

Abstract
Background & Aims:
Progressive familial intrahepatic cholestasis type 3 (PFIC3) is a rare lethal autosomal recessive liver disorder caused by loss-of-function variations of the ABCB4 gene, encoding a phosphatidylcholine transporter (ABCB4/MDR3). Currently, no effective treatment exists for PFIC3 outside of liver transplantation.
Methods:
We have produced and screened chemically and genetically modified mRNA variants encoding human ABCB4 (hABCB4 mRNA) encapsulated in lipid nanoparticles (LNPs). We examined their pharmacological effects in a cell-based model and in a new in vivo mouse model resembling human PFIC3 as a result of homozygous disruption of the Abcb4 gene in fibrosis-susceptible BALB/c.Abcb4−/− mice.
Results:
We show that treatment with liver-targeted hABCB4 mRNA resulted in de novo expression of functional hABCB4 protein and restored phospholipid transport in cultured cells and in PFIC3 mouse livers. Importantly, repeated injections of the hABCB4 mRNA effectively rescued the severe disease phenotype in young Abcb4−/− mice, with rapid and dramatic normalisation of all clinically relevant parameters such as inflammation, ductular reaction, and liver fibrosis. Synthetic mRNA therapy also promoted favourable hepatocyte-driven liver regeneration to restore normal homeostasis, including liver weight, body weight, liver enzymes, and portal vein blood pressure.
Conclusions:
Our data provide strong preclinical proof-of-concept for hABCB4 mRNA therapy as a potential treatment option for patients with PFIC3.
Keywords: Cholangiopathy, Cholangitis, Congenital biliary cirrhosis, Gene therapy, Liver fibrosis
Lay summary:
This report describes the development of an innovative mRNA therapy as a potential treatment for PFIC3, a devastating rare paediatric liver disease with no treatment options except liver transplantation. We show that administration of our mRNA construct completely rescues severe liver disease in a genetic model of PFIC3 in mice.
Graphical abstrac
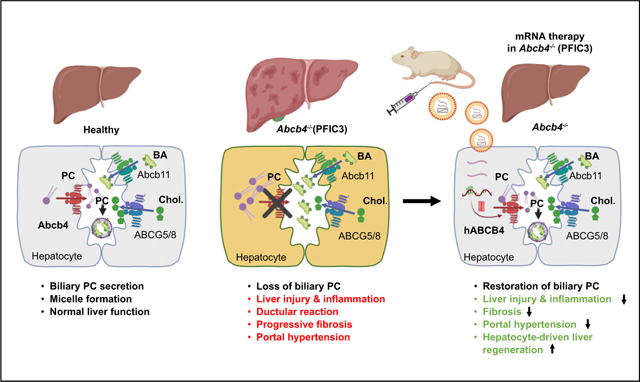
Introduction
Progressive familial intrahepatic cholestasis 3 (PFIC3) is a rare lethal autosomal recessive liver disorder, characterised by early onset of persistent cholestasis that progresses to liver cirrhosis and failure during early childhood.1–3 The disease is caused by loss-of-function variations of the ABCB4 (also called MDR3 in humans and MDR2 in rodents) gene, which encodes a liverspecific phosphatidylcholine (PC) transporter specifically localised in the canalicular membrane of hepatocytes.4 ABCB4/MDR3 flops PC from the inner to the outer leaflet of the canalicular membrane of hepatocytes, where PC is secreted into the bile for forming mainly PC-cholesterol vesicles and some mixed bile salt micelles.5 ABCB4 defects have been functionally characterised,6 with multiple variations resulting in low levels of biliary PC, leading to a lack of biliary vesicles and destabilised micelles, as well as damage of hepatocytes and cholangiocytes caused by the relatively high concentration of biliary bile salts.7 In contrast to other PFIC-1 and PFIC-2 disease types, patients with PFIC3 have elevated serum markers of cholestasis, proliferation of bile ducts (ductular reaction), inflammatory infiltrate, and progressive biliary fibrosis.8
Currently, therapeutic options for patients with PFIC3 are extremely limited. Ursodeoxycholic acid (UDCA) has been generally used in treating various cholestatic disorders through multiple mechanisms, including stimulation of hepatobiliary secretion and detoxification of bile salts,9 and shown to improve liver function and clinical symptoms in a subset of affected PFIC3 patients.10 However, children with ABCB4 variations, which result in no expression of ABCB4 at the canalicular membrane of hepatocytes, develop severe liver disease and are refractory to UDCA treatment.11 Thus, liver transplantation remains the only curative therapeutic option and improves cholestasis and clinical symptoms in 75–100% of PIFC3 patients throughout a short-term follow-up of 3–5 years.12 However, donor liver organ shortage and lifetime burden of immunosuppressive therapy limits this treatment option for this devastating condition. Thus, alternative therapeutic approaches that slow down or halt progressive liver disease in PFIC3 are urgently needed.
Although protein replacement therapies (such as enzyme replacement therapy [ERT]) have been proven to be successful for some inherited metabolic diseases such as lysosomal storage diseases,13 this strategy does not appear to be feasible for mutations involved in intracellular membrane-bound proteins. Recently, gene replacement therapies based on viral vector delivery technology have shown promising efficacy targeting genetic diseases caused by a deficiency of a broader range of proteins, including membrane-bound proteins such as ABCB4.14–16 However, inherent risks remain in terms of insertional mutagenesis (e.g. genotoxicity) and immunogenicity. To overcome many of these concerns, a new class of mRNA-based therapies may offer unique therapeutic advantages for a variety of human diseases including infectious diseases, cancer, and inherited metabolic liver diseases.17–19 In the present study, we aimed to develop and test an mRNA-based therapy for the treatment of PFIC3. We have produced and screened a series of chemically and genetically modified mRNA variants encoding human ATP-binding cassette 4 (hABCB4), and examined their pharmacological effects in a cell-based model and an in vivo mouse model recapitulating human PFIC3 phenotypes as a result of homozygous disruption of the ABCB4 gene. Our data show that administration of hepatocyte-delivered codon-optimised hABCB4 mRNA resulted in de novo expression of the functional hABCB4 protein and restored phospholipid transport in cultured cells and in livers of a PFIC3 mouse disease model. Repeated injections of hABCB4 mRNA construct effectively rescued severe disease phenotype in Abcb4−/− mice and restored normal homeostasis by promoting hepatocyte-driven liver regeneration, with rapid and dramatic normalisation of all clinically relevant disease parameters such as inflammation, ductular reaction, and fibrosis.
Materials and methods
All animals were housed in a specific pathogen-free environment with a 12-h light/dark cycle and permitted ad libitum consumption of water and a standard chow diet unless otherwise stated. All mouse experiments were approved by the IACUC of the Beth Israel Deaconess Medical Center (158–2008, 004–2012, 010–2015).
BALB/c.Abcb4−/− mouse model of PFIC3
Mice with homozygous deletion of the Abcb4 gene (also known as MDR2, mouse orthologue of human MDR3) on the fibrosis-susceptible BALB/c background, with congenital severe progressive, early-onset biliary fibrosis with portal hypertension developing around weaning, were generated and characterised as reported previously.20 Lipid nanoparticle (LNP)-encapsulated modified human ABCB4 mRNA (or eGFP mRNA as control) at 1 mg/kg was injected using a 30G needle syringe via a tail vein once into 4-week-old male age-matched BALB/c.Abcb4−/−, or repeatedly (5 doses total over 2 weeks, once every 3–4 days, rotating injection side each time) and the phenotypes were analysed 12 h after the last injection, unless specified otherwise (see Video S1 for the technical procedure).
Synthetic human mRNA-LNP formulations
Complete N1-methylpseudouridine or 5-methoxyuridine substituted mRNA was synthesised in vitro from a linearised DNA template containing the coding sequence, 5’ and 3’ untranslated regions, and a poly-A tail, as previously described.17 The final mRNA contains an N7-methylated cap to promote efficient mRNA translation. After purification, the mRNA was diluted in citrate buffer to the desired concentration and frozen.17 The open reading frame sequences and chemistry of the lead hABCB4v6-mRNA and eGFP-mRNA used in this study can be found in Tables S1 and S2.
Synthetic mRNA-LNP formulations were prepared by ethanol drop nanoprecipitation as previously described.21 Briefly, heptadecan-9-yl 8-((2-hydroxyeth yl)(8-(nonyloxy)-8-oxooctyl) amino)octanoate, dipalmitoylphosphatidylcholine, cholesterol, and 1,2-dimyristoyl-glycero-3-methoxypolyethylene glycol-2000 were dissolved in ethanol and combined with acidified mRNA (sodium acetate, pH 5) at a ratio of 3:1 (aqueous:ethanol). Formulations were concentrated using Amicon ultra-centrifugal filters (EMD Millipore), passed through a 0.22 lm filter, and stored at 4°C until use. All formulations were tested for particle size, RNA encapsulation, and endotoxin and were found to be between 80 and 100 nm in size, with greater than 90% encapsulation, and <10 EU/ml endotoxin.
Transient transfection studies
Human embryonic kidney (HEK293) cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and maintained under standard recommended conditions (EMEM supplied with 10% of FBS). The cell line has been previously used in studying the expression and transporting activities of ABCB4 in cell-based models, based on its low endogenous level of ABCB4 protein and high efficiency in transient transfection studies.22 One day before transfection, 0.4–4 million cells were seeded into 1 well of a 6-well plate or 100-mm tissue culture dish, resulting in ~70% confluency on the day of transfection. Cells were transfected with 1–3 μg of mRNA using lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were harvested and processed for whole cell lysates for protein analysis. Under some circumstances, 24 h after transfection, the cell culture medium was replaced with fresh serum-free medium containing 0.02% BSA and 0.5 mM taurocholate and cells were cultured for an additional 4 h before the medium (1 ml) was collected for lipid extraction and phospholipid analysis.
Collection of hepatic bile samples
Mice were anaesthetised by initial injection of ketamine 300 mg/ kg plus xylazine 30 mg/kg i.p. and maintained under narcosis by administration of ketamine 150 mg/kg every 20–30 min until completion of the study. Under deep anaesthesia, the abdominal cavity was opened through a midline approach. The common bile duct located below the liver was ligated with a 3–0 suture. The gallbladder was identified, and a ligature (3–0 suture) was placed loosely below the entrance of the cystic duct. The gallbladder was punctured with the microscissors, and a microcatheter inserted and affixed using the provisionally placed suture to the established gallbladder fistula. After bile flow through the microcatheter was confirmed, the abdominal cavity was closed, and bile collected for up to 2 h on a thermal pad. At the completion of study, mice were euthanised by exsanguination and tissues collected for analysis.
Direct invasive PVP measurements
Portal venous pressure (PVP) was measured at the study endpoint as previously established.20 Briefly, 1.2-Fr high-fidelity pressure catheter (Scisense, London, ON, Canada) was inserted into portal vein of an anaesthetised mouse after midline abdominal incision. Pressure signals were recorded at 2 kHz for 5 min, and analysed using PowerLab software chart 5.5.6 (ADInstruments, Colorado Springs, CO, USA), followed by cardiac puncture/exsanguination and tissue collection for analysis.
Protein expression by immunoblotting analysis
hABCB4 protein expression levels in cell lysates or tissue membrane preparations were determined by standard immunoblot analysis or by the Simple Western systems (Sally Sue, Protein Simple, San Jose, CA, USA). For cell lysates preparation, cultured HEK293 cells were collected in PBS and solubilised with 0.5% Triton X-100 in the presence of a cocktail of protease inhibitors (Catalogue #87786, ThermoFisher Scientific) and Benzonase endonuclease (Catalogue #1.01654.0001, EMD Millipore, 5 U/μl final). For membrane preparations from mouse tissues (liver, lung, kidney, spleen, brain, and heart), the Mem-PER plus membrane protein extraction kit (Thermo Scientific, Catalogue #89842) was used according to the manufacturer’s instructions. The total protein concentration in test samples was determined using the Pierce® BCA Protein Assay kit (Thermo Scientific). For immunoblot analysis, samples (containing 50–100 μg of total protein) were separated by 4–12% SDS-PAGE and transferred to nitrocellulose membranes (iBlot2, Invitrogen) using standard procedures (the Method P0 as recommended by the manufacturer). Membrane fractions were first incubated with the Odyssey® Blocking Buffer (Part # 927–40000, Li-Cor, Lincoln, NE, USA) for a minimum of 1 h, followed by probing with either the antiABCB4 antibody (PII26, C-219, or HPA053288), or other corresponding primary antibody, as detailed in Table S3 (0.2–1 lg/ml) for at least 1 h at room temperature. Following incubation with the infrared (IR)-labelled goat anti-rabbit secondary antibody (IRDye® 800CW, Li-Cor), IR-intensity signals (corresponding to protein expression levels) were assessed using the imaging instrument Odyssey CLx (Li-Cor). For the Simple Western system-based analysis, cell lysates were first separated by capillary electrophoresis based on the size of the proteins, and proteins were probed with the primary ABCB4 antibody (LS-Bio357461) and secondary antibodies as recommended by the manufacturer (DM-001, Protein Simple) followed by fluorescence-based detection and quantification of the protein of interest.
LC/MS analysis of phospholipid species
HEK293 cells were subcultured in poly-L-Lys-coated 6-well plates in MEM supplemented with 10% FBS. Twenty-four hours after transfection, the medium was replaced by Phenol Red-free MEM medium containing 0.02% BSA and 0.5 mM taurocholate for an additional 4 h, as previously reported.22 Total bile lipids were extracted from 1 ml medium by the Bligh and Dyer method.23 Bile samples were mixed with 10 volumes of methanol, followed by a brief centrifugation (5,000×g, 10 min) to remove any trace protein or other insoluble debris. In selected experiments, the samples prepared from normal bile from wild-type (WT; Abcb4+/+) mice were further diluted 10-fold with HPLC mobile phase solution before analysis. Finally, the samples were analysed by LC/MS using a 1290 Infinity UHPLC coupled with a 6530 Q-TOF (Agilent Technologies, Santa Clara, CA, USA). Chromatographic separation was performed using an Ace Excel 2 C18 column, 2 μm, 2.1×150 mm, maintained at 60°C. Following injection of 10 μl of deproteinised supernatant, an optimised phospholipid elution gradient was delivered at 0.6 ml/min using solutions of (A) 50% acetonitrile and 0.1% formic acid (v/v) in water, and (B) 10% acetonitrile and 0.1% formic acid (v/v) in isopropanol, at the following proportions and time points: 50–95% B, 0–10 min. The column was then flushed with 95% B for 3 min and finally re-equilibrated with 50% B for 2 min. MS spectra were acquired in positive ionisation mode. PC species 34:1 (m/z 760.585) was used as a representative molecule to monitor phospholipid levels in cell culture media and bile. Under certain circumstances, the PC species 34:2 (m/z 758.569) was also used to monitor the phospholipid level in bile.
IHC and immunofluorescence analysis
Immunohistochemistry (IHC), H&E, and connective tissue (Sirius Red) staining were performed in formalin-fixed, paraffin-embedded liver sections, as previously described.24 The primary antibodies used in this study are summarised in Table S3. For immunofluorescence staining, frozen liver tissues (6 mm thick) were fixed with acetone and blocked by 5% BSA. Primary antibodies were visualised via conjugation to Alexa Fluor 488 and Alexa Fluor 592 antibodies (Molecular Probes, Life Technologies, Eugene, OR, USA). Nuclei were counterstained blue with DAPI. Images were documented with an Axiovert 200M Apotome wide-field microscope and Axiovision software version 4.6 (Zeiss, Thornwood, NY, USA).
Quantitative reverse transcription-PCR
Relative mRNA levels were quantified in total liver RNA by quantitative reverse transcription-PCR (qRT-PCR) on a Light-Cycler 1.5 instrument (Roche, Mannheim, Germany) using the TaqMan method, as described in detail.25,26 Sequences of primers and probes used in this study are summarised in Table S4.
Hepatic collagen content
Hepatic collagen content was determined biochemically via hydroxyproline quantification (μg per 100 mg of wet liver) in 200–300-mg liver samples from 2 different lobes (representing>20% of whole liver) after hydrolysis in 6N HCl for 16 h at 110°C, as described.25
Serum and bile chemistry
Serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and total bilirubin (TBIL) were measured using an automated Catalyst Dx Chemistry Analyzer (IDEXX Laboratories, Inc., Westbrook, ME, USA), according to the manufacturer’s recommendations. Total bile acids in serum and bile and free cholesterol in bile were analysed by the commercially available total bile acid assay and cholesterol quantification kits, respectively, which were purchased from Abcam (total bile acids, Catalogue #ab239702; cholesterol, #ab65359), following the manufacturer’s instructions.
Statistical analysis
Data are expressed as mean ± standard error of mean, and statistical analyses were performed using Graph-Pad Prism version 8.0 (GraphPad Software, San Diego, CA, USA). Multiple comparisons were performed by 1- or 2-way ANOVA as appropriate, followed by Dunnett’s post-test. Comparisons between 2 groups were performed using the Student t test. Differences among experimental groups with 2-tailed p values <0.05 were considered significant.
Data availability
All the data used to support the findings of this study are included within the article. Reagents, resources, and protocols are included in the Supplementary methods.
Results
Identification of optimised mRNA sequence encoding hABCB4
To explore the feasibility of re-expression of human full-length functional ABCB4 mRNA to rescue its inherited loss-of-function variations, we synthesised mRNAs carrying the natural coding sequence of WT human (hABCB4WT) or mouse (mABCB4 WT) ABCB4 protein. To test whether these constructs resulted in full-length human or mouse ABCB4 protein translation, protein expression was determined using human or mouse ABCB4 specific antibodies in HEK293 cells transiently transfected with the hABCB4WT or mABCB4WT mRNAs (Fig. 1A). This cell-based model has been previously used for evaluating the function of exogenously introduced ABCB4 protein and its variants.22 Western blotting analysis by P3-II26 (an antibody specific to human ABCB4 but not mouse ABCB4) or C219 (an antibody with cross-reactivity to both human and mouse ABCB4), showed that both mRNAs, when transfected in HEK293 cells, resulted in readily detectable expression of protein with the expected molecular weight of ~140 kDa as previously reported.22
Fig. 1. In vitro characterisation and screening of WT and codon-optimised hABCB4 mRNAs constructs in mammalian cells.

(A) Western blot of ABCB4 protein expression in HEK293 cells transfected with eGFP or the natural coding sequences encoding murine ABCB4 (mABCB4wt, C219 antibody), or human ABCB4 (hABCB4wt, P3-II26 antibody). (B) PC-transporting activity in mock-, eGFP, or hABCB4 mRNA transfected HEK293 cells. PC species 34:1 (m/z 760.2) in the cell culture medium was determined as described in the Materials and methods. (C) Capillary electrophoresis immunoblotting for hABCB4 protein 24 h after transfection with 13 codon-optimised hABCB4 mRNAs variants (V1–V13). Densitometry analysis after normalisation to β-actin is shown below. (D) Enhanced PC-transporting activity by the codon-optimised hABCB4v6 mRNA (as denoted by the arrow in C) in transfected HEK293 cells. Data are mean ± SEM (from 3–4 independent experiments). **p<0.01, ****p<0.0001 (ANOVA followed by Tukey’s test). hABCB4, human ATP-binding cassette 4; MM, molecular mass marker; PC, phosphatidylcholine; WT, wild-type.
In the subsequent experiments, we focused on hABCB4 mRNA as we aimed to develop the human version as a potential therapy for PFIC3. To determine whether hABCB4 mRNA produces a functional protein, we used LC/MS to measure whether HEK293 cells transiently transfected with hABCB4WT mRNA could secrete PC into the culture medium. As shown in Fig. 1B, at 24 h post-transfection, the cells transfected with hABCB4WT mRNA made a significant secretion of PC into culture medium when compared to either mock- or eGFP-transfected cells, confirming that functional hABCB4 protein expression was achieved via transient hABCB4 mRNA transfection in vitro.
To improve the expression level of hABCB4 protein from the mRNA, we used codon optimisation similar to previously described approaches widely used to enhance the protein expression for therapeutic purposes including mRNA-based therapeutic applications.27 We produced 13 independent codon-optimised hABCB4-mRNA variants (V1–V13) using a computational algorithm developed internally at Moderna and quantified the hABCB4 protein expression after a transient transfection in HEK293 cells. Most of the codon-optimised hABCB4-mRNA variants resulted in significant improvement in protein expression efficiency when compared with the WT sequence without codon optimisation (Fig. 1C). We identified the hABCB4v6-mRNA variant as the superior mRNA sequence with the highest expression and activity, as determined by a markedly increased level of PC secreted into medium by cells transfected with the hABCB4v6 mRNA in comparison with the WT sequence (Fig. 1D).
Efficacy of hABCB4v6 mRNA-LNP in a murine model of PFIC3 (BALB/c.Abcb4−/−)
The use of LNPs to deliver mRNA has been extensively studied. LNPs allow for efficient cargo delivery to hepatocytes in vivo.19,28,29 Through structure-based rational design and targeted screening in a variety of in vitro and in vivo models, we have developed LNP-based formulations for safe and effective in vivo delivery of our modified mRNA constructs.21 To assess whether functional hABCB4 could be re-expressed in vivo, we administered a single i.v. injection of LNP-encapsulated hABCB4v6 mRNA (or eGFP-encoding control mRNA, Ctrl) into mice bearing a homozygous disruption of endogenous Abcb4 gene (i.e. BALB/c.Abcb4−/− mice) and measured hepatic protein expression and biliary PC secretion. Mice were euthanised at 12 h post-injection and hABCB4 protein levels in enriched liver membrane preparations were determined by immunoblot analysis. As shown in Fig. 2A, expression of human ABCB4 protein was readily detected in the livers of BALB/c.Abcb4−/− mice at 12 h after i.v. injection of LNP-formulated hABCB4v6 (hABCB4v6-LNP). As expected,5 BALB/c.Abcb4−/− mice exhibited absence of PC in their bile (PC concentration of <1% of WT littermates’ levels). In contrast to an almost complete absence of PC (as represented by a PC species of 34:2) in bile of control BALB/c.Abcb4−/− mice administered with either PBS or eGFP-encoding mRNA, a single injection of the hABCB4v6-LNP resulted in a robust increase in bile PC at ~25% of normal (WT) level at 12 h post-injection (Fig. 2B). Importantly, similar increases in biliary PC were achieved in both sexes, along with expected improvement of other biliary physiology parameters such as biliary cholesterol secretion (Fig. S4 and Table S5). We have also characterised the kinetics of PC secretion into bile (0–96 h) in response to a single administration of hABCB4v6 mRNA-LNP (1 mg/kg) in BALB/c.Abcb4−/− mice, which showed marked increase in PC secretion between 6 and 48 h, peaking at 12 h (Fig. S1). Interestingly, in this experiment brief PC restoration (6–48 h) to merely 10% of WT level resulted in an accompanying decrease in serum ALT activity, which lasted from 12 h until at least 96 h post-injection (the latest time point studied, Fig. S1). Further, LC/MS analysis suggested that by just 12 h after hABCB4v6-LNP injection, treated Abcb4−/− mice had recovered not just a few specific species of PCs but rather a complete range of bile PC subspecies, with phospholipid profiles essentially identical to that of WT Abcb4+/+ mice (Fig. 2C). To further confirm physiological subcellular localisation to the canicular hepatocyte domain of the de novo hABCB4v6-LNP-driven expression of ABCB4 protein, we performed double-immunostaining for hABCB4 and ZO-1, a tight junction marker protein that is (although not a specific canalicular marker in a strict sense) highly expressed in the canalicular domain of hepatocytes.30 High resolution confocal microscopy analysis revealed a strong colocalisation between hABCB4 and ZO-1, confirming proper subcellular localisation of the exogenous hABCB4 to canalicular cytoplasm membrane domain of hepatocytes (Fig. 2D). No ectopic hABCB4 expression was detected in non-liver tissues (lung, kidney, spleen, brain, heart) in Abcb4-/mice of both sexes at functional peak (12 h) after hABCB4v6-LNP administration, as assessed by immunoblotting analysis of tissue lysates (Fig. S3).
Fig. 2. In vivo characterisation of codon-optimised human hABCB4v6 mRNA construct in BALB/c.Abcb4−/− mice.

BALB/c.Abcb4−/− mice received a single i.v. injection of eGFP (Ctrl) or hABCB4v6 mRNA encapsulated in LNP at 1.0 mg/kg and analysed 12 h later. (A) Western blot of hABCB4 protein expression in enriched liver membrane fraction, with Na+/K+ ATPase as an internal control (P3II-26 and Sc-1517 antibody recognises human and mouse ABCB4, respectively). (B) Biliary PC analysis in Abcb4−/− mice administered with PBS only (n = 3), eGFP mRNA (n = 4), or the hABCB4v6 mRNA (n = 4), and WT (Abcb4+/+) mice (n = 3). (C) Summed high-resolution LC/MS spectra of phospholipid molecular species in representative bile samples (n = 3). WT bile samples were diluted 10-fold relative to those from the BALB/c.Abcb4−/− mice for direct qualitative comparison. The relative PC profiles of WT mice and hABCB4v6-treated BALB/c.Abcb4−/− mice are virtually indistinguishable, suggesting that treatment resulted in recovery of a wide range of PC molecular species. (D) Confocal analysis of co-staining for hABCB4 (green) and ZO-1 (red), demonstrates correct (canalicular) localisation of hABCB4 protein expression in BALB/c.Abcb4−/− mice administered with the hABCB4v6 mRNA. Nuclei counterstained with DAPI (blue). hABCB4, human ATP-binding cassette 4; LNP, lipid nanoparticle; PC, phosphatidylcholine; WT, wild-type.
Repeat administration of hABCB4v6 mRNA-LNP leads to rapid improvement of body weight, hepatomegaly, and normalisation of serum chemistry in a BALB/c.Abcb4−/− mouse model of PFIC3
Given the need for chronic administration of hABCB4v6-LNP mRNA to treat PFIC3 patients, repeat dose studies were conducted to evaluate efficacy in male BALB/c.Abcb4−/− mice, starting from 4 weeks (the age of overt disease manifestation and onset of progressive fibrosis in this model20,25). Specifically, 3 groups of BALB/c.Abcb4−/− mice received 5 consecutive i.v. injections of vehicle (PBS), eGFP mRNA, or the hABCB4v6 mRNA at 1 mg/kg twice a week for a total course of 2 weeks (until mice reached adulthood at 6 weeks of age, Fig. 3A). Repeat administration of hABCB4v6 mRNA-LNP significantly improved the body weight of BALB/c.Abcb4−/− mice, effectively addressing ‘failure to thrive’ phenotype typically observed post-weaning in this model (Fig. 3B). Hepatomegaly was also dramatically improved (28.4% reduction of liver weight compared with eGFP controls), and even partially reversed (15.8% below pretreatment ‘4 w start’ control levels), but remained slightly elevated (at 17.6%) above normal, WT littermates’ level (Fig. 3C). Repeat injections of the hABCB4v6 mRNA-LNP also resulted in a robust increase in PC levels in bile, with a restoration of ~42% of PC levels observed in the WT mice (Fig. 3D). This was accompanied by notable improvement in the levels of canalicular expression of hABCB4 protein observed in BALB/c.Abcb4−/− hepatocytes after multiple injections of hABCB4v6 mRNA-LNP by confocal microscopy, compared with single administration, suggesting cumulative increase in functional protein expression with repeated construct delivery (Fig. S2). This was accompanied by striking improvement in serum biochemistry, with complete normalisation of serum ALT and ALP activities, as well as well total bile acid concentration (Fig. 3E–G).
Fig. 3. Repeated injections of hepatocyte-targeted hABCB4v6 mRNA restore biliary PC secretion in vivo and markedly improve hepatomegaly and liver injury in BALB/c.Abcb4−/− mice.

(A) Scheme of experimental design. Male 4-week-old BALB/c.Abcb4−/− mice received 5 injections of LNP-encapsulated hABCB4v6, eGFP mRNA (1 mg/kg, i.v. twice a week), or PBS (n = 12–15). Untreated 4-week-old BALB/c.Abcb4−/− mice are ‘start of treatment’ controls (4 w start, n = 10) and Abcb4+/+ WT (n = 5) littermates are healthy controls. (B) Body weight gains, % change to initial body weight at 4 weeks of age (*p<0.05 compared with the eGFP group, 2-way ANOVA with Bonferroni post-test). (C) Changes in liver weight and (D) biliary PC concentration in BALB/c.Abcb4−/− mice, expressed as % of WT control levels. Serum levels of (E) ALT, (F) ALP, and (G) total BA. Data are mean ± SEM (n = 7–15; WT, n = 5). **p<0.01, ***p<0.001 vs. eGFP group (ANOVA with Dunnett’s post-test). ALP, alkaline phosphatase; ALT, alanine aminotransferase; BA, bile acids; hABCB4, human ATP-binding cassette 4; MM, molecular mass marker; PC, phosphatidylcholine; WT, wild-type.
Importantly, repeated administration of hABCB4v6 mRNA-LNP constructs at 1 mg/kg was well tolerated, with no weight loss, deaths or other drug-related toxicity events noted. As there was no significant difference between PBS-treated and eGFP mRNA-LNP-treated groups in any studied parameters (Fig. 3), the PBS-treated group was not included in subsequent analyses.
Liver fibrosis progression is halted by repeat administration of hABCB4v6 mRNA-LNP in BALB/c.Abcb4−/− mice
While control eGFP-treated BALB/c.Abcb4−/− mice developed advanced periportal fibrosis with thick ‘onion skinning’ around enlarged bile ducts, occasional portal–portal bridging and moderate perisinusoidal collagen deposition, hABCB4v6 mRNA-LNP administration resulted in markedly diminished histological signs of periportal fibrosis (Fig. 4A). Quantitative collagen morphometry showed that hABCB4v6 mRNA-LNP reduced the collagen area by 80% compared with eGFP group; both histopathological examination and collagen morphometry comparison to ‘start of treatment’ controls suggested that no appreciable histological progression of fibrosis occurred in hABCB4v6 mRNA LNP-treated group (Fig. 4A and B). Hepatic collagen deposition assessed biochemically (via hydroxyproline determination) also significantly decreased by 56% in the hABCB4v6 mRNA-LNP group but were still significantly higher than those at start of treatment (Fig. 4C). Importantly, PVP, measured invasively at the study endpoint was significantly reduced by hABCB4v6 mRNA-LNP treatment compared with the eGFP group (an average 1.81 mmHg reduction, p = 0.0004), and was not different compared with ‘4 w start’ pretreatment control levels (6.83 ± 0.31 mmHg compared with 4 w start 6.94 ± 0.43 mmHg, p = 0.853, not significant, Fig. 4D). Finally, key profibrogenic transcript levels that are massively upregulated in BALB/c.Abcb4-/mutant livers (procollagen α1(I), TGFβ1, TIMP-1, MMP-2,25 ITGB626) were suppressed in the livers of BALB/c.Abcb4−/− mice treated with hABCB4v6 mRNA to the levels near normal (healthy WT) levels. In contrast, putatively profibrolytic (e.g. mediating collagen breakdown) genes MMP-3, −924,25 were diminished compared with the eGFP group but remained ~5-fold elevated compared with the healthy WT control group (p = 0.0048 and 0.0061, respectively, Fig. 4E and F). Taken together, these results suggest that rapid progression of congenital biliary-type fibrosis was completely halted in BALB/c.Abcb4−/− mice by repeated delivery of ABCB4v6 mRNA-LNP. Importantly, similar improvement in biliary physiology (Table S6) and antifibrotic efficacy of repeated ABCB4v6 mRNA-LNP administration was achieved in female Abcb4−/− mice (Fig. S5).
Fig. 4. Repeated hABCB4v6 mRNA administration prevents liver fibrosis progression in BALB/c.Abcb4−/− mice.

(A) Representative low-magnification images of connective tissue staining (Sirius Red, ×50, scale bar 200 μm). Blow-up magnification of portal tract matched by the calibre of portal vein in hABCB4v6 mRNA-treated mice and eGFP controls (×200, portal vein marked by the asterisk). (B) Collagen morphometry (% of Picrosirius Red area, n = 3/5/10/11 per bar, average of 10 random HPF per animal). (C) Hepatic collagen content as determined biochemically via hydroxyproline (n = 10–15 for BALB/c.Abcb4−/− mice, n = 5 for WT). (D) Portal venous pressure, as determined via direct cannulation of portal vein with microtip pressure monitor at study endpoint. (E,F) Hepatic transcript levels of key profibrogenic molecules COLIA1, TGFβ1, TIMP-1, and ITGB6 (E) and of matrix-degrading enzymes (MMP-2, −3, −9, −12) (F) in BALB/c.Abcb4−/− mice treated with control eGFP or hABCB4v6 mRNA (n = 7–9 per group). Data are mean ± SEM (all animals are males, t test p value as indicated on each graph). See also Figure S6 for additional bile duct morphology images. hABCB4, human ATP-binding cassette 4; HPF, high-power field; WT, wild-type.
Non-activation of HSCs/myofiboblasts and resolution of ‘reactive ducts’ phenotype in livers of BALB/c.Abcb4−/− mice with hABCB4v6 mRNA-LNP treatment
Profibrogenic hepatic stellate cells (HSCs) activation, trans-differentiation into myofibroblasts and their expansion,31 and proliferation of cholangiocytes/adult hepatic progenitors (collectively termed ‘ductular reaction’) are key cellular processes driving (biliary) liver fibrosis progression, which were further analysed in situ via IHC followed by quantitative morphometric analysis. eGFP-treated BALB/c.Abcb4−/− controls demonstrated massive HSC activation along the expanding portal tracts and fibrotic septa, as revealed by IHC for an HSC activation marker, with 13-fold increase in a-SMA-positive area compared with healthy WT controls (Fig. 5A and B). In hABCB4v6 mRNA-LNP-treated BALB/c.Abcb4−/− mice, a remarkable decrease in a-SMA positivity was observed, with only few α-SMA-positive HSCs scattered throughout the liver lobule. Quantitatively, the α-SMA+ area was comparable with that of healthy WT mice (Fig. 5A and B). Pronounced ductular reaction (visualised by ductal cell marker CK-19 and hepatic progenitor cell marker integrin avb632 in injured bile ducts, duct-like proliferations (pseudo-ducts) and occasional single cells) was observed in eGFP-treated BALB/c.Abcb4−/− mice (6-fold and 7-fold increase in CK19- and avb6positive cells compared with WT, respectively, Fig. 5A, C and D), whereas immunopositivity for both ductal cell markers was drastically diminished by repeated hABCB4v6 mRNA-LNP and did not differ quantitatively from healthy WT controls (Fig. 5A–D). Serial sections depicting dramatic changes in bile duct morphology in large portal tracts after treatment are shown in Figure S6. Altogether, these data indicate that repeated hABCB4v6 mRNA-LNP injections resulted in prevention of HSC/myofibroblasts activation and expansion, and complete resolution of the ‘reactive ducts’ phenotype.
Fig. 5. Prevention of HSC activation and myofibroblast expansion, normalisation of ‘reactive ducts’ phenotype in livers of BALB/c.Abcb4−/− mice with hABCB4v6 mRNA treatment.

(A) Representative images of immunostaining for HSC/myofibroblast activation marker α-SMA (upper row, ×50, scale bar 200 μm), ductular reaction markers αKβ9 (middle row, ×200, scale bar 50 μm) and integrin avb6 (lower row, ×200, scale bar 50 μm) with morphometric quantification (B–D). Data are mean ± SEM (n = 4/7/7 individual animals per bar, average of 10 random HPF analysed per animal). The p value indicated as compared with eGFP group (ANOVA). hABCB4, human ATP-binding cassette 4; HPF, high-power field; HSC, hepatic stellate cell; WT, wild-type.
hABCB4v6 mRNA-LNP administration leads to rapid resolution of peribiliary inflammatory infiltrates and promotes hepatocyte-driven liver regeneration in BALB/c.Abcb4−/− mice
Next, we characterised the impact of hABCB4v6 mRNA on inflammation and hepatocyte regeneration in the liver of BALB/c.Abcb4−/− mice. H&E stain demonstrated that livers of BALB/c.Abcb4−/− mice developed pronounced characteristic portal inflammatory infiltrates, portal tract expansion caused by bile duct hyperplasia and fibrosis with early septal formation. Administration of hABCB4v6 mRNA-LNP resulted in remarkable reversal of liver portal inflammation and bile duct hyperplasia and return to nearly normal liver architecture like that of healthy WT mice (Fig. 6A, upper row). Inflammatory cell infiltrates were further characterised by immunostaining for markers of resident macrophages (F4/80) and infiltrating monocytes (CD11b). In livers of BALB/c.Abcb4−/− mice, large numbers of F4/80- and CD11b-positive cells accumulated along the portal tracts compared with WT mice. hABCB4v6 mRNA-LNP effectively suppressed the infiltration of F4/80-positive macrophages, which reversed to a normal, perisinusoidal distribution pattern of Kupffer cells in WT mice (Fig. 6A, middle row). Infiltrating CD11b-positive monocytes numbers dropped drastically in hABCB4v6 mRNA-LNP-treated livers, with few cells occasionally detected without clear association with bile ducts, similarly to a nearly absent CD11b staining in healthy livers (Fig. 6A).
Fig. 6. Resolution of inflammatory infiltrates and improved hepatocyte-driven liver regeneration in BALB/c.Abcb4−/− mice treated with hABCB4v6 mRNA.

(A) Representative images (×200) of H&E (upper row), F4/80 (middle row) and CD11b (lower row). (B) Representative images of IHC for nuclear cell proliferation marker Ki67 staining (×200). Arrows show Ki-67+ replicating hepatocyte nuclei. (C) Quantification of ductal cells vs. hepatocyte replication in situ. Data are average of at least 10 random HFP/animal analysed at 200× magnification, mean ± SEM (n = 5 mice per group, t test p value as indicated). Scale bar, 50 μm. Portal vein indicated by asterisks. hABCB4, human ATP-binding cassette 4; IHC, immunohistochemistry; WT, wild-type.
Simultaneously with these signs of resolution of inflammation, predominant cell proliferation within hepatic epithelium shifted from cholangiocyte lineage to hepatocytes. Thus, as detected via Ki-67 staining, hepatocytes replication notably increased in hABCB4v6-LNP-treated livers, in contrast to eGFP-treated BALB/c.Abcb4−/− controls, in which replication was observed predominantly in ductal cells (Fig. 6B and C). This suggests that resolution of inflammation, ductular reaction, and fibrosis achieved by hABCB4v6 mRNA-LNP therapy promoted normal, hepatocyte-driven liver regeneration, as opposed to pathological, HPC-driven regeneration in the settings of proinflammatory and pro-fibrotic milieu.33
Discussion
PFIC3 is a devastating congenital paediatric liver disease, with severe clinical manifestation and rapid progression to cirrhosis, with no effective therapy except for liver transplantation. Considering that mRNA therapy can be used to rescue the genetic deficiency via introducing the normal protein for enhancing the expression of specific proteins via systemic administration, an approach that is being currently tested in multiple mRNA-based clinical cancer trials,17–19 we hypothesised that it could be applied to therapeutically re-express functional ABCB4 protein in the liver. Furthermore, codon optimisation and protein engineering approaches could be used in the design of the mRNA production to improve protein expression and functions. In the present study, we demonstrate the remarkable efficacy of mRNA therapy as a potential treatment for PFIC3 using the Abcb4-null mouse model. Administration of novel hABCB4 mRNA via lipofectamine-based transient transfection to cultured cells or encapsulated in LNPs to mice resulted in de novo expression of functional hABCB4 protein and restored phospholipid transport. Repeated injections of the hABCB4 mRNA completely rescued the severe disease phenotype in juvenile Abcb4−/− mice, with rapid and dramatic normalisation of all clinically relevant parameters such as inflammation, ductular reaction, and fibrosis.
Homozygous disruption of the Abcb4 gene, originally performed in FVB mice results in a complete absence of PC from bile,5 which leads to congenital cholestasis, liver injury, and progressive fibrosis,25,34 recapitulating the genetic defect and main morphological features of liver disease in human PFIC3.4,35 However, the cross-species fidelity to human PFIC3 has been questioned because of the relatively mild disease in mice and slow progression of fibrosis.36 It appears that this could be explained in part by the less hydrophobic (and thus less toxic) nature of bile acid composition in mice compared with humans, that is, predominant hydrophilic muricholic acid in mice vs. cholic acid in humans,37 as feeding low doses of cholic acid led to aggravation of liver disease in Abcb4−/− mice.36 However, our recent studies into genetic susceptibility of inbred mouse strains to fibrosis showed that mild disease in original FVB.Abcb4−/− was as a result of its genetic background resistance to fibrosis; accordingly, transfer of the mutation onto a fibrosis-susceptible background resulted in severe, early-onset portal hypertension and fibrosis progressing at an over 3-fold faster rate and reaching end-stage disease around 7 months of age in BALB/c.Abcb4−/− mice.20 In the present study, we used our BALB/c.Abcbc4−/− mouse model to investigate the efficacy of hABCB4 mRNA therapy starting from very young age of 4 weeks (1 week post-weaning, the earliest age tail vein injections are technically feasible), as the model most closely recapitulating severe liver disease phenotype and timing of human PFIC3, usually manifesting shortly after birth and progressing to end stage before adulthood.3 We postulated that the rapid development of liver fibrosis during young age in this mouse model presents a unique tool and intervention window to assess the pharmacological effect of mRNA-based therapy for PFIC3.
We have previously developed LNP-based formulations for safe and effective in vivo delivery of our modified mRNA constructs for the treatment of otherwise intractable inherited errors in metabolisms.19,21,28,29 We have demonstrated that stabilised mRNA by genetic and chemical modification can use the cellular translational and trafficking machineries to express functional proteins in specialised spaces such as subcellular and membrane domains. In the present study, we sought to develop an mRNA-based therapy targeting PFIC3 by re-expressing ABCB4, the affected transporter protein that is localised in the canalicular domain of hepatocytes and intractable through traditional ERT. Using computational algorithms and cell-based screening tools, we have identified the hABCB4v6-mRNA variant as the best mRNA sequence with respect to protein expression and activity (Fig. 1D). A single i.v. injection of LNP-encapsulated hABCB4v6 mRNA into BALB/c.Abcb4−/− mice resulted in a robust increase in hepatic PC output into bile reaching 10–25% of normal (WT) PC level 12 h after injection (Fig. 2B), which coincided with significant and persistent improvement in the liver injury marker ALT for up to 96 h (Fig. S1). Repeat injections of the LNP-encapsulated hABCB4v6 mRNA further increased biliary PC concentrations to about ~42% of normal levels (Fig. 3D), thereby completely rescuing the PFIC3-like disease phenotype and preventing liver fibrosis in BALB/c.Abcb4−/− mice (Figs. 3–6). To the best of our knowledge, this is the first time that a ‘minimum’ of clinically meaningful hepatic PC output restoration by Abcb4-targeted gene therapy has been experimentally established. Thus, based on our data, biochemical response can be expected via increase of biliary PC to merely 10% of normal levels (lowered transaminases, Fig. S1), while complete resolution of virtually all disease parameters including liver injury, inflammation, and fibrosis can be achieved via increase to 42% of normal PC level (Figs. 3–6), allowing us to propose a fractional restoration within 10–42% of normal range of bile PC as a practical target for a therapeutic intervention. This is considerably lower than levels that could have been extrapolated from disease-free heterozygous (Abcb4+/−) mice (with 60% of PC level,5 or prior successful gene therapy attempts with 70–100% of PC level),14–16 and has important practical implication for gene therapy of PFIC3. Importantly, physiological subcellular localisation of hABCB4 in the canalicular membrane of hepatocytes was confirmed via the double-immunostaining for hABCB4 and ZO-1,30 confirming proper subcellular localisation of the exogenous hABCB4 in the canalicular cytoplasm membrane domain of hepatocytes (Fig. 2D).
It is not entirely surprising, at least theoretically, that re-expression of ABCB4 in the liver leads to a dramatic correction of multiple aspects of monogenic disease in Abcb4−/− mice. However, there are significant challenges present in our ‘highbar’ preclinical system utilising severely fibrotic BALB/c.Mdr2−/−: first, substantial pre-existing fibrosis with portal hypertension that is already present at the beginning of treatment, that is, at the age of 4 weeks,20 and second, the context of actively growing liver, both of which can impede efficiency of gene therapy.38 Recently, Aronson et al.,15 utilising 10-week-old C57BL/6.Abcb4−/− mice, and Weber et al.,14 studying 7-week-old FVB.Abcb4−/− mice, reported that an adeno-associated virus (AAV)-mediated gene therapy resulted in long-term correction of PFIC3-like disease, including fibrosis. Our study is distinct in demonstrating that another, mRNA-based approach can achieve nearly complete rescue of the disease phenotype in both juvenile mice of both sexes and in the settings of more severe disease course (Abcb4−/− strain on BALB/c background are more fibrotic than either FVB20 or C57BL/6 strain39). Moreover, we show that administration of hABCB4v6 mRNA-LNP prevented progressive portal hypertension (Fig. 4D), one of the best predictors of clinical progression in human cirrhosis,40,41 an important feature that has not been evaluated in prior gene therapy attempts for PFIC3. However, while almost all other parameters (serum chemistry, inflammation, ductular reaction, and collagen morphometry, Figs. 3–6) normalised to below ‘start of treatment’ control levels, suggesting disease reversal, portal pressure remained at the pretreatment level (Fig. 4D). With existing data, it is difficult to unequivocally conclude whether reversal of portal pressure represents a challenging therapeutic target, or simply requires a longer time to completely regress to its baseline. Indeed, a careful and detailed analysis of liver regeneration suggests that although injury, inflammation, and fibrogenesis were completely abrogated by the treatment, the process of remodelling and regeneration switched from cholangioctyte/progenitor cell- to hepatocyte-driven, but was still ongoing at the study endpoint (Fig. 6B and C).
mRNA therapy is an emerging and distinct therapeutic approach that has several unique advantages as compared with other nucleic acid-based therapies. Firstly, mRNA can exert its function once it reaches the cytoplasm and it does not need to enter the nucleus. Secondly, mRNA is active transiently and is eventually completely degraded via physiological metabolic pathways; therefore, allowing precise dosing control in a repeated fashion compared to traditional gene therapy approaches. Thirdly, mRNA-based therapeutics do not integrate into the genome and do not incur the risks of insertional mutagenesis.42 Additionally, mRNA therapy may prove to be superior in paediatric diseases such as PFIC3, as we observed robust phenotype correction in juvenile mice, whereas earlier animal studies suggested low persistence AAV-mediated transgene in developing liver.38 Moreover, the production of mRNA can now be scaled up for clinical use, and thus mRNA-based therapeutics might be envisioned as a promising option for severe genetic disorders such as PFIC3, which have very limited treatment options. An important limitation of the mRNA therapy approach, such as less durable expression, compared with DNA-based approaches, and necessity of repeated parenteral dosing for sustaining the therapeutic efficacy, must also be noted.
Potential applications of ABCB4 mRNA therapy are not limited to PFIC3, as multiple independent studies linked variations in ABCB4 to a spectrum of liver disorders, such as low phospholipid-associated cholelithiasis, parenteral nutrition-induced cholestasis, sepsis-associated cholestasis, and bile duct injury after liver transplantation.43,44 Several reports pointed to association of heterozygous ABCB4 variations in patients with cryptogenic cirrhosis.1,45,46 A recent large-scale population-level genome-wide association study in 2,636 Icelanders has further corroborated the significant association of ABCB4 with chronic liver disease.47 It also appears that the common ABCB4 variant p.T175A, which is not known to result in defective PC translocation, is linked to increased liver stiffness in patients with non-alcoholic fatty liver disease.48 The role of the ABCB4 variation in the development of drug-induced liver injury remains unclear, although several clinical studies have indicated a potential association in cholestatic liver injury induced by antibiotics, chemotherapy drugs, and psychotropic drugs, which might be related to the partial, functional ABCB4 deficit.49,50 These observations call for exploration of therapeutic feasibility of ABCB4 overexpression approaches in experimental models of non-PFIC3 liver diseases where functional deficit of ABCB4 is thought to contribute to disease pathogenesis.51
In summary, we report a novel mRNA-based, hepatocyte-delivered therapeutic system that completely prevents progression of liver disease in a clinically relevant model with a particular focus on a treatment scenario in young BALB/c.Abcb4-/mice. Our results suggest that such mRNA therapy holds promise to provide PFIC3 patients with a safe, effective, and reliable treatment as an alternative to liver transplantation and supports further evaluation in clinical trials.
Supplementary Material
Highlights.
Synthetic liver-targeted hABCB4 mRNA therapy was designed for PFIC3, a devastating rare paediatric liver disease.
Single injection restores hepatocyte ABCB4 expression and biliary phosphatidylcholine secretion in a genetic model of PFIC3.
Repeated administration rapidly and completely rescues PFIC3 disease in young Abcb4−/− mice.
Treatment ameliorated liver injury, inflammation, ductular reaction, fibrosis, portal hypertension and ‘failure to thrive’.
Financial support
This work was supported in part by research grants from Moderna Inc., PSC Partners for Cure Canada, and an institutional grant from the Department of Medicine, Beth Israel Deaconess Medical Center to YVP. GW is a recipient of a fellowship from China Postdoctoral Science Foundation (2020M673014).
Abbreviations
- AAV
adeno-associated virus
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ERT
enzyme replacement therapy
- hABCB4
human ATP-binding cassette 4
- HSCs
hepatic stellate cells
- IHC
immunohistochemistry
- IR
Infrared
- LNPs
lipid nanoparticles
- PC
phosphatidylcholine
- PFIC3
progressive familial intrahepatic cholestasis type 3
- PVP
portal venous pressure
- qRT-PCR
quantitative reverse transcription PCR
- TBIL
total bilirubin
- UDCA
ursodeoxycholic acid
Footnotes
Data availability statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2020.12.010.
Conflicts of interest
JC, JZ, LY, AF, VP, SG, SH, CL, PF, PHG, and PM are employees of, and receive salary, stock, and stock options from, Moderna, Inc.
Please refer to the accompanying ICMJE disclosure forms for further details.
References
Author names in bold designate shared co-first authorship
- [1].Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology 2001;120:1448–1458. [DOI] [PubMed] [Google Scholar]
- [2].Reichert MC, Lammert F. ABCB4 gene aberrations in human liver disease: an evolving spectrum. Semin Liver Dis 2018;38:299–307. [DOI] [PubMed] [Google Scholar]
- [3].Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects. Semin Liver Dis 2010;30:134–146. [DOI] [PubMed] [Google Scholar]
- [4].de Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci U S A 1998;95:282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993;75:451–462. [DOI] [PubMed] [Google Scholar]
- [6].Delaunay JL, Durand-Schneider AM, Dossier C, Falguieres T, Gautherot J, Davit-Spraul A, et al. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016;63:1620–1631. [DOI] [PubMed] [Google Scholar]
- [7].Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology 2006;130:908–925. [DOI] [PubMed] [Google Scholar]
- [8].Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis 2011;31:3–10. [DOI] [PubMed] [Google Scholar]
- [9].Beuers U. Drug insight: mechanisms and sites of action of ursodeoxycholic acid in cholestasis. Nat Clin Pract Gastroenterol Hepatol 2006;3:318–328. [DOI] [PubMed] [Google Scholar]
- [10].Colombo C, Vajro P, Degiorgio D, Coviello DA, Costantino L, Tornillo L, et al. Clinical features and genotype-phenotype correlations in children with progressive familial intrahepatic cholestasis type 3 related to ABCB4 mutations. J Pediatr Gastroenterol Nutr 2011;52:73–83. [DOI] [PubMed] [Google Scholar]
- [11].Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology 1997;25:519–523. [DOI] [PubMed] [Google Scholar]
- [12].Hori T, Egawa H, Takada Y, Ueda M, Oike F, Ogura Y, et al. Progressive familial intrahepatic cholestasis: a single-center experience of livingdonor liver transplantation during two decades in Japan. Clin Transpl 2011;25:776–785. [DOI] [PubMed] [Google Scholar]
- [13].Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 2015;66:471–486. [DOI] [PubMed] [Google Scholar]
- [14].Weber ND, Odriozola L, Martinez-Garcia J, Ferrer V, Douar A, Benichou B, et al. Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat Commun 2019;10:5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Aronson SJ, Bakker RS, Shi X, Duijst S, Ten Bloemendaal L, de Waart DR, et al. Liver-directed gene therapy results in long-term correction of progressive familial intrahepatic cholestasis type 3 in mice. J Hepatol 2019;71:153–162. [DOI] [PubMed] [Google Scholar]
- [16].Siew SM, Cunningham SC, Zhu E, Tay SS, Venuti E, Bolitho C, et al. Prevention of Cholestatic liver disease and reduced tumorigenicity in a murine model of PFIC type 3 using hybrid AAV-piggyBac gene therapy. Hepatology 2019;70:2047–2061. [DOI] [PubMed] [Google Scholar]
- [17].Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Modified mRNA vaccines protect against Zika virus Infection. Cell 2017;169:176. [DOI] [PubMed] [Google Scholar]
- [18].Hewitt SL, Bai A, Bailey D, Ichikawa K, Zielinski J, Karp R, et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36c, and OX40L mRNAs. Sci Transl Med 2019;11:eaat9143. [DOI] [PubMed] [Google Scholar]
- [19].Cao J, An D, Galduroz M, Zhuo J, Liang S, Eybye M, et al. mRNA therapy improves metabolic and behavioral abnormalities in a murine model of citrin deficiency. Mol Ther 2019;27:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ikenaga N, Liu SB, Sverdlov DY, Yoshida S, Nasser I, Ke Q, et al. A new Mdr2(−/−) mouse model of sclerosing cholangitis with rapid fibrosis progression, early-onset portal hypertension, and liver cancer. Am J Pathol 2015;185:325–334. [DOI] [PubMed] [Google Scholar]
- [21].Sabnis S, Kumarasinghe ES, Salerno T, Mihai C, Ketova T, Senn JJ, et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther 2018;26:1509–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Morita SY, Kobayashi A, Takanezawa Y, Kioka N, Handa T, Arai H, et al. Bile salt-dependent efflux of cellular phospholipids mediated by ATP binding cassette protein B4. Hepatology 2007;46:188–199. [DOI] [PubMed] [Google Scholar]
- [23].Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911–917. [DOI] [PubMed] [Google Scholar]
- [24].Popov Y, Sverdlov DY, Bhaskar KR, Sharma AK, Millonig G, Patsenker E, et al. Macrophage-mediated phagocytosis of apoptotic cholangiocytes contributes to reversal of experimental biliary fibrosis. Am J Physiol Gastrointest Liver Physiol 2010;298:G323–G334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/−mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol 2005;43:1045–1054. [DOI] [PubMed] [Google Scholar]
- [26].Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol 2008;48:453–464. [DOI] [PubMed] [Google Scholar]
- [27].Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med 2014;20:604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].An D, Schneller JL, Frassetto A, Liang S, Zhu X, Park JS, et al. Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep 2017;21:3548–3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang L, Berraondo P, Jerico D, Guey LT, Sampedro A, Frassetto A, et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat Med 2018;24:1899–1909. [DOI] [PubMed] [Google Scholar]
- [30].Mottino AD, Cao J, Veggi LM, Crocenzi F, Roma MG, Vore M. Altered localization and activity of canalicular Mrp2 in estradiol-17beta-Dglucuronide-induced cholestasis. Hepatology 2002;35:1409–1419. [DOI] [PubMed] [Google Scholar]
- [31].Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016;63:217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kuramitsu K, Sverdlov DY, Liu SB, Csizmadia E, Burkly L, Schuppan D, et al. Failure of fibrotic liver regeneration in mice is linked to a severe fibrogenic response driven by hepatic progenitor cell activation. Am J Pathol 2013;183:182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mauad TH, van Nieuwkerk CM, Dingemans KP, Smit JJ, Schinkel AH, Notenboom RG, et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am J Pathol 1994;145:1237–1245. [PMC free article] [PubMed] [Google Scholar]
- [35].Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis 2001;21:551–562. [DOI] [PubMed] [Google Scholar]
- [36].Van Nieuwkerk CM, Elferink RP, Groen AK, Ottenhoff R, Tytgat GN, Dingemans KP, et al. Effects of ursodeoxycholate and cholate feeding on liver disease in FVB mice with a disrupted mdr2 P-glycoprotein gene. Gastroenterology 1996;111:165–171. [DOI] [PubMed] [Google Scholar]
- [37].Fickert P, Wagner M. Biliary bile acids in hepatobiliary injury – what is the link? J Hepatol 2017;67:619–631. [DOI] [PubMed] [Google Scholar]
- [38].Bortolussi G, Zentillin L, Vanikova J, Bockor L, Bellarosa C, Mancarella A, et al. Life-long correction of hyperbilirubinemia with a neonatal liverspecific AAV-mediated gene transfer in a lethal mouse model of CriglerNajjar Syndrome. Hum Gene Ther 2014;25:844–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peng ZW, Rothweiler S, Wei G, Ikenaga N, Liu SB, Sverdlov DY, et al. The ectonucleotidase ENTPD1/CD39 limits biliary injury and fibrosis in mouse models of sclerosing cholangitis. Hepatol Commun 2017;1:957–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ripoll C, Groszmann RJ, Garcia-Tsao G, Bosch J, Grace N, Burroughs A, et al. Hepatic venous pressure gradient predicts development of hepatocellular carcinoma independently of severity of cirrhosis. J Hepatol 2009;50:923–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Abraldes JG, Tarantino I, Turnes J, Garcia-Pagan JC, Rodes J, Bosch J. Hemodynamic response to pharmacological treatment of portal hypertension and long-term prognosis of cirrhosis. Hepatology 2003;37:902–908. [DOI] [PubMed] [Google Scholar]
- [42].Sahin U, Kariko K, Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov 2014;13:759–780. [DOI] [PubMed] [Google Scholar]
- [43].Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis 2007;27:77–98. [DOI] [PubMed] [Google Scholar]
- [44].Gonzales E, Davit-Spraul A, Baussan C, Buffet C, Maurice M, Jacquemin E. Liver diseases related to MDR3 (ABCB4) gene deficiency. Front Biosci (Landmark Ed 2009;14:4242–4256. [DOI] [PubMed] [Google Scholar]
- [45].Ziol M, Barbu V, Rosmorduc O, Frassati-Biaggi A, Barget N, Hermelin B, et al. ABCB4 heterozygous gene mutations associated with fibrosing cholestatic liver disease in adults. Gastroenterology 2008;135:131–141. [DOI] [PubMed] [Google Scholar]
- [46].Hakim A, Zhang X, DeLisle A, Oral EA, Dykas D, Drzewiecki K, et al. Clinical utility of genomic analysis in adults with idiopathic liver disease. J Hepatol 2019;70:1214–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet 2015;47:435–444. [DOI] [PubMed] [Google Scholar]
- [48].Krawczyk M, Rau M, Grunhage F, Schattenberg JM, Bantel H, Pathil A, et al. The ABCB4 p.T175A variant as potential modulator of hepatic fibrosis in patients with chronic liver diseases: looking beyond the cholestatic realm. Hepatology 2017;66:666–667. [DOI] [PubMed] [Google Scholar]
- [49].Lang C, Meier Y, Stieger B, Beuers U, Lang T, Kerb R, et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics 2007;17:47–60. [DOI] [PubMed] [Google Scholar]
- [50].Smith AJ,vanHelvoort A,vanMeer G,Szabo K,Welker E,Szakacs G,et al.MDR3 P-glycoprotein, a phosphatidylcholine translocase, transports several cytotoxic drugs and directly interacts with drugs as judged by interference with nucleotide trapping. J Biol Chem 2000;275:23530–23539. [DOI] [PubMed] [Google Scholar]
- [51].Trauner M, Fickert P, Wagner M. MDR3 (ABCB4) defects: a paradigm for the genetics of adult cholestatic syndromes. Semin Liver Dis 2007;27:77–98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data used to support the findings of this study are included within the article. Reagents, resources, and protocols are included in the Supplementary methods.