

Abstract
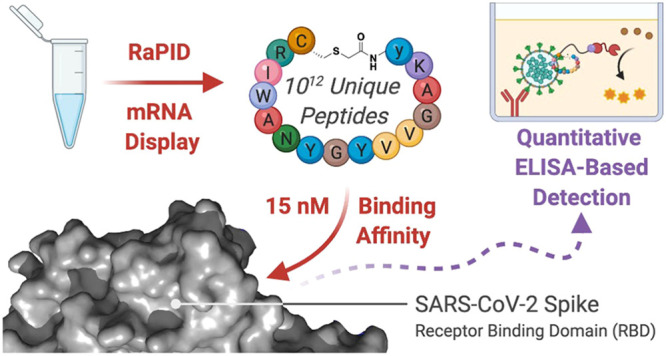
The COVID-19 pandemic, caused by SARS-CoV-2, has led to substantial morbidity, mortality, and disruption globally. Cellular entry of SARS-CoV-2 is mediated by the viral spike protein, and affinity ligands to this surface protein have the potential for applications as antivirals and diagnostic reagents. Here, we describe the affinity selection of cyclic peptide ligands to the SARS-CoV-2 spike protein receptor binding domain (RBD) from three distinct libraries (in excess of a trillion molecules each) by mRNA display. We identified six high affinity molecules with dissociation constants (KD) in the nanomolar range (15–550 nM) to the RBD. The highest affinity ligand could be used as an affinity reagent to detect the spike protein in solution by ELISA, and the cocrystal structure of this molecule bound to the RBD demonstrated that it binds to a cryptic binding site, displacing a β-strand near the C-terminus. Our findings provide key mechanistic insight into the binding of peptide ligands to the SARS-CoV-2 spike RBD, and the ligands discovered in this work may find future use as reagents for diagnostic applications.
Short abstract
We describe the discovery of high affinity ligands to the SARS-CoV-2 spike protein receptor binding domain using RaPID mRNA display and demonstrate their utility for viral detection by ELISA.
Introduction
The COVID-19 pandemic, caused by infection with the severe acute respiratory syndrome related coronavirus-2 (SARS-CoV-2), has led to widespread morbidity and mortality and has resulted in crippling effects on the global economy. At the time of writing, there had been >150 million confirmed cases of COVID-19 and >3.2 million deaths resulting from SARS-CoV-2 infections. Since the first reported case of COVID-19 in the province of Hubei, China in December 2019, there has been an intense global research effort centered on the development of an effective vaccine for the control of the disease. For example, two mRNA vaccines (BNT162b2 and mRNA-1273, from Pfizer/BioNTech and Moderna, respectively) and two adenovirus-vectored vaccines (ChAdOx1 nCoV-19 and Ad26.COV2.S, from Oxford University/AstraZeneca and Janssen, respectively) very recently developed are now in clinical use.1−3 In addition to the requirement for a vaccine, effective antiviral drugs are also critically needed for COVID-19 treatment; these drugs will be particularly important for use in unvaccinated individuals, in cases where the efficacy of a vaccine wanes or the virus develops resistance to vaccine-induced responses.4
While several candidate antiviral molecules are already in clinical trials, none were developed specifically for treatment of SARS-CoV-2 infection. Moreover, even the most promising of these, including remdesivir, hydroxychloroquine, lopinavir-ritonavir, and type I interferon therapy, have been deemed ineffective or only modestly efficacious in clinical trials.5−13 Thus, in addition to investigations into the repurposing of existing drugs, there is an urgent need for the development of novel molecules that specifically target SARS-CoV-2 for the treatment of COVID-19. These molecules may also find use for immediate deployment in situations where SARS-CoV-2 becomes endemic, including for pre-exposure prophylaxis in high-risk populations, or for further SARS-CoV outbreaks, e.g., through future zoonoses.
The critical initiating step in viral infection involves entry of SARS-CoV-2 into human cells, a process that is mediated by interaction between the receptor binding domain (RBD) of the viral spike protein with cell-surface angiotensin converting enzyme 2 (ACE2) (Figure 1).14 Crystal structures of RBD–ACE2 complexes have elucidated the mechanism of this interaction and shown that the RBD binding site on ACE2 is located on an N-terminal helical region, distinct from the catalytic site (Figure 1).15,16 Importantly, it has recently been demonstrated that blocking this interaction (through addition of either soluble RBD protein or soluble ACE2 protein) potently inhibits viral infection of cultured cells in a dose dependent manner.17−19 These data provided early evidence that the RBD–ACE2 interaction was a valid target for the development of novel antivirals against SARS-CoV-2 infection. Several approaches have since been adopted for the discovery of novel RBD binding ligands, including full antibodies,20−28 as well as antibody fragments such as VH domains,29,30 nanobodies,31−35 and monobodies.36 In addition, engineered ACE2 mimetics/decoy receptors,37−39de novo designed miniproteins,40,41 peptide-based ACE2 mimetics,42,43 and synthetic peptide libraries44 have also been reported that are capable of binding the SARS-CoV-2 spike RBD.
Figure 1.
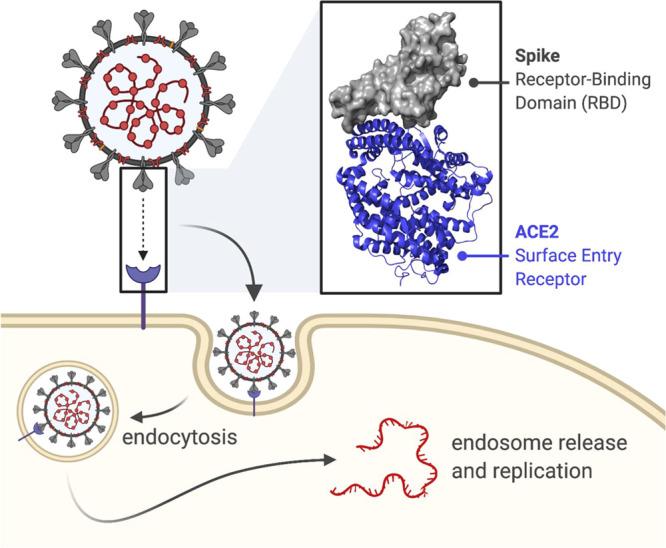
Cartoon depicting the interaction between the SARS-CoV-2 trimeric spike protein and the human ACE2 receptor, a key step in viral entry (endocytic entry route shown). Inset: crystal structure (PDB: 6LZG) showing the interaction between the N-terminal helix of ACE2 (blue) and the SARS-CoV-2 spike RBD (gray).
Macrocyclic peptides are a class of molecules demonstrated to be highly effective at disrupting protein–protein interactions, particularly in cases such as the spike–ACE2 interaction where a defined binding pocket is lacking.45−54 In this work, we explored this chemotype for the development of SARS-CoV-2 RBD-binding molecules that block the spike–ACE2 interaction, with a view to discovering novel inhibitors of viral entry. To discover novel cyclic peptides, we employed cyclic peptide mRNA display, an approach that enables the generation of libraries of >1012 macrocyclic peptides that can be subsequently selected against the target of interest, in our case the RBD of the spike protein of SARS-CoV-2 (Figure 1).
Results and Discussion
To identify ligands to the SARS-CoV-2 RBD, we performed three parallel affinity selections using very high diversity macrocyclic peptide libraries (Figure 2A). Two of these were genetically reprogrammed Random nonstandard Peptide Integrated Discovery (RaPID) libraries, comprising thioether-closed macrocyclic peptides (one initiated with N-chloroacetyl-l-tyrosine and one initiated with N-chloroacetyl-d-tyrosine). The third comprised disulfide-closed macrocyclic peptides. In each case, a semirandom DNA library was transcribed into mRNA, followed by covalent ligation to puromycin and in vitro translation to yield a cyclic peptide-mRNA fusion library in excess of 1012 unique molecules. Following counter selection (to remove streptavidin ligands), each library was panned against biotinylated SARS-CoV-2 RBD immobilized on streptavidin beads, and an enriched DNA library was recovered by RT-PCR. After seven iterative rounds of this process, the final DNA library was sequenced to identify peptide ligands predicted to bind to SARS-CoV-2 RBD with high affinity (see the Supporting Information). From this sequencing, we chose nine diverse and enriched peptides for further evaluation: three l-tyrosine initiated, three d-tyrosine initiated, and three disulfide closed cyclic peptides (Figure 2B).
Figure 2.

A) Schematic of the cyclic peptide mRNA display technology used. DNA libraries incorporating 4–15 randomized NNS (N = A, C, G, or T; S = C or G) codons were transcribed into mRNA, covalently ligated to puromycin (to allow conjugation between each mRNA and its cognate peptide), translated in in vitro reactions, and reverse transcribed to afford very high diversity (>1012 molecules) peptide-mRNA:cDNA libraries. Iterative rounds of affinity selection against the recombinant SARS-CoV-2 RBD protein followed by recovery of the DNA by PCR and resynthesis of the peptide-mRNA:cDNA library were conducted to enrich for SARS-COV-2 RBD ligands. In two libraries, the initiating N-formylmethionine residue was genetically reprogrammed to N-chloroacetyl-l-tyrosine or N-chloroacetyl-d-tyrosine (dY), which spontaneously cyclize to a downstream cysteine residue to form a thioether. A third library included an additional cysteine residue affording cyclic peptides through disulfide formation. B) Sequence alignment of the nine enriched unique peptide sequences from each library chosen for further characterization.
The nine target cyclic peptides 1–9 were subsequently synthesized by solid-phase peptide synthesis (SPPS). Specifically, the target peptide sequences were first assembled on Rink amide resin using Fmoc-strategy SPPS (Scheme 1). For peptides 1–6, the N-termini were derivatized with chloroacetic acid (Scheme 1A), while the N-terminal methionine was N-acetylated in 7–9 (Scheme 1B). Each of the peptides was subsequently cleaved from resin with concomitant side chain deprotection by treatment with an acidic cocktail. It should be noted that, despite significant optimization of the solid-phase synthesis, the precursor linear peptides to 5 and 7 were generated with significant sequence truncations (as judged by LC-MS analysis after the cleavage step); these peptides were also poorly soluble in both aqueous media and organic solvents. We therefore chose to resynthesize these two sequences with a hexaethylene glycol solubility tag on the C-terminus. Given that the RaPID peptides were panned on the RBD bearing a large mRNA tag on the C-terminus, we were confident that this modification would not influence the binding affinity to the RBD55,56 (Scheme 1). For the thioether-linked peptides 1–6, the linear peptide precursors were cyclized by treatment with N,N-diisopropylethylamine in DMSO or acetonitrile/water mixtures (depending on the solubility of the linear peptides, see the Supporting Information). In contrast, the disulfide-linked cyclic peptides 7–9 were generated through oxidation of the linear cleaved peptides by incubating in aqueous ammonium bicarbonate. Purification of each of the macrocyclic peptide targets by reverse-phase HPLC afforded 1–9 in 2–14% yield over the iterative SPPS and cyclization steps.
Scheme 1. General Scheme Depicting the Synthetic Route to A) Thioether Cyclic Peptide Targets 1–6 and B) Disulfide Linked Cyclic Peptide Targets 7–9.

With the synthetic cyclic peptides 1–9 in hand, we next assessed the ability of each to bind to the RBD of the SARS-CoV-2 spike protein using surface plasmon resonance (SPR). Briefly, RBD was expressed and purified before immobilizing onto a CM5 SPR chip using 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS) chemistry according to the manufacturer’s protocol (see the Supporting Information). Of the nine cyclic peptides screened, the six N-chloroacetylated peptides all exhibited high affinity for the RBD with dissociation constants (KD) in the nanomolar range (KD = 15–550 nM, Table 1, see the Supporting Information for SPR sensorgrams). The highest affinity of these was peptide 4, with an observed KD = 15 nM against RBD and a markedly low rate of dissociation (koff = 1.4 × 10–3 s–1). Interestingly, this molecule is a higher affinity cyclic analogue of a linear RBD ligand recently identified through an affinity selection-mass spectrometry approach.44 Peptide 5 was also notable in the series with a KD = 76 nM against the RBD. The disulfide-closed peptides showed not only very high apparent RBD affinity but also strong nonspecific binding (data not shown). Based on these data, we selected peptides 4 and 5 for assessment of antiviral activity against SARS-CoV-2.
Table 1. Binding Kinetics and Dissociation Constants (KD) for the Nine SARS-CoV-2 RBD Cyclic Peptide Ligands Synthesized and Evaluated against the RBD by SPRa.

* peptides contain C-terminal hexaethylene glycol moieties; # refractive index values high on sensorgram; NSB = nonspecific binding (see the Supporting Information for sensorgrams).
The antiviral activity of 4 and 5 was assessed in VeroE6 cells using high content fluorescence microscopy. Briefly, the cyclic peptides and SARS-CoV-2 were preincubated for 1 h and then coincubated with VeroE6 cells for 72 h (MOI 0.5) within 384-well plates. SARS-CoV-2 infection under these conditions leads to viral cytopathic effects (CPEs) and cell loss that is correlated to the level of available infectious virus in the initial inoculum. Addition of inhibitors that bind the RBD of the spike protein, thus preventing interactions with cell surface ACE2, e.g., monoclonal antibodies, reduces CPE/cell loss in a dose dependent manner.20−22 To enumerate the reduction of CPE in high content, live cell nuclei were stained using Hoechst 33342 (NucBlue), and then the entire 384-well plate was imaged (see the Supporting Information). Nuclei were then counted in high content using InCarta image analysis software used to give a quantitative measure of the cytopathic effect (see the Supporting Information). Unfortunately, despite their nM affinities for RBD, neither 4 nor 5 showed antiviral activity against SARS-CoV-2 up to a concentration of 10 μM. Additionally, no inhibition was seen in a SARS-CoV-2 pseudovirion neutralization assay at peptide concentrations of up to 50 μM (see the Supporting Information). These data were corroborated by biolayer interferometry (BLI) experiments, in which 4 and 5 were unable to disrupt the interaction between ACE2 and SARS-CoV-2 RBD (see the Supporting Information).
We next sought to understand the mechanism by which peptide 4 binds to the RBD. To this end, we obtained crystals of the RBD–4 complex and solved the structure of the complex by X-ray crystallography to a resolution of 3.96 Å (Figure 3; PDB code 7L4Z). Despite the relatively low resolution, we could identify the binding site of peptide 4, which lay near the N- and C-termini of the RBD (Figure 3B and Figure S7) and close to a recently discovered fatty acid binding pocket (Figure 3C).57 Unexpectedly, the peptide binds to a cryptic region of the RBD by displacing a C-terminal β-strand of the domain (523TVCG526) with residues 5VVYG8 of the peptide, which adopt essentially the same backbone conformation and make corresponding interactions with the RBD. Interestingly, a similar motif is observed in a linear RBD peptide ligand (1TVFG4; Figure 3D) that was previously identified by affinity selection-mass spectrometry by Pentelute and co-workers.44 Ala mutagenesis of this 13-residue linear peptide highlighted the importance of the motif, particularly residues Val2 and Gly4, which are also present in the sequences of both the RBD C-terminus and peptide 4. It is therefore tempting to speculate that a similar mode of binding for the RBD would be observed for the linear peptide that was discovered through an independent affinity selection method.
Figure 3.
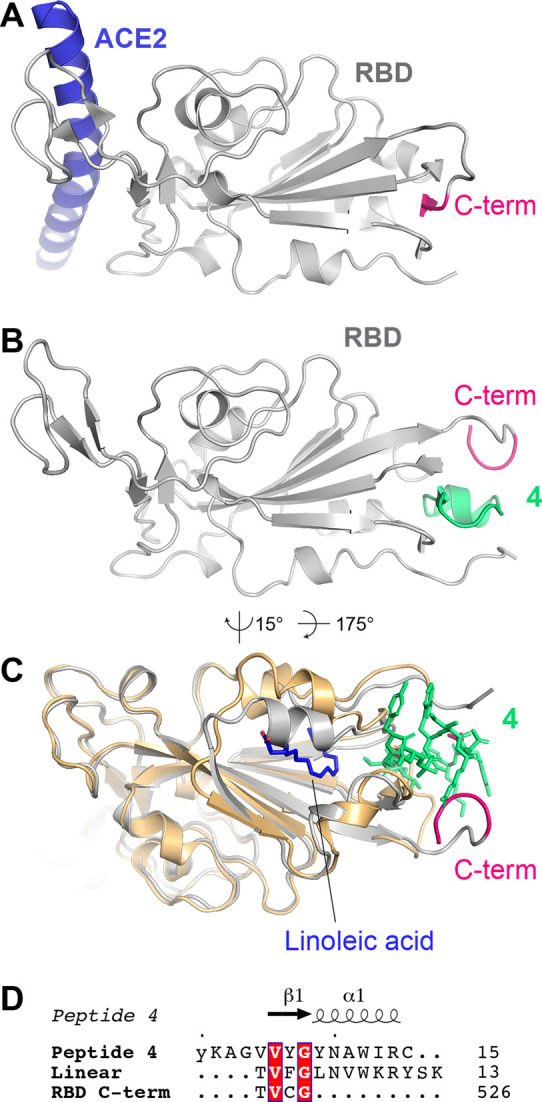
A) Structure of ACE2 (only helix 1 shown; blue) in complex with the spike RBD (gray; PDB ID 6M0J). The C-terminal β-strand (523TVCG526) is highlighted in pink. B) Structure of the spike RBD in complex with peptide 4 (green). C) Structure of RBD-4 (gray, with 4 shown in green sticks) superimposed onto the spike–linoleic acid complex (PDB code 6ZB4; shown in gold cartoon and blue sticks, respectively). Only the RBD is shown for clarity. D) Alignment of peptide 4, linear RBD-binding peptide ligand,44 and spike RBD C-terminal β-strand. The most C-terminal amino acid shown from the peptide/RBD is numbered on the right.
The peptide-binding site is distal from the ACE2-binding interface (Figure 3A), providing a rationale for the lack of antiviral activity. Within the context of the full-length spike protein, peptide binding would only be achieved if the C-terminal β-strand of the RBD undergoes a similar rearrangement to what we observe in our RBD-4 crystal structure. Moreover, conformational changes in the regions directly flanking the RBD domain would also be required, as the helix formed by peptide 4 residues 9YNAWIR14 would otherwise clash with these regions.
Despite the lack of antiviral activity of the RBD-binding cyclic peptides, we rationalized that the affinity of the peptides could instead prove useful for detection of the spike protein. In order to assess this possibility, we developed an ELISA-based detection assay for the full spike ectodomain and the spike RBD. Peptides 4 and 5 were first resynthesized bearing a C-terminal biotin tag (see the Supporting Information). ELISA plates were first coated with BD-218, a neutralizing monoclonal antibody identified from a COVID-19 patient.28 Either the spike ectodomain trimer or RBD alone was captured and then detected with biotinylated 4 or 5 and HRP-conjugated streptavidin (Figure 4A). In a second strategy, ELISA plates were directly coated with the spike or RBD protein, and an antigen was detected as above (Figure 4B). Gratifyingly, detection of both the RBD and spike ectodomain was possible with biotinylated 4 and 5; however, 5 had substantial nonspecific binding, as evidenced by detection of an unrelated protein, bovine serum albumin (BSA). In further quantitative assays, biotinylated 4 allowed detection of the spike ectodomain trimer at concentrations as low as 31.25 ng mL–1 (see Figure 4C and the Supporting Information).
Figure 4.
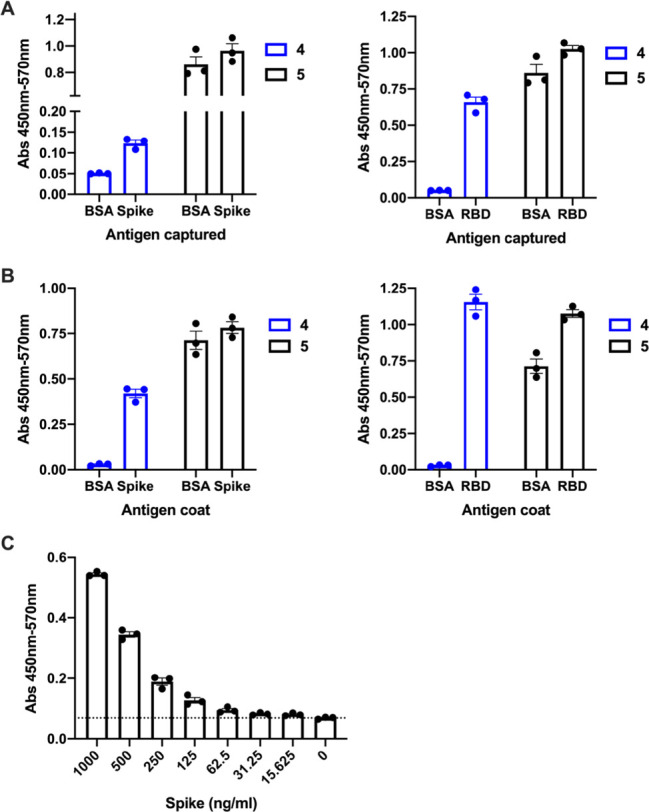
Detection of the spike ectodomain or the spike RBD from SARS-CoV-2 using biotinylated variants of 4 and 5 by ELISA. A) Antigen capture ELISA. The plate was coated with antibody BD-218 and blocked with BSA, and then the spike or RBD (1 μg mL–1) was captured and detected with biotinylated 4 and 5. B) Direct ELISA. The plate was coated with the spike ectodomain or RBD (1 μg mL–1) and blocked with BSA, and viral antigen was detected with biotinylated 4 and 5. C) Quantitative direct ELISA. The plate was coated with the spike protein (dilution series 1000–15.625 ng/mL) and blocked with BSA, and viral antigen was detected with biotinylated 4. Individual data points for technical triplicates are shown, with SEM.
Interestingly, although peptide 4 could differentially detect the spike when the latter was immobilized either by direct adsorption or indirectly via BD-218 antibody capture (Figure 4), we were unable to detect an interaction between 4 and the spike using SPR under the conditions used (see the Supporting Information). Together, these data suggest that the spike protein may undergo conformational changes that expose the peptide-binding site when directly or indirectly immobilized in the ELISA assays but not when covalently linked to the sensor used for SPR analysis. However, these data show that 4 may therefore be useful for detection of the full-length spike using ELISA-based approaches in future applications.
Conclusions
In summary, we have shown the utility of cyclic peptide mRNA display for the discovery of high affinity ligands to a viral glycoprotein, specifically the spike protein of SARS-CoV-2. We identified six peptides that bind to the RBD of the SARS-CoV-2 spike protein with nanomolar dissociation constants, including two with KD values <100 nM. While these peptides were unable to neutralize SARS-CoV-2 infection of cells, we identified a 15-residue cyclic peptide (4) which was capable of selectively and quantitatively detecting the SARS-CoV-2 spike or RBD protein in an ELISA-based detection assay. Moreover, we solved the structure of 4 in complex with the SARS-CoV-2 spike RBD. The unexpected binding mode of the cyclic peptide adjacent to the N- and C-termini of the domain suggests that significant structural reorganization of the spike protein would be necessary to accommodate the ligand. The binding of the ligand distal to the ACE2 binding site of the RBD explains the lack of antiviral activity of the cyclic peptide. Importantly, while there have been more than 120 structures of RBD–ligand complexes deposited in the protein data bank over the past 12 months, this represents the first structure of a peptide complexed with the domain, and the first structure of a ligand bound in this cryptic site. This binding mode is likely made possible by the small size of cyclic peptide 4 that enables it to access the compact site on the RBD. A similar binding mode would be difficult to achieve with ACE2 mimics, antibody and antibody fragments that have been the primary focus of RBD- and spike-binding ligands to date. Future work in our laboratories will capitalize on the unique binding mode discovered here by investigating cyclic peptide 4 (and analogues thereof) as easily accessible and cheap reagents for use in rapid, reproducible, and simple ELISA-based quantitation of the virally expressed SARS-CoV-2 spike protein in patient or environmental samples. Furthermore, the location of this new binding site adjacent to a recently discovered fatty acid binding pocket on the SARS-CoV-2 RBD raises the interesting possibility of designing lipid-linked variants of 4 (Figure 3C).57 This hybridization approach may lead to molecules that possess improved affinity for the spike RBD, as well as antiviral activity.
Methods
No unexpected or unusually high safety hazards were encountered during the course of this research. All experimental methods can be found in the Supporting Information.
Acknowledgments
We would like to acknowledge funding from the National Health and Medical Research Council (Investigator Grant APP1174941 to R.J.P.) and a seed grant from the Drug Discovery Initiative and the Marie Bashir Institute at the University of Sydney. We would also like to thank Dr. Luke Dowman for producing Figure 1 included in this manuscript. We thank the beamline scientists at the Australian Synchrotron for their assistance with data collection. This research was undertaken using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. Cyclic peptide display screening and SPR were supported by Sydney Analytical, The University of Sydney, and sequencing was conducted at the Ramaciotti Centre for Genomics, University of New South Wales.
Glossary
Abbreviations
- ACE2
angiotensin converting enzyme 2
- BLI
biolayer interferometry
- BSA
bovin serum albumin
- COVID-19
coronavirus disease 19
- CPE
cytopathic effect(s)
- EDC
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- ELISA
enzyme-linked immunosorbent assay
- KD
dissociation constant
- mRNA
messenger ribonucleic acid
- NHS
N-hydroxysuccinimide
- PDB
protein data bank
- PCR
polymerase chain reaction
- RBD
receptor binding domain
- SARS-CoV-2
severe acute respiratory syndrome coronavirus-2
- SPR
surface plasmon resonance
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01708.
Materials and methods, peptide characterization, supporting crystallographic data, and supplementary figures [peptides 1–9 and biotinylated 4 and 5 (structures, analytical HPLC trace, and ESI MS], SPR and BLI sensorgrams, SARS-CoV-2 and SARS-CoV-2 pseudovirus neutralization assays, quantitative detection of SARS-CoV-2 spike protein and RBD by ELISA, and difference Fo–Fc density between N- and C-termini of RBD (PDF)
Author Contributions
∇ A.N., C.F., and M.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Knoll M. D.; Wonodi C. Oxford-AstraZeneca COVID-19 vaccine efficacy. Lancet 2021, 397 (10269), 72–74. 10.1016/S0140-6736(20)32623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack F. P.; Thomas S. J.; Kitchin N.; Absalon J.; Gurtman A.; Lockhart S.; Perez J. L.; Pérez Marc G.; Moreira E. D.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voysey M.; Clemens S. A. C.; Madhi S. A.; Weckx L. Y.; Folegatti P. M.; Aley P. K.; Angus B.; Baillie V. L.; Barnabas S. L.; Bhorat Q. E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397 (10269), 99–111. 10.1016/S0140-6736(20)32661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; VanBlargan L. A.; Rothlauf P. W.; Bloyet L.-M.; Chen R. E.; Stumpf S.; Zhao H.; Errico J. M.; Theel E. S.; Ellebedy A. H.; Fremont D. H.; Diamond M. S.; Whelan S. P. J. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29 (3), 477–488.e4. 10.1016/j.chom.2021.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; et al. Remdesivir for the Treatment of Covid-19 — Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang D.; Du G.; Du R.; Zhao J.; Jin Y.; Fu S.; Gao L.; Cheng Z.; Lu Q. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395 (10236), 1569–1578. 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulware D. R.; Pullen M. F.; Bangdiwala A. S.; Pastick K. A.; Lofgren S. M.; Okafor E. C.; Skipper C. P.; Nascene A. A.; Nicol M. R.; Abassi M.; et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. N. Engl. J. Med. 2020, 383, 517–525. 10.1056/NEJMoa2016638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skipper C. P.; Pastick K. A.; Engen N. W.; Bangdiwala A. S.; Abassi M.; Lofgren S. M.; Williams D. A.; Okafor E. C.; Pullen M. F.; Nicol M. R.; et al. Hydroxychloroquine in nonhospitalized adults with early COVID-19: a randomized trial. Ann. Intern. Med. 2020, 173 (8), 623–631. 10.7326/M20-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalcanti A. B.; Zampieri F. G.; Rosa R. G.; Azevedo L. C.; Veiga V. C.; Avezum A.; Damiani L. P.; Marcadenti A.; Kawano-Dourado L.; Lisboa T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N. Engl. J. Med. 2020, 383 (21), 2041–2052. 10.1056/NEJMoa2019014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horby P. W.; Mafham M.; Bell J. L.; Linsell L.; Staplin N.; Emberson J.; Palfreeman A.; Raw J.; Elmahi E.; Prudon B.; et al. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 2020, 396 (10259), 1345–1352. 10.1016/S0140-6736(20)32013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.; Wang Y.; Wen D.; Liu W.; Wang J.; Fan G.; Ruan L.; Song B.; Cai Y.; Wei M.; et al. A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer-Smadja N.; Yazdanpanah Y. Nebulised interferon beta-1a for patients with COVID-19. Lancet Respir. Med. 2021, 9 (2), 122–123. 10.1016/S2213-2600(20)30523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G. The role of type I interferons in the pathogenesis and treatment of COVID-19. Front. Immunol. 2020, 11, 595739. 10.3389/fimmu.2020.595739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Wan Y.; Luo C.; Ye G.; Geng Q.; Auerbach A.; Li F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (21), 11727–11734. 10.1073/pnas.2003138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181 (4), 894–904. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J.; Ye G.; Shi K.; Wan Y.; Luo C.; Aihara H.; Geng Q.; Auerbach A.; Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581 (7807), 221–224. 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteil V.; Kwon H.; Prado P.; Hagelkrüys A.; Wimmer R. A.; Stahl M.; Leopoldi A.; Garreta E.; Del Pozo C. H.; Prosper F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181 (4), 905–913. 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoufaly A.; Poglitsch M.; Aberle J. H.; Hoepler W.; Seitz T.; Traugott M.; Grieb A.; Pawelka E.; Laferl H.; Wenisch C.; et al. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8 (11), 1154–1158. 10.1016/S2213-2600(20)30418-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai W.; He L.; Zhang X.; Pu J.; Voronin D.; Jiang S.; Zhou Y.; Du L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17 (6), 613–620. 10.1038/s41423-020-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi X.; Yan R.; Zhang J.; Zhang G.; Zhang Y.; Hao M.; Zhang Z.; Fan P.; Dong Y.; Yang Y.; et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science 2020, 369 (6504), 650–655. 10.1126/science.abc6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Wang P.; Nair M. S.; Yu J.; Rapp M.; Wang Q.; Luo Y.; Chan J. F.-W.; Sahi V.; Figueroa A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584 (7821), 450–456. 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Barnes C. O.; Jette C. A.; Abernathy M. E.; Dam K.-M. A.; Esswein S. R.; Gristick H. B.; Malyutin A. G.; Sharaf N. G.; Huey-Tubman K. E.; Lee Y. E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N. C.; Yuan M.; Liu H.; Lee C.-C. D.; Zhu X.; Bangaru S.; Torres J. L.; Caniels T. G.; Brouwer P. J.; Van Gils M. J.; et al. An alternative binding mode of IGHV3–53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 2020, 33 (3), 108274. 10.1016/j.celrep.2020.108274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S.; Cao Y.; Zhu Q.; Yu P.; Qi F.; Wang G.; Du X.; Bao L.; Deng W.; Zhu H.; et al. Structurally resolved SARS-CoV-2 antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy. Cell 2020, 183 (4), 1013–1023. 10.1016/j.cell.2020.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreye J.; Reincke S. M.; Kornau H.-C.; Sánchez-Sendin E.; Corman V. M.; Liu H.; Yuan M.; Wu N. C.; Zhu X.; Lee C.-C. D.; et al. A therapeutic non-self-reactive SARS-CoV-2 antibody protects from lung pathology in a COVID-19 hamster model. Cell 2020, 183 (4), 1058–1069. 10.1016/j.cell.2020.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C. O.; West A. P.; Huey-Tubman K.; Hoffmann M. A.; Sharaf N. G.; Hoffman P. R.; Koranda N.; Gristick H. B.; Gaebler C.; Muecksch F.; et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell 2020, 182 (4), 828–842. 10.1016/j.cell.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D.; Park Y.-J.; Beltramello M.; Walls A. C.; Tortorici M. A.; Bianchi S.; Jaconi S.; Culap K.; Zatta F.; De Marco A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Su B.; Guo X.; Sun W.; Deng Y.; Bao L.; Zhu Q.; Zhang X.; Zheng Y.; Geng C.; et al. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 2020, 182 (1), 73–84. 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken C. J.; Lim S. A.; Solomon P.; Rettko N. J.; Nguyen D. P.; Zha B. S.; Schaefer K.; Byrnes J. R.; Zhou J.; Lui I.; et al. Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2. Nat. Chem. Biol. 2021, 17, 113–121. 10.1038/s41589-020-00679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Schäfer A.; Kulkarni S. S.; Liu X.; Martinez D. R.; Chen C.; Sun Z.; Leist S. R.; Drelich A.; Zhang L.; et al. High potency of a bivalent human VH domain in SARS-CoV-2 animal models. Cell 2020, 183 (2), 429–441. 10.1016/j.cell.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof M.; Faust B.; Saunders R. A.; Sangwan S.; Rezelj V.; Hoppe N.; Boone M.; Billesbølle C. B.; Puchades C.; Azumaya C. M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370 (6523), 1473–1479. 10.1126/science.abe3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Nambulli S.; Xiao Z.; Liu H.; Sang Z.; Duprex W. P.; Schneidman-Duhovny D.; Zhang C.; Shi Y. Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Science 2020, 370 (6523), 1479–1484. 10.1126/science.abe4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J.; Le Bas A.; Ruza R. R.; Duyvesteyn H. M.; Mikolajek H.; Malinauskas T.; Tan T. K.; Rijal P.; Dumoux M.; Ward P. N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27 (9), 846–854. 10.1038/s41594-020-0469-6. [DOI] [PubMed] [Google Scholar]
- Dong J.; Huang B.; Wang B.; Titong A.; Kankanamalage S. G.; Jia Z.; Wright M.; Parthasarathy P.; Liu Y. Development of humanized tri-specific nanobodies with potent neutralization for SARS-CoV-2. Sci. Rep. 2020, 10, 17806. 10.1038/s41598-020-74761-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye G.; Gallant J. P.; Massey C.; Shi K.; Tai W.; Zheng J.; Odle A. E.; Vickers M. A.; Shang J.; Wan Y. The Development of a Novel Nanobody Therapeutic for SARS-CoV-2. BioRxiv 2020, 10.1101/2020.11.17.386532. [DOI] [Google Scholar]
- Kondo T.; Iwatani Y.; Matsuoka K.; Fujino T.; Umemoto S.; Yokomaku Y.; Ishizaki K.; Kito S.; Sezaki T.; Hayashi G.; Murakami H. Antibody-like proteins that capture and neutralize SARS-CoV-2. Science Adv. 2020, 6 (42), eabd3916 10.1126/sciadv.abd3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow A.; Glasgow J.; Limonta D.; Solomon P.; Lui I.; Zhang Y.; Nix M. A.; Rettko N. J.; Zha S.; Yamin R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (45), 28046–28055. 10.1073/pnas.2016093117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L.; Bi W.; Wang X.; Xu W.; Yan R.; Zhang Y.; Zhao K.; Li Y.; Zhang M.; Bao X.; et al. Engineered Trimeric ACE2 Binds and Locks” Three-up” Spike Protein to Potently Inhibit SARS-CoVs and Mutants. Cell Res. 2021, 31, 98–100. 10.1038/s41422-020-00438-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi Y.; Suzuki T.; Arimori T.; Ikemura N.; Kirita Y.; Ohgitani E.; Mazda O.; Motooka D.; Nakamura S.; Matsuura Y. High affinity modified ACE2 receptors prevent SARS-CoV-2 infection. BioRxiv 2020, 10.1101/2020.09.16.299891. [DOI] [Google Scholar]
- Cao L.; Goreshnik I.; Coventry B.; Case J. B.; Miller L.; Kozodoy L.; Chen R. E.; Carter L.; Walls A. C.; Park Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370 (6515), 426–431. 10.1126/science.abd9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsky T. W.; Vergara R.; Codina N.; Nelson J. W.; Walker M. J.; Su W.; Barnes C. O.; Hsiang T.-Y.; Esser-Nobis K.; Yu K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370 (6521), 1208–1214. 10.1126/science.abe0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Pearce R.; Zhang Y. De novo design of protein peptides to block association of the SARS-CoV-2 spike protein with human ACE2. Aging 2020, 12 (12), 11263–11276. 10.18632/aging.103416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Pomplun S.; Loftis A. R.; Loas A.; Pentelute B. L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 spike RBD. BioRxiv 2020, 10.1101/2020.03.19.999318. [DOI] [Google Scholar]
- Pomplun S.; Jbara M.; Quartararo A. J.; Zhang G.; Brown J. S.; Lee Y.-C.; Ye X.; Hanna S.; Pentelute B. L. De Novo Discovery of High Affinity Peptide Binders for the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2021, 7 (1), 156–163. 10.1021/acscentsci.0c01309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7 (7), 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Yudin A. K. Macrocycles: lessons from the distant past, recent developments, and future directions. Chem. Sci. 2015, 6 (1), 30–49. 10.1039/C4SC03089C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda E.; Nordgren I. K.; Male A. L.; Lawrence C. E.; Hoakwie F.; Cuda F.; Court W.; Fox K. R.; Townsend P. A.; Packham G. K. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J. Am. Chem. Soc. 2013, 135 (28), 10418–10425. 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini A.; Cendron L.; Chen S.; Touati J.; Winter G.; Zanotti G.; Heinis C. Bicyclic peptide inhibitor reveals large contact interface with a protease target. ACS Chem. Biol. 2012, 7 (5), 817–821. 10.1021/cb200478t. [DOI] [PubMed] [Google Scholar]
- Heinis C. Drug discovery: tools and rules for macrocycles. Nat. Chem. Biol. 2014, 10 (9), 696–698. 10.1038/nchembio.1605. [DOI] [PubMed] [Google Scholar]
- Qian Z.; Dougherty P. G.; Pei D. Targeting intracellular protein-protein interactions with cell-permeable cyclic peptides. Curr. Opin. Chem. Biol. 2017, 38, 80–86. 10.1016/j.cbpa.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Cheng K.; Yin H. Targeting protein- protein interfaces using macrocyclic peptides. Biopolymers 2015, 104 (4), 310–316. 10.1002/bip.22625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawatha M.; Rogers J. M.; Bonn S. M.; Livneh I.; Lemma B.; Mali S. M.; Vamisetti G. B.; Sun H.; Bercovich B.; Huang Y.; Ciechanover A.; Fushman D.; Suga H.; Brik A. De novo macrocyclic peptides that specifically modulate Lys48-linked ubiquitin chains. Nat. Chem. 2019, 11 (7), 644–652. 10.1038/s41557-019-0278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y.; Shoji I.; Miyagawa S.; Kawakami T.; Katoh T.; Goto Y.; Suga H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18 (12), 1562–1570. 10.1016/j.chembiol.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Johansen-Leete J.; Passioura T.; Foster S. R.; Bhusal R. P.; Ford D. J.; Liu M.; Jongkees S. A.; Suga H.; Stone M. J.; Payne R. J. Discovery of Potent Cyclic Sulfopeptide Chemokine Inhibitors via Reprogrammed Genetic Code mRNA Display. J. Am. Chem. Soc. 2020, 142 (20), 9141–9146. 10.1021/jacs.0c03152. [DOI] [PubMed] [Google Scholar]
- Bashiruddin N. K.; Hayashi M.; Nagano M.; Wu Y.; Matsunaga Y.; Takagi J.; Nakashima T.; Suga H. Development of cyclic peptides with potent in vivo osteogenic activity through RaPID-based affinity maturation. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (49), 31070–31077. 10.1073/pnas.2012266117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K.; Passioura T.; Sato H.; Ito K.; Furuhashi H.; Umitsu M.; Takagi J.; Kato Y.; Mukai H.; Warashina S.; et al. Macrocyclic peptide-based inhibition and imaging of hepatocyte growth factor. Nat. Chem. Biol. 2019, 15 (6), 598–606. 10.1038/s41589-019-0285-7. [DOI] [PubMed] [Google Scholar]
- Toelzer C.; Gupta K.; Yadav S. K.; Borucu U.; Davidson A. D.; Williamson M. K.; Shoemark D. K.; Garzoni F.; Staufer O.; Milligan R. J. S.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370 (6517), 725–730. 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.