

ABSTRACT
Chlamydiae are pathogenic intracellular bacteria that cause a wide variety of diseases throughout the globe, affecting the eye, lung, coronary arteries and female genital tract. Rather than by direct cellular toxicity, Chlamydia infection generally causes pathology by inducing fibrosis and scarring that is largely mediated by host inflammation. While a robust immune response is required for clearance of the infection, certain elements of that immune response may also damage infected tissue, leading to, in the case of female genital infection, disease sequelae such as pelvic inflammatory disease, infertility and ectopic pregnancy. It has become increasingly clear that the components of the immune system that destroy bacteria and those that cause pathology only partially overlap. In the ongoing quest for a vaccine that prevents Chlamydia-induced disease, it is important to target mechanisms that can achieve protective immunity while preventing mechanisms that damage tissue. This review focuses on mouse models of genital Chlamydia infection and synthesizes recent studies to generate a comprehensive model for immunity in the murine female genital tract, clarifying the respective contributions of various branches of innate and adaptive immunity to both host protection and pathogenic genital scarring.
This review summarizes mouse model studies that significantly contributed to our current understanding of the protective and pathogenic immune responses taking place during genital Chlamydia infections.
INTRODUCTION
Chlamydia species cause a plethora of clinical syndromes comprising a significant public health burden worldwide. Different species and serovars of Chlamydia display tropism for different tissues underlying their respective disease phenotypes. Chlamydia pneumoniae causes an extremely common atypical pneumonia and is linked with the development of atherosclerosis and coronary artery disease (reviewed in Campbell and Kuo 2004; Porritt and Crother 2016). Certain serovars of Chlamydia trachomatis are tropic for ocular tissues, comprising the leading cause of infectious blindness worldwide (reviewed in Taylor et al. 2014). Chlamydia trachomatis serovars that display tropism for genital tissues cause the most common sexually-transmitted bacterial infections in the US (Braxton et al. 2018). Of the sexually-transmitted serovars, L serovars cause lymphogranuloma venereum (LGV), an infection of lymph nodes as well as genital and rectal tissue (reviewed in Mabey and Peeling 2002). Chlamydia trachomatis serovars D–K establish infection inside genital epithelial cells, cause pathology mostly in women, and can be asymptomatic or cause pelvic pain and mucopurulent cervical discharge. Repeated or chronic infection can lead to scarring of the upper genital tract, increasing the risk of ectopic pregnancy and infertility (reviewed in Brunham and Paavonen 2020). All these species of Chlamydia infect epithelial cells, inside which they replicate and persist (reviewed in Elwell, Mirrashidi and Engel 2016). The infected epithelium activates mechanisms of cell-autonomous immunity, undergoes cellular changes and produces proinflammatory cytokines, which recruit and activate innate immune cells such as neutrophils, macrophages, dendritic cells and potentially tissue-resident lymphoid cells (reviewed in Finethy and Coers 2016). As the infection proceeds, antigen-presenting cells activate adaptive immunity, leading to production of anti-Chlamydia antibodies by B cells and influx of Chlamydia-specific CD4 and CD8 T cells into infected tissue (reviewed in Poston and Darville 2018). The result is an inflammatory environment that contains and frequently clears the infection but also damages the infected tissue.
There has been significant controversy regarding which of these responses to Chlamydia infection mediate pathology. It was initially thought that the antigen-specific responses—mediated by T cells, Chlamydia-specific antibodies or even autoreactive antibodies resulting from molecular mimicry to Chlamydia Hsp60—resulted in tissue scarring and pathology (reviewed in Brunham and Peeling 1994). The contributions of antibodies to pathology have largely been discredited (Ness et al. 2008), with more focus placed on inflammatory signaling derived from infected epithelial cells and subsequent recruitment of innate immune cells, such as neutrophils, and induction of various T cell responses (reviewed in Darville and Hiltke 2010).
The studies regarding chlamydial pathogenesis in humans have been extremely informative in correlating certain immune responses with pathology. Mechanistic studies, however, require animal models, and while non-human primate models have occasionally been used (Bell et al. 2011), chlamydial disease has been studied mainly in mice and guinea pigs. Amongst these two models the immunological response in the guinea pig may better recapitulate that observed in humans (Rank and Sanders 1992; Rank, Bowlin and Kelly 2000), but genetic tractability and reagent availability make the mouse the most useful model to examine specific immune mechanisms on a granular level. After treatment with medroxyprogesterone to halt uterine epithelial cell shedding in estrus, Chlamydia infection can successfully be established in the murine genital tract via the intravaginal, intrauterine, or transcervical routes, depending on the Chlamydia strain (Tuffrey and Taylor-Robinson 1981; Gondek et al. 2012).
Different Chlamydia species display tropism for different host species, demonstrating evolutionary adaptation to varying host immune mechanisms. Chlamydia trachomatis mouse pneumonitis (MoPn) strain, now commonly referred to as Chlamydia muridarum, is the mouse-adapted Chlamydia species (Barron et al. 1981), subverts murine cell-autonomous immune mechanisms (Nelson et al. 2005; Coers et al. 2008), and establishes a self-resolving 4–8-week-long genital infection in mouse hosts. Human-pathogenic C. trachomatis serovars, in contrast, are rapidly cleared from the mouse female genital tract (Fig. 1) and fail to establish productive infections or induce pathogenic immune responses (Perry, Feilzer and Caldwell 1997). Likewise, C. muridarum is highly susceptible to the human cell-autonomous immunity and is therefore nonpathogenic in human hosts (Haldar et al. 2016). A primary C. muridarum genital infection is able to elicit robust genital pathology in mice. Significant inflammatory infiltrate predominated by neutrophils in the oviduct lumen (pyosalpinx) is readily detected in C. muridarum-infected mice at 14dpi and 21dpi, and resolves by 28dpi. Beginning at 28dpi and at later timepoints, glandular duct blockade causes uterine horn dilation; and tissue fibrosis and occlusion of the oviduct lumen cause oviduct dilation (hydrosalpinx) (Shah et al. 2005; Sun et al. 2015). These representations of genital scarring are associated with infertility in primary murine C. muridarum infection. Humans, on the other hand, seem to require secondary C. trachomatis infections to drive significant pathology (reviewed in Brunham and Paavonen 2020). The specific immune mechanisms that mediate human pathology, however, have not been fully elucidated. Mouse models using C. muridarum can therefore be used to implicate specific pathogenic immune mechanisms which can then be investigated further for their roles in human disease.
Figure 1.
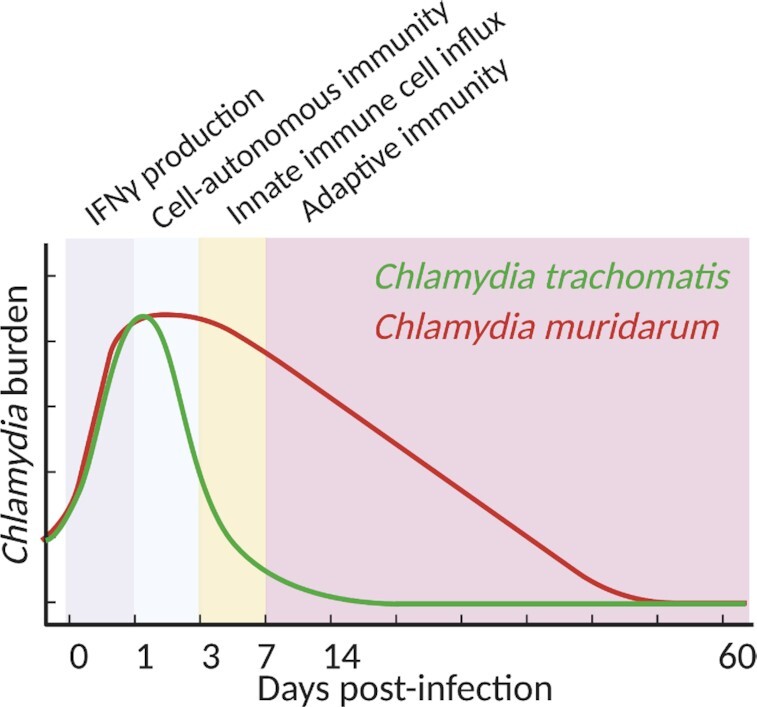
Kinetics of bacterial burden in murine genital Chlamydia infection. Prior to activation of cell-autonomous immunity, C. trachomatis and C. muridarum grow unchecked inside genital epithelial cells. Innate IFNγ production induces expression of the IRG system, resulting in destruction of C. trachomatis but not C. muridarum. CD4 T cells ultimately clear genital C. muridarum infections after 4–7 weeks. Figure created with Biorender.com.
Chlamydia infection induces an array of immune responses, beginning with the infected epithelial cell, followed by activation and recruitment of innate immune cells such as neutrophils, macrophages and potentially tissue-resident lymphoid cells within the first week after infection, leading into subsequent cellular and humoral adaptive immunity (reviewed in Finethy and Coers 2016; Poston and Darville 2018). Not all of these responses are important for clearing the infection, nor does each of them cause tissue damage and subsequent scarring. An ideal vaccine would induce immune mechanisms that specifically destroy bacteria, resulting in protective immunity, without activating immune mechanisms that may cause tissue damage and disease sequelae. In this review, we describe the components of the anti-Chlamydia immune response during primary mouse female genital tract infection, parsing the respective contributions of branches of genital immunity to bacterial clearance and immunopathology.
Murine innate immunity controls C. trachomatis but not C. muridarum genital infection
Cell-autonomous immunity to Chlamydia infection in mouse cells
Once Chlamydia enter epithelial cells, they form a membrane-bound inclusion body inside which they replicate prior to lysing or extruding from infected cells (reviewed in Hybiske and Stephens 2008). The first approximately 24 h of murine genital Chlamydia infection is marked by rapid bacterial growth as the infection proceeds unchecked (Rank, Bowlin and Kelly 2000). During this time, bacteria are sensed by various cellular pattern-recognition receptors including Toll-like receptors 2 and 3 (TLR2 and TLR3) (Darville et al. 2003; Derbigny, Kerr and Johnson 2005; Derbigny et al. 2010, 2012; Yu et al. 2016), nucleotide-binding oligomerization domain-containing protein 1 (NOD1) (Welter-Stahl et al. 2006), as well as the nucleic acid sensors cyclic GMP–AMP synthase (cGAS), and stimulator of interferon genes (STING) (Prantner, Darville and Nagarajan 2010; Barker et al. 2013; Zhang et al. 2014a, 2017). In days 1–3 after infection, bacterial sensing leads to genital production of the cytokine gamma-interferon (IFNγ; Rasmussen et al. 1997), which induces cell-autonomous defense programs in both infected and non-infected cells (reviewed in Finethy and Coers 2016). Cell-autonomous immunity to intracellular bacterial pathogens such as Chlamydia is mediated in part by the immunity-related GTPase (IRG) family of proteins (reviewed in Pilla-Moffett et al. 2016), members of which localize to Chlamydia inclusions and trigger the lytic destruction of targeted inclusions (Coers et al. 2008; Haldar et al. 2013, 2015).
IFNγ treatment of murine cells is sufficient for robust restriction of C. trachomatis growth in non-immune cells such as mouse embryonic fibroblasts (MEFs) (Bernstein-Hanley et al. 2006; Coers et al. 2008), and indeed IFNγ production in the genital tract coincides with a decrease in C. trachomatis burden after the first day post-infection (dpi). However, the mouse-adapted pathogen C. muridarum is able to subvert the IRG system and evade IFNγ-mediated restriction in non-immune cells (Coers et al. 2008) corresponding with a further increase in bacterial burden in the female genital tract through 6 dpi (Perry, Feilzer and Caldwell 1997; Coers et al. 2011; Gondek et al. 2012). For this reason, mice are readily vaginally infected with C. muridarum as it can ascend to the uterus and oviducts, whereas C. trachomatis must be instilled transcervically into the uterus in order to establish infection in IRG-competent animals.
The importance and early sources of interferon-γ
As infection proceeds, macrophage and neutrophil populations begin to expand within infected tissue (Morrison and Morrison 2000; Rank, Bowlin and Kelly 2000; Lijek et al. 2018). Both cell types are able to phagocytose and destroy extracellular bacteria, but generally play different roles in immune responses to bacterial infection. Macrophages are the primary innate immune cell type in defense against intracellular bacterial pathogens, and once activated by IFNγ possess potent antimicrobial properties (reviewed in Mosser and Zhang 2008). Indeed, IFNγ priming further stimulates the ability of mouse bone marrow derived phagocytes to clear C. trachomatis or C. muridarum infections via complex mechanisms including perforin-2 (Fields et al. 2013), components of the autophagy pathway (Radomski et al. 2017), and phagolysosomal nitric oxide production (Rajaram and Nelson 2015; Nagarajan et al. 2018). Similar to epithelial cells, macrophage cytokine responses to Chlamydia are mediated by TLR2 (Darville et al. 2003) and STING (Prantner, Darville and Nagarajan 2010). Additionally, Chlamydia infection can activate the NLRP3 and AIM2 inflammasomes, as well as the noncanonical caspase11 inflammasome, leading to pyroptosis and production of pro-inflammatory cytokines (Prantner et al. 2009; Finethy et al. 2015; Chen et al. 2017; Webster et al. 2017; Allen et al. 2019).
Macrophages can display varying ontogenies and phenotypes (reviewed in Ginhoux and Guilliams 2016) and no genetic deletion or antibody depletion studies have effectively demonstrated the in vivo importance of macrophages to restriction of Chlamydia growth. Nonetheless, the aforementioned in vitro macrophage studies suggest that they do play important roles in vivo, both directly killing extracellular Chlamydia and potentiating immune responses through production of cytokines. However, the effectiveness of macrophage immunity varies with the Chlamydia species used. Whereas innate immunity, including macrophage-mediated processes, is sufficient to sterilize murine C. trachomatis infection in vivo, C. muridarum is able to overcome innate immune clearance in mouse models. Escape from destruction inside macrophages via inhibition of phagosome–lysosome fusion (Rajaram and Nelson 2015), combined with its ability to subvert epithelial cell-autonomous immunity (reviewed in Finethy and Coers 2016), explains how C. muridarum is able to establish a successful genital infection in mice.
The anti-Chlamydia activities of both macrophages and infected epithelial cells are potentiated by IFNγ (Johansson et al. 1997). Whereas wildtype mice demonstrate decreased C. trachomatis burden from 3 to 6 dpi, C. trachomatis-infected mice lacking IFNγ or the IRG family members Irgm1 and Irgm3, or wildtype mice infected with C. muridarum, demonstrate a comparable increase in burden through 6 dpi (Gondek et al. 2012). These findings beautifully illustrate that while IFNγ in the first week after infection is sufficient and necessary to restrict C. trachomatis growth in an IRG-dependent manner, it is insufficient to clear C. muridarum infection within that timeframe.
IFNγ-producing CD4 T cells (Th1 cells) are the major producers of IFNγ in the latter stages of infection (Nagarajan et al. 2011), but more controversy surrounds the source(s) of IFNγ in the first several days after infection. There are few tissue resident immune cell types known to secrete IFNγ that have the potential to respond quickly to genital Chlamydia infection. The most obvious is the natural killer (NK) cell, the most prominent subpopulation of innate lymphoid cells (ILCs) whose functions include the induction of cytotoxicity in infected cells and the production of cytokines such as IFNγ (reviewed in Zhang and Huang 2017). NK cells are present in the genital tract early after infection, and administration of anti-NK1.1 or anti-asialo-GM1 depleting antibodies eliminate IFNγ-producing cells in the iliac lymph nodes of C. muridarum-infected mice (Tseng and Rank 1998). However, these depletion schemes also eliminate other ILC populations that may produce significant amounts of IFNγ. Notably, IFNγ-producing ILC1s are important sources of IFNγ in other mucosal tissues such as the gut, as are ‘ex-ILC3s,’ i.e. IL-17-producing non-cytotoxic ILCs that convert into IFNγ-producing ILCs (reviewed in Artis and Spits 2015; Vivier et al. 2018). While the physical presence of these alternative ILC subpopulations in the murine genital tract has not been demonstrated, mice deficient for ILCs, as achieved by genetic deletion of the IL-7 receptor or deletion of both Rag2 and the IL-2 receptor common γ-chain (γc), were unable to control early C. trachomatis growth in an IFNγ-dependent manner (Xu et al. 2020). These findings suggest that ILC1 and/or ex-ILC3 populations in the genital tract may be responsible for the production of innate IFNγ prior to Th1 cell influx, and that this innate production of IFNγ is sufficient to control early C. trachomatis growth. It is possible that other innate-like cell types, such as γδT cells, may provide a redundant early source of genital IFNγ, and additional studies are warranted to evaluate these genital ILC populations and the mechanisms of their activation during C. trachomatis infection.
Although C. muridarum is able to evade IFNγ-mediated restriction in epithelial cells and macrophages, innate IFNγ does nonetheless appear to play a pivotal role in preventing C. muridarum dissemination from the female genital tract into other tissues. Earlier studies using polyclonal stocks of the C. muridarum Nigg strain demonstrated prolonged vaginal shedding of bacteria, increased bacterial dissemination throughout the course of the infection and rare instances of lethality in vaginally infected IFNγ-deficient mice. Wildtype mice on the other hand survived infection, displayed minimal bacterial dissemination and cleared the vaginal infection within approximately 4 weeks (Cotter et al. 1997; Perry, Feilzer and Caldwell 1997). These findings were essentially confirmed in subsequent studies, with the notable distinction that the use of the more virulent C. muridarum Weiss strain (Sturdevant and Caldwell 2014) or hypervirulent plaque-purified clonal isolates of the Nigg strain (Poston et al. 2018; Mercado et al. 2020) resulted in more pervasive lethality in IFNγ-deficient mice. The systemic and sometimes lethal infection of vaginally inoculated IFNγ-deficient mice is not seen in mice lacking conventional αβ T cells (Mercado et al. 2020), suggesting that ILC-derived innate IFNγ plays an important role in preventing C. muridarum dissemination and host survival. In support of this conclusion the total elimination of ILCs in Rag2-/γc-deficient mice led to greater C. muridarum dissemination, accelerated weight loss and an earlier onset of death compared to infected Rag1-deficient mice (Mercado et al. 2020). Altogether, these findings suggest that innate IFNγ rather than Th1-derived IFNγ is responsible for limiting early C. muridarum dissemination and subsequent lethality, although Th1 cells are clearly important for ultimate clearance of the genital infection (Mercado et al. 2020). The mechanisms by which innate IFNγ prevents C. muridarum dissemination remain unclear. Additional studies are needed to parse the distinct roles of innate and Th1-derived IFNγ in defense against C. muridarum infection.
Neutrophils play a minor role in defense against Chlamydia
Apart from IFNγ-responsive macrophages, neutrophils are the other major innate immune cell type that responds early in genital Chlamydia infection. Neutrophils are present at low levels in uninfected tissues and are recruited to infected tissues from the bloodstream (reviewed in Ley et al. 2018; Petri and Sanz 2018). Similar to macrophages, neutrophils are voracious phagocytes and are able to destroy engulfed bacteria using reactive nitrogen and oxygen species. Neutrophils possess additional functions that are effective against extracellular pathogens: in a specialized form of programmed cell death, neutrophils release cytoplasmic and nuclear contents such as DNA and oxygen radicals, forming a neutrophil extracellular trap (NET) that is toxic to microbes and host tissue alike. Neutrophils therefore comprise the primary effector immune cell type in infection with extracellular pathogens (reviewed in Ley et al. 2018), with their roles in infection with an intracellular pathogen such as Chlamydia less established.
Neutrophils are present in Chlamydia-infected genital tissue as early as 24 hpi (Rank et al. 2011) and have been shown to destroy extracellular Chlamydia in vitro (Naglak, Morrison and Morrison 2017). Protective roles for neutrophils in vivo vary with the Chlamydia species used. Antibody depletion experiments have shown that neutrophils may play a role in defense against C. muridarum within the first 2 days of infection (Rank et al. 2011) but not throughout the course of infection (Lee et al. 2010; Frazer et al. 2011). For defense against C. trachomatis mouse infection, on the other hand, neutrophils appear to be dispensable (Lijek et al. 2018). Interestingly, electron micrographs of infected genital epithelia have shown neutrophils entering epithelial cells to engulf whole inclusions or extruding infected epithelial cells into the uterine lumen (Rank et al. 2011). While these observations suggest novel mechanisms for protective functions of neutrophils in defense against intracellular pathogens, their contribution to in vivo immunity has not been shown. Because C. muridarum is able to survive inside murine epithelial cells, it is quite possible that neutrophils function predominantly in the destruction of extracellular C. muridarum, thus limiting cell-to-cell spread. Neutrophils would therefore be largely dispensable in defense against C. trachomatis, which is readily destroyed by immune-primed murine epithelial cells. Alternatively, Chlamydia may have evolved mechanisms to interfere with neutrophil function or recruitment (Dolat and Valdivia 2020). Altogether, neutrophils appear to play only a minor protective role in genital Chlamydia infection.
Collectively, there are three phases of the early (pre-adaptive) immune response to genital Chlamydia infection (Fig. 1). In the first day of infection Chlamydia growth proceeds unchecked prior to activation of cell-autonomous immune defenses elicited by IFNγ priming. Unprimed infected epithelial cells sense intracellular Chlamydia via TLR2, TLR3, NOD1 and STING, secrete proinflammatory cytokines and activate an unknown cellular source—likely to be ILCs and/or γδT cells—to produce IFNγ. In response to IFNγ, the IRG system is induced in mouse epithelial cells, leading to an initial restriction of the growth of C. trachomatis but not C. muridarum. Macrophages and neutrophils then respond, killing extracellular and possibly intracellular Chlamydia and further secreting cytokines via activation of the same pattern-recognition receptors as in epithelial cells as well as inflammasomes. These functions are sufficient to clear C. trachomatis infection, but because C. muridarum is able to survive inside cytokine-primed epithelial cells as well as macrophages, C. muridarum infection persists beyond innate immunity and requires an adaptive immune response for clearance.
Adaptive immunity provides sustained IFNγ production to clear an established Chlamydia infection
Th1 cells comprise the crucial element of adaptive immune responses
While the innate response restricts Chlamydia growth in the genital tract in the first week of infection and limits dissemination to other tissues, dendritic cells in genital tissue ingest Chlamydia antigens, migrate to the draining iliac lymph nodes and present these antigens via the major histocompatibility complexes (MHC) I and II to naïve T cells (Su et al. 1998; Marks et al. 2007). The corresponding lymphocyte responses consist predominantly of Th1- and Th17-polarized CD4 T cells as well as CD8 T cells (Ramsey and Rank 1991; Igietseme et al. 1993, et al. 1994; Roan and Starnbach 2006; Roan et al. 2006; Coers et al. 2011; Gondek et al. 2012; Li and McSorley 2013; Vicetti Miguel et al. 2016), which migrate to the genital tract in a CXCR3- and CCR5-dependent manner (Olive, Gondek and Starnbach 2011; Lijek et al. 2018) to execute different effector functions. Genital CD4 and CD8 lymphocyte numbers peak at 14 dpi and remain elevated for weeks after the resolution of infection (Morrison and Morrison 2000; Gondek et al. 2012). While B cells do respond to Chlamydia infection, they are likely dispensable for clearance of primary infection from the genital tract (Ramsey, Soderberg and Rank 1988; Johansson et al. 1997), and instead produce antibodies which aid in preventing bacterial dissemination to distant tissues (Li and McSorley 2013; Poston et al. 2018; Malaviarachchi et al. 2020). Additionally, B cells have a protective role in reinfection (Su et al. 1997; reviewed in Li and McSorley 2015), possibly by production of Chlamydia-specific IgA in genital secretions (Shillova et al. 2020). However, the preponderance of evidence suggests that the primary adaptive cell type responsible for defense against primary Chlamydia infection is the Th1 CD4 T cell.
Th1 cells differentiate in the draining lymph nodes in response to the cytokine IL-12 and develop into IFNγ-secreting effector cells (reviewed in Zhu and Paul 2010). Mouse models deficient for all T cells (Rank, Soderberg and Barron 1985), αβT cells (Perry, Feilzer and Caldwell 1997), CD4, MHC class II (Morrison, Feilzer and Tumas 1995), IL-12 (Perry, Feilzer and Caldwell 1997), or IFNγ (Rank et al. 1992; Cotter et al. 1997; Johansson et al. 1997; Perry, Feilzer and Caldwell 1997; Ito and Lyons 1999; Perry et al. 1999a) all demonstrate increased susceptibility to primary C. muridarum infection. Furthermore, adoptive transfer of Chlamydia-specific Th1 cells confers protection against C. muridarum in nude recipient mice (Igietseme et al. 1993), and against C. trachomatis infection in IFNγ-deficient recipient mice (Gondek, Roan and Starnbach 2009; Gondek et al. 2012). With respect to C. trachomatis, the IFNγ produced by Th1 cells is not necessary for clearance, as mice lacking all T and B cells are able to clear C. trachomatis infection spontaneously (Sturdevant and Caldwell 2014), demonstrating that the innate sources of IFNγ are sufficient to induce cell-autonomous destruction of C. trachomatis in epithelial cells and macrophages. Interestingly, mice deficient for IRG-mediated epithelial cell-autonomous immunity are able to spontaneously clear C. trachomatis infection after a 2-week delay, correlated with an increase in Th1 responses (Coers et al. 2011) indicating that sustained IFNγ production is able to drive clearance of Chlamydia infection via macrophages, neutrophils and lymphocytes. Likewise, as C. muridarum is able to evade the IRG system and persist beyond the early innate phases of the immune response, strong Th1 responses are required to successfully clear the infection by 28–45 dpi. Indeed, in mice deficient for TCRβ C. muridarum does not disseminate to other tissues but bacterial growth and shedding in the genital tract continues unchecked at 60 dpi (Mercado et al. 2020), confirming that T cell responses are required for clearance of C. muridarum infection in the genital tract even if they are dispensable for controlling C. muridarum dissemination.
There is little evidence supporting protective roles for other populations of T cells in primary Chlamydia infection in mouse models. Th2 cells normally function in immunity to helminth infections and play important roles in allergic responses (reviewed in Zhu and Paul 2010). Th17 cells are important for defense against extracellular pathogens, as the primary Th17 cytokine IL-17 largely functions to promote neutrophil recruitment and activity in infected tissue (Li, Casanova and Puel 2018). Adoptive transfer of Chlamydia-specific Th2- or Th17-polarized cells fails to confer any level of protection against C. trachomatis (Hawkins, Rank and Kelly 2002; Gondek, Roan and Starnbach 2009). Furthermore, transfer of Th2 cells actually worsens Chlamydia infection, likely by inhibiting protective Th1 responses (Andrew et al. 2013), and increased Th2 responses in IL-12-deficient mice are associated with increased rates of hydrosalpinx (Zhang et al. 2014b). Interestingly, IL17−/− mice infected with C. muridarum demonstrate decreased and shortened bacterial shedding, correlated with decreased neutrophil and macrophage influx into the oviducts (Andrew et al. 2013). Another study showed that IL17ra−/− mice demonstrate partially impaired Th1 immunity, but wildtype levels of bacterial burden in C. muridarum infection (Scurlock et al. 2011), demonstrating an indirect role for Th17 cells in supporting protective Th1 responses. On the whole, however, the evidence demonstrates that Th17 cells, similar to neutrophils, play a minor role in protection against primary female genital Chlamydia infection.
CD8 T cells support protective immunity via production of IFNγ
CD8 T cells are important in defense against intracellular bacterial and viral infections and function to produce cytokines such as IFNγ and tumor necrosis factor α (TNFα) as well as to identify and destroy infected host cells (reviewed in Zhang and Bevan 2011). CD8 T cells are indeed induced in genital Chlamydia infection and present in the genital tract (Roan and Starnbach 2006). It is therefore surprising that they appear to be largely dispensable for defense against primary Chlamydia infection. Adoptive transfer of Chlamydia-specific CD8 T cells has shown protection against dissemination in C. trachomatis infection (Starnbach, Bevan and Lampe 1994), and either mild protection when transferred to athymic nude mice (Igietseme et al. 1994) or no protection when transferred to naive wildtype mice (Su and Caldwell 1995) in genital C. muridarum infection. A more recent study demonstrated no difference in the course of C. muridarum genital infection or dissemination to gut tissues in CD8-deficient mice compared to wildtype mice (Xie et al. 2020). While CD8 T cells are able to lyse infected cells in vitro (Beatty and Stephens 1994), their protective function in vivo appears to be associated with or dependent upon the secretion of IFNγ (Igietseme et al. 1994; Starnbach, Bevan and Lampe 1994; Lampe et al. 1998) rather than cytotoxic activity (Perry et al. 1999b). They are therefore mostly redundant to Th1 cells and play only a minor supporting role in defense against primary Chlamydia infection.
It is clear that protection against female genital Chlamydia infection in mouse models depends primarily upon IFNγ. Innate IFNγ, likely produced by ILCs and/or γδT cells, provides sufficient stimulation to infected cells to clear C. trachomatis infection and assists in preventing dissemination in C. muridarum infection. After the first week post-infection, adaptive immunity takes over the role of producing IFNγ. Th1 cells produce the majority of IFNγ with minor support from CD8 T cells. Lymphocytes persist in the genital tract for weeks after inoculation and are able to spontaneously clear C. muridarum infection in wildtype mice after 28–45 days (Fig. 1).
Determinants and mechanisms of genital pathology in Chlamydia infection
Factors underlying the differences in pathogenicity of Chlamydia strains
Elimination of the bacteria is achieved via a multifaceted immune response, yet at the same time it is also the components of the immune system that largely mediate tissue damage and pathologic sequelae. Understanding this double-edged sword of the immune response to genital Chlamydia infection is therefore crucial to design treatments and vaccines that prevent pathology. A plethora of mouse studies targeting various molecular and cellular mechanisms of disease have shed light on this issue, although much remains to be understood. Chlamydia trachomatis serovar L2 causes LGV in human hosts, and while it comprises an excellent infection model in murine hosts to study the immune mechanisms that are important in limiting bacterial growth, it does not cause pronounced genital pathology in immunocompetent mice (Lijek et al. 2018). Mouse studies examining immunopathology have therefore generally employed C. muridarum or C. trachomatis serovar D, which mirror the disease pathology observed in the human female genital tract, with genital scarring leading to adhesions, hydrosalpinx, chronic pelvic inflammation and infertility (reviewed in Darville and Hiltke 2010).
In total, two main factors underlie these strain-specific differences in pathogenicity. First, C. muridarum undergoes sustained replication in the mouse host, resulting in a productive and persistent infection, while C. trachomatis is cleared within 14 dpi. The short-lived presence of proinflammatory bacterial ligands in C. trachomatis infection may simply be insufficient to induce a robustly pathogenic immune response; indeed, in mice treated with IFNγ-depleting antibodies, C. trachomatis serovar L2 establishes a prolonged high-burden infection and pathology ensues (Zhong et al. 1989). Furthermore, mice deficient for Chlamydia sensor TLR2 (Darville et al. 2003) or LPS sensor and inflammasome component caspase11 (Allen et al.2019) both demonstrate equivalent C. muridarum growth compared to wildtype mice but develop significantly less genital pathology. These findings suggest that the presence of bacterial ligands, regardless of the strain, drives pathogenic inflammation.
Second, there may be bacterial strain-specific factors that directly mediate tissue pathology independent of bacterial growth. While the genomes of human and rodent Chlamydia species share approximately 99% synteny, stark genomic diversity is found in a 50 kb ‘plasticity zone’ within the Chlamydia chromosome (Read et al. 2000) that has been the focus of much study. Within the plasticity zone, C. muridarum and C. trachomatis serovar D harbor genes encoding putative cytotoxins that bear resemblance to Clostridium toxin B. In contrast to C. muridarum and C. trachomatis serovar D, C. trachomatis serovar L2 does not express a functional cytotoxin (Belland et al. 2001; Carlson et al. 2004; Thalmann et al. 2010). Analysis of C. muridarum plasticity zone mutants demonstrated wildtype bacterial growth with decreased genital pathology (Rajaram et al. 2015), suggesting that the putative cytotoxin may underlie strain-specific differences in pathogenicity. Additionally, C. muridarum and many strains of C. trachomatis harbor a small plasmid that is associated with virulence in primate ocular models (Kari et al. 2011) and murine genital models (O'Connell et al. 2007; Frazer et al. 2011; Lei et al. 2014; Sigar et al. 2014). The plasmid contains the virulence factor Pgp3 that inhibits bacterial killing by cationic antimicrobial peptides (Hou et al. 2015, 2019), is essential for the establishment of persistence (Yang et al. 2020), comprises an immunodominant antigen in infected women (Chen et al. 2010a), and is required for C. muridarum-induced genital pathology in mice (Lei et al. 2014; Liu et al. 2014; Yang et al. 2020). Altogether, the capacity of a Chlamydia strain to induce genital pathology appears to depend upon its ability to persist in genital tissue and its intrinsic ability to induce tissue damage based on the presence of one or more putative toxins.
Neutrophils, Th17 cells and CD8 T cells are pivotal drivers of genital pathology
The combination of bacterial ligands, acting through immune recognition by TLR2, inflammasomes and possibly other sensors such as STING, as well as toxin-mediated tissue damage induce inflammatory responses that act as the main drivers of genital pathology in Chlamydia infection. A study comparing C. muridarum infection in 11 mouse strains revealed varying susceptibilities to hydrosalpinx that did not correlate strain-specific variation in bacterial burden (Chen et al. 2014), demonstrating that host-mediated responses to infection drive pathology. The various components of the immune response to genital Chlamydia infection execute their antimicrobial functions with distinct mechanisms that each have a different potential for collateral damage to host tissue. Genetic deletion or antibody-blocking and -depleting experiments have examined the relative contribution to pathology of many immune signaling molecules and cell types (Table 1).
Table 1.
Host factors implicated in pathogenesis in murine Chlamydia infection. Comprehensive summary of in vivo Chlamydia infection models that examined inflammation and pathology. Listed are the target host factors; a brief description of the model design; the Chlamydia strain and substrain used (when available); the relevant phenotypes of the listed model and the reference for the experiments.
| Host factor | Model design | Chlamydia strain | Phenotype (compared to wt) | Citation |
|---|---|---|---|---|
| Cytokines | ||||
| IL-1 | IL1R −/− | C. muridarum (Nigg) | Increased bacterial burden, decreased pathology | Nagarajan et al. (2012) |
| IL1Ra −/− (IL-1R antagonist) | C. muridarum (Nigg) | Decreased burden, enhanced pathology | Nagarajan et al. (2012) | |
| IL1A −/− | C. muridarum (Nigg) | Decreased pathology | Gyorke et al. (2020) | |
| IL1B −/− | C. muridarum (Nigg) | Decreased pathology | Prantner et al. (2009); Gyorke et al. (2020) | |
| IL-6 | IL6 −/− | C. muridarum (Nigg clone CMG13.32.1) | Increased burden, decreased pathology | Sun et al. (2017) |
| IL-17 | IL17 −/− | C. muridarum (Weiss) | Decreased burden, decreased pathology | Andrew et al. (2013) |
| IL-12 | IL12p35 −/−, IL12p40−/− | C. muridarum (Nigg) | Increased pathology | Chen et al. (2013) |
| Type I IFN | IFNAR −/− | C. muridarum (Nigg) | Decreased burden, decreased pathology | Nagarajan et al. (2008) |
| C5 | C5 −/− | C. muridarum (Nigg) | Decreased pathology | Yang et al. (2014) |
| TNFα | TNFA −/- | C. muridarum (Nigg) | Decreased pathology | Murthy et al. (2011) |
| TNFAR1 −/- | C. muridarum (Nigg) | Decreased pathology | Dong et al. (2014) | |
| IFNγ | IFNG −/− | C. muridarum (Nigg 1942) | Increased pathology | Murthy et al. (2007); Scurlock et al. (2011) |
| Signaling molecules | ||||
| MyD88 | MyD88 −/− | C. muridarum (Nigg) | Increased uterine inflammation and hydrosalpinx | Chen et al. (2010b); Nagarajan et al. (2011) |
| STING | STING gt/gt | C. muridarum (Nigg) | Increased incidence of bilateral hydrosalpinx | Zhang et al. (2017) |
| TLR2 | TLR2 −/− | C. muridarum (Nigg) | Decreased pathology | Darville et al. (2003) |
| TLR3 | TLR3 −/− | C. muridarum (Nigg) | Increased burden, increased pathology | Carrasco et al. (2018) |
| CD28 | CD28 −/−, CD80/86−/− | C. muridarum (Nigg) | Decreased cytokine production and pathology | Chen et al. (2009) |
| CD40 | CD40 −/−, CD40L−/− | C. muridarum (Nigg) | Decreased IFNγ production, increased burden | |
| Inflammasomes | ||||
| Caspase 11 | Casp11 −/− | C. muridarum (Nigg) | Increased burden, decreased pathology | Allen et al. (2019) |
| Casp1/Casp11−/− | C. muridarum | Decreased pathology | Cheng et al. (2008) | |
| Casp1/Casp11−/− | C. trachomatis L2 | Increased fertility in infected mice | Igietseme et al. (2013) | |
| Pan-Caspase inhibitor | C. trachomatis L2 | Increased fertility in infected mice | Igietseme et al. (2013) | |
| Cell effector molecules | ||||
| MMPs | MMP chemical inhibitor | C. muridarum | Decreased ascension to upper genital tract, decreased pathology | Imtiaz et al. (2006) |
| MMP9 | MMP9 −/− | C. muridarum (Weiss) | Decreased ascension to upper genital tract, decreased pathology | Imtiaz et al. (2007) |
| iNOS (RNS) | NOS2 −/− | C. muridarum (Weiss) | Increased pathology | Ramsey et al. (2001a) |
| Phagocyte oxidase | P47phox -/− | C. muridarum (Weiss—Ramsey et al.; Nigg3 clone CMG0.1.1–Dai et al.) | Decreased pathology | Ramsey et al. (2001b); Dai et al. (2016a) |
| Perforin | Perforin −/− | C. muridarum (Nigg) | Decreased pathology | Murthy et al. (2011) |
| TAP1 | TAP1 −/− | C. muridarum (Nigg) | Decreased pathology | Murthy et al. (2011) |
| Chemokine receptors | ||||
| CXCR2 | CXCR2 −/− | C. muridarum (Weiss) | Decreased pathology | Lee et al. (2010) |
| CXCR3 | CXCR3 −/− | C. muridarum (Nigg) | Delayed bacterial clearance, decreased pathology | Jiang et al. (2017) |
| Anti-CXCR3 antibody depletion | C. trachomatis D | Decreased T cell infiltrate, decreased pathology | Xie et al. (2003) | |
| CCR7 | CCR7 −/− | C. muridarum (Weiss) | Decreased burden, decreased pathology | Li et al. (2017) |
| Cell types | ||||
| Neutrophils | Anti-GR1 antibody depletion | C. trachomatis D | Decreased neutrophil and monocyte infiltrate, decreased pathology | Lijek et al. (2018) |
| Anti-Ly6G antibody depletion | C. trachomatis D | Decreased neutrophil infiltrate, decreased pathology | Lijek et al. (2018) | |
| Mcl1-LysM-Cre (absence of mature neutrophils) | C. muridarum (03DC39) | Increased burden, decreased pathology | Zortel et al. (2018) | |
| CD8+ T cells | CD8 −/− | C. muridarum (Nigg) | Decreased pathology | Murthy et al. (2011) |
| Anti-CD8 antibody depletion | C. muridarum (Nigg—Murthy et al.) (Nigg3 clone G13.32.1–Yu et al.) | Decreased pathology | Murthy et al. (2011); Yu et al. (2019) | |
| OT-I (ovalbumin-specific CD8 T cells) | C. muridarum (Nigg) | Decreased pathology | Manam, Nicholson and Murthy (2013); Vlcek et al. (2016) | |
| CD8-derived TNFα | Adoptive transfer of TNFA−/− CD8 T cells | C. muridarum (Nigg) | Decreased pathology compared to transfer of wt CD8+ T cells | Murthy et al. (2011) |
| CD4+ T cells | Anti-CD4 antibody depletion in CD8−/− mice | C. muridarum (Nigg3 clone G13.32.1) | Decreased pathology | Yu et al. (2019) |
| Regulatory T cells | Anti-CD25 antibody depletion | C. muridarum | Decreased pathology | Moore-Connors et al. (2013) |
Relative to other components of innate immunity, neutrophils are by far the most implicated in inducing immunopathology. Chlamydia muridarum strain-specific induction of neutrophils (Frazer et al. 2011) as well as mouse strain-specific duration of early pyosalpinx (Zhang et al. 2014a) are associated with long-lasting hydrosalpinx. Depletion of neutrophils results in decreased pathology in genital infection with C. trachomatis (Lijek et al. 2018) or C. muridarum (Lee et al. 2010; Zortel et al. 2018). Mice deficient for certain neutrophil effectors such as MMP9 (Imtiaz et al. 2007) and phagocyte oxidase (Ramsey et al. 2001a; Dai et al. 2016b), as well as signaling mediated by neutrophil-derived cytokines such as TNFα (Murthy et al. 2011; Dong et al. 2014) and IL-1 (Nagarajan et al. 2012), all demonstrated reduced pathology in C. muridarum infection. IL-1 is produced in two forms: IL-1α is produced by epithelial and hematopoietic cells in Chlamydia-infected genital tracts (Gyorke et al. 2020) and IL-1β is produced by hematopoietic cells such as macrophages and neutrophils upon inflammasome activation (Prantner et al. 2009). Both forms of IL-1 act on the IL-1 receptor and function to potentiate pro-inflammatory cytokine programs that result in further recruitment of neutrophils and tissue damage (reviewed in Dinarello 2018). Indeed, mice lacking IL-1α, IL-1β or the IL-1 receptor demonstrate decreased neutrophil recruitment and genital scarring upon genital C. muridarum infection, with minimal effects on bacterial burden (Prantner et al. 2009; Nagarajan et al. 2012; Gyorke et al. 2020).
Neutrophils are unlikely to be the sole contributor to genital pathology, as pathology was only partially reduced in all of these studies. Macrophages, which have a prominent role in host-protective immunity, produce MMPs, TNFα (Kol et al. 1998) and IL-1β (Finethy et al. 2015; Allen et al. 2019) in Chlamydia infection, and therefore also have the potential to cause tissue damage. Further studies are warranted to identify the protective and pathogenic contributions of specific macrophage populations in genital Chlamydia infection.
Just as the various subsets of CD4 T cells play different roles in clearance of Chlamydia infection, they also likely make different contributions to pathogenesis. Th1 cells, through their production of IFNγ, limit genital bacterial growth and are essential for clearance of C. muridarum infection. Th1 cells therefore play an almost exclusively host-protective role. Th17 cells, on the other hand, appear to play a more pathogenic role. This should not be surprising, as their signature cytokine, IL-17, functions mainly through neutrophils, which are largely pathogenic. Indeed, IL17RA−/− mice displayed decreased neutrophil influx in C. muridarum-infected genital tracts (Scurlock et al. 2011), and Th17-mediated immunity was correlated with increased genital pathology in a reinfection model using C. trachomatis serovar D (Vicetti Miguel et al. 2016). However, IL17−/− mice infected with C. muridarum demonstrated no difference in C. muridarum bacterial burden but increased genital pathology at 35 dpi (Andrew et al. 2013), suggesting a protective role for IL-17. It has been proffered that Th17 cells support Th1 responses (Scurlock et al. 2011), and further studies are warranted to better characterize the roles of Th17 cells in Chlamydia infection outside of their role in neutrophil recruitment.
Whereas CD8 T cells play a modest and largely redundant protective role in Chlamydia infection via their production of IFNγ, a plethora of studies have demonstrated their potent pathogenicity. Both genetic deficiency of CD8 or antibody-mediated depletion of CD8 T cells in wildtype mice reduced genital pathology in C. muridarum infection. Adoptive transfer of wildtype CD8 T cells into CD8−/− mice restored pathology (Murthy et al. 2011). Chlamydia muridarum infection in OT-I transgenic mice, in which all CD8 T cells are specific for ovalbumin, induced no pathology (Manam, Nicholson and Murthy 2013), yet adoptive transfer of wildtype CD8 T cells again restored pathology in these mice (Vlcek et al. 2016), implying that Chlamydia-specific CD8 T cells are required for pathology. Additional studies indicated that CD8 T cells both produce and respond to TNFα during the immunopathogenesis of genital C. muridarum infections. Whereas TNFA−/− mice displayed no pathology in C. muridarum infection, transfer of wildtype CD8 T cells restored pathology; further, TNFA−/− CD8 T cells did not restore pathology in CD8−/− recipient mice (Murthy et al. 2011), indicating that production of TNFα by CD8 T cell is important for pathogenesis. Notably, CD8 T cells were required to express TNFR2 (tumor necrosis factor receptor 2), in order to promote pathology in C. muridarum-infected mice. Because TNFα-producing, Chlamydia-specific wildtype CD8 T cells were unable to restore pathology in TNFR1−/− mice (Manam et al. 2015), these data in aggregate suggest that TNFR2 signaling in CD8 T cells and TNFR1 signaling in other cell types are responsible for TNFα-mediated pathology associated with C. muridarum infections. Interestingly, depletion of CD8+ T cells from OT-I mice resulted in an increase in pathology in C. muridarum infection, and adoptive transfer of OT-I CD8+ T cells suppressed pathology in recipient wildtype or CD8−/− mice (Xie et al. 2020). These findings suggest that, while Chlamydia-specific CD8+ T cells certainly promote pathogenesis, non-specific CD8+ T cells may paradoxically suppress pathology. Further studies are necessary to elucidate the underlying mechanisms.
Intestinal colonization promotes immunopathogenesis of genital Chlamydia infection
While components of both innate and adaptive immunity can contribute to tissue damage and subsequent scarring, recent studies have implicated immune reactions in distant tissues in genital pathogenesis. Chlamydia muridarum and C. trachomatis serovars D–K—the serovars most responsible for genital pathology in their respective host species—are not known to cause gastrointestinal illness in immunocompetent mice or humans, but can colonize the gut in both species (Yeruva et al. 2013; Gratrix et al. 2014, 2015, 2016; Peters et al. 2014; Craig et al. 2015; Musil et al. 2016). Indeed, C. muridarum inoculated via the genital tract can spread hematogenously to the mouse gut (Zhang et al. 2015; Dai et al. 2016a; Wang et al. 2016). Recent studies have demonstrated that genital infection with an attenuated C. muridarum mutant deficient for gut colonization is unable to induce hydrosalpinx unless wildtype C. muridarum is inoculated intragastrically (Tian et al. 2020). Since intragastric inoculation of wildtype C. muridarum does not result in dissemination to the genital tract or genital pathology on its own (Wang et al. 2016), these findings suggest that some component of the immune system is educated in the gut to generate a pathogenic immune response in the Chlamydia-infected genital tract, giving rise to a ‘two-hit’ model for Chlamydial pathogenesis (Zhong 2018). The nature of this mysterious gut-genital immune axis and its relevance to human disease remains to be fully elucidated.
Relevance to human infection
Genetic variants in specific immune pathways identified in humans, as well as observed immune responses, have been shown to correlate with either protection from infection or susceptibility to pathology, and the conclusions from these human cohort studies largely agree with the findings from more tractable mouse models. For example, polymorphisms in bacteria-sensing pattern-recognition receptors correlate with increased rates of tubal factor infertility in women with prior C. trachomatis exposure (den Hartog et al. 2006), cervical cell production of IFNγ and IL-12 positively correlates with fertility in C. trachomatis seropositive women, and production of IL-1β, IL-6 and IL-8 correlates with infertility (Agrawal et al. 2009). Moreover, cervical mucus neutrophils (Peipert et al. 1996) and markers of neutrophil activation correlate with endometritis (Wiesenfeld et al. 2002) in C. trachomatis-infected women. Unsurprisingly, robust T cell responses to Chlamydia antigens correlate with tubal factor infertility in women with prior C. trachomatis infection (Kinnunen et al. 2003; Tiitinen et al. 2006; Srivastava et al. 2008). However, strong IFNγ responses in T cells stimulated with Chlamydia antigens correlate with protection against C. trachomatis infection (Cohen et al. 2005), and conversely weak IFNγ T cell responses correlate with increased susceptibility (Debattista et al. 2002). Polymorphisms in TNFA are correlated with increased rates of tubal factor infertility in women who had been infected with C. trachomatis (Ohman et al. 2009). Finally, polymorphisms in HLA-A31, an HLA allele corresponding to CD8 T cell activation, are also correlated with C. trachomatis pelvic inflammatory disease (Kimani et al. 1996). These correlative studies have demonstrated that, for the most part, the different mechanisms of the human immune response to genital Chlamydia infection play similarly protective or pathogenic roles as in the mouse system.
CONCLUSION
The inflammatory response to genital Chlamydia infection is truly a double-edged sword: it is required for clearance of the infection but also induces tissue pathology that can cause infertility. The wealth of studies targeting individual components of the immune response have demonstrated that IFNγ-mediated responses, including cell-autonomous immunity and Th1 cells, are protective, whereas neutrophils, Th17 cells and CD8 T cells are largely dispensable for immunity and instead contribute to tissue pathology. After disruption of the epithelial barrier due to neutrophil-derived MMPs or reactive oxygen species, or CD8 T cell-derived TNFα, wound-healing fibrogenic responses are required to repair the damage, resulting in scarred genital tissue and disease sequelae (Fig. 2). While the murine host demonstrates slightly different immune characteristics compared to humans, and while human Chlamydia species have undoubtedly evolved adaptions specifically tailored for colonization of their natural host, mouse models of infection are nonetheless highly informative, shaping our understanding of the mammalian immune response to Chlamydia infections and providing insights for the development of human vaccines.
Figure 2.

Course of immune responses during murine genital Chlamydia infection. (A) Infectious elementary bodies (EBs) ascend through the cervix into the uterine horns and potentially to the oviducts and ovaries. EBs enter epithelial cells, establish membrane-bound inclusion bodies, and differentiate into replicative reticular bodies. (B) Bacteria are sensed by epithelial cell pattern recognition receptors, and epithelial cells produce cytokines and chemokines, including IL-1α, IL-6 and IL-8, to activate nearby immune cells. (C) Chlamydia extrude inclusions from infected cells or lyse infected cells, as bacteria spread to neighboring cells. (D) Tissue-resident immune cells (such as ILCs or γδT cells) produce IFNγ, which activates epithelial cell-autonomous immunity. C. trachomatis is effectively cleared by cell-autonomous immunity, but C. muridarum infection persists. (E) Innate immune cells such as macrophages and neutrophils accumulate in the genital tract, engulfing and destroying extracellular bacteria. Neutrophils comprise the most prominent source of matrix metalloproteases (MMPs) and reactive oxygen species, which contribute to tissue damage. (F) Chlamydia-specific lymphocytes traffic to the genital mucosa. Th1 cells sustain production of IFNγ, driving clearance of Chlamydia infection. Th17 cells produce IL-17, strengthening pathogenic neutrophil responses. CD8+ T cells produce TNFα, further driving tissue damage. (G) Wound-healing responses repair damaged tissue, resulting in pathogenic fibrosis and genital scarring. Figure created with Biorender.com.
Contributor Information
Jacob Dockterman, Department of Immunology, Duke University Medical Center, Durham, NC 22710, USA.
Jörn Coers, Department of Immunology, Duke University Medical Center, Durham, NC 22710, USA; Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, NC 22710, USA.
Conflicts of Interest
None declared.
REFERENCES
- Agrawal T, Vats V, Wallace PKet al. Recruitment of myeloid and plasmacytoid dendritic cells in cervical mucosa during Chlamydia trachomatis infection. Clin Microbiol Infect. 2009;15:50–9. [DOI] [PubMed] [Google Scholar]
- Allen J, Gyorke CE, Tripathy MK, et al. Caspase-11 contributes to oviduct pathology during genital Chlamydia infection in mice. Infect Immun. 2019;87. DOI: 10.1128/IAI.00262-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew DW, Cochrane M, Schripsema JHet al. The duration of Chlamydia muridarum genital tract infection and associated chronic pathological changes are reduced in IL-17 knockout mice but protection is not increased further by immunization. PLoS One. 2013;8:e76664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. [DOI] [PubMed] [Google Scholar]
- Barker JR, Koestler BJ, Carpenter VKet al. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio. 2013;4:e00018–00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron AL, White HJ, Rank RGet al. A new animal model for the study of Chlamydia trachomatis genital infections: infection of mice with the agent of mouse pneumonitis. J Infect Dis. 1981;143:63–6. [DOI] [PubMed] [Google Scholar]
- Beatty PR, Stephens RS. CD8+ T lymphocyte-mediated lysis of Chlamydia-infected L cells using an endogenous antigen pathway. J Immunol. 1994;153:4588–95. [PubMed] [Google Scholar]
- Belland RJ, Scidmore MA, Crane DDet al. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc Natl Acad Sci U S A. 2001;98:13984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JD, Bergin IL, Schmidt Ket al. Nonhuman primate models used to study pelvic inflammatory disease caused by Chlamydia trachomatis. Infect Dis Obstet Gynecol. 2011;2011:675360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein-Hanley I, Coers J, Balsara ZRet al. The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci U S A. 2006;103:14092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braxton J, Davis DW, Emerson Bet al. Sexually Transmitted Disease Surveillance 2017, Centers for Disease Control and Prevention; 2018. DOI: 10.15620/cdc.59237. [Google Scholar]
- Brunham RC, Paavonen J. Reproductive system infections in women: upper genital tract, fetal, neonatal and infant syndromes. Pathog Dis. 2020;78. [DOI] [PubMed] [Google Scholar]
- Brunham RC, Peeling RW. Chlamydia trachomatis antigens: role in immunity and pathogenesis. Infect Agents Dis. 1994;3:218–33. [PubMed] [Google Scholar]
- Campbell LA, Kuo CC. Chlamydia pneumoniae–an infectious risk factor for atherosclerosis?. Nat Rev Microbiol. 2004;2:23–32. [DOI] [PubMed] [Google Scholar]
- Carlson JH, Hughes S, Hogan Det al. Polymorphisms in the Chlamydia trachomatis cytotoxin locus associated with ocular and genital isolates. Infect Immun. 2004;72:7063–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco SE, Hu S, Imai DMet al. Toll-like receptor 3 (TLR3) promotes the resolution of Chlamydia muridarum genital tract infection in congenic C57BL/6N mice. PLoS One. 2018;13:e0195165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Lei L, Lu Cet al. Characterization of Pgp3, a Chlamydia trachomatis plasmid-encoded immunodominant antigen. J Bacteriol. 2010a;192:6017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Shivshankar P, Li Zet al. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect Immun. 2008;76:515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang H, Zhou Zet al. Chlamydial induction of hydrosalpinx in 11 strains of mice reveals multiple host mechanisms for preventing upper genital tract pathology. PLoS One. 2014;9:e95076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Cheng W, Shivshankar Pet al. Distinct roles of CD28- and CD40 ligand-mediated costimulation in the development of protective immunity and pathology during Chlamydiamuridarum urogenital infection in mice. Infect Immun. 2009;77:3080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Lei L, Chang Xet al. Mice deficient in MyD88 develop a Th2-dominant response and severe pathology in the upper genital tract following Chlamydia muridarum infection. J Immunol. 2010b;184:2602–10. [DOI] [PubMed] [Google Scholar]
- Chen L, Lei L, Zhou Zet al. Contribution of interleukin-12 p35 (IL-12p35) and IL-12p40 to protective immunity and pathology in mice infected with Chlamydia muridarum. Infect Immun. 2013;81:2962–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu X, Yu Xet al. Chlamydia muridarum Infection of macrophages stimulates IL-1beta secretion and cell death via activation of Caspase-1 in an RIP3-independent manner. Biomed Res Int. 2017;2017:1592365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coers J, Bernstein-Hanley I, Grotsky Det al. Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol. 2008;180:6237–45. [DOI] [PubMed] [Google Scholar]
- Coers J, Gondek DC, Olive AJet al. Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog. 2011;7:e1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CR, Koochesfahani KM, Meier ASet al. Immunoepidemiologic profile of Chlamydia trachomatis infection: importance of heat-shock protein 60 and interferon- gamma. J Infect Dis. 2005;192:591–9. [DOI] [PubMed] [Google Scholar]
- Cotter TW, Ramsey KH, Miranpuri GSet al. Dissemination of Chlamydia trachomatis chronic genital tract infection in gamma interferon gene knockout mice. Infect Immun. 1997;65:2145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AP, Kong FY, Yeruva Let al. Is it time to switch to doxycycline from azithromycin for treating genital chlamydial infections in women? Modelling the impact of autoinoculation from the gastrointestinal tract to the genital tract. BMC Infect Dis. 2015;15:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Tang L, Chen Jet al. The p47phox deficiency significantly attenuates the pathogenicity of Chlamydia muridarum in the mouse oviduct but not uterine tissues. Microbes Infect. 2016a;18:190–8. [DOI] [PubMed] [Google Scholar]
- Dai J, Zhang T, Wang Let al. Intravenous inoculation with Chlamydia muridarum leads to a long-lasting infection restricted to the gastrointestinal tract. Infect Immun. 2016b;84:2382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darville T, Hiltke TJ. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis. 2010;201 Suppl 2:S114–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darville T, O'Neill JM, Andrews CW Jr.et al. Toll-like receptor-2, but not Toll-like receptor-4, is essential for development of oviduct pathology in chlamydial genital tract infection. J Immunol. 2003;171:6187–97. [DOI] [PubMed] [Google Scholar]
- Debattista J, Timms P, Allan Jet al. Reduced levels of gamma-interferon secretion in response to chlamydial 60 kDa heat shock protein amongst women with pelvic inflammatory disease and a history of repeated Chlamydia trachomatis infections. Immunol Lett. 2002;81:205–10. [DOI] [PubMed] [Google Scholar]
- den Hartog JE, Ouburg S, Land JAet al. Do host genetic traits in the bacterial sensing system play a role in the development of Chlamydia trachomatis-associated tubal pathology in subfertile women?. BMC Infect Dis. 2006;6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbigny WA, Johnson RM, Toomey KSet al. The Chlamydia muridarum-induced IFN-beta response is TLR3-dependent in murine oviduct epithelial cells. J Immunol. 2010;185:6689–97. [DOI] [PubMed] [Google Scholar]
- Derbigny WA, Kerr MS, Johnson RM. Pattern recognition molecules activated by Chlamydia muridarum infection of cloned murine oviduct epithelial cell lines. J Immunol. 2005;175:6065–75. [DOI] [PubMed] [Google Scholar]
- Derbigny WA, Shobe LR, Kamran JCet al. Identifying a role for Toll-like receptor 3 in the innate immune response to Chlamydia muridarum infection in murine oviduct epithelial cells. Infect Immun. 2012;80:254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281:8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolat L, Valdivia RH. An endometrial organoid model of Chlamydia-epithelial and immune cell interactions. bioRxiv. 2020. DOI: 2020.2007.2029.226969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Liu Y, Chang Xet al. Signaling via tumor necrosis factor receptor 1 but not Toll-like receptor 2 contributes significantly to hydrosalpinx development following Chlamydia muridarum infection. Infect Immun. 2014;82:1833–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell C, Mirrashidi K, Engel J. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol. 2016;14:385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields KA, McCormack R, de Armas LRet al. Perforin-2 restricts growth of Chlamydia trachomatis in macrophages. Infect Immun. 2013;81:3045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Coers J. Sensing the enemy, containing the threat: cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol Rev. 2016. DOI: 10.1093/femsre/fuw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Jorgensen I, Haldar AKet al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect Immun. 2015. DOI: 10.1128/IAI.00856-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer LC, O'Connell CM, Andrews CW Jr.et al. Enhanced neutrophil longevity and recruitment contribute to the severity of oviduct pathology during Chlamydia muridarum infection. Infect Immun. 2011;79:4029–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44:439–49. [DOI] [PubMed] [Google Scholar]
- Gondek DC, Olive AJ, Stary Get al. CD4+ T cells are necessary and sufficient to confer protection against Chlamydia trachomatis infection in the murine upper genital tract. J Immunol. 2012;189:2441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek DC, Roan NR, Starnbach MN. T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol. 2009;183:1313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratrix J, Brandley J, Dane Met al. A retrospective review of treatment failures using Azithromycin and Doxycycline in the treatment of rectal Chlamydia infections in women and men who have sex with men. Sex Transm Dis. 2016;43:110–2. [DOI] [PubMed] [Google Scholar]
- Gratrix J, Singh AE, Bergman Jet al. Evidence for increased Chlamydia case finding after the introduction of rectal screening among women attending 2 Canadian sexually transmitted infection clinics. Clin Infect Dis. 2015;60:398–404. [DOI] [PubMed] [Google Scholar]
- Gratrix J, Singh AE, Bergman Jet al. Prevalence and characteristics of rectal chlamydia and gonorrhea cases among men who have sex with men after the introduction of nucleic acid amplification test screening at 2 Canadian sexually transmitted infection clinics. Sex Transm Dis. 2014;41:589–91. [DOI] [PubMed] [Google Scholar]
- Gyorke CE, Kollipara A, Allen Jtet al. IL-1alpha is essential for oviduct pathology during genital Chlamydial infection in mice. J Immunol. 2020, 205, 3037–49. [DOI] [PubMed] [Google Scholar]
- Haldar AK, Foltz C, Finethy Ret al. Ubiquitin systems mark pathogen-containing vacuoles as targets for host defense by guanylate binding proteins. Proc Natl Acad Sci U S A. 2015;112:E5628–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar AK, Piro AS, Finethy Ret al. Chlamydia trachomatis is resistant to inclusion ubiquitination and associated host defense in gamma interferon-primed human epithelial cells. mBio. 2016;7. DOI: 10.1128/mBio.01417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar AK, Saka HA, Piro ASet al. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog. 2013;9:e1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RA, Rank RG, Kelly KA. A Chlamydia trachomatis-specific Th2 clone does not provide protection against a genital infection and displays reduced trafficking to the infected genital mucosa. Infect Immun. 2002;70:5132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Dong X, Yang Zet al. Chlamydial plasmid-encoded virulence factor Pgp3 neutralizes the antichlamydial activity of human cathelicidin LL-37. Infect Immun. 2015;83:4701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Sun X, Dong Xet al. Chlamydial plasmid-encoded virulence factor Pgp3 interacts with human cathelicidin peptide LL-37 to modulate immune response. Microbes Infect. 2019;21:50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Exit strategies of intracellular pathogens. Nat Rev Microbiol. 2008;6:99–110. [DOI] [PubMed] [Google Scholar]
- Igietseme JU, Magee DM, Williams DMet al. Role for CD8+ T cells in antichlamydial immunity defined by Chlamydia-specific T-lymphocyte clones. Infect Immun. 1994;62:5195–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igietseme JU, Omosun Y, Partin Jet al. Prevention of Chlamydia-induced infertility by inhibition of local caspase activity. J Infect Dis. 2013;207:1095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igietseme JU, Ramsey KH, Magee DMet al. Resolution of murine chlamydial genital infection by the adoptive transfer of a biovar-specific, Th1 lymphocyte clone. Reg Immunol. 1993;5:317–24. [PubMed] [Google Scholar]
- Imtiaz MT, Distelhorst JT, Schripsema JHet al. A role for matrix metalloproteinase-9 in pathogenesis of urogenital Chlamydia muridarum infection in mice. Microbes Infect. 2007;9:1561–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz MT, Schripsema JH, Sigar IMet al. Inhibition of matrix metalloproteinases protects mice from ascending infection and chronic disease manifestations resulting from urogenital Chlamydia muridarum infection. Infect Immun. 2006;74:5513–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito JI, Lyons JM. Role of gamma interferon in controlling murine chlamydial genital tract infection. Infect Immun. 1999;67:5518–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Maxion H, Champion CIet al. Expression of CXCR3 on adaptive and innate immune cells contributes oviduct pathology throughout Chlamydia muridarum Infection. J Mucosal Immunol Res. 2017;1:104. [PMC free article] [PubMed] [Google Scholar]
- Johansson M, Schon K, Ward Met al. Genital tract infection with Chlamydia trachomatis fails to induce protective immunity in gamma interferon receptor-deficient mice despite a strong local immunoglobulin A response. Infect Immun. 1997;65:1032–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari L, Whitmire WM, Olivares-Zavaleta Net al. A live-attenuated chlamydial vaccine protects against trachoma in nonhuman primates. J Exp Med. 2011;208:2217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimani J, Maclean IW, Bwayo JJet al. Risk factors for Chlamydia trachomatis pelvic inflammatory disease among sex workers in Nairobi, Kenya. J Infect Dis. 1996;173:1437–44. [DOI] [PubMed] [Google Scholar]
- Kinnunen A, Surcel HM, Halttunen Met al. Chlamydia trachomatis heat shock protein-60 induced interferon-gamma and interleukin-10 production in infertile women. Clin Exp Immunol. 2003;131:299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol A, Sukhova GK, Lichtman AHet al. Chlamydial heat shock protein 60 localizes in human atheroma and regulates macrophage tumor necrosis factor-alpha and matrix metalloproteinase expression. Circulation. 1998;98:300–7. [DOI] [PubMed] [Google Scholar]
- Lampe MF, Wilson CB, Bevan MJet al. Gamma interferon production by cytotoxic T lymphocytes is required for resolution of Chlamydia trachomatis infection. Infect Immun. 1998;66:5457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Schripsema JH, Sigar IMet al. A role for CXC chemokine receptor-2 in the pathogenesis of urogenital Chlamydia muridarum infection in mice. FEMS Immunol Med Microbiol. 2010;60:49–56. [DOI] [PubMed] [Google Scholar]
- Lei L, Chen J, Hou Set al. Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect Immun. 2014;82:983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Hoffman HM, Kubes Pet al. Neutrophils: new insights and open questions. Sci Immunol. 2018;3. DOI: 10.1126/sciimmunol.aat4579. [DOI] [PubMed] [Google Scholar]
- Li J, Casanova JL, Puel A. Mucocutaneous IL-17 immunity in mice and humans: host defense vs. excessive inflammation. Mucosal Immunol. 2018;11:581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijek RS, Helble JD, Olive AJet al. Pathology after Chlamydia trachomatis infection is driven by nonprotective immune cells that are distinct from protective populations. Proc Natl Acad Sci U S A. 2018. DOI: 10.1073/pnas.1711356115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LX, Labuda JC, Imai DMet al. CCR7 deficiency allows accelerated clearance of Chlamydia from the female reproductive tract. J Immunol. 2017;199:2547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LX, McSorley SJ. A re-evaluation of the role of B cells in protective immunity to Chlamydia infection. Immunol Lett. 2015;164:88–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LX, McSorley SJ. B cells enhance antigen-specific CD4 T cell priming and prevent bacteria dissemination following Chlamydia muridarum genital tract infection. PLoS Pathog. 2013;9:e1003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Huang Y, Yang Zet al. Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun. 2014;82:5327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabey D, Peeling RW. Lymphogranuloma venereum. Sex Transm Infect. 2002;78:90–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviarachchi PA, Mercado MAB, McSorley SJet al. Antibody, but not B-cell-dependent antigen presentation, plays an essential role in preventing Chlamydia systemic dissemination in mice. Eur J Immunol. 2020;50:676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manam S, Nicholson BJ, Murthy AK. OT-1 mice display minimal upper genital tract pathology following primary intravaginal Chlamydia muridarum infection. Pathog Dis. 2013;67:221–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manam S, Thomas JD, Li Wet al. Tumor Necrosis Factor (TNF) receptor superfamily member 1b on CD8+ T cells and TNF receptor superfamily member 1a on non-CD8+ T cells contribute significantly to upper genital tract pathology following chlamydial infection. J Infect Dis. 2015;211:2014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks E, Verolin M, Stensson Aet al. Differential CD28 and inducible costimulatory molecule signaling requirements for protective CD4+ T-cell-mediated immunity against genital tract Chlamydia trachomatis infection. Infect Immun. 2007;75:4638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado MAB, Du W, Malaviarachchi PAet al. Innate IFNγ is essential for systemic Chlamydia control while CD4 T cell-dependent IFNγ production is highly redundant in the female reproductive tract. bioRxiv. 2020; DOI: 2020.2008.2026.269480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore-Connors JM, Fraser R, Halperin SAet al. CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 responses and genital tract inflammation upon intracellular Chlamydia muridarum infection. J Immunol. 2013;191:3430–9. [DOI] [PubMed] [Google Scholar]
- Morrison RP, Feilzer K, Tumas DB. Gene knockout mice establish a primary protective role for major histocompatibility complex class II-restricted responses in Chlamydia trachomatis genital tract infection. Infect Immun. 1995;63:4661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SG, Morrison RP. In situ analysis of the evolution of the primary immune response in murine Chlamydia trachomatis genital tract infection. Infect Immun. 2000;68:2870–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DM, Zhang X. Activation of murine macrophages. Curr Protoc Immunol. Chapter 14: Unit 14 12, 2008. DOI: 10.1002/0471142735.im1402s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy AK, Chambers JP, Meier PAet al. Intranasal vaccination with a secreted chlamydial protein enhances resolution of genital Chlamydia muridarum infection, protects against oviduct pathology, and is highly dependent upon endogenous gamma interferon production. Infect Immun. 2007;75:666–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy AK, Li W, Chaganty BKet al. Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital Chlamydia muridarum infection. Infect Immun. 2011;79:2928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musil K, Currie M, Sherley Met al. Rectal chlamydia infection in women at high risk of chlamydia attending Canberra Sexual Health Centre. Int J STD AIDS. 2016;27:526–30. [DOI] [PubMed] [Google Scholar]
- Nagarajan UM, Prantner D, Sikes JDet al. Type I interferon signaling exacerbates Chlamydia muridarum genital infection in a murine model. Infect Immun. 2008;76:4642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan UM, Sikes J, Prantner Det al. MyD88 deficiency leads to decreased NK cell gamma interferon production and T cell recruitment during Chlamydia muridarum genital tract infection, but a predominant Th1 response and enhanced monocytic inflammation are associated with infection resolution. Infect Immun. 2011;79:486–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan UM, Sikes JD, Yeruva Let al. Significant role of IL-1 signaling, but limited role of inflammasome activation, in oviduct pathology during Chlamydia muridarum genital infection. J Immunol. 2012;188:2866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan UM, Tripathy M, Kollipara Aet al. Differential signaling pathways are initiated in macrophages during infection depending on the intracellular fate of Chlamydia spp. Immunol Cell Biol. 2018;96:246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naglak EK, Morrison SG, Morrison RP. Neutrophils are central to antibody-mediated protection against genital Chlamydia. Infect Immun. 2017;85. DOI: 10.1128/IAI.00409-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Virok DP, Wood Het al. Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A. 2005;102:10658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RB, Soper DE, Richter HEet al. Chlamydia antibodies, chlamydia heat shock protein, and adverse sequelae after pelvic inflammatory disease: the PID Evaluation and Clinical Health (PEACH) Study. Sex Transm Dis. 2008;35:129–35. [DOI] [PubMed] [Google Scholar]
- O'Connell CM, Ingalls RR, Andrews CW Jr.et al. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol. 2007;179:4027–34. [DOI] [PubMed] [Google Scholar]
- Ohman H, Tiitinen A, Halttunen Met al. Cytokine polymorphisms and severity of tubal damage in women with Chlamydia-associated infertility. J Infect Dis. 2009;199:1353–9. [DOI] [PubMed] [Google Scholar]
- Olive AJ, Gondek DC, Starnbach MN. CXCR3 and CCR5 are both required for T cell-mediated protection against C. trachomatis infection in the murine genital mucosa. Mucosal immunology. 2011;4:208–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peipert JF, Boardman L, Hogan JWet al. Laboratory evaluation of acute upper genital tract infection. Obstet Gynecol. 1996;87:730–6. [DOI] [PubMed] [Google Scholar]
- Perry LL, Feilzer K, Caldwell HD. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol. 1997;158:3344–52. [PubMed] [Google Scholar]
- Perry LL, Feilzer K, Hughes Set al. Clearance of Chlamydia trachomatis from the murine genital mucosa does not require perforin-mediated cytolysis or Fas-mediated apoptosis. Infect Immun. 1999a;67:1379–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry LL, Su H, Feilzer Ket al. Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J Immunol. 1999b;162:3541–8. [PubMed] [Google Scholar]
- Peters RP, Dubbink JH, van der Eem Let al. Cross-sectional study of genital, rectal, and pharyngeal Chlamydia and gonorrhea in women in rural South Africa. Sex Transm Dis. 2014;41:564–9. [DOI] [PubMed] [Google Scholar]
- Petri B, Sanz MJ. Neutrophil chemotaxis. Cell Tissue Res. 2018;371:425–36. [DOI] [PubMed] [Google Scholar]
- Pilla-Moffett D, Barber MF, Taylor GAet al. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol. 2016. DOI: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt RA, Crother TR. Chlamydia pneumoniae Infection and Inflammatory Diseases. For Immunopathol Dis Therap. 2016;7:237–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston TB, Darville T. Chlamydia trachomatis: protective adaptive responses and prospects for a vaccine. Curr Top Microbiol Immunol. 2018;412:217–37. [DOI] [PubMed] [Google Scholar]
- Poston TB, O'Connell CM, Girardi Jet al. T cell-independent gamma interferon and B cells cooperate to prevent mortality associated with disseminated Chlamydia muridarum genital tract infection. Infect Immun. 2018;86. DOI: 10.1128/IAI.00143-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Darville T, Nagarajan UM. Stimulator of IFN gene is critical for induction of IFN-beta during Chlamydia muridarum infection. J Immunol. 2010;184:2551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Darville T, Sikes JDet al. Critical role for interleukin-1beta (IL-1beta) during Chlamydia muridarum genital infection and bacterial replication-independent secretion of IL-1beta in mouse macrophages. Infect Immun. 2009;77:5334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomski N, Kagebein D, Liebler-Tenorio Eet al. Mito-xenophagic killing of bacteria is coordinated by a metabolic switch in dendritic cells. Sci Rep. 2017;7:3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Giebel AM, Toh Eet al. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun. 2015;83:2870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Nelson DE. Chlamydia muridarum infection of macrophages elicits bactericidal nitric oxide production via reactive oxygen species and Cathepsin B. Infect Immun. 2015;83:3164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Miranpuri GS, Sigar IMet al. Chlamydia trachomatis persistence in the female mouse genital tract: inducible nitric oxide synthase and infection outcome. Infect Immun. 2001a;69:5131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Rank RG. Resolution of chlamydial genital infection with antigen-specific T-lymphocyte lines. Infect Immun. 1991;59:925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Sigar IM, Rana SVet al. Role for inducible nitric oxide synthase in protection from chronic Chlamydia trachomatis urogenital disease in mice and its regulation by oxygen free radicals. Infect Immun. 2001b;69:7374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KH, Soderberg LS, Rank RG. Resolution of chlamydial genital infection in B-cell-deficient mice and immunity to reinfection. Infect Immun. 1988;56:1320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Bowlin AK, Kelly KA. Characterization of lymphocyte response in the female genital tract during ascending Chlamydial genital infection in the guinea pig model. Infect Immun. 2000;68:5293–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Ramsey KH, Pack EAet al. Effect of gamma interferon on resolution of murine chlamydial genital infection. Infect Immun. 1992;60:4427–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Sanders MM. Pathogenesis of endometritis and salpingitis in a guinea pig model of chlamydial genital infection. Am J Pathol. 1992;140:927–36. [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Soderberg LS, Barron AL. Chronic chlamydial genital infection in congenitally athymic nude mice. Infect Immun. 1985;48:847–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG, Whittimore J, Bowlin AKet al. In vivo ultrastructural analysis of the intimate relationship between polymorphonuclear leukocytes and the chlamydial developmental cycle. Infect Immun. 2011;79:3291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SJ, Eckmann L, Quayle AJet al. Secretion of proinflammatory cytokines by epithelial cells in response to Chlamydia infection suggests a central role for epithelial cells in chlamydial pathogenesis. J Clin Invest. 1997;99:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen Cet al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Gierahn TM, Higgins DEet al. Monitoring the T cell response to genital tract infection. Proc Natl Acad Sci U S A. 2006;103:12069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roan NR, Starnbach MN. Antigen-specific CD8+ T cells respond to Chlamydia trachomatis in the genital mucosa. J Immunol. 2006;177:7974–9. [DOI] [PubMed] [Google Scholar]
- Scurlock AM, Frazer LC, Andrews CW Jr.et al. Interleukin-17 contributes to generation of Th1 immunity and neutrophil recruitment during Chlamydia muridarum genital tract infection but is not required for macrophage influx or normal resolution of infection. Infect Immun. 2011;79:1349–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah AA, Schripsema JH, Imtiaz MTet al. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm Dis. 2005;32:49–56. [DOI] [PubMed] [Google Scholar]
- Shillova N, Howe SE, Hyseni Bet al. Chlamydia-specific IgA secretion in the female reproductive tract induced via per-oral immunization confers protection against primary Chlamydia challenge. Infect Immun. 2020. DOI: 10.1128/IAI.00413-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigar IM, Schripsema JH, Wang Yet al. Plasmid deficiency in urogenital isolates of Chlamydia trachomatis reduces infectivity and virulence in a mouse model. Pathog Dis. 2014;70:61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P, Jha R, Bas Set al. In infertile women, cells from Chlamydia trachomatis infected sites release higher levels of interferon-gamma, interleukin-10 and tumor necrosis factor-alpha upon heat-shock-protein stimulation than fertile women. Reprod Biol Endocrinol. 2008;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starnbach MN, Bevan MJ, Lampe MF. Protective cytotoxic T lymphocytes are induced during murine infection with Chlamydia trachomatis. J Immunol. 1994;153:5183–9. [PubMed] [Google Scholar]
- Sturdevant GL, Caldwell HD. Innate immunity is sufficient for the clearance of Chlamydia trachomatis from the female mouse genital tract. Pathog Dis. 2014;72:70–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Caldwell HD. CD4+ T cells play a significant role in adoptive immunity to Chlamydia trachomatis infection of the mouse genital tract. Infection & Immunity. 1995;63:3302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Feilzer K, Caldwell HDet al. Chlamydia trachomatis genital tract infection of antibody-deficient gene knockout mice. Infect Immun. 1997;65:1993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Messer R, Whitmire Wet al. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable Chlamydiae. J Exp Med. 1998;188:809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Tian Q, Wang Let al. IL-6-mediated signaling pathways limit Chlamydia muridarum infection and exacerbate its pathogenicity in the mouse genital tract. Microbes Infect. 2017;19:536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Yang Z, Zhang Het al. Chlamydia muridarum induction of glandular duct dilation in mice. Infect Immun. 2015;83:2327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HR, Burton MJ, Haddad Det al. Trachoma. Lancet. 2014;384:2142–52. [DOI] [PubMed] [Google Scholar]
- Thalmann J, Janik K, May Met al. Actin re-organization induced by Chlamydia trachomatis serovar D–evidence for a critical role of the effector protein CT166 targeting Rac. PLoS One. 2010;5:e9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Zhou Z, Wang Let al. Gastrointestinal coinfection promotes Chlamydial pathogenicity in the genital tract. Infect Immun. 2020;88. DOI: 10.1128/IAI.00905-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiitinen A, Surcel HM, Halttunen Met al. Chlamydia trachomatis and chlamydial heat shock protein 60-specific antibody and cell-mediated responses predict tubal factor infertility. Hum Reprod. 2006;21:1533–8. [DOI] [PubMed] [Google Scholar]
- Tseng CT, Rank RG. Role of NK cells in early host response to chlamydial genital infection. Infect Immun. 1998;66:5867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuffrey M, Taylor-Robinson D. Progesterone as a key factor in the development of a mouse model for genital-tract infection with Chlamydia trachomatis. FEMS Microbiol Lett. 1981;12:111–5. [Google Scholar]
- Vicetti Miguel RD, Quispe Calla NE, Pavelko SDet al. Intravaginal Chlamydia trachomatis challenge infection elicits TH1 and TH17 immune responses in mice that promote pathogen clearance and genital tract damage. PLoS One. 2016;11:e0162445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Artis D, Colonna Met al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–66. [DOI] [PubMed] [Google Scholar]
- Vlcek KR, Li W, Manam Set al. The contribution of Chlamydia-specific CD8(+) T cells to upper genital tract pathology. Immunol Cell Biol. 2016;94:208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang Q, Zhang Tet al. The Chlamydia muridarum organisms fail to auto-inoculate the mouse genital tract after colonization in the gastrointestinal tract for 70 days. PLoS One. 2016;11:e0155880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Brode S, Ellis Let al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog. 2017;13:e1006383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter-Stahl L, Ojcius DM, Viala Jet al. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol. 2006;8:1047–57. [DOI] [PubMed] [Google Scholar]
- Wiesenfeld HC, Heine RP, Krohn MAet al. Association between elevated neutrophil defensin levels and endometritis. J Infect Dis. 2002;186:792–7. [DOI] [PubMed] [Google Scholar]
- Xie JH, Nomura N, Lu Met al. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol. 2003;73:771–80. [DOI] [PubMed] [Google Scholar]
- Xie L, He C, Chen Jet al. Suppression of Chlamydial pathogenicity by nonspecific CD8(+) T lymphocytes. Infect Immun. 2020;88. DOI: 10.1128/IAI.00315-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Su X, Zhao Yet al. Innate lymphoid cells are required for endometrial resistance to Chlamydia trachomatis infection. Infect Immun. 2020;88. DOi: 10.1128/IAI.00152-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kari L, Lei Let al. Chlamydia trachomatis plasmid gene protein 3 is essential for the establishment of persistent infection and associated immunopathology. mBio. 2020;11. DOi: 10.1128/mBio.01902-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Conrad T, Zhou Zet al. Complement factor C5 but not C3 contributes significantly to hydrosalpinx development in mice infected with Chlamydia muridarum. Infect Immun. 2014;82:3154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeruva L, Spencer N, Bowlin AKet al. Chlamydial infection of the gastrointestinal tract: a reservoir for persistent infection. Pathog Dis. 2013;68:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Lin H, Xie Let al. Chlamydia muridarum induces pathology in the female upper genital tract via distinct mechanisms. Infect Immun. 2019;87. DOI: 10.1128/IAI.00145-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Xiao L, Lin Let al. STAT3-mediated TLR2/4 pathway upregulation in an IFN-gamma-induced Chlamydia trachomatis persistent infection model. Pathog Dis. 2016;74. DOI: 10.1093/femspd/ftw076. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou Z, Chen Jet al. Lack of long-lasting hydrosalpinx in A/J mice correlates with rapid but transient chlamydial ascension and neutrophil recruitment in the oviduct following intravaginal inoculation with Chlamydia muridarum. Infect Immun. 2014a;82:2688–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Huang Y, Gong Set al. In vivo and ex vivo imaging reveals a long-lasting Chlamydial infection in the mouse gastrointestinal tract following genital tract inoculation. Infect Immun. 2015;83:3568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang B. The development and diversity of ILCs, NK cells and their relevance in health and diseases. Adv Exp Med Biol. 2017;1024:225–44. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Shao L, Li Xet al. Uterotubal junction prevents chlamydial ascension via innate immunity. PLoS One. 2017;12:e0183189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yeruva L, Marinov Aet al. The DNA sensor, cyclic GMP-AMP synthase, is essential for induction of IFN-beta during Chlamydia trachomatis infection. J Immunol. 2014b;193:2394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G. Chlamydia spreading from the genital tract to the gastrointestinal tract - a two-hit hypothesis. Trends Microbiol. 2018;26:611–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong GM, Peterson EM, Czarniecki CWet al. Role of endogenous gamma interferon in host defense against Chlamydia trachomatis infections. Infect Immun. 1989;57:152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Paul WE. Heterogeneity and plasticity of T helper cells. Cell Res. 2010;20:4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zortel T, Schmitt-Graeff A, Kirschnek Set al. Apoptosis modulation in the immune system reveals a role of neutrophils in tissue damage in a murine model of Chlamydial genital infection. J Infect Dis. 2018;217:1832–43. [DOI] [PubMed] [Google Scholar]