

Abstract
Emerging SARS-CoV-2 strains, B.1.1.7 and B.1.351, from the UK and South Africa, respectively, show decreased neutralization by monoclonal antibodies and convalescent or vaccinee sera raised against the original wild-type virus, and are thus of clinical concern. However, the neutralization potency of two antibodies, 1–57 and 2–7, which target the receptor-binding domain (RBD) of the spike, was unaffected by these emerging strains. Here, we report cryo-EM structures of 1–57 and 2–7 in complex with spike, revealing each of these antibodies to utilize a distinct mechanism to bypass or accommodate RBD mutations. Notably, each antibody represented an immune response with recognition distinct from those of frequent antibody classes. Moreover, many epitope residues recognized by 1–57 and 2–7 were outside hotspots of evolutionary pressure for ACE2 binding and neutralizing antibody escape. We suggest the therapeutic use of antibodies, such as 1–57 and 2–7, which target less prevalent epitopes, could ameliorate issues of monoclonal antibody escape.
Keywords: B.1.351 and B.1.1.7 variants, COVID-19, cryo-EM, low-frequency immune response, neutralizing antibody, receptor-binding domain, SARS-CoV-2
Graphical abstract
Cerutti et al. report the structural characterization of two potent neutralizing antibodies, 1–57 and 2–7, bound to the SARS-CoV-2 spike. The cryo-EM structures elucidate distinct mechanisms to accommodate the RBD mutations observed in the B.1.1.7 and B.1.351 variants. Both antibodies represent low-frequency immune responses and their use as therapeutics is suggested.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent for coronavirus disease 2019 (COVID-19), emerged in 2019, rapidly establishing an ongoing worldwide pandemic with over 100 million infected and over 2 million dead at the time of writing (Callaway et al., 2020; Cucinotta and Vanelli, 2020; Dong et al., 2020). The possible emergence of mutant strains of SARS-CoV-2 with improved transmissibility, virulence, or ability to evade human immunity has been a major concern (Baric, 2020). The SARS-CoV-2 lineage known as B.1.1.7 emerged in September 2020 in South East England, quickly becoming the dominant variant in the UK, and subsequently spreading to over 50 countries, potentially due to enhanced transmissibility (Rambaut et al., 2020). The B.1.1.7 strain contains the early spike mutation D614G, now common to most SARS-CoV-2 lineages, as well as eight additional spike mutations, including two deletions (69-70del and 144del) in the N-terminal domain (NTD), a single mutation (N501Y) in receptor-binding domain (RBD) and (A570D) in SD1, two mutations (P681H and T716I) near the furin cleavage site, and two mutations in S2 (S982A and D1118H). Another emerging lineage, SARS-CoV-2 B.1.351, appeared late in 2020 in Eastern Cape, South Africa (Tegally et al., 2020), and also became dominant locally, again raising the possibility of increased transmissibility. B.1.351 contains nine spike mutations in addition to D614G, including a cluster of mutations (e.g., 242-244del and R246I) in NTD, one mutation (A701V) near the furin cleavage site, and three mutations (K417N, E484K, and N501Y) in RBD.
Recent studies have shown that some of these new variants impede the function of some SARS-CoV-2 neutralizing antibodies, with most NTD-directed neutralizing antibodies showing a near complete loss of potency against either the B.1.351 or B.1.1.7 strains (Wang et al., 2021b; Wibmer et al., 2021). With respect to RBD-directed antibodies, however, while the most prevalent classes of multi-donor RBD-directed antibodies, originating from the VH3-53/66 and VH1-2 genes (Banach et al., 2021; Rapp et al., 2021; Yuan et al., 2020), are generally inhibited by mutations in RBD (K417N and N501Y for VH3-53/66 class antibodies and E484K for VH1-2 class antibodies) (Wang et al., 2021b). Fortunately, many RBD-directed antibodies not from these frequent classes retain their activity.
In a companion manuscript (Wang et al., 2021b), we screened monoclonal antibodies and found two super potent RBD-directed antibodies, 1–57 and 2–7 (0.008 and 0.003 μg/mL of live virus neutralization at half-maximal inhibitory concentrations for 1–57 and 2–7, respectively), whose binding competed with that of the ACE2 receptor (Liu et al., 2020), but whose neutralization was unaffected by the emerging B.1.351 and B.1.1.7 strains (Wang et al., 2021b). To understand how these antibodies accommodate the mutations present in B.1.351 and B.1.1.7 strains, we determined their cryoelectron microscopy (cryo-EM) structures in complex with spike. We used the 52 currently known complex structures of RBD-directed neutralizing antibody to create a residue-level map of antibody recognition frequency. This analysis suggested mutations in RBD to emerge under evolutionary pressure from both antibody escape and binding to the ACE2 receptor on host cells. We suggest that neutralizing antibodies, such as 1–57 and 2–7, that target epitopes with few selection hotspots may result in less viral escape and lead to more effective therapeutic strategies.
Results
Antibody 1–57 utilizes a hydrophilic pocket to accommodate mutation E484K in emerging strains
From complexes of SARS-CoV-2 spike—stabilized by 2P mutations (Wrapp et al., 2020)—with the antigen-binding fragment (Fab) of antibody 1–57, we collected single-particle data on a Titan Krios microscope, yielding a cryo-EM reconstruction to an overall resolution of 3.42 Å (Figures 1 A, S1, and S2; Table S1). A single conformational state with three Fabs per trimer, each bound to an RBD in the “down” conformation, was identified. Recognition of RBD by 1–57 was dominated by the heavy chain, which buried 533.7 Å2 surface area, with a smaller 223.3 Å2 contribution by the light chain. The 21 amino acid long CDR H3 formed a β hairpin stabilized by a disulfide bond between Cys100a and Cys100f (Kabat numbering scheme [Wu and Kabat, 1970]) provided the primary interactions with RBD, with additional contributions from CDR L1 and L2 (Figure 1C, left panel). CDR H3 formed mainly hydrophobic contacts with RBD residues Tyr351, Leu452, Phe490, and Leu492. The light chain contributed to RBD recognition mainly through hydrogen bonds formed between the hydroxyl groups of Ser30, Tyr32, and Ser53 in CDR L1 and L2, and residues Gln493, Ser494, and Gln498 in RBD, respectively (Figure 1C, right panel). Antibody 1–57 also displayed a small quaternary interaction, as Asn100c on the tip of CDR H3 hydrogen bonds with both Thr470 in a neighboring RBD and glycan N165 in a neighboring NTD (Figure 1C, middle panel).
Figure 1.

Antibody 1–57 utilizes a hydrophilic pocket to accommodate mutation E484K in emerging strains
(A) Cryo-EM reconstruction for spike complex with antibody 1–57 from two orthogonal views; a single conformation with all RBDs down is observed. NTD is shown in orange, RBD in green, glycans in red, antibody heavy chain in blue, and light chain in gray.
(B) Domain level view of 1–57 in complex with RBD, with the emerging mutants highlighted in red.
(C) Details of antibody 1–57 recognition of RBD showing the overall interface (left panel), recognition by CDR H3 (middle panel), and recognition by CDR L1 and L2 (right panel). CDR H1, H2, H3 are colored in shades of blue; CDR L1, L2, and L3 are colored in shades of gray. E484 is highlighted in bright red (right panel). Nitrogen atoms are colored in blue, oxygen atoms in red; hydrogen bonds (distance <3.2 Å) are represented as dashed lines.
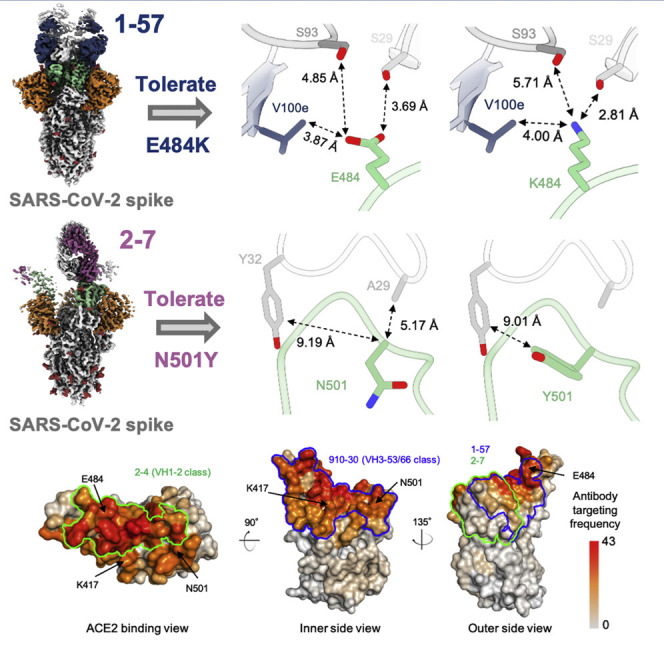
(D) Expanded view of the E484 environment at the interface with 1–57 (left panel) and modeling of K484 (right panel) suggest a mechanism of antibody 1–57 accommodation of the E484K mutation; colored as in (B).
With respect to the three RBD mutations in the UK and South Africa strains, only residue E484K of the South African strain was near the binding site of 1–57 (Figure 1B). Despite its proximity to the epitope, however, Glu484 did not interact significantly with 1–57, as the amino acids in the immediate vicinity—Val100e from the heavy chain, and Ser29 and Ser93 from the light chain—were too distant (Figure 1D, left panel). Structural modeling of the E484K mutation showed that a Lys residue was geometrically compatible with 1–57 binding; modeling of K484 with a high-frequency rotamer showed that the distance between the amino group of K484 and the side chain of Ser29 was compatible with a hydrogen bond (2.81 Å) (Figure 1D, right panel).
Structural basis of antibody 2–7 accommodation of mutation N501Y in emerging strains
Cryo-EM analysis of the Fab from antibody 2–7 in complex with SARS-CoV-2 spike produced a reconstruction with three Fabs bound to a single spike, and was refined to an overall resolution of 3.72 Å (Figures 2A, S1, and S3; Table S1). Only one conformation was observed, with Fabs bound to two RBDs in the “up” conformation and one in the “down” conformation. Due to extensive conformational heterogeneity, the up RBDs could not be resolved to high resolution, thus the down RBD was the focus of structural analysis.
Figure 2.

Structural basis of antibody 2–7 accommodation of mutation N501Y in emerging strains
(A) Cryo-EM reconstruction for spike complex with antibody 2–7 from two orthogonal views; a single conformation with one RBD down and two RBDs up is observed. NTD is shown in orange, RBD in green, glycans in red, antibody heavy chain in magenta, and light chain in gray.
(B) Domain level view of 2–7 in complex with RBD, with the emerging mutants highlighted in red.
(C) Details of antibody 2–7 recognition of RBD showing the overall interface (left panel), recognition by CDR H2 (middle panel), and recognition by CDR L1 and L3 (right panel). CDR H1, H2, and H3 are colored in shades of magenta; CDR L1, L2, and L3 are colored in shades of gray. N501 is highlighted in bright red (right panel).
(D) Expanded view of the N501 environment at the interface with 2–7 (left panel) and modeling of Y501 (right panel) suggest a mechanism of antibody 2–7 accommodation of the N501Y mutation; colored as in (B).
Recognition of RBD by antibody 2–7 was dominated by interactions proximal to the RBD loops formed by residues 438–451 and 495–502 (Figure 2C, left panel). CDR H2 formed an extensive network of hydrogen bonds and salt bridges (Figure 2C, middle panel). RBD residue Lys444 formed salt bridges with Fab heavy chain residues Asp56 and Asp54. In addition, CDR H2 residues Asp54, Tyr52, and Arg58 formed hydrogen bonds with RBD residues Asn450, the backbone amine of Val445, and the backbone carbonyl of Gly447, respectively. Val445 at the apex of loop 438–451 was at the center of a hydrophobic pocket, which included Pro499 on RBD that accommodated several residues on the heavy chain (Trp47, Leu50, Tyr52, and Ile97) and one on the light chain (Tyr91). Light chain residue Tyr32 also formed a hydrogen bond with the RBD via residue N440 (Figure 2C, right panel).
With respect to the three mutated positions in the UK and South Africa variants, antibody 2–7 bound near only N501, but the side chain of N501 pointed away from the antibody (Figures 2B and 2D). While some conformational change of the 495–502 loop would be expected in the context of the N501Y mutation, this loop contributed only 225 Å2 out of 736 Å2 and contained few residues that form significant interactions with the Fab.
Genetic and epitope similarities to other SARS-CoV-2 neutralizing antibodies
The heavy chain of 1–57 derived from the VH3-72∗01 gene with a CDR H3 of 21 amino acids, and the light chain was from KV3-20∗01, with a 9 residue CDR L3 (Figure S1). Antibody 2–7 utilized VH2-5∗02 with an 11 amino acid CDR H3, and LV2-14∗01 with a 9 residue CDR L3. Both antibodies thus utilized heavy chain genes with relatively low frequencies in SARS-CoV-2-specific neutralizing antibody repertoire (Rapp et al., 2021), suggesting that antibodies with genetic features similar to 1–57 and 2–7 may not appear frequently in human response to SARS-CoV-2.
To assess the neutralization mechanism for antibodies 1–57 and 2–7 we compared the structure of the ACE2:RBD complex (Lan et al., 2020) with the structures of 1–57 and 2–7 aligned on RBD: the structural superposition shows that binding of these two antibodies is not compatible with receptor binding, as major clashes would result (Figure S4A). In addition, we performed ACE2 competition ELISA assays for both antibodies binding to SARS-CoV-2 spike, which confirmed that binding of ACE2 is progressively depleted as the concentration of 1–57 and 2–7 increases (Figure S4B).
To understand whether the binding orientations of antibodies 2–7 and 1–57 were common among RBD-directed antibodies, we superposed structures of RBDs from the 52 RBD-directed antibody complex structures deposited in the PDB and measured root-mean-square deviations (RMSDs) between antibodies. Overall, clustering of the 52 antibodies using pairwise RMSD showed that antibodies 1–57 and 2–7 did not cluster with the most frequent VH3-53 and VH1-2 antibody classes (Figure 3 A). The binding orientation of antibody 1–57 was similar to antibodies P2B-2F6 and CV07-270 (Figure S4C). Antibody 2–7 was grouped with REGN10987 (Figure S4D), which was a component of a SARS-CoV-2 therapeutic cocktail (Hansen et al., 2020). However, these antibodies utilize paratopes different from 1–57 and 2–7 for RBD recognition, probably because each antibody has a unique genetic origin.
Figure 3.

Antibodies 2–7 and 1–57 exemplify rare responses, suggesting that mutations against these antibodies have low selection pressure
(A) Analysis of 52 known RBD-directed neutralizing antibodies indicates that 2–7 and 1–57 approach RBD with angles distinct from prevalent antibody classes.
(B) Per residue frequency recognized by the 52 antibodies. VH1-2 and VH3-53 antibody classes recognize RBD residues with high targeting frequency; 1–57 and 2–7 recognize RBD residues with low targeting frequencies.
See also Figure S4.
During in vivo viral infection, epitopes frequently targeted by the antibody may impose strong selection pressure for viral escape. We used the 52 neutralizing antibodies in the PDB to estimate the frequencies of the epitopes of 1–57 and 2–7 being recognized by assuming that these antibodies recapitulate in vivo recognition frequencies of RBD epitopes. In brief, for each antibody, we identified epitope residues and calculated the frequency of each RBD residue being recognized by antibody. The analysis revealed that the epitope residues of 1–57 and 2–7 showed lower antibody recognition frequencies (about 11.2 and 19.8 antibodies per residue on average for 2–7 and 1–57, respectively) compared with those targeted by the prevalent antibody classes (about 27.4 and 25.9 antibodies per residue on average for VH1-2 and VH3-53 class, respectively, Figure 3B), suggesting that 1–57 and 2–7 epitopes are relatively less-targeted antigenic sites.
Antibody escape mutations may result from ACE2 binding and antibody escape selection pressure
Analysis of RBD mutations observed in circulating SARS-CoV-2 sequences revealed many emerging mutations (Figure 4 A). To understand whether the emerging mutations affect antibodies 1–57 and 2–7 recognition, we analyzed structural locations of the top 10 most frequent RBD mutations. We found that, while antibodies 1–57 and 2–7 are not affected by the three mutations observed in the UK and South Africa strains, several RBD mutations may affect antibodies 1–57 and 2–7. For example, mutations at position 452 and 494 were within the antibody 1–57 binding footprint (Figure 4B). Mutations at position 439 may affect 2–7 binding. However, further experiments will be required to assess such effects.
Figure 4.

Prevalent emerging mutations appear to arise at epitopes of prevalent neutralizing response, suggesting that resistance mutants might arise less frequently to rare responses, such as antibodies 1–57 and 2–7
(A) Most frequent mutations and positions observed in circulating SARS-CoV-2 strains.
(B) Location of prevalent RBD mutations and antibody footprints.
(C) Correlation between per residue antibody recognition frequency and the top 50 RBD position mutations.
(D) Correlation between mutation effect on ACE2 binding and its frequency. The blue dashed line, R value, and p value represent the fit after removing the two “outliers,” K417N and F486L mutations.
See also Figure S4.
Studies have proposed that many SARS-CoV-2 escape mutations are selected by neutralizing antibodies (Baum et al., 2020; Ku et al., 2021). However, it is unclear whether the observed antibody-resistant mutations are all the results of antibody selection. To shed light on this, we calculated correlations between positional mutation frequency and antibody recognition frequency by assuming that the estimated per RBD residue targeting frequency reflects the in vivo frequency of antibody recognition and that the most frequently targeted RBD residues may undergo strong selection pressure to mutate. To our surprise, we only observed a very weak correlation (Figure 4C, r = 0.23, p = 0.1), albeit that some of the most frequently targeted residues do have higher mutation frequencies. Because RBD is always under selection pressure from ACE2 binding for entry (Starr et al., 2020), we next examined whether the emerging RBD mutations were selected by enhancing ACE2 binding. We calculated the correlation between the frequencies of RBD mutations and their normalized effects on ACE2 binding affinity obtained from deep mutational scanning data (Starr et al., 2020), which showed a significant correlation (Figure 4D, Pearson's r = 0.34, p = 0.015). The correlation coefficient could be further increased significantly when removing two “outlier” mutations, K417N and F486L, both of which reduce ACE2 binding affinity significantly (Figure 4D). Further analysis of the antibody escape mutations showed that the frequent antibody escape mutations we observed may arise from selection advantage for both ACE2 binding and antibody escape. For example, the most frequent N501Y could improve the binding affinity of ACE2 as well as impair recognition of the most frequent VH3-53 class antibodies (Wang et al., 2021b). The E484K mutation—detected in B.1.351 and recently emerging strains P.1 and P.2 (Faria et al., 2021)—impairs recognition of numerous antibodies (Wang et al., 2021a) and also weakly improves ACE2 binding affinity. However, the frequent mutation K417N resulted in escape of the prevalent VH3-53-derived antibody class (Wang et al., 2021b), and may be selected predominantly by antibody pressure. Despite the ability of 1–57 and 2–7 to still potently neutralize emerging strains that carry N501Y, K417N, and E484K mutations, we observed that individual mutations at 452, 494, 477, and 439 (Figure 4B)—which may affect 1–57 and 2–7 recognition—do not or only weakly enhance ACE2 binding (Bayarri-Olmos et al., 2021; Lopez et al., 2021; Starr et al., 2020; Thomson et al., 2020; Tian et al., 2021; Zahradník et al., 2021) (Figure S4E). Thus, these mutations may undergo weak selection pressure for enhancing ACE2 binding affinity.
Our analysis suggests that potent antibodies 2–7 and 1–57 may not put much selection pressure on circulating strains, thus resistance mutants to these two antibodies are less likely to arise. Overall, due to the diversity of emerging strains, different antibody cocktails become more and more important, and 2–7 and 1–57 are decent candidates for therapeutic development.
Discussion
The emergence of mutant SARS-CoV-2 strains and their impact on the function of neutralizing antibodies has become a major concern. The B.1.1.7 (UK) and B.1.351 (South Africa) variants contain mutations that evade the most frequently elicited classes of RBD-directed neutralizing antibodies from the VH1-2 (B.1.351) and VH3-53/66 classes (B.1.1.7) (Ku et al., 2021; Wang et al., 2021b). Both antibody classes recognize predominantly the RBM, which is mutated convergently in many emerging viral lineages (e.g., K417T, E484K, and N501Y in both B.1.351 and B.1.1.28 [Brazil]). In this study, we report the structures and accommodation mechanisms for two potent SARS-CoV-2 neutralizing antibodies, 1–57 and 2–7, which are not impaired by the mutations in the B.1.1.7 and B.1.351 lineages.
Quantifying epitope-targeting frequency is important for understanding mechanisms of antibody-induced viral escape. Despite an algorithm having been developed to estimate linear epitope-targeting frequency by serum antibodies in a high-throughput way (Shrock et al., 2020), it is still difficult to measure the epitope-targeting frequency of antibodies recognizing three-dimensional epitopes. Methods, such as competition ELISA, can only reveal frequencies of targeted epitope regions with no residue-level information. Here, we use a method to roughly estimate residue-level frequencies for antibody recognition of RBD based on the 52 currently known atomic-level structures of neutralizing antibody-RBD complexes. These antibodies, identified in separate studies of infected donors from around the world (Barnes et al., 2020; Hansen et al., 2020; Lv et al., 2020; Pinto et al., 2020) were generally chosen for study due to their neutralization potency. Overall, these studies identified antibodies mainly targeting RBD (Brouwer et al., 2020; Shi et al., 2020; Tortorici et al., 2020; Wu et al., 2020), with some NTD-directed neutralizers (Cerutti et al., 2021; Chi et al., 2020), with antibodies of the same class often identified in multiple studies. The residue-level antibody-interaction frequencies we present for RBD are likely to represent a reasonable estimate of relative frequencies, but only for the most potent and most frequent classes. Thus, the calculated residue-level antibody-interaction frequency may not reflect epitope-targeting frequencies of weakly neutralizing or non-neutralizing antibodies, which are a significant portion of antibody response to SARS-CoV-2 (Liu et al., 2020; Robbiani et al., 2020). It is also unclear whether weakly neutralizing or non-neutralizing antibodies impose selection pressure on RBD epitopes. Therefore, the weak correlation between antibody-interaction frequency and mutation frequency could be due to multiple confounding factors not included. Nonetheless, many of the identified antibody-interaction hotspots are within or close to the ACE2 binding site. Linear-peptide epitopes around these hotspots are also recognized by numerous serum antibodies (Shrock et al., 2020), confirming that the hotspots we identified are frequently targeted.
The study of mechanisms of mutant accommodation by antibodies 1–57 and 2–7 shed insights on effective therapeutic strategies. Studies have shown that persistent SARS-CoV-2 infection can last for months in immunocompromised human individuals (Avanzato et al., 2020; Choi et al., 2020). During such a long acute infection period, the arms race between the immune system and virus results in many viral mutations to be accumulated in the spike to escape antibody neutralization. Because the human neutralizing antibody response frequently and convergently targets the ACE2 receptor-binding region on RBD, there is strong positive selection pressure to mutate this region. In the meantime, our results showed that the most frequent mutations tend to enhance ACE2 binding, suggesting additional selection pressure underlying SARS-CoV-2 evolution. Because most RBD mutations are detrimental to ACE2 binding (Starr et al., 2020), the virus may have a limited mutation space for escaping antibody neutralization. This may explain the rapid spread of convergent RBD mutations in different emerging SARS-CoV-2 lineages (e.g., E484K mutation in Brazilian P.1, SA B.1.351, and US B.1.526 lineages) (Sabino et al., 2021; West et al., 2021). For therapeutics, antibodies that can either tolerate mutations in the ACE2-binding region or recognize epitopes outside this region are critical for protection. Here, 1–57 and 2–7 bind epitopes that are not predominantly focused on the ACE2-binding region and also accommodate RBD mutations. A residue-level interaction frequency analysis of RBD interaction with all currently known RBD-directed neutralizing antibodies of structure revealed them to represent relatively low-frequency antibody response, which may not form strong selection pressure on the epitope regions. Therapeutics developed from such neutralizing antibodies could lead to persistent protection for a longer period of time.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 1-57 | Liu et al., 2020 | N/A |
| 2-7 | Liu et al., 2020 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 spike protein | Wrapp et al., 2020 | N/A |
| Polyethylenimine | Polysciences | Cat# 24765-2 |
| Freestyle 293 Expression Media | Thermo Scientific | Cat# 12338-026 |
| Expi293 Expression Media | Thermo Scientific | Cat# A14635 |
| Opti-MEM™ Reduced Serum Media | Thermo Scientific | Cat# 31985-070 |
| IMAC Sepharose 6 Fast Flow | GE Healthcare | Cat# 17092109 |
| Tris Base | Thermo Scientific | Cat# BP152-5 |
| Sodium Chloride | Thermo Scientific | Cat# S271-10 |
| Imidazole | ACROS | Cat# 301870025 |
| HEPES | Sigma | Cat# H3375 |
| Critical commercial assays | ||
| FuGENE 6 | Promega | Cat# E2691 |
| Strep-Tactin XT Superflow 50% | Zymo research | Cat# P2004-1-5 |
| Spin Miniprep Kit | Qiagen | Cat# 27106 |
| Hispeed Plasmid Maxi Kit | Qiagen | Cat# 12663 |
| HisTrap Fast Flow | GE Healthcare | Cat# 17-0921-09 |
| Ni-NTA Agarose | Thermo Scientific | Cat# R90115 |
| Pierce™ Fab Preparation Kit | Thermo Scientific | Cat# 44985 |
| Superdex 200 Increase 10/300 GL | Cytiva | Cat# 28990945 |
| Deposited data | ||
| Cryo-EM structure of neutralizing antibody 1-57 in complex with prefusion SARS-CoV-2 spike glycoprotein | This paper | PDB: 7LS9 |
| Cryo-EM map of neutralizing antibody 1-57 in complex with prefusion SARS-CoV-2 spike glycoprotein | This paper | EMDB: EMD-23506 |
| Cryo-EM structure of the SARS-CoV-2 spike glycoprotein bound to Fab 2-7 | This paper | PDB: 7LSS |
| Cryo-EM map of the SARS-CoV-2 spike glycoprotein bound to Fab 2-7 | This paper | EMDB: EMD-23507 |
| Experimental models: Cell lines | ||
| FreeStyle 293-F | Thermo Scientific | Cat# R79007 |
| Expi293F Cells | Thermo Scientific | Cat# A14635 |
| Recombinant DNA | ||
| pCMV3-SARS-CoV-2-spike | Dr. Peihui Wang, Shandong University, China | N/A |
| gWiz-blank | Aldevron | Cat# 5009 |
| Software and algorithms | ||
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| cryoSPARC | Punjani et al., 2017 | https://cryosparc.com |
| Leginon | Suloway et al., 2005 | https://sbgrid.org/software/titles/leginon |
| Molprobity | Davis et al., 2004 | http://molprobity.biochem.duke.edu |
| Phenix | Adams et al., 2004 | https://www.phenix-online.org |
| The PyMOL Molecular Graphics System, Version 2.0 | Schrödinger, LLC | https://pymol.org/2/support.html#page-top |
| SAbPred | Dunbar et al., 2016 | http://opig.stats.ox.ac.uk/webapps/newsabdab/sabpred |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF Chimera X | Pettersen et al., 2021 | https://www.cgl.ucsf.edu/chimerax/ |
| GraphPad Prism Software | GraphPad Prism Software, Inc. | N/A |
| PDBePISA | Krissinel and Henrick, 2007 | https://www.ebi.ac.uk/pdbe/pisa/ |
| The PyMol Molecular Graphics System, v1.8.6 | Schrödinger, LLC | https://pymol.org/2/ |
| IgBLAST-1.16 | Ye et al., 2013 | https://www.ncbi.nlm.nih.gov/igblast/ |
| IMGT | IMGT | http://www.imgt.org/ |
| Python v3.8.3 | Python | https://www.python.org/ |
| The R Project for Statistical Computing | R Core Team | https://www.r-project.org/ |
| R bio3d package | Grant et al., 2006 | http://thegrantlab.org/bio3d/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lawrence Shapiro (lss8@columbia.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The cryo-EM structures have been deposited to the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (RCSB PDB). Cryo-EM structural models and maps for antibodies 1-57 and 2-7 in complex with SARS-CoV-2 spike have been deposited in the PDB and EMDB with accession codes PDB 7LS9, EMD-23506, and PDB 7LSS, EMD-23507, respectively.
Experimental model and subject details
Cell lines
FreeStyle 293-F (cat# R79007), Expi293F cells (cat# A14635) were from Thermo Fisher Scientific. FreeStyle 293-F cells and were cultured in serum-free FreeStyle 293 Expression Medium (GIBCO, cat# 12338026) at 37°C, 10% CO2, 115 rpm. Expi293F cells were cultured in Expi293 Expression Medium (GIBCO, cat# A14635) at 37°C, 8% CO2, 125 rpm. Cell lines were not specifically authenticated; cell lines were of female origin.
Method details
SARS-CoV-2 spike expression and purification
SARS-CoV-2 S2P spike was produced as described in (Wrapp et al., 2020). Protein expression was carried out in Human Embryonic Kidney (HEK) 293 Freestyle cells (Invitrogen) in suspension culture using serum-free media (Invitrogen) by transient transfection using polyethyleneimine (Polysciences). Cell growths were harvested four days after transfection, and the secreted protein was purified from supernatant by nickel affinity chromatography using Ni-NTA IMAC Sepharose 6 Fast Flow resin (GE Healthcare) followed by size exclusion chromatography on a Superdex 200 column (GE Healthcare) in 10 mM Tris, 150 mM NaCl, pH 7.4.
Production of 1-57 and 2-7 Fab
Monoclonal antibody 1-57 was expressed and purified as Fab: VHCH1 with a C-terminal His-tag (His8) and LC were constructed separately into the gWiz expression vector, and then co-transfected and expressed in Expi293. Five days after transfection, supernatants were harvested and 1-57 Fab was purified by nickel affinity chromatography using Ni-NTA agarose (Invitrogen cat# R90115).
Monoclonal antibody 2-7 was expressed and purified as described in (Liu et al., 2020). Fab fragment was produced by digestion of IgG with immobilized papain at 37°C for 3 hrs in 50 mM phosphate buffer, 120 mM NaCl, 30 mM cysteine, 1 mM EDTA, pH 7. The resulting Fab was purified from Fc by affinity chromatography on protein A.
Fab purity was analyzed by SDS-PAGE; all Fabs were buffer-exchanged into 10 mM Tris, 150 mM, pH 7.4 for cryo-EM experiments.
Cryo-EM samples preparation
The final sample for EM analysis of the 1-57 in complex with SARS-CoV-2 S2P spike was produced by mixing the Fab and spike in a 1:9 molar ratio, with a final trimer concentration of 0.33 mg/mL, followed by incubation on ice for 1 hr. The final buffer was 10 mM sodium acetate, 150 mM NaCl, 0.005% (w/v) n-Dodecyl β-D-maltoside, pH 4.5. Cryo-EM grids were prepared by applying 2 μL of sample to a freshly glow-discharged carbon-coated copper grid (CF 1.2/1.3 300 mesh); the sample was vitrified in liquid ethane using a Vitrobot Mark IV with a wait time of 30 s and a blot time of 3 s.
The final sample for EM analysis of Fab 2-7 in complex with SARS-CoV-2 S2P spike was produced by mixing the Fab and spike in a 1:9 molar ratio, with a final trimer concentration of 0.66 mg/mL, followed by incubation on ice for 1 hr. The final puffer was 10 mM sodium acetate, 150 mM NaCl, 0.005% (w/v) n-Dodecyl β-D-maltoside, pH 5.5. Cryo-EM grids were prepared by applying 2 μL of sample to a freshly plasma-cleaned carbon-coated copper grid (CF 1.2/1.3 300 mesh); the sample was vitrified in liquid ethane using a Leica EMGP with a wait time of 15 s and a blot time of 1.5 s.
Cryo-EM data collection, processing and structure refinement
For 1-57, cryo-EM data were collected using the Leginon software (Suloway et al., 2005) installed on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3-BioQuantum direct detection device. The total dose was fractionated for 3 s over 60 raw frames. Motion correction, CTF estimation, particle extraction, 2D classification, ab initio model generation, 3D refinements and local resolution estimation for all datasets were carried out in cryoSPARC 2.15 (Punjani et al., 2017); particles were picked using Topaz (Bepler et al., 2019). The final 3D reconstruction was obtained using non-uniform refinement with C3 symmetry.
SARS CoV-2 S2P spike density was modeled using PDB entry 7L2E (Cerutti et al., 2021), as initial template. The initial model for 1-57 Fab variable region was obtained using the SAbPred server (Dunbar et al., 2016).
For 2-7, cryo-EM data were collected as described for 1-57, except that the total electron flux was fractionated over 2 s with 40 total frames. Data processing was also performed as described above. The final reconstruction was obtained using non-uniform refinement with C1 symmetry, followed by local refinement of the ‘down’ RBD and Fab. The SARS-CoV-2 S2P spike density was modeled using PDB entry 6XEY (Liu et al. 2020) as an initial template. A homology model for the 2-7 Fab variable region was obtained using Schrodinger Release 2020-2: BioLuminate (Zhu et al., 2014).
Automated and manual model building were iteratively performed using real space refinement in Phenix (Adams et al., 2004) and Coot (Emsley and Cowtan, 2004) respectively. For 2-7, ISOLDE v1.1 (Croll, 2018) was also used to interactively refine the structure. Half maps were provided to Resolve Cryo-EM tool in Phenix to support manual model building. Geometry validation and structure quality assessment were performed using EMRinger (Barad et al., 2015) and Molprobity (Davis et al., 2004). Map-fitting cross correlation (Fit-in-Map tool) and figures preparation were carried out using PyMOL and UCSF Chimera (Pettersen et al., 2004) and Chimera X (Pettersen et al., 2021). A summary of the cryo-EM data collection, reconstruction and refinement statistics is shown in Table S1.
Clustering of published RBD-directed antibodies
Information of published SARS-CoV-2 neutralizing antibodies were obtained from CoV-AbDab database (Raybould et al., 2021), the structure for each antibody was download from PBD. For each pair of antibodies, the RBDs were superimposed and the RMSD of Cα between antibody variable domains were calculated. The heavy and light chain sequence alignment were performed using the online muscle program included in bio3d package (Grant et al., 2006). RMSD of Cα were then calculated based on the sequence alignment by an in-house python script, clustering of the RMSD matrix was performed by hclust package in R.
Calculation of antibody targeting frequency for RBD
The epitope RBD-directed antibodies were determined by PISA with the default parameters (Krissinel and Henrick, 2007), the RBD residues with non-zero BSA value were considering as epitope residue. For each residue on RBD, the number of contact antibodies was counted as the frequency of antibody recognition. The antibody targeting frequency was set as b factor of RBD and displayed by Pymol 2.3.2 (DeLano, 2002).
Calculation of positional mutation frequency and correlations
The positional mutation frequency were calculated based on the SARS-CoV2 spike sequences deposited in GISAID at Jan 23rd, 2021 (Elbe and Buckland-Merrett, 2017). Briefly, spike sequences were aligned pairwise with Wuhan-Hu-01 strain as reference, the mutation frequency for each position were calculated as the total mutations divided by total number of deposited sequences, the low quality ‘X’ residue were not counted as mutations. Normalized effects on ACE2 binding affinity for each mutation on RBD were download from the source data (Starr et al., 2020). The r value and p value for correlations were calculated by cor.test function in R 4.0.3.
ACE2 competition ELISA assay
For the ACE2 competition ELISA, 100 ng of ACE2 protein (Abcam) was immobilized overnight on plates at 4°C. The unbound ACE2 was washed away by PBST (0.5% Tween-20 in PBS) and then the plates were blocked. After washing, 50 μL dilution buffer containing 100 ng of S trimer was added into each well, followed by additional 50 μL of serially diluted competitor antibodies and incubation at 37°C for 1 h. The ELISA plates were washed 4 times with PBST and then 100 μL of 2000-fold diluted anti-strep-HRP (Millipore Sigma) was added into each well for another 1 h at 37°C. The plates were then washed and developed with TMB (3,3',5,5'-tetramethylbenzidine) and absorbance was read at 450 nm after stopping the reaction. For all the competition ELISA experiments, the relative binding of ACE2 to the S trimer in the presence of competitors was normalized by comparing to competitor-free controls.
Quantification and statistical analysis
The statistical analyses for the pseudovirus neutralization assessments were performed using GraphPad Prism. Cryo-EM data were processed and analyzed using cryoSPARC. Cryo-EM and crystallographic structural statistics were analyzed using Phenix, Molprobity, EMringer and Chimera. The correlations were performed in R. Statistical details of experiments are described in method details or figure legends.
Acknowledgments
A portion of cryo-EM data collection was performed at the National Center for Cryo-EM Access and Training and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the NIH Common Fund Transformative High Resolution Cryo- Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and NY State Assembly. We thank R. Grassucci, Y.-C. Chi, and Z. Zhang from the Cryo-EM Center at Columbia University, for assistance with additional cryo-EM data collection. Support for this work was also provided by the Intramural Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, NIH, and by the Samuel Yin, Pony Ma, Peggy & Andrew Cherng, Brii Bioscieces, Jack Ma Foundation, JBP Foundation, Carol Ludwig, and Roger & David Wu, COVID-19 Fast Grants, the Self Graduate Fellowship Program, and NIH grants DP5OD023118, R21AI143407, and R21AI144408.
Author contributions
G.C. and M.R. determined the cryo-EM structures of 1–57 and 2–7. Y.G. performed bioinformatics analysis. F.B., E.R.R., and J.B. produced spike and Fabs. J.Y., P.W., L.L., Y.H., and D.D.H. contributed unpublished data related to the neutralization of 1–57 and 2–7 and also produced IgGs and Fabs for these antibodies. D.D.H. supervised IgG production and neutralization assay. P.D.K. contributed to bioinformatics and structural analysis. Z.S. supervised bioinformatics analysis. L.S. supervised structural determinations and led the overall project. G.C., M.R., Y.G., P.D.K., Z.S., and L.S. wrote the manuscript, with all comments from all authors.
Declaration of interests
D.D.H., Y.H., J.Y., L.L., and P.W. are inventors of a patent describing some of the antibodies reported on here.
Published: June 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.05.014.
Supplemental information
References
- Adams P.D., Gopal K., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Pai R.K., Read R.J., Romo T.D., et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- Avanzato V.A., Matson M.J., Seifert S.N., Pryce R., Williamson B.N., Anzick S.L., Barbian K., Judson S.D., Fischer E.R., Martens C., et al. Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell. 2020;183:1901–1912.e9. doi: 10.1016/j.cell.2020.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banach B.B., Cerutti G., Fahad A.S., Shen C.-H., de Souza M.O., Katsamba P.S., Tsybovsky Y., Wang P., Nair M.S., Huang Y., et al. Paired heavy and light chain signatures contribute to potent SARS-CoV-2 neutralization in public antibody responses. bioRxiv. 2021 doi: 10.1101/2020.12.31.424987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad B.A., Echols N., Wang R.Y., Cheng Y., DiMaio F., Adams P.D., Fraser J.S. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baric R.S. Emergence of a highly fit SARS-CoV-2 variant. N. Engl. J. Med. 2020;383:2684–2686. doi: 10.1056/NEJMcibr2032888. [DOI] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A., Fulton B.O., Wloga E., Copin R., Pascal K.E., Russo V., Giordano S., Lanza K., Negron N., Ni M., et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science. 2020;369:1014–1018. doi: 10.1126/science.abd0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayarri-Olmos R., Rosbjerg A., Johnsen L.B., Helgstrand C., Bak-Thomsen T., Garred P., Skjoedt M.O. The SARS-CoV-2 Y453F mink variant displays a pronounced increase in ACE-2 affinity but does not challenge antibody neutralization. J. Biol. Chem. 2021:100536. doi: 10.1016/j.jbc.2021.100536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T., Morin A., Rapp M., Brasch J., Shapiro L., Noble A.J., Berger B. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods. 2019;16:1153–1160. doi: 10.1038/s41592-019-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer P.J.M., Caniels T.G., van der Straten K., Snitselaar J.L., Aldon Y., Bangaru S., Torres J.L., Okba N.M.A., Claireaux M., Kerster G., et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643–650. doi: 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E., Cyranoski D., Mallapaty S., Stoye E., Tollefson J. The coronavirus pandemic in five powerful charts. Nature. 2020;579:482–483. doi: 10.1038/d41586-020-00758-2. [DOI] [PubMed] [Google Scholar]
- Cerutti G., Guo Y., Zhou T., Gorman J., Lee M., Rapp M., Reddem E.R., Yu J., Bahna F., Bimela J., et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe. 2021;29:819–833.e7. doi: 10.1016/j.chom.2021.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X., Yan R., Zhang J., Zhang G., Zhang Y., Hao M., Zhang Z., Fan P., Dong Y., Yang Y., et al. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science. 2020:eabc6952. doi: 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi B., Choudhary M.C., Regan J., Sparks J.A., Padera R.F., Qiu X., Solomon I.H., Kuo H.H., Boucau J., Bowman K., et al. Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N. Engl. J. Med. 2020;383:2291–2293. doi: 10.1056/NEJMc2031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta D., Vanelli M. WHO declares COVID-19 a pandemic. Acta Biomed. 2020;91:157–160. doi: 10.23750/abm.v91i1.9397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. DeLano Scientific; 2002. The PyMOL Molecular Graphics System. [Google Scholar]
- Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar J., Krawczyk K., Leem J., Marks C., Nowak J., Regep C., Georges G., Kelm S., Popovic B., Deane C.M. SAbPred: a structure-based antibody prediction server. Nucleic Acids Res. 2016;44:W474–W478. doi: 10.1093/nar/gkw361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Faria N.R., Mellan T.A., Whittaker C., Claro I.M., Candido D.D.S., Mishra S., Crispim M.A.E., Sales F.C., Hawryluk I., McCrone J.T., et al. medRxiv; 2021. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B.J., Rodrigues A.P., ElSawy K.M., McCammon J.A., Caves L.S. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E., Fulton B.O., Yan Y., Koon K., Patel K., et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020 doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Ku Z., Xie X., Davidson E., Ye X., Su H., Menachery V.D., Li Y., Yuan Z., Zhang X., Muruato A.E., et al. Molecular determinants and mechanism for antibody cocktail preventing SARS-CoV-2 escape. Nat. Commun. 2021;12:469. doi: 10.1038/s41467-020-20789-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F., Sahi V., Figueroa A., et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Lopez E., Haycroft E.R., Adair A., Mordant F.L., O’Neill M.T., Pymm P., Redmond S., Gherardin N.A., Wheatley A.K., Juno J.A., et al. Simultaneous evaluation of antibodies that inhibit SARS-CoV-2 RBD variants with a novel competitive multiplex assay. medRxiv. 2021 doi: 10.1101/2021.03.20.21254037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z., Deng Y.Q., Ye Q., Cao L., Sun C.Y., Fan C., Huang W., Sun S., Sun Y., Zhu L., et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science. 2020;369:1505–1509. doi: 10.1126/science.abc5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D.L., Volz E. virological.org; 2020. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations.https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 [Google Scholar]
- Rapp M., Guo Y., Reddem E.R., Yu J., Liu L., Wang P., Cerutti G., Katsamba P., Bimela J.S., Bahna F.A., et al. Modular basis for potent SARS-CoV-2 neutralization by a prevalent VH1-2-derived antibody class. Cell Rep. 2021;35:108950. doi: 10.1016/j.celrep.2021.108950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould M.I.J., Kovaltsuk A., Marks C., Deane C.M. CoV-AbDab: the coronavirus antibody database. Bioinformatics. 2021;37:734–735. doi: 10.1093/bioinformatics/btaa739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani D.F., Gaebler C., Muecksch F., Lorenzi J.C.C., Wang Z., Cho A., Agudelo M., Barnes C.O., Gazumyan A., Finkin S., et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–442. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino E.C., Buss L.F., Carvalho M.P.S., Prete C.A., Jr., Crispim M.A.E., Fraiji N.A., Pereira R.H.M., Parag K.V., da Silva Peixoto P., Kraemer M.U.G., et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397:452–455. doi: 10.1016/S0140-6736(21)00183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020 doi: 10.1038/s41586-41020-42381-y. [DOI] [PubMed] [Google Scholar]
- Shrock E., Fujimura E., Kula T., Timms R.T., Lee I.H., Leng Y., Robinson M.L., Sie B.M., Li M.Z., Chen Y., et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science. 2020;370:eabd4250. doi: 10.1126/science.abd4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T.N., Greaney A.J., Hilton S.K., Ellis D., Crawford K.H.D., Dingens A.S., Navarro M.J., Bowen J.E., Tortorici M.A., Walls A.C., et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295–1310.e20. doi: 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C.S., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 doi: 10.1101/2020.12.21.20248640. [DOI] [Google Scholar]
- Thomson E.C., Rosen L.E., Shepherd J.G., Spreafico R., da Silva Filipe A., Wojcechowskyj J.A., Davis C., Piccoli L., Pascall D.J., Dillen J., et al. The circulating SARS-CoV-2 spike variant N439K maintains fitness while evading antibody-mediated immunity. bioRxiv. 2020 doi: 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F., Tong B., Sun L., Shi S., Zheng B., Wang Z., Dong X., Zheng P. Mutation N501Y in RBD of spike protein strengthens the interaction between COVID-19 and its receptor ACE2. bioRxiv. 2021 doi: 10.1101/2021.02.14.431117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.A., Beltramello M., Lempp F.A., Pinto D., Dang H.V., Rosen L.E., McCallum M., Bowen J., Minola A., Jaconi S., et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science. 2020;370:950–957. doi: 10.1126/science.abe3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Casner R.G., Nair M.S., Wang M., Yu J., Cerutti G., Liu L., Kwong P.D., Huang Y., Shapiro L., et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe. 2021;29:747–751.e4. doi: 10.1016/j.chom.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Nair M.S., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature. 2021;593:130–135. doi: 10.1038/s41586-021-03398-2. [DOI] [PubMed] [Google Scholar]
- West A.P., Barnes C.O., Yang Z., Bjorkman P.J. SARS-CoV-2 lineage B.1.526 emerging in the New York region detected by software utility created to query the spike mutational landscape. bioRxiv. 2021 doi: 10.1101/2021.02.14.431043. [DOI] [Google Scholar]
- Wibmer C.K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Lambson B.E., Vermeulen M., van den Berg K., Rossouw T., Boswell M., et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. bioRxiv. 2021 doi: 10.1038/s41591-021-01285-x. [DOI] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T.T., Kabat E.A. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J. Exp. Med. 1970;132:211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368:1274–1278. doi: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Ma N., Madden T.L., Ostell J.M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res 41. 2013:34–40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Liu H., Wu N.C., Lee C.D., Zhu X., Zhao F., Huang D., Yu W., Hua Y., Tien H., et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369:1119–1123. doi: 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahradník J., Marciano S., Shemesh M., Zoler E., Chiaravalli J., Meyer B., Dym O., Elad N., Schreiber G. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. bioRxiv. 2021 doi: 10.1101/2021.01.06.425392. [DOI] [Google Scholar]
- Zhu K., Day T., Warshaviak D., Murrett C., Friesner R., Pearlman D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins. 2014;82:1646–1655. doi: 10.1002/prot.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM structures have been deposited to the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (RCSB PDB). Cryo-EM structural models and maps for antibodies 1-57 and 2-7 in complex with SARS-CoV-2 spike have been deposited in the PDB and EMDB with accession codes PDB 7LS9, EMD-23506, and PDB 7LSS, EMD-23507, respectively.