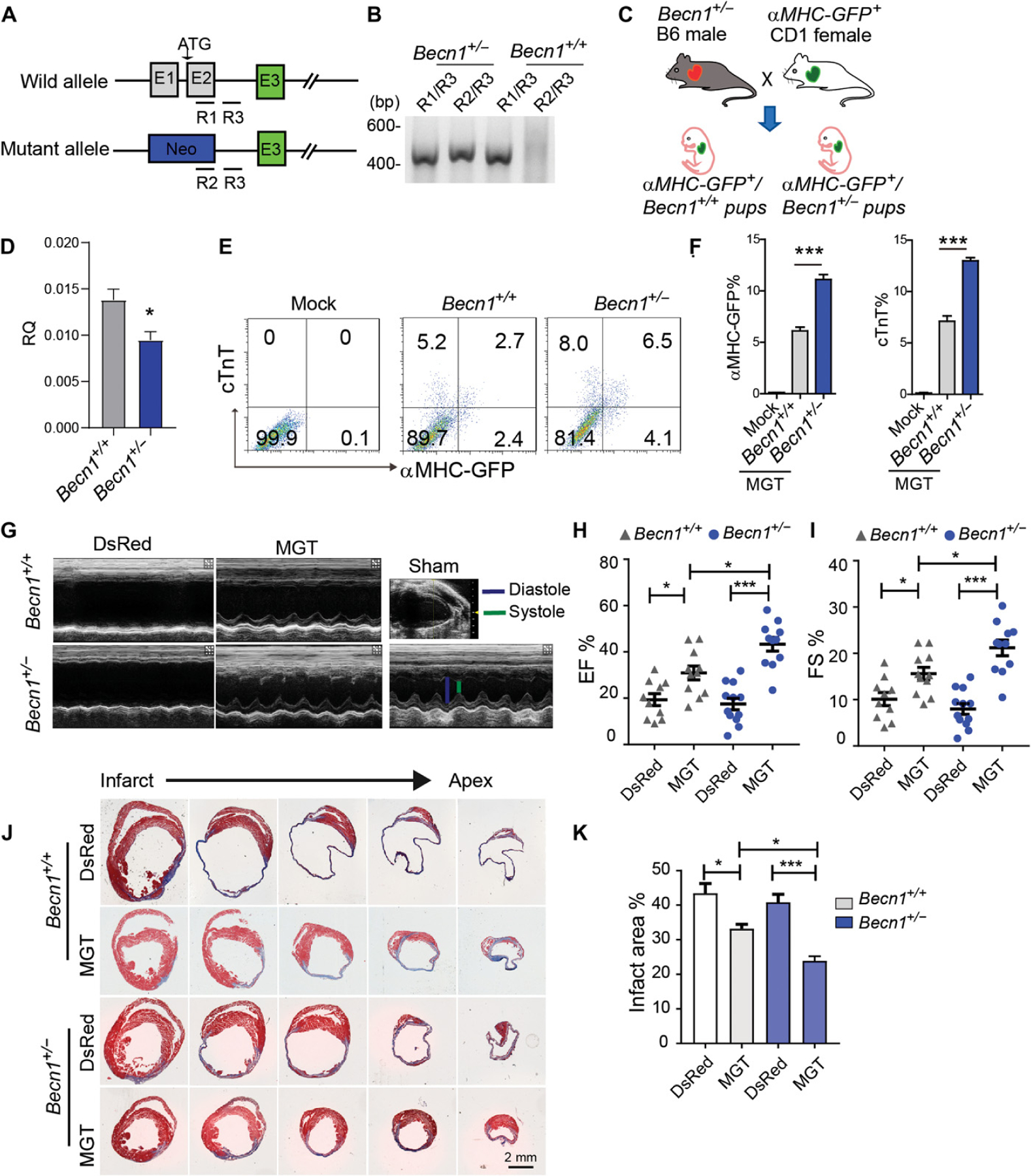
(A) Schematic of Becn1 heterozygous mutant mouse. R1 to R3 indicate the location of PCR primers for genotyping. (B) Representative genotyping results from wild-type (Becn1+/+) or Becn1 heterozygous (Becn1+/−) mice. PCR primer pairs are listed above the gel plot. (C) Schematic of breeding strategy to cross αMHC-GFP reporter line with Becn1 mutant mouse line. (D) Relative expression of Becn1 in heterozygous ExCFs determined by qPCR. (E and F) Representative flow plots (E) and quantification data (F) for αMHC-GFP+ and cTnT+ cells 10 days after transduction of MGT to Becn1+/− and Becn1+/+ CFs. (G) Echocardiography to measure ventricular contractility 4 weeks after MI. (H and I) Quantification of 4-week ejection fraction (EF) (H) and FS (I) as absolute values is shown. (J and K) Representative histological heart sections with Masson trichrome staining and quantification (K) showing the areas of fibrosis among retroviral DsRed or G, M, and T delivered Becn1 mutant or wild-type mice. All experiments were repeated at least three times. Mean values from technical triplicates [except n = 4 for (F) and n = 5 to 10 in each group for (H) to (K)] were used for statistics. Groups were compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Two-way ANOVA followed by Tukey post hoc test was used for (H), (I), and (K). Error bars indicate means ± SEM; *P < 0.05 and ***P < 0.001.
We next sought to determine in vivo iCM reprogramming in Becn1+/− mice given the promising ex vivo data using isolated Becn1+/− CFs. The Becn1+/− mice and Becn1+/+ littermate controls at the age of 10 to 12 weeks in C57BL/6 background were randomly grouped. Retrovirus encoding DsRed or MGT was injected intramyocardially immediately after permanent ligation of the left ascending coronary artery (12, 15). No significant difference in heart function was observed between Becn1+/− and Becn1+/+ mice before myocardial injury (fig. S5D). Consistent with literature, MGT enhanced cardiac function compared to DsRed control in both Becn1+/− and Becn1+/+ groups 4 weeks after MI (Fig. 4, G to I). The Becn1+/− reprogrammed mice exhibited more improved heart function with significantly higher ejection fraction and fraction shortening (FS) values (P < 0.001) than those of the Becn1+/+ reprogrammed mice. We further quantified the scar size using Masson’s Trichrome staining on sections from multiple layers of left ventricles from Becn1+/− and Becn1+/+ with or without MGT. Consistent with echocardiography data, although MGT treatment led to a reduced scar in both Becn1+/− and Becn1+/+ infarcted hearts, a further decreased scar area and more profound regenerated muscles along the border zone were observed in Becn1+/− reprogrammed infarcted hearts (Fig. 4, J to K). Together, our study using a murine genetic model demonstrates that Becn1 heterozygosity resulted in further improvements in MGT-induced iCM generation and enhancement in heart function after MI.
Autophagy-independent role of Becn1 during iCM reprogramming
The data, thus far, have demonstrated that Becn1 functions to suppress CM fate acquisition during iCM reprogramming, which appears to differ from the other canonical autophagic factors we tested. We thus sought to understand the molecular mechanisms underlying how Becn1 functions to regulate iCM identity. We determined the time point when the shBecn1-treated group started to show an enhancement in iCM induction by quantifying the percentage of αMHC-GFP+ and cTnT+ iCMs over several days during reprogramming (Fig. 5A). Enhanced iCM reprogramming by shBecn1 became pronounced at day 5 and onward (Fig. 5B and fig. S6, A and B). We also performed gene expression analysis of a panel of cardiac and fibrotic genes. On day 3, most of the genes showed similar expression. Starting from day 5, shBecn1 resulted in higher expression of cardiac genes encoding sarcomeric protein (Actc1), gap junction (Gja1), and ion channel (Scn5a, Kcna5) proteins, and lower expression of fibroblast-related genes (Postn, Tcf21, Col1a2, and Col1a3; Fig. 5C). Likewise, Western blot analysis manifested similar up-regulation of cardiac marker αMHC-GFP and endogenous αActinin expression from day 5 (Fig. 5D and fig. S6C). We evaluated the expression of M, G, and T at both mRNA and protein levels and found that expression was not affected by shBecn1 (Fig. 5, C and D). Next, to define the time window required for Becn1 knockdown, we introduced shBecn1 to CFs at different time points after MGT transduction (Fig. 5E). An elevated percentage of αMHC-GFP+ and cTnT+ cells was observed only when shBecn1 was introduced within 4 days after MGT induction (Fig. 5F). Our results together suggest that shBecn1 was required at the early stage of reprogramming to promote iCM induction (days 1 to 4); thus, Becn1 likely functions to suppress the establishment of CM fate.
Fig. 5. Becn1 inhibits iCM conversion independent of autophagy.

(A) Schematic of experimental design to determine the temporal requirement of Becn1 knockdown in cardiac reprogramming. (B) Quantification of αMHC-GFP% and cTnT% on reprogramming day 10 as designed in (A). (C) qPCR of gene expression at different time points upon MGT transduction and Becn1 knockdown. Values were normalized to uninfected control on day 3 (mock). (D) Western blot analysis of αMHC-GFP, Mef2c, Gata4, and Tbx5 expression. β-Actin serves as a loading control. (E) Schematic of experiment design to determine the time window of Becn1 knockdown. (F) Fold change (FC) of αMHC-GFP% and cTnT% in iCMs as described in (E). (G and H) Representative images showing GFP-LC3 puncta formation (G) and quantification (H) in iCMs transduced with shNT or shBecn1 on reprogramming day 5. Lentiviral GFP-LC3 constructs were transduced into iCMs on reprogramming day 1. (I and J) Representative Western blot (I) and quantification (J) showing the autophagy flux in iCMs treated with shNT or shBecn1 from reprogramming day 3 to day 14. Sample on the indicated dates were treated with or without CQ for 4 hours. β-Actin serves as loading control. wo/CQ, without CQ treatment; w/CQ, with CQ treatment. (K) Representative images showing autophagosome (yellow) and autolysosome (red) in iCMs transduced with mRFP-GFP-LC3 construct. shRNAs targeting Becn1 or control shRNA (shNT) were introduced into iCMs on day 1, and images were taken on reprogramming day 5 after cells were treated with or without CQ for 4 hours. (L) Quantification of autophagosome ratio in (K). (M and N) FACS analysis (M) and quantification (N) of iCMs upon lentiviral delivery of shNT, shBecn1, shAtg5, and shBecn1 plus shAtg5 on reprogramming day 10. All experiments were repeated at least three times. Mean values from technical triplicates [except n = 10 to 15 for (H) and (L)] were used for statistics. Groups were compared using one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. Error bars indicate means ± SEM; **P < 0.01 and ***P < 0.001.
We next sought to evaluate the effect of Becn1 knockdown on autophagy during iCM reprogramming. At baseline, loss of Becn1 minimally affected GFP-LC3 puncta formation (Fig. 5, G and H). We further evaluated autophagy flux to confirm the limited role of Becn1 on autophagy during iCM fate conversion (early stage of reprogramming). Loss of Becn1 marginally affected LC3-II expression from reprogramming day 3 to day 7, while it markedly reduced LC3-II from reprogramming day 10 to day 14 (Fig. 5, I and J). A similar effect of shBecn1 on LC3-II expression was observed when the reprogramming culture was treated with CQ treatment. At the transcriptional level, we observed a transient up-regulation of genes that regulates autophagy initiation (Vps34 and Ambra1), autophagosome elongation (Atg5, Atg7, and Lc3b), and expansion (Atg16 and Atg4b) in MGT-transduced cells at early stage of reprogramming followed by gradual down-regulation to basal expression (fig. S6D). Depletion of Becn1 had little effect on the expression of Atgs at early time points (D3 and/or D5) but significantly reduced Atgs expression from D7 to D14 (fig. S6D). Of note, Becn1 knockdown did not change Ambra1 and Atg12 expression. Together, these data indicate that Becn1 knockdown did not affect autophagy at early stage of reprogramming (when iCM fate is determined) but reduced autophagy at late stage when iCM cell fate is already established.
To further confirm Becn1’s autophagy-independent role, we performed an mRFP-GFP-LC3 dual fluorescent assay and noted that Becn1 knockdown did not significantly affect autophagosome formation and autophagy flux at early reprogramming days (Fig. 5, K and L). In parallel, we performed an epistasis experiment to determine the relationship between Becn1 and Atg5-mediated autophagy (Fig. 2, I to L). To this end, we knocked down Atg5 in shBecn1 iCMs to test whether enhanced reprogramming could be abolished by blocking autophagy through depleting Atg5. Simultaneous loss of both Becn1 and Atg5 still exhibited higher reprogramming efficiency compared to control (Fig. 5, M and N, and fig. S6, E and F), suggesting an alternative autophagy-independent pathway by which Becn1 regulates iCM induction. These results suggest that the repressive role of Becn1 on iCM induction is independent from its well-established role in autophagy.
Becn1 modulates transcriptome shift during early stage of reprogramming without direct alteration of chromatin accessibility
We and others demonstrated that epigenetic repatterning is a prerequisite for successful iCM induction (13, 18, 21). The shBecn1-mediated enhancement of iCM induction prompted us to evaluate whether loss of Becn1 affected epigenetic landscape. ATAC-seq (assay for transposase-accessible chromatin using sequencing) provides a rapid and sensitive technique to assess genome-wide epigenetic alterations by mapping chromatin accessibility (48). We and others have shown that epigenetic repatterning occurs as early as reprogramming day 3 (13, 18, 24). Therefore, we generated ATAC-Seq data from day 3 shNT- and shBecn1-treated MGT- or control LacZ-transduced fibroblasts. We interrogated the enrichment of ATAC-seq reads at +1 and −1 kb around transcription start sites. Comparing MGT-iCMs with LacZ-transduced cells, we found that MGT-iCMs gained multiple open chromatin regions compared to LacZ-transduced cells (Fig. 6A and fig. S7A), consistent with our previous observation that epigenetic repatterning occurs as early as day 3 (24). In total, 22,262 high-confidence open chromatin regions were identified when comparing MGT-shNT iCMs with LacZ-shNT cells, and 11,401 open regions were found in MGT-shBecn1 iCMs when compared to LacZ-shBecn1 cells (Fig. 6B). However, MGT-shBecn1 iCMs exhibited minimal differences that did not reach statistical significance when compared with MGT-shNT cells. MGT-shBecn1 iCMs had the same number of open chromatin regions as the MGT-shNT cells (Fig. 6, A and B).
Fig. 6. Loss of Becn1 minimal affected early chromatin accessibility of iCMs but altered transcriptomic profile at later stage.

(A) Signal intensity (top) and heatmap (bottom) of ATAC-seq signals in cells treated with LacZ + shNT, LacZ + shBecn1, MGT + shNT, and MGT+shBecn1 and collected 3 days after viral transduction. Average reads from a 2-kb region across the transcriptional start sites (TSSs) were used. The color in heatmap represents the intensity of chromatin accessibility. (B) Differential analysis of ATAC-seq peaks between LacZ versus MGT or shNT versus shBecn1 samples. Yellow indicates LacZ-transduced shNT or shBecn1 cells, and green indicates the MGT-transduced cells. Two-way arrows indicate the samples for differential analysis. The number of differential regions is shown. Gain, numbers of sites that gain chromatin accessibility; Loss, numbers of sites that lose chromatin accessibility. (C) Hierarchical clustering and heatmap showing the expression of cardiac and fibrotic genes in samples as in (A) at reprogramming day 3 and day 5. (D and E) GSEA analysis shows enrichment of indicated gene sets in MGT + shBecn1 cells compared to MGT + shNT cells on reprogramming day 3 (D) and day 5 (E). (F and G) Overlap of up-regulated (F) band down-regulated (G) differentially expressed genes (DEGs) induced by Becn1 knockdown between MGT and LacZ-transduced cells on reprogramming day 5. (H and I) GO analysis of up-regulated (H) and down-regulated (I) DEGs using gene list from (F) and (G), respectively. (J) qPCR analysis of indicated gene expression at different time points of reprogramming. All experiments were repeated at least three times. Mean values from technical triplicates were used for statistics. Two-tailed unpaired t test was used for (J) comparing shBecn1 versus shNT at each time point. Error bars indicate means ± SEM; **P < 0.01 and ***P < 0.001.
Next, by using GREAT (genomic regions enrichment of annotation tool) (49), we associated the enhancer and promoter regions to genes and obtained the Gene Ontology (GO) terms. Analyzing GO terms with P value <0.05, we identified 19 enriched GO terms related to metabolism and cell division when comparing MGT-shNT with LacZ-shNT groups (fig. S7B). In comparison, 33 enriched GO terms were found when comparing MGT-shBecn1 with LacZ-shBecn1 groups (fig. S7C). Except for eight GO terms shared by both comparisons, we found that the GO terms specifically enriched in MGT-shBecn1 group were associated with “myofibril assembly,” “adherent junction organization,” and “regulation of tight junction,” suggesting the enhanced fibroblast-to-myocyte conversion. This observation is consistent with our earlier molecular characterizations. We next analyzed the regions that lost chromatin accessibility. Few shared closed regions were present among the following pairwise comparisons: MGT-shNT versus MGT-shBecn1, MGT-shNT versus LacZ-shNT, and MGT-shBecn1 versus LacZ-shBecn1. Thus, we could not find enriched GO terms using the regions defined (Fig. 6B). Collectively, these results indicate that MGT transduction led to major changes in chromatin accessibility, but loss of Becn1 marginally altered chromatin status during iCM fate conversion in day 3 samples.
In parallel, we performed RNA sequencing (RNA-seq) to profile transcriptome changes between day 3 and day 5 shNT- or shBecn1-treated reprogramming and control cells (fig. S8A). To explore global transcriptome differences among samples, we first performed principal components analysis (PCA) and unsupervised hierarchical clustering. PCA plot and heatmap of sample-to-sample distances showed that LacZ- and MGT-transduced cells were separated from each other on PC1 (principal component) dimension, which accounted for 77% of total variance (fig. S8, B and C). To confirm the global transcriptome differences, we analyzed the expression of cardiac and fibroblast signature genes from all samples. Heatmap showed that MGT transduction led to a global up-regulation of cardiac genes and down-regulation of fibroblast genes, which was further enhanced by Becn1 knockdown (Fig. 6C). Next, we profiled the differentially expressed genes (DEGs) at each time point (table S1). Gene set enrichment analysis (GSEA) revealed that shBecn1 significantly up-regulated gene sets related to myogenesis, such as those involved in the formation of contractile fiber and myofibril assembly and down-regulated gene sets related to cell proliferation and inflammation (Fig. 6, D and E). GSEA demonstrated additional enrichment plots of Wnt/β-catenin signaling in shBecn1 iCMs.
To identify altered molecular features of reprogramming cells resulting from the loss of Becn1, we conducted pairwise comparison between shBecn1- and shNT-treated iCMs and then referenced data obtained from pairwise comparison between shBecn1- and shNT-treated LacZ-transduced control cells with a focus on DEGs that showed at least twofold changes (P < 0.05) in expression. On reprogramming day 3, shBecn1 led to up-regulation of 107 genes in iCMs (iCM-up) and down-regulation of 225 genes (iCM-down; fig. S8D). We then performed GO enrichment analysis on iCM-up and iCM-down genes, respectively. Because of the limited number of iCM-up genes, we did not identify significantly enriched GO terms for the iCM-up genes. In contrast, iCM-down genes were related to cytokinesis, cell division, and cell cycle, consistent with what we and others have demonstrated: that inhibition of proliferation is a prerequisite for iCM reprogramming (fig. S8E) (19, 24, 50). On reprogramming day 5, we observed 652 genes that were differentially overexpressed after Becn1 knockdown, 456 of which were iCM-up genes (Fig. 6, F and G). GO analysis indicated that iCM-up genes were clustered to groups regulating heart development, cardiac muscle contraction, protein phosphorylation, and regulation of gene expression (Fig. 6H). A total of 651 iCM-down genes were uncovered, and most of them were classified into immune regulation, cell matrix, and adhesion regulation (Fig. 6I). It is worth noting that GSEA analysis and GO analysis both identified a GO annotation “negative regulation of Wnt signaling pathway” (Fig. 6, E and I). We further confirmed the GSEA and GO data analysis by examining the DEG expression corresponding to GO terms “phosphorylation” (up-regulated), “regulation of gene expression” (up-regulated), “regulation of cell cycle” (down-regulated), and “regulation of immune response” (down-regulated; fig. S8, F to I). RT-PCR analysis was also performed to validate the expression of the representative genes from reprogramming day 3 to day 14, including serine/threonine-protein kinase Srpk3; transcription factors and cofactors including Lef1 (Lymphoid enhancer-binding factor 1), Cited1 (Cbp/p300-interacting trans-activator 1), and Mycn (N-myc proto-oncogene protein); Wnt inhibitors such as Sfrp1 (Secreted frizzled-related protein 1), Dkk3 (Dickkopf-related protein 3), and Dact3 (Dapper homolog 3); and immune response genes such as Toll-like receptor 8 and tumor necrosis factor–α (Fig. 6J).
Together, our data indicate that knocking down Becn1 led to global transcriptome changes in reprogramming cells. In addition to the expected alterations in the expression of cardiac and fibroblast genes, shBecn1 also resulted in changes in the expression of genes involved in several different biological/molecular processes, such as cell cycle, protein phosphorylation, gene expression, immune response, and Wnt signaling pathway.
Becn1 negatively regulates Wnt signaling to modulate iCM induction
To identify downstream functional mediators of Becn1 during iCM reprogramming, we performed a loss-of-function screen targeting 17 top DEGs that we identified from RNA-seq and were further verified by quantitative PCR (qPCR). For each candidate, a pool of shRNAs (four to five independent shRNA constructs) targeting different regions within the gene was used, and knockdown efficiency was validated (fig. S9A). Knockdown of four up-regulated genes and three down-regulated genes resulted in attenuated or enhanced iCM generation, respectively (Fig. 7A and fig. S9B). We decide to continue with epistatic rescue experiment using four functionally important up-regulated genes: serine/threonine-protein kinase Pak6 [P21 (RAC1) activated kinase 6], transcription factors Foxa2 and Lef1, and transcription regulator Bas2a (bromodomain adjacent to zinc finger domain protein 2A, an essential component of the nucleolar remodeling complex that mediates silencing of a fraction of ribosomal DNA).
Fig. 7. Becn1 knockdown activated Wnt/β-catenin signaling through regulating the expression of Lef1 and Wnt inhibitors.
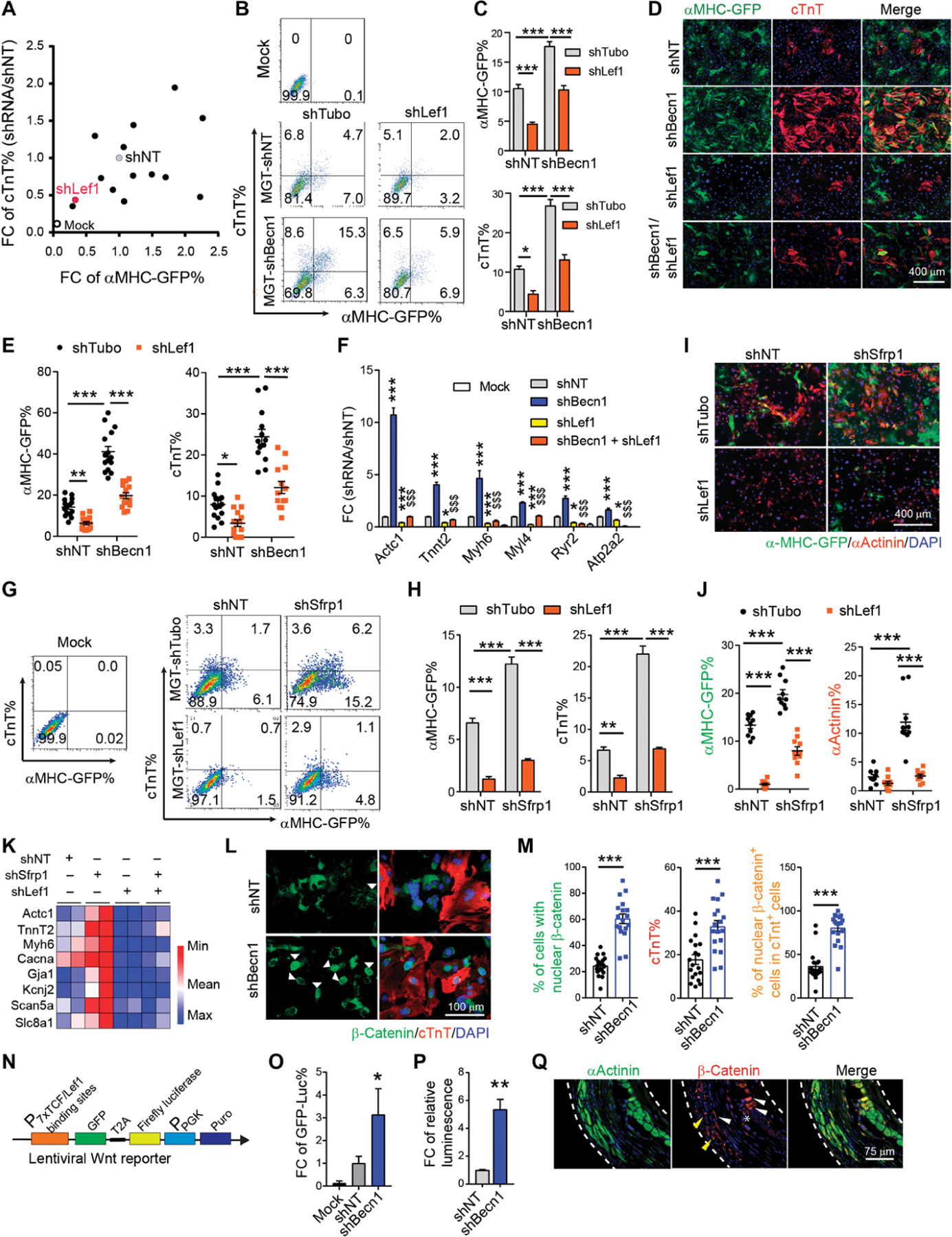
(A) shRNA screen for identifying Becn1 downstream effectors. 2D scatterplot showing fold change (FC) in the percentage of αMHC-GFP+ and cTnT+ cells on freshly isolated CFs. (B and C) Representative flow cytometry plots (B) and quantification (C) of αMHC-GFP+ and cTnT+ cells 10 days after MGT and indicated lentiviral shRNA transduction on freshly isolated CFs. (D and E) Representative images (D) and quantification (E) of cells transduced with MGT followed by shRNAs targeting Becn1, Lef1, and both on reprogramming day 14. (F) qPCR for cardiac gene expression in uninfected (mock) or MGT-infected iCMs treated with shNT, shBecn1, shLef1, or shBecn1 + shLef1. *, compared to the MGT+shNT group; $, compared to the MGT + shBecn1 group. (G and H) Representative flow cytometry plots (G) and quantification (H) of αMHC-GFP+ and cTnT+ cells 10 days after MGT transduction and after infection of shRNAs targeting NT, Sfrp1, Lef1, and Sfrp1 + Lef1 on freshly isolated CFs. (I and J) Representative images (I) and quantification (J) of cells as in (G). (K) Heatmap of cardiac gene expression in cells as in (G). (L and M) Representative images (L) and quantification (M) showing nuclear translocation of β-catenin (green) in iCMs positive for cTnT (red). (N) Schematic of lentiviral Wnt reporter construct harboring both GFP and firefly luciferase expression. (O) Quantification of flow analysis showing the FC of GFP-luciferase+ percentage on freshly isolated wild-type CD1 CFs transduced with Wnt reporter on day 14. Cells in the mock group were not infected by any kind of virus. Values were normalized to the MGT + shNT group. (P) Quantification of relative luminescence in cells as in (O). (Q) Representative ICC images showing cellular localization of β-catenin in hearts of Becn1+/− mice 8 weeks after MI and MGT delivery. White dashed line, area of infarct zone; yellow arrowhead, intercalated discs; white arrowhead, cytoplasmic localization; star, nuclear localization. All experiments were repeated at least three times. Mean values from technical triplicates [except n = 10 to 20 for (E), (J), and (M)] were used for statistics. Groups were compared using one-way ANOVA with Tukey’s multiple comparisons test for multiple groups and two-way ANOVA followed by Tukey post hoc test was used for (C), (E), (H), and (J). Error bars indicate means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001; $$$P < 0.001.
To this end, we introduced shRNAs against the four genes together with shBecn1 to MGT-infected CFs. Only Lef1 depletion blocked shBecn1-induced enhancement of iCM induction (Fig. 7, B and C, and fig. S9C), suggesting that other factors (Foxa2, Pak6, and Baz2a) are potentially essential for iCM reprogramming but are not directly involved in shBecn1-mediated improvement of iCM induction. We conducted ICC and quantified the number of αMHC-GFP+ and cTnT+ iCMs transduced with lentiviral shRNAs targeting Becn1 and Lef1 separately or simultaneously. Compared to the shNT control group, Becn1 deficiency consistently gave rise to an increased number of iCMs, whereas Lef1 ablation significantly decreased the number of αMHC-GFP+ or cTnT+ iCMs. Lef1 deficiency further blunted the enhanced conversion of CFs to iCMs induced by Becn1 depletion (Fig. 7, D and E). In accordance with flow and ICC results, the expression of sarcomere and ion channel markers (Actc1, TnnT2, Myh6, and Ryr2) showed similar changes in their expression upon double knockdown of Lef1 and Becn1 (Fig. 7F). On the basis of these observations, our results indicate that Lef1 mediated the repressive effect of Becn1 on iCM generation.
Among the three down-regulated genes, Sfrp1 encodes proteins that inhibit the Wnt pathway through direct binding to Wnt molecules (51). Similar to shBecn1, knockdown of Sfrp1 resulted in a greater number of iCMs with enhanced expression of cardiac genes (Fig. 7, G to K). We next simultaneously knocked down Sfrp1 and Lef1 in reprogramming fibroblasts. Ablation of Lef1 remarkably decreased the enhanced iCM conversion induced by shSfrp1 (Fig. 7, G to K). Together these data suggest that Lef1 is a critical factor that regulates CF to iCM cell fate conversion. Lef1 has been known for its essential role in the activation of Wnt/β-catenin downstream genes (52). In the absence of nuclear β-catenin, Lef1 acts as a transcriptional repressor through direct binding with Groucho/TLE molecules (52). The presence and binding of extracellular Wnt to its receptors stabilizes β-catenin, which, in turn, translocates into nucleus, where it interacts with Lef1 and activates gene expression (fig. S9D). We thus examined the translocation of β-catenin in shNT- and shBecn1-treated reprogramming cells. About 20 to 40% of shNT-transduced reprogramming cells exhibited nuclear localization of β-catenin, whereas the percentage in shBecn1-treated reprogramming cells increased to 60 to 80% (Fig. 7, L and M). Costaining of β-catenin with αActinin indicated that about 80 to 100% of cTnT+ cells in shBecn1 group showed nuclear retention of β-catenin, much higher than the percentage in the shNT group (about 40% of cTnT+ cells), indicating that shBecn1 promoted β-catenin translocation from cytoplasm to nucleus in reprogramming cells.
Given the observation that shBecn1 treatment enhanced Lef1 expression and β-catenin translocation to nucleus, we asked whether enhanced β-catenin nuclear translocation could increase Wnt signaling activation. We used a polycistronic Wnt reporter construct harboring GFP and firefly luciferase expression driven by a promoter containing specific T cell factor/lymphoid enhancer factor binding sequences (53) (Fig. 7N). We introduced lentivirus encoding this reporter construct together with shRNAs and MGT into fibroblasts. We evaluated GFP reporter expression (GFP-Luc) by flow cytometry and found that the percentage of GFP+ cells was increased three- to fivefold after Becn1 knockdown (Fig. 7O). Meanwhile, we performed Luciferase assays and calculated relative luminescence per cell. Compared to shNT control iCMs, shBecn1-iCMs exhibited a five- to sevenfold increase in the luciferase activity (Fig. 7P). These data demonstrate that Becn1 ablation led to elevated Lef1 expression and its higher transcriptional activity. Together, our results indicate that Becn1 functions to regulate Lef1 expression and Wnt antagonists to repress iCM generation.
Last, we asked whether Wnt signaling was also involved in Becn1-mediated in vivo cardiac reprogramming. To address this question, we performed immunofluorescence staining to examine the cellular localization of β-catenin in wild-type and Becn1+/− mice after MI and viral injection of reprogramming factors as shown in Fig. 4. Previous studies have demonstrated the intercalated disc staining of β-catenin in healthy adult heart (54, 55). We found similar localized staining pattern of β-catenin to the adherent junctions of cTnT+ cardiomyocytes at the distal zone and border zone (fig. S10, A to C). At the infarct zone, β-catenin expression was marginally detected in DsRed-treated control or Becn1+/− hearts (fig. S10D). In MGT-treated control hearts, β-catenin mainly exhibited cytoplasmic expression in iCMs, whereas in MGT-treated Becn1+/− mice, similar cytoplasmic localization of β-catenin was observed, but we also saw localized β-catenin at the cell-cell junction site of iCMs, indicating the maturation/integration of Becn1+/− iCMs into the surrounding myocardium. Some other iCMs demonstrated nuclear localization of β-catenin (Fig. 7Q), which may represent a transitional state that functions to accelerate iCM induction in vivo, consistent with what we observed from in vitro experiments.
Becn1 suppresses iCM conversion through the PI3K complex downstream of ULK1
To study how Becn1 regulates Wnt signaling pathway, we sought to identify endogenous Becn1 interaction proteins during iCM reprogramming. We conducted coimmunoprecipitation (Co-IP) assays with anti-Becn1 or anti–immunoglobulin G (IgG) control antibody in MGT-transduced CFs. Precipitated proteins were further processed for mass spectrometric (MS) analysis, and the experiments were repeated twice (table S5). We identified 130 candidate proteins that exhibited at least a twofold increase in Becn1 IP sample compared to that from IgG control, including the documented Becn1-binding partners NEDD4 (E3 ubiquitin-protein ligase NEDD4) (56) and 14–3-3 proteins (Ywhaz) (57) (Fig. 8A and table S2). To understand the biological processes involving these enriched candidates, we performed GO analysis and identified that these candidates were categorized in pathways regulating cell adhesion, cytoskeleton organization and nucleation, endocytosis, and macroautophagy (Fig. 8B). Core components of the autophagic PI3K III complex, including Vps34, Vps15, Atg14, Nrbf2, and Becn1 itself (58–60), were significantly enriched in the anti-Becn1 IP group. MS results were further confirmed by probing Vps34, Atg14, and Becn1 in an anti-Becn1 Co-IP product (Fig. 8, C and D).
Fig. 8. Becn1 suppresses iCM conversion by interacting with the PI3K complex downstream of ULK1.
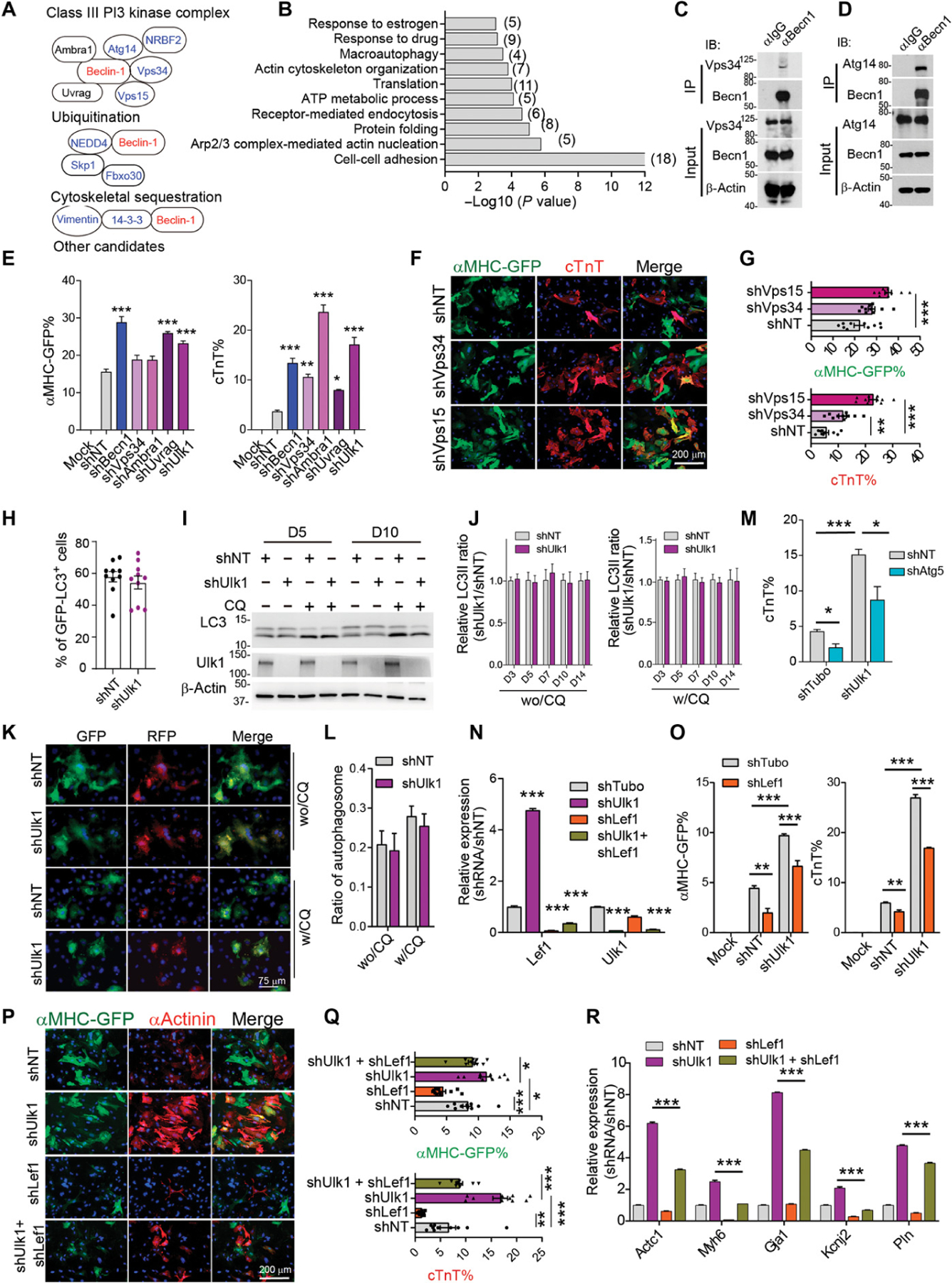
(A) Known Becn1-interacting protein identified from mass spectral analysis. (B) Enriched GO terms from potential Becn1 binding partners identified by mass spectral analysis. (C and D) Representative Western blot analysis showing the immunoprecipitation of Becn1 with Vps34 (C) and Atg14 (D) in iCMs 5 days after MGT transduction. (E) Quantification of flow analysis showing the percentage of αMHC-GFP+ and cTnT+ cells 10 days after MGT transduction and after infection of indicated shRNAs targeting the PI3K III complex. (F and G) Representative images (F) and quantification (G) of cells transduced with MGT followed by infection of shRNAs targeting NT, Vps35, and Vps15 on reprogramming day 14. (H) Quantification of GFP-LC3 puncta formation in iCMs treated with shUlk1 or shNT at reprogramming day 5. (I) Representative Western blot showing the autophagy flux in iCMs treated with shUlk1 or control shNT on reprogramming day 5 and day 10. (J) Quantification of autophagy flux detected by Western blot in iCMs with Ulk1 knockdown from day 3 to day 14. wo/CQ, without CQ treatment; w/CQ, with CQ treatment (20 µm) for 4 hours. (K and L) Representative images (K) and quantification (L) showing the formation of autophagosomes in iCMs upon Ulk1 knockdown in the presence or absence of CQ treatment. (M) Quantification of flow analysis showing the percentage of cTnT+ cells 10 days after MGT plus indicated shRNA transduction. (N) qPCR analysis of Lef1 and Ulk1 expression in cells as in cells treated with indicated shRNA. (O) Quantification of FACS analysis showing the percentage of αMHC-GFP+ and cTnT+ cells 10 days after MGT plus indicated shRNAs transduction. (P and Q) Representative (P) and quantification (Q) of ICC images showing the percentage of αMHC-GFP+ and cTnT+ cells 14 days after MGT and indicated shRNA transduction. (R) qPCR analysis of indicated cardiac gene expression in cells treated as in (P). All experiments were repeated at least three times. Mean values from technical triplicates [except n = 10 to 15 for (G), (J), (M), and (Q)] were used for statistics. Groups were compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups and two-way ANOVA followed by Tukey post hoc test was used for (K) and (O). Error bars indicate means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001.
To uncover the functional importance of the candidates that potentially bind to Becn1 during iCM reprogramming, we performed another loss-of-function screen targeting 22 genes that are representative of each biological GO process. We introduced lentiviruses encoding corresponding shRNAs to MGT-transduced CFs and evaluated their impact on iCM reprogramming. Ablations of genes involved in cell adhesion (Ywhaz, Lrrfip1, and Lrrfip2), Arp2/3 (actin-related protein 2/3 complex subunit 2) complex–mediated actin nucleation (Arp2 and Arp3), cytoskeleton organization (Nexn, Flii, and Ppp12a1), ubiquitylation (Nedd4 and Foxo30), and other unclassified genes (Gdi2 and Pawr) had minimal effects on iCM generation (fig. S11A). Knockdown of Ehd4 [EH (Eps15 homology) domain–containing protein 4] that plays a role in early endosomal transport (61, 62) significantly reduced the percentage of reprogrammed cells. In contrast, knocking down pre-mRNA alternative splicing regulator Ybx1 (nuclease-sensitive element-binding protein 1) with a knockdown efficiency reaching more than 70% increased reprogramming efficiency by two- to threefold (fig. S11, A and B). Prompted by their high enrichment from mass spectrometry and further validated binding by Co-IP experiment, we focused on the PI3K complex components and designed shRNAs targeting Vps34, Vps15, and other core components Ambra1 and Uvrag. Knocking down Vps34, Vps15, Ambra1, and Uvrag with more than 80% knockdown efficiency (fig. S11C) resulted in enhanced iCM induction, closely resembling the loss of Becn1 phenotype (Fig. 8, E to G, and fig. S11D).
We next determined whether an upstream regulator of the PI3K III complex also regulated iCM reprogramming. ULK1 has been reported to phosphorylate Becn1 and activate proautophagic VPS34 complexes (33). We thus transduced several different fibroblasts with MGT along with shRNAs targeting Ulk1. Ablation of Ulk1 with validated shRNAs significantly enhanced the yield and quality of iCMs regardless of which types of fibroblasts were used (Fig. 8E and fig. S11, E to N). To determine whether ULK1 played an autophagic role during reprogramming, we first evaluated the activity of ULK1 during reprogramming by measuring the expression of phosphorylated form of ULK1 and its substrate Becn1 and Atg14 (33, 63, 64) and found no change during early stage of reprogramming (fig. S12, A to I). In addition, Ulk1 ablation barely affected LC3-II expression, GFP-LC3 puncta formation, autophagy flux, and autophagosome formation (Fig. 8, H to J), and it did not alter mitophagy (fig. S11, J to Q). Furthermore, shUlk1-mediated enhanced iCM conversion was not completely blocked by interrupting autophagy with shAtg5 treatment (Fig. 8M). These data suggest that the Ulk1-Becn1 axis functions, at least partially, independent of autophagy during iCM conversion.
Last, we determined whether the Ulk1-Becn1-Lef1 regulatory axis functioned epistatically during iCM generation. Knockdown of Ulk1 alone gave rise to a five- to sixfold increase in Lef1 expression, mimicking the effect of shBecn1 on Lef1 expression (Fig. 8N). We then simultaneously knocked down Ulk1 and Lef1 in CFs after MGT transduction: shUlk1-enhanced iCM induction was attenuated when Lef1 was depleted (Fig. 8, N to R). Together, our data suggest that Ulk1 acts as an upstream regulator of Becn1 to modulate Lef1 expression and facilitate Becn1 to represses iCM conversion.
DISCUSSION
In this study, we demonstrated that autophagy is activated and required during direct cardiac reprogramming. Enhanced autophagy further increased iCM reprogramming efficiency. Knocking down Atg5 significantly blocked autophagy induction and led to attenuated iCM induction, suggesting an Atg5-dependent autophagic regulation during iCM generation. In contrast, the well-known autophagic factor Becn1 seemed to function as a barrier to iCM formation. Knocking down Becn1 led to enhanced quality and quantity of iCM, whereas in a mouse model, loss of one copy of Becn1 resulted in further improved MGT-induced enhancement in heart function and reduction in scar size after MI. Through a series of epistatic analyses, we established an autophagy-independent role of Becn1 in inducing myocyte identity. To explore the underlying mechanisms, we found that Becn1 depletion led to global transcriptome changes in reprogramming cells. In particular, Becn1 depletion resulted in enhanced activation of canonical Wnt/β-catenin through up-regulation of Lef1, suppression of Wnt inhibitors and enhanced β-catenin nuclear translocation. We also performed endogenous Becn1 immunoprecipitation followed by MS analyses and shRNA functional screening to further elucidate the molecular mechanisms by which Becn1 regulates Wnt signaling. We identified components in the classical PI3K complex that functioned similarly to Becn1 and demonstrated that the upstream factor ULK1 regulates Becn1-Lef1 signaling (fig. S13).
Autophagy has mostly been considered as a recycling process to maintain cellular homeostasis by promoting basal turnover of long-lived proteins and organelles. On the other hand, autophagy provides prosurvival signaling pathways in response to a wide variety of stresses such as starvation, ischemia, and infection (65). Recent studies have shown that autophagy plays critical roles in somatic reprogramming, ESC differentiation, and maintenance of cellular stemness (29–31, 66, 67). Our previous work demonstrated a deterministic trajectory of iCM reprogramming that requires removal of the starting fibrotic features and establishment of CM-related structures and metabolic program in the cells (24). Induction of autophagy during iCM fate conversion potentially facilitates the recycling of cellular components and provides new building blocks to remodel the cell’s architecture. Consistent with this idea, we observed engulfed mitochondria in the autophagosome around reprogramming day 7 (Fig. 1C), suggesting an intermediate stage of organelle turnover during subcellular reconstruction. Furthermore, pharmacological manipulation of autophagy by rapamycin and torin remarkably increased iCM reprogramming efficiency, indicating that selectively boosting autophagy may enhance iCM fate transition. It remains elusive and will be interesting to explore whether MGT-induced autophagy also functions in other aspects such as mitochondrial dynamics to combat cellular stress including apoptosis, oxidative stress, and epithelial-mesenchymal transition for successful cell fate determination and maintenance.
Dysregulation of Becn1 has been associated with many cardiovascular diseases (68). Becn1 overexpression in cardiomyocytes attenuated pathologic remodeling, whereas heterozygous Becn1 deletion attenuated pressure overload–induced cardiac hypertrophy (69) Adult mice with systemic heterozygous disruption of Becn1 display significantly reduced autophagy and develop cardiac dysfunction after starvation (70). The molecular mechanism underlying how Becn1 regulates cardiac function remains elusive. Our study has demonstrated that Becn1 deficiency led to higher iCM reprogramming efficiency, highlighting the role of Becn1 in iCM fate acquisition. Our findings also demonstrate a link between Becn1 and cardiac transcription profile through Wnt/β-catenin signaling. Knockdown of Becn1 led to an increased expression of Lef1 from reprogramming day 5, suppressed expression of Wnt inhibitors including Sfrp1/2, and translocation of β-catenin into nucleus that subsequently transactivated downstream gene expression. Knockdown of Lef1 hindered iCM conversion and blocked shBecn1-induced increase in iCM generation. Wnt/β-catenin signaling has been reported to play a biphasic role during cardiac specification and development (71). Canonical Wnt signaling controls CM proliferation in vivo and promotes proliferation of human ESC-derived CMs in vitro (72, 73). These observations, together with our study, highlight a critical role of Wnt/β-catenin signaling for cardiomyocyte fate determination and specification. In addition, our study identified a previously unidentified signaling pathway by which ULK1 regulates Becn1, interacting with the PI3K complex to repress Lef1, which provides further insights into the role of Wnt/β-catenin in controlling cell fate.
Although we show here the activation of autophagy during MGT-mediated cardiac reprogramming and provide evidence that Becn1 down-regulation resulted in enhanced direct cardiac reprogramming possibly through Wnt pathway, we acknowledge several inevitable limitations and alternative interpretations of our data in this study. First, the precise molecular mechanisms underlying how autophagy is induced by MGT transduction remain to be further elucidated. It has been shown that during iPSC generation, reprogramming factors seemed to repress mTORC1 (mammalian target of rapamycin complex 1) signaling, thus activating autophagy (30, 31); however, autophagy in direct reprogramming, in particular, cardiac reprogramming, has not been investigated. Integrative analysis using iCM transcriptomic (24) and epigenomic (46, 74) profiles may help to identify candidates that potentially activate autophagy during iCM reprogramming. We performed in vivo reprogramming using a Becn1 haploinsufficiency genetic model and observed further improved heart function and reduced scar size upon cardiac injury. This phenotype may not solely be due to a loss of Becn1 in CFs or the improved conversion of fibroblasts into iCMs. Future work using fibroblast-specific Becn1 knockout mice coupled with genetic lineage tracing may help further delineate how Becn1 functions in CFs and their fate conversion to myocytes in diseased hearts. In addition, although we demonstrated that Becn1 inhibits Wnt signaling to regulate iCM conversion, it remains unknown whether Becn1 directly or indirectly regulates Wnt/β-catenin signaling pathway and, if indirectly, what the functional mediators are. It is still unclear how the Becn1-containing PI3K complex inhibits the expression of Lef1, thereby negatively regulating transcriptional landscape of iCMs. Further exploration on these questions will shed light on how Becn1 and its involved pathway regulate direct cardiac reprogramming.
In summary, our study demonstrated autophagy-dependent and -independent pathways that regulate iCM fate acquisition. We identified Becn1 as a cellular barrier to iCM fate induction and found a previously unrecognized regulatory mechanism involving the ULK1–PI3K complex–Wnt/β-catenin signaling network. Our study thus provides further mechanistic insights into the molecular basis of fate conversion from fibroblast to cardiomyocyte.
MATERIALS AND METHODS
Study design
This study aimed to determine whether autophagy is activated during iCM reprogramming and the role of autophagic factor Becn1 in this process. Autophagy was assessed using Western blot showing LC3II conversion, transmission electron microscopy, and fluorescent GFP-LC3 and mRFP-GFP-LC3 puncta formation assays. The reprogramming efficiency in vitro was evaluated by flow cytometry, RT-qPCR, and ICC. The functionality of iCMs generated was examined by spontaneous beating assay, calcium oscillation assay, and MEA. To investigate the molecular mechanisms that Becn1 inhibits iCM reprogramming, we performed bulk RNA-seq, ATAC-seq, immunoprecipitation, and mass spectrometry. Last, to determine whether Becn1 deficiency affects heart function upon retroviral MGT injection, we performed permanent ligation of the left anterior descending artery (LAD) with Becn1-haplodeficient mice and littermate controls. Echocardiography was performed to evaluate heart function, and Masson Trichrome staining was used to quantify scar size. For in vitro experiments, at least biological triplicates were used in each single experiment, unless otherwise mentioned. Animal experiments were approved by the Institutional Animal Care and Use Committee at the University of North Carolina (UNC). Animals were randomized into groups receiving different viral injection after MI. Animals with FS% value of more than 70 (indicating no or small injury) after MI were excluded. The investigators were blinded to sample allocation in the study whenever possible. Some data were excluded from data analysis due to technical flaws. All primary data are reported in data file S1.
Mouse strains and plasmids
Transgenic mice of CD1 background that contain αMHC promoter-driven GFP were described previously (13). Becn1+/− mice on a C57BL/6 background were a gift from J. Taylor’s laboratory and were originally purchased from the Jackson Laboratory. Animal care was performed in accordance with the guidelines established by UNC, Chapel Hill. The empty pMXs-puro retroviral vectors were purchased from Cell Biolabs, and they harbor a partial lacZ sequence as a stuffer in between the 5′ and 3′ cloning sites. They were thereby referred to as RV (retroviral)–lacZ in this manuscript. pMXs retroviral vectors containing mouse Gata4, Mef2c, Tbx5, polycistronic MGT, and DsRed were described previously (11, 13). For gene overexpression, lentiviral backbone construct pLenti-GFP was purchased from Addgene (#17448) and was digested with Bam HI and Sal I for subsequent molecular cloning. Mouse wtLef1 was amplified from mouse spleen lymphocyte complementary DNA and was assembled into digested pLenti-GFP backbone by the Gibson Assembly Cloning Kit (NEB). The control pLenti-lacZ vector was cloned by replacing the GFP insert in pLenti-GFP with the partial lacZ sequence from pMXs-puro using Bam HI/Sal I digestion. Cloning primers are listed in table S3. Other constructs, including pMXs-IP-EGFP-mAtg5 (#38196) and pXL010-Wnt dual (GFP-Fire) reporter (#40588), were all purchased from Addgene. The nontargeting shNT pLKO.1-Scramble plasmid was a gift from Q. Zhang’s laboratory, UNC (75). Constructs carrying mRFP-GFP-LC3 and GFP-LC3 were gifted from J. Taylor’s laboratory and were cloned into plenti-CMV-puro backbone using Bam HI/Sal I restriction sites. All other shRNAs (pLKO.1 vector based, MISSION shRNA glycerol stock) were purchased from Sigma-Aldrich, and their TRC numbers are listed in table S4. All constructs used in this study were sequenced.
For shRNA experiments, because oligo #1 and #4 targeting Becn1 exhibited the highest cTnT+ ratio, we used a combination of #1 and #4 for all the experiments, unless otherwise mentioned. We used pooled shAtg5 #2 and #5 for the experiments, unless otherwise mentioned.
Cell culture and viral production
Human embryonic kidney (HEK) 293T cells (American Type Culture Collection) and platE cells (Cell Biolabs) were cultured in Dulbecco’s minimal essential medium (DMEM; high glucose) supplemented with 2 mM L-glutamine, 10% fetal bovine serum (FBS), 1× nonessential amino acids (NEAA), penicillin (100 U ml−1), and streptomycin (100 µg ml−1; all from Invitrogen). One day before transfection, cells were seeded at a density of 4 million to 5 million cells per 10-cm dish. For lentivirus production, 10 µg of viral constructs, 7 µg of psPAX2 (Addgene), and 3 µg of pMD2.G were cotransfected to 293T cells with Nanofect (Alstem) according to the manufacturer’s instructions. For murine MGT retrovirus production, the platE cells were transfected with 20-µg pMx-based plasmid with NanoFect or Lipofectamine 2000 (NEB). Culture supernatants were harvested at 48 and 72 hours after transfection, filtered through 45-µm pore size filters (Thermo Fisher Scientific), and incubated with polyethylene glycol (PEG)–4000 solution (8% final concentration) overnight at 4°C. The next day, the mixture was spun down at 3800 rpm for 30 min at 4°C, and the pelleted virus was resuspended with 100 µl of DMEM per 10-cm dish. For human MGT retrovirus production, the 293T cells were transduced with NanoFect together with 5 µg of Gag/pol, 2.5 µg of pCMV–vesicular stomatitis virus glycoprotein (Addgene #8454), and 15 µg of pMxs-hMGT as we previously described (46). Retrovirus was freshly used to infect target cells, and lentivirus can be used either freshly or frozen at −80°C until further use.
Reprogramming murine fibroblasts into iCMs
Murine fibroblasts were seeded at a density of 2 × 104 (without lentivirus inoculation) or 5 × 104 (with lentivirus inoculation) cells per well of a 24-well plate in IMDM medium containing 20% FBS, penicillin (100 U ml−1), and streptomycin (100 µg ml−1) on day −1 (D−1). Medium was changed to iCM medium [10% FBS in DMEM/M199 (4:1)] on the next day supplemented with Polybrene at a final concentration of 4 µg/ml (D0). Freshly resuspended retrovirus (20 µl) was added to the cells (one well of a 24-well plate, d0), and lentivirus (10 µl) was added 24 hours after retroviral infection (D1). At 72 hours after retrovirus infection (D3), iCM media containing puromycin (2 µg/ml) were added to cells to select virus-infected cells for the following 3 days. Medium was replaced by regular iCM medium since D6 and was changed every other day. On D14, iCM medium was changed to StemPro-34 SF medium (SPF34, Gibco) supplemented with GlutaMAX (Gibco; 10 µl/ml), ascorbic acid (Sigma-Aldrich; 50 µg/ml), recombinant human vascular endothelial growth factor 165 (VEGF165; R&D Systems; 5 ng/ml), recombinant human fibroblast growth factor (FGF) basic 146 amino acids (R&D Systems; 10 ng/ml), and recombinant human FGF10 (R&D Systems; 50 ng/ml). Medium was changed every 3 to 4 days. The number of spontaneously beating cells was counted under an EVOS microscope (40×) at 4 to 6 weeks.
Reprogramming human fibroblasts into iCMs
Human H9F fibroblasts (46) were cultured in IMDM medium containing 20% FBS supplemented with penicillin (100 U ml−1) and streptomycin (100 µg ml−1). One day before reprogramming, 4 × 104 H9Fs per well were seeded onto a SureCoat (Fisher Cellutron)–coated 24-well plate. Cells were infected with 10 µl of retrovirus encoding hMGT in iCM media containing polybrene (8 µg/ml). Lentivirus encoding indicated that shRNAs (4 µl) were delivered into cells 24 hours after hMGT transduction. Puromycin (1 µg/ml) was added to cultures 3 days after hMGT transduction, and media were replaced every 3 days.
Flow cytometry and immunofluorescence staining
Immunofluorescence staining and flow cytometry were performed as previously described (18). Primary antibodies (table S5) were used at the following dilutions: rabbit anti-GFP (Invitrogen, A11122; 1:500), chicken anti-GFP (Abcam, ab13970; 1:1500), anti-αActinin (Sigma-Aldrich, A7811; 1:500), anti-Cx43 (Sigma-Aldrich, C6219; 1:200), anti-Lamp1 (Abcam, ab25245; 1:200), and anti–β-catenin (Abcam, ab16051; 1:200). Secondary antibodies including Alexa Fluor 488–conjugated donkey anti-rabbit IgG, Alexa Fluor 488–conjugated donkey anti-chicken IgG, Alexa Fluor 647–conjugated donkey anti-mouse IgG, cyanine Cy3-conjugated donkey anti-rabbit IgG, and cyanine Cy3-conjugated donkey anti-mouse IgG were all from Jackson ImmunoResearch Inc. Images were captured using EVOS FL Auto Cell Imaging System (Life Technologies). For quantification, 10 to 20 images were randomly taken under ×10 or ×20 magnifications at the same exposure setting and then counted in a double-blinded way. For flow cytometry, iCMs on d10 were harvested by trypsin digestion at 37°C for 5 min. Cells were fixed, permeabilized, probed for αMHC-GFP and cTNT, and then analyzed on a BD Accuri C6 or Cyan flow cytometer. FlowJo software (Tree Star) was used to analyze flow cytometry data.
Microelectrode array
To characterize the electrophysiological properties of the iCMs, an MEA recording system (Multichannel Systems, Reutlingen, Germany) was used. The MEAs have 60 microelectrodes, with a diameter of 30 µm, positioned on an 8 × 8 grid with 200-µm spacing. Freshly isolated neonatal FBs (fibroblasts) were plated on gelatin-coated MEA culture plates and were transduced with MGT retrovirus in the presence of shRNA targeting Becn1 or nontargeting (NT) control. The reprogramming was performed as aforementioned. Standard measurements of field potential were performed at 5 kHz in culture medium at 37°C (76). Data acquisition and analysis were finished using MC_Rack software. The number of spikes recorded from electrodes showing extracellular electrograms was calculated in a time frame of 30 s.
Luciferase assay
Lentivirus harboring pXL010-Wnt dual (GFP-Fire) reporter was prepared as mentioned above. On reprogramming day 10, iCMs transduced with shBcn1 or shNT were rinsed in phosphate-buffered saline (PBS) and were singularized using 0.05% trypsin. Luciferase expression was quantified using the Bright-Glo Luciferase Assay (Promega) according to the manufacturer’s instructions. Plates were read on GloMAX (Promega). Expression was normalized to total live single cell number determined by manually counting using Trypan Blue solution 0.4% (Sigma-Aldrich).
Mouse MI model and in vivo delivery of retrovirus
Animal protocol was approved by institutional guidelines [UNC-Chapel Hill DAML (Division of Laboratory Animal Medicine)], and all procedures conformed to the National Institutes of Health (NIH) guidelines and DLAM guidelines. MI was induced to Becn1+/− mutant or Becn1+/+ littermate control mice that were 10 to 12 weeks old. Both male and female mice were used and randomly assigned to receive reprogramming factors or DsRed control retrovirus. Before surgery, mice were anesthetized with ketamine (100 mg/kg) plus xylazine (10 mg/kg) and ventilated with a MiniVent Type 845 ventilator. After exposing the heart, MI was performed by permanent ligation of the LAD with a 7–0 prolene suture as described previously (12, 15). Retroviruses expressing M, G, and T/DsRed (for tracing M-, G-, and T-infected cells) and DsRed alone (negative control) were generated and subjected to standard ultracentrifugation to reach a viral titer about 1 × 1010 plaque-forming units per milliliter. In addition, 10 µl of virus plus 10 µl of PBS supplemented with polybrene (8 µg/ml) was injected into the myocardium along the boundary between the infarct zone and border zone based on the blanched infarct area immediately after coronary artery occlusion (12, 15). Mice were allowed to recover on the heating pad after closing the chest. MI, echocardiography, and histology were performed in a double-blinded manner.
Statistical analysis
Unless otherwise stated, values are expressed as average ± SEM of multiple biologically independent samples and were statistically analyzed by GraphPad Prism 7 software. Statistical tests performed include Student’s t test (if only two conditions were tested) and one-way (for experiments containing three or more conditions) or two-way analysis of variance (ANOVA), followed by Tukey post hoc test (for experiments comparing multiple treatment groups with each other). Generally, a P value of <0.05 was considered statistically significant (*), a P value of <0.01 was considered highly significant (**), and a P value of <0.001 was considered strongly significant (***). All data are representative of multiple repeated experiments.
Supplementary Material
Data file S1. Primary data.
Table S3. List of qPCR primers and cloning primers used in this study.
Table S4. Sequence information for shRNA clones used in this study.
Table S5. List of antibodies used for immunofluorescent staining, immunoblotting, and flow cytometry.
Fig. S1. Autophagy is activated during iCM reprogramming.
Fig. S2. Atg5 and Atg7 knockdown impaired iCM reprogramming.
Fig. S3. Atg5-dependent autophagy induction during iCM reprogramming.
Fig. S4. Knockdown of Becn1 enhanced iCM reprogramming.
Fig. S5. Characterization of heart structure and cardiac function in young Becn1+/− mice.
Fig. S6. Characterization of Becn1 during reprogramming.
Fig. S7. Transduction of MGT reprogramming factors changed chromatin accessibility but not Becn1 expression.
Fig. S8. Transcriptomic alternations induced by Becn1 knockdown.
Fig. S9. Validation and confirmation of shRNA screen.
Fig. S10. Localization of β-catenin in injured heart.
Fig. S11. Identification of Becn1 interaction partners and characterization of Ulk1 in iCM reprogramming.
Fig. S12. Ulk1 activity and mitophagy remain unchanged during reprogramming.
Fig. S13. Schematic representation of autophagy-dependent and -independent mechanisms of iCM induction.
Fig. S14. Unprocessed original scans of western blots.
References (77–85)
Table S2. MS analysis of Becn1 interaction proteins.
Acknowledgments:
We thank the UNC HTSF Core for all sequencing work and assistance on bioinformatics analysis, the UNC FLOW Core for assistance on flow cytometry, UNC Michael Hooker Proteomics Core for mass spectrometry sample processing and data analysis, and UNC Microscopy Services Laboratory for TEM experiments. We thank M. Combs, Q. Zhu, and R. Lu for data consultation. We thank members of the Qian and Liu laboratories for helpful discussions and critical reviews of the manuscript.
Funding:
This study was supported by American Heart Association (AHA) Career Development Award 18CDA34110340 to L.W.; NIH/NHLBI R01HL139880 and HL139976 to J.L.; and AHA 18TPA34180058, NIH/NHLBI R01HL128331, and R01HL144551 to L.Q.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. The RNA-seq and ATAC-seq datasets reported in this paper have been deposited in the Gene Expression Omnibus with accession GSE153560.
REFERENCES AND NOTES
- 1.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S, Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S, Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Song G, Pacher M, Balakrishnan A, Yuan Q, Tsay H-C, Yang D, Reetz J, Brandes S, Dai Z, Pützer BM, Araúzo-Bravo MJ, Steinemann D, Luedde T, Schwabe RF, Manns MP, Schöler HR, Schambach A, Cantz T, Ott M, Sharma AD, Direct reprogramming of hepatic myofibroblasts into hepatocytes in vivo attenuates liver fibrosis. Cell Stem Cell 18, 797–808 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Han DW, Greber B, Wu G, Tapia N, Araúzo-Bravo MJ, Ko K, Bernemann C, Stehling M, Schöler HR, Direct reprogramming of fibroblasts into epiblast stem cells. Nat. Cell Biol 13, 66–71 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, Hui L, Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 475, 386–389 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Szabo E, Rampalli S, Risueño RM, Schnerch A, Mitchell R, Fiebig-Comyn A, Levadoux-Martin M, Bhatia M, Direct conversion of human fibroblasts to multilineage blood progenitors. Nature 468, 521–526 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M, Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karow M, Sánchez R, Schichor C, Masserdotti G, Ortega F, Heinrich C, Gascón S, Khan MA, Lie DC, Dellavalle A, Cossu G, Goldbrunner R, Götz M, Berninger B, Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 11, 471–476 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Qian L, Berry EC, Fu J.-d., Ieda M, Srivastava D, Reprogramming of mouse fibroblasts into cardiomyocyte-like cells in vitro. Nat. Protoc 8, 1204–1215 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Mathison M, Singh VP, Gersch RP, Ramirez MO, Cooney A, Kaminsky SM, Chiuchiolo MJ, Nasser A, Yang J, Crystal RG, Rosengart TK, “Triplet” polycistronic vectors encoding Gata4, Mef2c, and Tbx5 enhances postinfarct ventricular functional improvement compared with singlet vectors. J. Thorac. Cardiovasc. Surg 148, 1656–1664.e2 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, Bursac N, Liu J, Qian L, Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res 116, 237–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu J.-d., Srivastava D, In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485, 593–598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D, Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song K, Nam Y-J, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN, Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485, 599–604 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma H, Wang L, Yin C, Liu J, Qian L, In vivo cardiac reprogramming using an optimal single polycistronic construct. Cardiovasc. Res 108, 217–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohamed TMA, Stone NR, Berry EC, Radzinsky E, Huang Y, Pratt K, Ang Y-S, Yu P, Wang H, Tang S, Magnitsky S, Ding S, Ivey KN, Srivastava D, Chemical enhancement of in vitro and in vivo direct cardiac reprogramming. Circulation 135, 978–995 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, Nishiyama T, Kaneda R, Fukuda T, Takeda S, Tohyama S, Hashimoto H, Kawamura Y, Goshima N, Aeba R, Yamagishi H, Fukuda K, Ieda M, MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. EMBO J 33, 1565–1581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z, Chen O, Zheng M, Wang L, Zhou Y, Yin C, Liu J, Qian L, Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res 16, 507–518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y, Wang L, Liu Z, Alimohamadi S, Yin C, Liu J, Qian L, Comparative gene expression analyses reveal distinct molecular signatures between differentially reprogrammed cardiomyocytes. Cell Rep 20, 3014–3024 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyamoto K, Akiyama M, Tamura F, Isomi M, Yamakawa H, Sadahiro T, Muraoka N, Kojima H, Haginiwa S, Kurotsu S, Tani H, Wang L, Qian L, Inoue M, Ide Y, Kurokawa J, Yamamoto T, Seki T, Aeba R, Yamagishi H, Fukuda K, Ieda M, Direct in vivo reprogramming with Sendai virus vectors improves cardiac function after myocardial infarction. Cell Stem Cell 22, 91–103.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu J-D, Wang GG, Liu J, Qian L, Bmi1 is a key epigenetic barrier to direct cardiac reprogramming. Cell Stem Cell 18, 382–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H, Morales MG, Hashimoto H, Dickson ME, Song K, Ye W, Kim MS, Niederstrasser H, Wang Z, Chen B, Posner BA, Bassel-Duby R, Olson EN, ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression. Genes Dev 31, 1770–1783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou H, Dickson ME, Kim MS, Bassel-Duby R, Olson EN, Akt1/protein kinase B enhances transcriptional reprogramming of fibroblasts to functional cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A 112, 11864–11869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Wang L, Welch JD, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, Yin C, Shen W, Prins JF, Liu J, Qian L, Single-cell transcriptomics reconstructs fate conversion from fibroblast to cardiomyocyte. Nature 551, 100–104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abad M, Hashimoto H, Zhou H, Morales MG, Chen B, Bassel-Duby R, Olson EN, Notch inhibition enhances cardiac reprogramming by increasing MEF2C transcriptional activity. Stem Cell Rep 8, 548–560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Lei I, Karatas H, Li Y, Wang L, Gnatovskiy L, Dou Y, Wang S, Qian L, Wang Z, Targeting Mll1 H3K4 methyltransferase activity to guide cardiac lineage specific reprogramming of fibroblasts. Cell Discov 2, 16036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DCO, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC, Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev 90, 1383–1435 (2010). [DOI] [PubMed] [Google Scholar]
- 28.McPhee CK, Logan MA, Freeman MR, Baehrecke EH, Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 465, 1093–1096 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, Liu K, Cao N, Nie B.-m., Zhu S.-y., Xu S, Li K, Wei W.-g., Wu Y, Guan K.-l., Ding S, Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol 17, 1379–1387 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Wu Y, Li Y, Zhang H, Huang Y, Zhao P, Tang Y, Qiu X, Ying Y, Li W, Ni S, Zhang M, Liu L, Xu Y, Zhuang Q, Luo Z, Benda C, Song H, Liu B, Lai L, Liu X, Tse H-F, Bao X, Chan W-Y, Esteban MA, Qin B, Pei D, Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat. Cell Biol 17, 715–725 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z, Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 13, 617–625 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B, Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol 72, 8586–8596 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell RC, Tian Y, Yuan H, Park HW, Chang Y-Y, Kim J, Kim H, Neufeld TP, Dillin A, Guan K-L, ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol 15, 741–750 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, Cecconi F, mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol 15, 406–416 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Jin M, Zhu Z, Wang H, Yu J, Li Y, Hao Y, Choi A, Ke H, Ma D, Yuan J, Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yue Z, Jin S, Yang C, Levine AJ, Heintz N, Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. U.S.A 100, 15077–15082 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, Levine B, Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Arozena AA, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, Agarwal R, Aghi MK, Agnello M, Agostinis P, Aguilar PV, Aguirre-Ghiso J, Airoldi EM, Ait-Si-Ali S, Akematsu T, Akporiaye ET, Al-Rubeai M, Albaiceta GM, Albanese C, Albani D, Albert ML, Aldudo J, Algül H, Alirezaei M, Alloza I, Almasan A, Almonte-Beceril M, Alnemri ES, Alonso C, Altan-Bonnet N, Altieri DC, Alvarez S, Alvarez-Erviti L, Alves S, Amadoro G, Amano A, Amantini C, Ambrosio S, Amelio I, Amer AO, Amessou M, Amon A, An Z, Anania FA, Andersen SU, Andley UP, Andreadi CK, Andrieu-Abadie N, Anel A, Ann DK, Anoopkumar-Dukie S, Antonioli M, Aoki H, Apostolova N, Aquila S, Aquilano K, Araki K, Arama E, Aranda A, Araya J, Arcaro A, Arias E, Arimoto H, Ariosa AR, Armstrong JL, Arnould T, Arsov I, Asanuma K, Askanas V, Asselin E, Atarashi R, Atherton SS, Atkin JD, Attardi LD, Auberger P, Auburger G, Aurelian L, Autelli R, Avagliano L, Avantaggiati ML, Avrahami L, Awale S, Azad N, Bachetti T, Backer JM, Bae D-H, Bae J-S, Bae O-N, Bae SH, Baehrecke EH, Baek S-H, Baghdiguian S, Bagniewska-Zadworna A, Bai H, Bai J, Bai X-Y, Bailly Y, Balaji KN, Balduini W, Ballabio A, Balzan R, Banerjee R, Bánhegyi G, Bao H, Barbeau B, Barrachina MD, Barreiro E, Bartel B, Bartolomé A, Bassham DC, Bassi MT, Bast RC Jr., Basu A, Batista MT, Batoko H, Battino M, Bauckman K, Baumgarner BL, Bayer KU, Beale R, Beaulieu J-F, Beck GR Jr., Becker C, Beckham JD, Bédard P-A, Bednarski PJ, Begley TJ, Behl C, Behrends C, Behrens GM, Behrns KE, Bejarano E, Belaid A, Belleudi F, Bénard G, Berchem G, Bergamaschi D, Bergami M, Berkhout B, Berliocchi L, Bernard A, Bernard M, Bernassola F, Bertolotti A, Bess AS, Besteiro S, Bettuzzi S, Bhalla S, Bhattacharyya S, Bhutia SK, Biagosch C, Bianchi MW, Biard-Piechaczyk M, Billes V, Bincoletto C, Bingol B, Bird SW, Bitoun M, Bjedov I, Blackstone C, Blanc L, Blanco GA, Blomhoff HK, Boada-Romero E, Böckler S, Boes M, Boesze-Battaglia K, Boise LH, Bolino A, Boman A, Bonaldo P, Bordi M, Bosch J, Botana LM, Botti J, Bou G, Bouché M, Bouchecareilh M, Boucher M-J, Boulton ME, Bouret SG, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady N, Braga VM, Brancolini C, Braus GH, Bravo-San Pedro JM, Brennan LA, Bresnick EH, Brest P, Bridges D, Bringer M-A, Brini M, Brito GC, Brodin B, Brookes PS, Brown EJ, Brown K, Broxmeyer HE, Bruhat A, Brum PC, Brumell JH, Brunetti-Pierri N, Bryson-Richardson RJ, Buch S, Buchan AM, Budak H, Bulavin DV, Bultman SJ, Bultynck G, Bumbasirevic V, Burelle Y, Burke RE, Burmeister M, Bütikofer P, Caberlotto L, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calatayud S, Camougrand N, Campanella M, Campbell GR, Campbell M, Campello S, Candau R, Caniggia I, Cantoni L, Cao L, Caplan AB, Caraglia M, Cardinali C, Cardoso SM, Carew JS, Carleton LA, Carlin CR, Carloni S, Carlsson SR, Carmona-Gutierrez D, Carneiro LA, Carnevali O, Carra S, Carrier A, Carroll B, Casas C, Casas J, Cassinelli G, Castets P, Castro-Obregon S, Cavallini G, Ceccherini I, Cecconi F, Cederbaum AI, Ceña V, Cenci S, Cerella C, Cervia D, Cetrullo S, Chaachouay H, Chae H-J, Chagin AS, Chai C-Y, Chakrabarti G, Chamilos G, Chan EY, Chan MT, Chandra D, Chandra P, Chang C-P, Chang RC-C, Chang TY, Chatham JC, Chatterjee S, Chauhan S, Che Y, Cheetham ME, Cheluvappa R, Chen C-J, Chen G, Chen G-C, Chen G, Chen H, Chen JW, Chen J-K, Chen M, Chen M, Chen P, Chen Q, Chen Q, Chen S-D, Chen S, Chen SS-L, Chen W, Chen W-J, Chen WQ, Chen W, Chen X, Chen Y-H, Chen Y-G, Chen Y, Chen Y, Chen Y, Chen Y-J, Chen Y-Q, Chen Y, Chen Z, Chen Z, Cheng A, Cheng CH, Cheng H, Cheong H, Cherry S, Chesney J, Cheung CHA, Chevet E, Chi HC, Chi S-G, Chiacchiera F, Chiang H-L, Chiarelli R, Chiariello M, Chieppa M, Chin L-S, Chiong M, Chiu GN, Cho D-H, Cho S-G, Cho WC, Cho Y-Y, Cho Y-S, Choi AM, Choi E-J, Choi E-K, Choi J, Choi ME, Choi S-I, Chou T-F, Chouaib S, Choubey D, Choubey V, Chow K-C, Chowdhury K, Chu CT, Chuang T-H, Chun T, Chung H, Chung T, Chung Y-L, Chwae Y-J, Cianfanelli V, Ciarcia R, Ciechomska IA, Ciriolo MR, Cirone M, Claerhout S, Clague MJ, Clària J, Clarke PG, Clarke R, Clementi E, Cleyrat C, Cnop M, Coccia EM, Cocco T, Codogno P, Coers J, Cohen EE, Colecchia D, Coletto L, Coll NS, Colucci-Guyon E, Comincini S, Condello M, Cook KL, Coombs GH, Cooper CD, Cooper JM, Coppens I, Corasaniti MT, Corazzari M, Corbalan R, Corcelle-Termeau E, Cordero MD, Corral-Ramos C, Corti O, Cossarizza A, Costelli P, Costes S, Cotman SL, Coto-Montes A, Cottet S, Couve E, Covey LR, Cowart LA, Cox JS, Coxon FP, Coyne CB, Cragg MS, Craven RJ, Crepaldi T, Crespo JL, Criollo A, Crippa V, Cruz MT, Cuervo AM, Cuezva JM, Cui T, Cutillas PR, Czaja MJ, Czyzyk-Krzeska MF, Dagda RK, Dahmen U, Dai C, Dai W, Dai Y, Dalby KN, Valle LD, Dalmasso G, D’Amelio M, Damme M, Darfeuille-Michaud A, Dargemont C, Darley-Usmar VM, Dasarathy S, Dasgupta B, Dash S, Dass CR, Davey HM, Davids LM, Dávila D, Davis RJ, Dawson TM, Dawson VL, Daza P, de Belleroche J, de Figueiredo P, de Figueiredo RCBQ, de la Fuente J, De Martino L, De Matteis A, De Meyer GR, De Milito A, De Santi M, de Souza W, De Tata V, De Zio D, Debnath J, Dechant R, Decuypere J-P, Deegan S, Dehay B, Bello BD, Del Re DP, Delage-Mourroux R, Delbridge LM, Deldicque L, Delorme-Axford E, Deng Y, Dengjel J, Denizot M, Dent P, Der CJ, Deretic V, Derrien B, Deutsch E, Devarenne TP, Devenish RJ, Bartolomeo SD, Daniele ND, Domenico FD, Nardo AD, Paola SD, Pietro AD, Renzo LD, Antonio AD, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dickey CA, Dickson RC, Diederich M, Digard P, Dikic I, Dinesh-Kumar SP, Ding C, Ding W-X, Ding Z, Dini L, Distler JH, Diwan A, Djavaheri-Mergny M, Dmytruk K, Dobson RCJ, Doetsch V, Dokladny K, Dokudovskaya S, Donadelli M, Dong XC, Dong X, Dong Z, Donohue TM Jr., Doran KS, D’Orazi G, Dornnd GW, Dosenko V, Dridi S, Drucker L, Du J, Du L-L, Du L, du Toit A, Dua P, Duan L, Duann P, Dubey VK, Duchen MR, Duchosal MA, Duez H, Dugail I, Dumit VI, Duncan MC, Dunlop EA, Dunn WA Jr., Dupont N, Dupuis L, Durán RV, Durcan TM, Duvezin-Caubet S, Duvvuri U, Eapen V, Ebrahimi-Fakhari D, Echard A, Eckhart L, Edelstein CL, Edinger L, Eichinger L, Eisenberg T, Eisenberg-Lerner A, Eissa NT, El-Deiry WS, El-Khoury V, Elazar Z, Eldar-Finkelman H, Elliott CJ, Emanuele E, Emmenegger U, Engedal N, Engelbrecht A-M, Engelender S, Enserink JM, Erdmann R, Erenpreisa J, Eri R, Eriksen JL, Erman A, Escalante R, Eskelinen E-L, Espert L, Esteban-Martínez L, Evans TJ, Fabri M, Fabrias G, Fabrizi C, Facchiano A, Færgeman NJ, Faggioni A, Fairlie WD, Fan C, Fan D, Fan J, Fang S, Fanto M, Fanzani A, Farkas T, Faure M, Favier FB, Fearnhead H, Federici M, Fei E, Felizardo TC, Feng H, Feng Y, Feng Y, Ferguson TA, Fernández ÁF, Fernandez-Barrena MG, Fernandez-Checa JC, Fernández-López A, Fernandez-Zapico ME, Feron O, Ferraro E, Ferreira-Halder CV, Fesus L, Feuer R, Fiesel FC, Filippi-Chiela EC, Filomeni G, Fimia GM, Fingert JH, Finkbeiner S, Finkel T, Fiorito F, Fisher PB, Flajolet M, Flamigni F, Florey O, Florio S, Floto RA, Folini M, Follo C, Fon EA, Fornai F, Fortunato F, Fraldi A, Franco R, Francois A, François A, Frankel LB, Fraser ID, Frey N, Freyssenet DG, Frezza C, Friedman SL, Frigo DE, Fu D, Fuentes JM, Fueyo J, Fujitani Y, Fujiwara Y, Fujiya M, Fukuda M, Fulda S, Fusco C, Gabryel B, Gaestel M, Gailly P, Gajewska M, Galadari S, Galili G, Galindo I, Galindo MF, Galliciotti G, Galluzzi L, Galluzzi L, Galy V, Gammoh N, Gandy S, Ganesan AK, Ganesan S, Ganley IG, Gannagé M, Gao F-B, Gao F, Gao J-X, Nannig LG, Véscovi EG, Garcia-Macía M, Garcia-Ruiz C, Garg AD, Garg PK, Gargini R, Gassen NC, Gatica D, Gatti E, Gavard J, Gavathiotis E, Ge L, Ge P, Ge S, Gean P-W, Gelmetti V, Genazzani AA, Geng J, Genschik P, Gerner L, Gestwicki JE, Gewirtz DA, Ghavami S, Ghigo E, Ghosh D, Giammarioli AM, Giampieri F, Giampietri C, Giatromanolaki A, Gibbings DJ, Gibellini L, Gibson SB, Ginet V, Giordano A, Giorgini F, Giovannetti E, Girardin SE, Gispert S, Giuliano S, Gladson CL, Glavic A, Gleave M, Godefroy N, Gogal RM Jr., Gokulan K, Goldman GH, Goletti D, Goligorsky MS, Gomes AV, Gomes LC, Gomez H, Gomez-Manzano C, Gómez-Sánchez R, Gonçalves DA, Goncu E, Gong Q, Gongora C, Gonzalez CB, Gonzalez-Alegre P, Gonzalez-Cabo P, González-Polo RA, Goping IS, Gorbea C, Gorbunov NV, Goring DR, Gorman AM, Gorski SM, Goruppi S, Goto-Yamada S, Gotor C, Gottlieb RA, Gozes I, Gozuacik D, Graba Y, Graef M, Granato GE, Grant GD, Grant S, Gravina GL, Green DR, Greenhough A, Greenwood MT, Grimaldi B, Gros F, Grose C, Groulx J-F, Gruber F, Grumati P, Grune T, Guan J-L, Guan K-L, Guerra B, Guillen C, Gulshan K, Gunst J, Guo C, Guo L, Guo M, Guo W, Guo X-G, Gust AA, Gustafsson ÅB, Gutierrez E, Gutierrez MG, Gwak H-S, Haas A, Haber JE, Hadano S, Hagedorn M, Hahn DR, Halayko AJ, Hamacher-Brady A, Hamada K, Hamai A, Hamann A, Hamasaki M, Hamer I, Hamid Q, Hammond EM, Han F, Han W, Handa JT, Hanover JA, Hansen M, Harada M, Harhaji-Trajkovic L, Harper JW, Harrath AH, Harris AL, Harris J, Hasler U, Hasselblatt P, Hasui K, Hawley RG, Hawley TS, He C, He CY, He F, He G, He R-R, He X-H, He Y-W, He Y-Y, Heath JK, Hébert M-J, Heinzen RA, Helgason GV, Hensel M, Henske EP, Her C, Herman PK, Hernández A, Hernandez C, Hernández-Tiedra S, Hetz C, Hiesinger PR, Higaki K, Hilfiker S, Hill BG, Hill JA, Hill WD, Hino K, Hofius D, Hofman P, Höglinger GU, Höhfeld J, Holz MK, Hong Y, Hood DA, Hoozemans JJ, Hoppe T, Hsu C, Hsu C-Y, Hsu L-C, Hu D, Hu G, Hu H-M, Hu H, Hu MC, Hu Y-C, Hu Z-W, Hua F, Hua Y, Huang C, Huang H-L, Huang K-H, Huang K-Y, Huang S, Huang S, Huang W-P, Huang Y-R, Huang Y, Huang Y, Huber TB, Huebbe P, Huh W-K, Hulmi JJ, Hur GM, Hurley JH, Husak Z, Hussain SN, Hussain S, Hwang JJ, Hwang S, Hwang TI, Ichihara A, Imai Y, Imbriano C, Inomata M, Into T, Iovane V, Iovanna JL, Iozzo RV, Ip NY, Irazoqui JE, Iribarren P, Isaka Y, Isakovic AJ, Ischiropoulos H, Isenberg JS, Ishaq M, Ishida H, Ishii I, Ishmael JE, Isidoro C, Isobe K-I, Isono E, Issazadeh-Navikas S, Itahana K, Itakura E, Ivanov AI, Iyer AKV, Izquierdo JM, Izumi Y, Izzo V, Jäättelä M, Jaber N, Jackson DJ, Jackson WT, Jacob TG, Jacques TS, Jagannath C, Jain A, Jana NR, Jang BK, Jani A, Janji B, Jannig PR, Jansson PJ, Jean S, Jendrach M, Jeon J-H, Jessen N, Jeung E-B, Jia K, Jia L, Jiang H, Jiang H, Jiang L, Jiang T, Jiang X, Jiang X, Jiang X, Jiang Y, Jiang Y, Jiménez A, Jin C, Jin H, Jin L, Jin M, Jin S, Jinwal UK, Jo E-K, Johansen T, Johnson DE, Johnson GV, Johnson JD, Jonasch E, Jones C, Joosten LA, Jordan J, Joseph A-M, Joseph B, Joubert AM, Ju D, Ju J, Juan H-F, Juenemann K, Juhász G, Jung HS, Jung JU, Jung Y-K, Jungbluth H, Justice MJ, Jutten B, Kaakoush NO, Kaarniranta K, Kaasik A, Kabuta T, Kaeffer B, Kågedal K, Kahana A, Kajimura S, Kakhlon O, Kalia M, Kalvakolanu DV, Kamada Y, Kambas K, Kaminskyy VO, Kampinga HH, Kandouz M, Kang C, Kang R, Kang T-C, Kanki T, Kanneganti T-D, Kanno H, Kanthasamy AG, Kantorow M, Kaparakis-Liaskos M, Kapuy O, Karantza V, Karim MR, Karmakar P, Kaser A, Kaushik S, Kawula T, Kaynar AM, Ke P-Y, Ke Z-J, Kehrl JH, Keller KE, Kemper JK, Kenworthy AK, Kepp O, Kern A, Kesari S, Kessel D, Ketteler R, do Carmo Kettelhut I, Khambu B, Khan MM, Khandelwal VK, Khare S, Kiang JG, Kiger AA, Kihara A, Kim AL, Kim H, Kim DR, Kim D-H, Kim EK, Kim HY, Kim H-R, Kim J-S, Kim JH, Kim JC, Kim JH, Kim KW, Kim MD, Kim M-M, Kim PK, Kim SW, Kim S-Y, Kim Y-S, Kim Y, Kimchi A, Kimmelman AC, Kimura T, King JS, Kirkegaard K, Kirkin V, Kirshenbaum LA, Kishi S, Kitajima Y, Kitamoto K, Kitaoka Y, Kitazato K, Kley RA, Klimecki WT, Klinkenberg M, Klucken J, Knævelsrud H, Knecht E, Knuppertz L, Ko J-L, Kobayashi S, Koch JC, Koechlin-Ramonatxo C, Koenig U, Koh YH, Köhler K, Kohlwein SD, Koike M, Komatsu M, Kominami E, Kong D, Kong HJ, Konstantakou EG, Kopp BT, Korcsmaros T, Korhonen L, Korolchuk VI, Koshkina NV, Kou Y, Koukourakis MI, Koumenis C, Kovács AL, Kovács T, Kovacs WJ, Koya D, Kraft C, Krainc D, Kramer H, Kravic-Stevovic T, Krek W, Kretz-Remy C, Krick R, Krishnamurthy M, Kriston-Vizi J, Kroemer G, Kruer MC, Kruger R, Ktistakis NT, Kuchitsu K, Kuhn C, Kumar AP, Kumar A, Kumar A, Kumar D, Kumar D, Kumar R, Kumar S, Kundu M, Kung H-J, Kuno A, Kuo S-H, Kuret J, Kurz T, Kwok T, Kwon TK, Kwon YT, Kyrmizi I, La Spada AR, Lafont F, Lahm T, Lakkaraju A, Lam T, Lamark T, Lancel S, Landowski TH, Lane DJR, Lane JD, Lanzi C, Lapaquette P, Lapierre LR, Laporte J, Laukkarinen J, Laurie GW, Lavandero S, Lavie L, La Voie MJ, Law BYK, Law HK-W, Law KB, Layfield R, Lazo PA, Cam LL, Le Roch KG, Stunff HL, Leardkamolkarn V, Lecuit M, Lee B-H, Lee C-H, Lee EF, Lee GM, Lee H-J, Lee H, Lee JK, Lee J, Lee J-H, Lee JH, Lee M, Lee M-S, Lee PJ, Lee SW, Lee S-J, Lee S-J, Lee SY, Lee SH, Lee SS, Lee S-J, Lee S, Lee Y-R, Lee YJ, Lee YH, Leeuwenburgh C, Lefort S, Legouis R, Lei J, Lei Q-Y, Leib DA, Leibowitz G, Lekli I, Lemaire SD, Lemasters JJ, Lemberg MK, Lemoine A, Leng S, Lenz G, Lenzi P, Lerman LO, Barbato DL, Leu JI-J, Leung HY, Levine B, Lewis PA, Lezoualc’h F, Li C, Li F, Li F-J, Li J, Li K, Li L, Li M, Li M, Li Q, Li R, Li S, Li W, Li W, Li X, Li Y, Lian J, Liang C, Liang Q, Liao Y, Liberal J, Liberski PP, Lie P, Lieberman AP, Lim HJ, Lim K-L, Lim K, Lima RT, Lin C-S, Lin C-F, Lin F, Lin F, Lin F-C, Lin K, Lin K-H, Lin P-H, Lin T, Lin W-W, Lin Y-S, Lin Y, Linden R, Lindholm D, Lindqvist LM, Lingor P, Linkermann A, Liotta LA, Lipinski MM, Lira VA, Lisanti MP, Liton PB, Liu B, Liu C, Liu C-F, Liu F, Liu H-J, Liu J, Liu J-J, Liu J-L, Liu K, Liu L, Liu L, Liu Q, Liu R-Y, Liu S, Liu S, Liu W, Liu X-D, Liu X, Liu X-H, Liu X, Liu X, Liu X, Liu Y, Liu Y, Liu Z, Liu Z, Liuzzi JP, Lizard G, Ljujic M, Lodhi IJ, Logue SE, Lokeshwar BL, Long YC, Lonial S, Loos B, López-Otín C, López-Vicario C, Lorente M, Lorenzi PL, Lõrincz P, Los M, Lotze MT, Lovat PE, Lu B, Lu B, Lu J, Lu Q, Lu S-M, Lu S, Lu Y, Luciano F, Luckhart S, Lucocq JM, Ludovico P, Lugea A, Lukacs NW, Lum JJ, Lund AH, Luo H, Luo J, Luo S, Luparello C, Lyons T, Ma J, Ma Y, Ma Y, Ma Z, Machado J, Machado-Santelli GM, Macian F, MacIntosh GC, MacKeigan JP, Macleod KF, MacMicking JD, MacMillan-Crow LA, Madeo F, Madesh M, Madrigal-Matute J, Maeda A, Maeda T, Maegawa G, Maellaro E, Maes H, Magariños M, Maiese K, Maiti TK, Maiuri L, Maiuri MC, Maki CG, Malli R, Malorni W, Maloyan A, Mami-Chouaib F, Man N, Mancias JD, Mandelkow E-M, Mandell MA, Manfredi AA, Manié SN, Manzoni C, Mao K, Mao Z, Mao Z-W, Marambaud P, Marconi AM, Marelja Z, Marfe G, Margeta M, Margittai E, Mari M, Mariani FV, Marin C, Marinelli S, Mariño G, Markovic I, Marquez R, Martelli AM, Martens S, Martin KR, Martin SJ, Martin S, Martin-Acebes MA, Martín-Sanz P, Martinand-Mari C, Martinet W, Martinez J, Martinez-Lopez N, Martinez-Outschoorn U, Martínez-Velázquez M, Martinez-Vicente M, Martins WK, Mashima H, Mastrianni JA, Matarese G, Matarrese P, Mateo R, Matoba S, Matsumoto N, Matsushita T, Matsuura A, Matsuzawa T, Mattson MP, Matus S, Maugeri N, Mauvezin C, Mayer A, Maysinger D, Mazzolini GD, McBrayer MK, Call KM, Cormick CM, McInerney GM, McIver SC, Kenna SM, McMahon JJ, McNeish IA, Mechta-Grigoriou F, Medema JP, Medina DL, Megyeri K, Mehrpour M, Mehta JL, Mei Y, Meier U-C, Meijer AJ, Meléndez A, Melino G, Melino S, de Melo EJT, Mena MA, Meneghini MD, Menendez JA, Menezes R, Meng L, Meng L-H, Meng S, Menghini R, Menko AS, Menna-Barreto RF, Menon MB, Meraz-Ríos MA, Merla G, Merlini L, Merlot AM, Meryk A, Meschini S, Meyer JN, Mi M-T, Miao C-Y, Micale L, Michaeli S, Michiels C, Migliaccio AR, Mihailidou AS, Mijaljica D, Mikoshiba K, Milan E, Miller-Fleming L, Mills GB, Mills IG, Minakaki G, Minassian BA, Ming X-F, Minibayeva F, Minina EA, Mintern JD, Minucci S, Miranda-Vizuete A, Mitchell CH, Miyamoto S, Miyazawa K, Mizushima N, Mnich K, Mograbi B, Mohseni S, Moita LF, Molinari M, Molinari M, Møller AB, Mollereau B, Mollinedo F, Mongillo M, Monick MM, Montagnaro S, Montell C, Moore DJ, Moore MN, Mora-Rodriguez R, Moreira PI, Morel E, Morelli MB, Moreno S, Morgan MJ, Moris A, Moriyasu Y, Morrison JL, Morrison LA, Morselli E, Moscat J, Moseley PL, Mostowy S, Motori E, Mottet D, Mottram JC, Moussa CE-H, Mpakou VE, Mukhtar H, Levy JMM, Muller S, Muñoz-Moreno R, Muñoz-Pinedo C, Münz C, Murphy ME, Murray JT, Murthy A, Mysorekar IU, Nabi IR, Nabissi M, Nader GA, Nagahara Y, Nagai Y, Nagata K, Nagelkerke A, Nagy P, Naidu SR, Nair S, Nakano H, Nakatogawa H, Nanjundan M, Napolitano G, Naqvi NI, Nardacci R, Narendra DP, Narita M, Nascimbeni AC, Natarajan R, Navegantes LC, Nawrocki ST, Nazarko TY, Nazarko VY, Neill T, Neri LM, Netea MG, Netea-Maier RT, Neves BM, Ney PA, Nezis IP, Nguyen HT, Nguyen HP, Nicot A-S, Nilsen H, Nilsson P, Nishimura M, Nishino I, Niso-Santano M, Niu H, Nixon RA, Njar VC, Noda T, Noegel AA, Nolte EM, Norberg E, Norga KK, Noureini SK, Notomi S, Notterpek L, Nowikovsky K, Nukina N, Nürnberger T, O’Donnell VB, O’Donovan T, O’Dwyer PJ, Oehme I, Oeste CL, Ogawa M, Ogretmen B, Ogura Y, Oh YJ, Ohmuraya M, Ohshima T, Ojha R, Okamoto K, Okazaki T, Oliver FJ, Ollinger K, Olsson S, Orban DP, Ordonez P, Orhon I, Orosz L, O’Rourke EJ, Orozco H, Ortega AL, Ortona E, Osellame LD, Oshima J, Oshima S, Osiewacz HD, Otomo T, Otsu K, Ou J-HJ, Outeiro TF, Ouyang D-Y, Ouyang H, Overholtzer M, Ozbun MA, Ozdinler PH, Ozpolat B, Pacelli C, Paganetti P, Page G, Pages G, Pagnini U, Pajak B, Pak SC, Pakos-Zebrucka K, Pakpour N, Palková Z, Palladino F, Pallauf K, Pallet N, Palmieri M, Paludan SR, Palumbo C, Palumbo S, Pampliega O, Pan H, Pan W, Panaretakis T, Pandey A, Pantazopoulou A, Papackova Z, Papademetrio DL, Papassideri I, Papini A, Parajuli N, Pardo J, Parekh VV, Parenti G, Park J-I, Park J, Park OK, Parker R, Parlato R, Parys JB, Parzych KR, Pasquet J-M, Pasquier B, Pasumarthi KB, Patschan D, Patterson C, Pattingre S, Pattison S, Pause A, Pavenstädt H, Pavone F, Pedrozo Z, Peña FJ, Peñalva MA, Pende M, Peng J, Penna F, Penninger JM, Pensalfini A, Pepe S, Pereira GJ, Pereira PC, Pérez-de la Cruz V, Pérez-Pérez ME, Pérez-Rodríguez D, Pérez-Sala D, Perier C, Perl A, Perlmutter DH, Perrotta I, Pervaiz S, Pesonen M, Pessin JE, Peters GJ, Petersen M, Petrache I, Petrof BJ, Petrovski G, Phang JM, Piacentini M, Pierdominici M, Pierre P, Pierrefite-Carle V, Pietrocola F, Pimentel-Muiños FX, Pinar M, Pineda B, Pinkas-Kramarski R, Pinti M, Pinton P, Piperdi B, Piret JM, Platanias LC, Platta HW, Plowey ED, Pöggeler S, Poirot M, Polčic P, Poletti A, Poon AH, Popelka H, Popova B, Poprawa I, Poulose SM, Poulton J, Powers SK, Powers T, Pozuelo-Rubio M, Prak K, Prange R, Prescott M, Priault M, Prince S, Proia RL, Proikas-Cezanne T, Prokisch H, Promponas VJ, Przyklenk K, Puertollano R, Pugazhenthi S, Puglielli L, Pujol A, Puyal J, Pyeon D, Qi X, Qian W-B, Qin Z-H, Qiu Y, Qu Z, Quadrilatero J, Quinn F, Raben N, Rabinowich H, Radogna F, Ragusa MJ, Rahmani M, Raina K, Ramanadham S, Ramesh R, Rami A, Randall-Demllo S, Randow F, Rao H, Rao VA, Rasmussen BB, Rasse TM, Ratovitski EA, Rautou P-E, Ray SK, Razani B, Reed BH, Reggiori F, Rehm M, Reichert AS, Rein T, Reiner DJ, Reits E, Ren J, Ren X, Renna M, Reusch JE, Revuelta JL, Reyes L, Rezaie AR, Richards RI, Richardson DR, Richetta C, Riehle MA, Rihn BH, Rikihisa Y, Riley BE, Rimbach G, Rippo MR, Ritis K, Rizzi F, Rizzo E, Roach PJ, Robbins J, Roberge M, Roca G, Roccheri MC, Rocha S, Rodrigues CM, Rodríguez CI, de Cordoba SR, Rodriguez-Muela N, Roelofs J, Rogov VV, Rohn TT, Rohrer B, Romanelli D, Romani L, Romano PS, Roncero MIG, Rosa JL, Rosello A, Rosen KV, Rosenstiel P, Rost-Roszkowska M, Roth KA, Roué G, Rouis M, Rouschop KM, Ruan DT, Ruano D, Rubinsztein DC, Rucker EB III, Rudich A, Rudolf E, Rudolf R, Ruegg MA, Ruiz-Roldan C, Ruparelia AA, Rusmini P, Russ DW, Russo GL, Russo G, Russo R, Rusten TE, Ryabovol V, Ryan KM, Ryter SW, Sabatini DM, Sacher M, Sachse C, Sack MN, Sadoshima J, Saftig P, Sagi-Eisenberg R, Sahni S, Saikumar P, Saito T, Saitoh T, Sakakura K, Sakoh-Nakatogawa M, Sakuraba Y, Salazar-Roa M, Salomoni P, Saluja AK, Salvaterra PM, Salvioli R, Samali A, Sanchez AM, Sánchez-Alcázar JA, Sanchez-Prieto R, Sandri M, Sanjuan MA, Santaguida S, Santambrogio L, Santoni G, Nunes Dos Santos C, Saran S, Sardiello M, Sargent G, Sarkar P, Sarkar S, Sarrias MR, Sarwal MM, Sasakawa C, Sasaki M, Sass M, Sato K, Sato M, Satriano J, Savaraj N, Saveljeva S, Schaefer L, Schaible UE, Scharl M, Schatzl HM, Schekman R, Scheper W, Schiavi A, Schipper HM, Schmeisser H, Schmidt J, Schmitz I, Schneider BE, Schneider EM, Schneider JL, Schon EA, Schönenberger MJ, Schönthal AH, Schorderet DF, Schröder B, Schuck S, Schulze RJ, Schwarten M, Schwarz TL, Sciarretta S, Scotto K, Scovassi AI, Screaton RA, Screen M, Seca H, Sedej S, Segatori L, Segev N, Seglen PO, Seguí-Simarro JM, Segura-Aguilar J, Seki E, Sell C, Seiliez I, Semenkovich CF, Semenza GL, Sen U, Serra AL, Serrano-Puebla A, Sesaki H, Setoguchi T, Settembre C, Shacka JJ, Shajahan-Haq AN, Shapiro IM, Sharma S, She H, Shen C-KJ, Shen C-C, Shen H-M, Shen S, Shen W, Sheng R, Sheng X, Sheng Z-H, Shepherd TG, Shi J, Shi Q, Shi Q, Shi Y, Shibutani S, Shibuya K, Shidoji Y, Shieh J-J, Shih C-M, Shimada Y, Shimizu S, Shin DW, Shinohara ML, Shintani M, Shintani T, Shioi T, Shirabe K, Shiri-Sverdlov R, Shirihai O, Shore GC, Shu C-W, Shukla D, Sibirny AA, Sica V, Sigurdson CJ, Sigurdsson EM, Sijwali PS, Sikorska B, Silveira WA, Silvente-Poirot S, Silverman GA, Simak J, Simmet T, Simon AK, Simon H-U, Simone C, Simons M, Simonsen A, Singh R, Singh SV, Singh SK, Sinha D, Sinha S, Sinicrope FA, Sirko A, Sirohi K, Sishi BJ, Sittler A, Siu PM, Sivridis E, Skwarska A, Slack R, Slaninová I, Slavov N, Smaili SS, Smalley KS, Smith DR, Soenen SJ, Soleimanpour SA, Solhaug A, Somasundaram K, Son JH, Sonawane A, Song C, Song F, Song HK, Song J-X, Song W, Soo KY, Sood AK, Soong TW, Soontornniyomkij V, Sorice M, Sotgia F, Soto-Pantoja DR, Sotthibundhu A, Sousa MJ, Spaink HP, Span PN, Spang A, Sparks JD, Speck PG, Spector SA, Spies CD, Springer W, Clair DS, Stacchiotti A, Staels B, Stang MT, Starczynowski DT, Starokadomskyy P, Steegborn C, Steele JW, Stefanis L, Steffan J, Stellrecht CM, Stenmark H, Stepkowski TM, Stern ST, Stevens C, Stockwell BR, Stoka V, Storchova Z, Stork B, Stratoulias V, Stravopodis DJ, Strnad P, Strohecker AM, Ström A-L, Stromhaug P, Stulik J, Su Y-X, Su Z, Subauste CS, Subramaniam S, Sue CM, Suh SW, Sui X, Sukseree S, Sulzer D, Sun F-L, Sun J, Sun J, Sun S-Y, Sun Y, Sun Y, Sun Y, Sundaramoorthy V, Sung J, Suzuki H, Suzuki K, Suzuki N, Suzuki T, Suzuki YJ, Swanson MS, Swanton C, Swärd K, Swarup G, Sweeney ST, Sylvester PW, Szatmari Z, Szegezdi E, Szlosarek PW, Taegtmeyer H, Tafani M, Taillebourg E, Tait SW, Takacs-Vellai K, Takahashi Y, Takáts S, Takemura G, Takigawa N, Talbot NJ, Tamagno E, Tamburini J, Tan C-P, Tan L, Tan ML, Tan M, Tan Y-J, Tanaka K, Tanaka M, Tang D, Tang D, Tang G, Tanida I, Tanji K, Tannous BA, Tapia JA, Tasset-Cuevas I, Tatar M, Tavassoly I, Tavernarakis N, Taylor A, Taylor GS, Taylor GA, Taylor JP, Taylor MJ, Tchetina EV, Tee AR, Teixeira-Clerc F, Telang S, Tencomnao T, Teng B-B, Teng R-J, Terro F, Tettamanti G, Theiss AL, Theron AE, Thomas KJ, Thomé MP, Thomes PG, Thorburn A, Thorner J, Thum T, Thumm M, Thurston TL, Tian L, Till A, Ting JP-Y, Titorenko VI, Toker L, Toldo S, Tooze SA, Topisirovic I, Torgersen ML, Torosantucci L, Torriglia A, Torrisi MR, Tournier C, Towns R, Trajkovic V, Travassos LH, Triola G, Tripathi DN, Trisciuoglio D, Troncoso R, Trougakos IP, Truttmann AC, Tsai K-J, Tschan MP, Tseng Y-H, Tsukuba T, Tsung A, Tsvetkov AS, Tu S, Tuan H-Y, Tucci M, Tumbarello DA, Turk B, Turk V, Turner RF, Tveita AA, Tyagi SC, Ubukata M, Uchiyama Y, Udelnow A, Ueno T, Umekawa M, Umemiya-Shirafuji R, Underwood BR, Ungermann C, Ureshino RP, Ushioda R, Uversky VN, Uzcátegui NL, Vaccari T, Vaccaro MI, Váchová L, Vakifahmetoglu-Norberg H, Valdor R, Valente EM, Vallette F, Valverde AM, Van den Berghe G, Van Den Bosch L, van den Brink GR, van der Goot FG, van der Klei IJ, van der Laan LJ, van Doorn WG, van Egmond M, van Golen KL, Van Kaer L, van Lookeren Campagne M, Vandenabeele P, Vandenberghe W, Vanhorebeek I, Varela-Nieto I, Vasconcelos MH, Vasko R, Vavvas DG, Vega-Naredo I, Velasco G, Velentzas AD, Velentzas PD, Vellai T, Vellenga E, Vendelbo MH, Venkatachalam K, Ventura N, Ventura S, Veras PS, Verdier M, Vertessy BG, Viale A, Vidal M, Vieira HLA, Vierstra RD, Vigneswaran N, Vij N, Vila M, Villar M, Villar VH, Villarroya J, Vindis C, Viola G, Viscomi MT, Vitale G, Vogl DT, Voitsekhovskaja OV, von Haefen C, von Schwarzenberg K, Voth DE, Vouret-Craviari V, Vuori K, Vyas JM, Waeber C, Walker CL, Walker MJ, Walter J, Wan L, Wan X, Wang B, Wang C, Wang C-Y, Wang C, Wang C, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H-J, Wang H, Wang H-G, Wang H, Wang H-D, Wang J, Wang J, Wang M, Wang M-Q, Wang P-Y, Wang P, Wang RC, Wang S, Wang T-F, Wang X, Wang X-J, Wang X-W, Wang X, Wang X, Wang Y, Wang Y, Wang Y, Wang Y-J, Wang Y, Wang Y, Wang YT, Wang Y, Wang Z-N, Wappner P, Ward C, McVey Ward D, Warnes G, Watada H, Watanabe Y, Watase K, Weaver TE, Weekes CD, Wei J, Weide T, Weihl CC, Weindl G, Weis SN, Wen L, Wen X, Wen Y, Westermann B, Weyand CM, White AR, White E, Whitton JL, Whitworth AJ, Wiels J, Wild F, Wildenberg ME, Wileman T, Wilkinson DS, Wilkinson S, Willbold D, Williams C, Williams K, Williamson PR, Winklhofer KF, Witkin SS, Wohlgemuth SE, Wollert T, Wolvetang EJ, Wong E, Wong GW, Wong RW, Wong VKW, Woodcock EA, Wright KL, Wu C, Wu D, Wu GS, Wu J, Wu J, Wu M, Wu M, Wu S, Wu WK, Wu Y, Wu Z, Xavier CP, Xavier RJ, Xia G-X, Xia T, Xia W, Xia Y, Xiao H, Xiao J, Xiao S, Xiao W, Xie C-M, Xie Z, Xie Z, Xilouri M, Xiong Y, Xu C, Xu C, Xu F, Xu H, Xu H, Xu J, Xu J, Xu J, Xu L, Xu X, Xu Y, Xu Y, Xu Z-X, Xu Z, Xue Y, Yamada T, Yamamoto A, Yamanaka K, Yamashina S, Yamashiro S, Yan B, Yan B, Yan X, Yan Z, Yanagi Y, Yang D-S, Yang J-M, Yang L, Yang M, Yang P-M, Yang P, Yang Q, Yang W, Yang WY, Yang X, Yang Y, Yang Y, Yang Z, Yang Z, Yao M-C, Yao PJ, Yao X, Yao Z, Yao Z, Yasui LS, Ye M, Yedvobnick B, Yeganeh B, Yeh ES, Yeyati PL, Yi F, Yi L, Yin X-M, Yip CK, Yoo Y-M, Yoo YH, Yoon S-Y, Yoshida K-I, Yoshimori T, Young KH, Yu H, Yu JJ, Yu J-T, Yu J, Yu L, Yu WH, Yu X-F, Yu Z, Yuan J, Yuan Z-M, Yue BY, Yue J, Yue Z, Zacks DN, Zacksenhaus E, Zaffaroni N, Zaglia T, Zakeri Z, Zecchini V, Zeng J, Zeng M, Zeng Q, Zervos AS, Zhang DD, Zhang F, Zhang G, Zhang G-C, Zhang H, Zhang H, Zhang H, Zhang H, Zhang J, Zhang J, Zhang J, Zhang J, Zhang J-P, Zhang L, Zhang L, Zhang L, Zhang L, Zhang M-Y, Zhang X, Zhang XD, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhang Y, Zhao M, Zhao W-L, Zhao X, Zhao YG, Zhao Y, Zhao Y, Zhao Y-X, Zhao Z, Zhao ZJ, Zheng D, Zheng X-L, Zheng X, Zhivotovsky B, Zhong Q, Zhou G-Z, Zhou G, Zhou H, Zhou S-F, Zhou X-J, Zhu H, Zhu H, Zhu W-G, Zhu W, Zhu X-F, Zhu Y, Zhuang S-M, Zhuang X, Ziparo E, Zois CE, Zoladek T, Zong W-X, Zorzano A, Zughaier SM, Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mizushima N, Yoshimori T, Levine B, Methods in mammalian autophagy research. Cell 140, 313–326 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC, Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet 36, 585–595 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, Hailey DW, Oorschot V, Klumperman J, Baehrecke EH, Lenardo MJ, Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leidal AM, Levine B, Debnath J, Autophagy and the cell biology of age-related disease. Nat. Cell Biol 20, 1338–1348 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N, The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Pyo J-O, Yoo S-M, Ahn H-H, Nah J, Hong S-H, Kam T-I, Jung S, Jung Y-K, Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun 4, 2300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hescheler J, Halbach M, Egert U, Lu ZJ, Bohlen H, Fleischmann BK, Reppel M, Determination of electrical properties of ES cell-derived cardiomyocytes using MEAs. J. Electrocardiol 37, 110–116 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Zhou Y, Liu Z, Welch JD, Gao X, Wang L, Garbutt T, Keepers B, Ma H, Prins JF, Shen W, Liu J, Qian L, Single-cell transcriptomic analyses of cell fate transitions during human cardiac reprogramming. Cell Stem Cell 25, 149–164.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen E-L, Mizushima N, Ohsumi Y, Cattoretti G, Levine B, Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest 112, 1809–1820 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ, ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109, 21.29.1–21.29.9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G, GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bektik E, Dennis A, Pawlowski G, Zhou C, Maleski D, Takahashi S, Laurita KR, Deschênes, Fu J-D, S-phase synchronization facilitates the early progression of induced-cardiomyocyte reprogramming through enhanced cell-cycle exit. Int. J. Mol. Sci 19, 1364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cruciat C-M, Niehrs C, Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol 5, a015081 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daniels DL, Weis WI, β-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol 12, 364–371 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Azarin SM, Lian X, Larson EA, Popelka HM, de Pablo JJ, Palecek SP, Modulation of Wnt/β-catenin signaling in human embryonic stem cells using a 3-D microwell array. Biomaterials 33, 2041–2049 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baurand A, Zelarayan L, Betney R, Gehrke C, Dunger S, Noack C, Busjahn A, Huelsken J, Taketo MM, Birchmeier W, Dietz R, Bergmann MW, β-catenin downregulation is required for adaptive cardiac remodeling. Circ. Res 100, 1353–1362 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Swope D, Cheng L, Gao E, Li J, Radice GL, Loss of cadherin-binding proteins β-catenin and plakoglobin in the heart leads to gap junction remodeling and arrhythmogenesis. Mol. Cell. Biol 32, 1056–1067 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Platta HW, Abrahamsen H, Thoresen SB, Stenmark H, Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J 441, 399–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J, Levine B, Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 338, 956–959 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T, Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol 11, 385–396 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Lu J, He L, Behrends C, Araki M, Araki K, Jun Wang Q, Catanzaro JM, Friedman SL, Zong W-X, Fiel MI, Li M, Yue Z, NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat. Commun 5, 3920 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z, Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol 11, 468–476 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ, EH domain proteins regulate cardiac membrane protein targeting. Circ. Res 107, 84–95 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Braun A, Pinyol R, Dahlhaus R, Koch D, Fonarev P, Grant BD, Kessels MM, Qualmann B, EHD proteins associate with syndapin I and II and such interactions play a crucial role in endosomal recycling. Mol. Biol. Cell 16, 3642–3658 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim J, Kundu M, Viollet B, Guan K-L, AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol 13, 132–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park J-M, Jung CH, Seo M, Otto NM, Grunwald D, Kim KH, Moriarity B, Kim Y-M, Starker C, Nho RS, Voytas D, Kim D-H, The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 12, 547–564 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murrow L, Debnath J, Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol 8, 105–137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P, Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Cho Y-H, Han K-M, Kim D, Lee J, Lee S-H, Choi K-W, Kim J, Han Y-M, Autophagy regulates homeostasis of pluripotency-associated proteins in hESCs. Stem Cells 32, 424–435 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Sciarretta S, Maejima Y, Zablocki D, Sadoshima J, The role of autophagy in the heart. Annu. Rev. Physiol 80, 1–26 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA, Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest 117, 1782–1793 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hariharan N, Maejima Y, Nakae J, Paik J, DePinho RA, Sadoshima J, Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res 107, 1470–1482 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gessert S, Kühl M, The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res 107, 186–199 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Ye B, Hou N, Xiao L, Xu Y, Boyer J, Xu H, Li F, APC controls asymmetric Wnt/β-catenin signaling and cardiomyocyte proliferation gradient in the heart. J. Mol. Cell. Cardiol 89, 287–296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buikema JW, Mady AS, Mittal NV, Atmanli A, Caron L, Doevendans PA, Sluijter JPG, Domian IJ, Wnt/β-catenin signaling directs the regional expansion of first and second heart field-derived ventricular cardiomyocytes. Development 140, 4165–4176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hashimoto H, Wang Z, Garry GA, Malladi VS, Botten GA, Ye W, Zhou H, Osterwalder M, Dickel DE, Visel A, Liu N, Bassel-Duby R, Olson EN, Cardiac reprogramming factors synergistically activate genome-wide cardiogenic stage-specific enhancers. Cell Stem Cell 25, 69–86.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, Geisen C, Chakraborty AA, Moslehi JJ, Smalley DM, Wei X, Chen X, Chen Z, Beres JM, Zhang J, Tsao JL, Brenner MC, Zhang Y, Fan C, DePinho RA, Paik J, Gygi SP, Kaelin WG Jr., Zhang Q, Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev 28, 1429–1444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zwi L, Caspi O, Arbel G, Huber I, Gepstein A, Park I-H, Gepstein L, Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 120, 1513–1523 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Wang L, Liu Z, Yin C, Zhou Y, Liu J, Qian L, Improved generation of induced cardiomyocytes using a polycistronic construct expressing optimal ratio of Gata4, Mef2c and Tbx5. J. Vis. Exp, e53426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup, The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T, deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin S-F, Palmieri C, Caldas C, Carroll JS, Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liao Y, Smyth GK, Shi W, featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 84.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Primary data.
Table S3. List of qPCR primers and cloning primers used in this study.
Table S4. Sequence information for shRNA clones used in this study.
Table S5. List of antibodies used for immunofluorescent staining, immunoblotting, and flow cytometry.
Fig. S1. Autophagy is activated during iCM reprogramming.
Fig. S2. Atg5 and Atg7 knockdown impaired iCM reprogramming.
Fig. S3. Atg5-dependent autophagy induction during iCM reprogramming.
Fig. S4. Knockdown of Becn1 enhanced iCM reprogramming.
Fig. S5. Characterization of heart structure and cardiac function in young Becn1+/− mice.
Fig. S6. Characterization of Becn1 during reprogramming.
Fig. S7. Transduction of MGT reprogramming factors changed chromatin accessibility but not Becn1 expression.
Fig. S8. Transcriptomic alternations induced by Becn1 knockdown.
Fig. S9. Validation and confirmation of shRNA screen.
Fig. S10. Localization of β-catenin in injured heart.
Fig. S11. Identification of Becn1 interaction partners and characterization of Ulk1 in iCM reprogramming.
Fig. S12. Ulk1 activity and mitophagy remain unchanged during reprogramming.
Fig. S13. Schematic representation of autophagy-dependent and -independent mechanisms of iCM induction.
Fig. S14. Unprocessed original scans of western blots.
References (77–85)
Table S2. MS analysis of Becn1 interaction proteins.