

Abstract
Small molecules that target the spliceosome SF3B complex are potent inhibitors of cancer cell growth. The compounds affect an early stage of spliceosome assembly when U2 snRNP first engages the branch point sequence of an intron. Employing an inactive herboxidiene analog (iHB) as a competitor, we investigated factors that influence inhibitor interactions with SF3B to inhibit pre-mRNA splicing in vitro. Order-of addition experiments show that inhibitor interactions are long lasting and affected by both temperature and the presence of ATP. Our data are also consistent with the idea that not all SF3B conformations observed in structural studies are conducive to productive inhibitor interactions. Notably, SF3B inhibitors do not impact an ATP-dependent rearrangement in U2 snRNP that exposes the branch binding sequence for base pairing. We also report extended structure activity relationship analysis of the splicing inhibitor herboxidiene. We identified features of the tetrahydropyran ring that mediate its interactions with SF3B and its ability to interfere with splicing. Analyzing our data in the context of recent structural models of SF3B interactions with inhibitors, our results lead us to extend the model for early spliceosome assembly and inhibitor mechanism. We postulate that interactions between a carboxylic acid substituent of herboxidiene and positively charged SF3B1 sidechains in the inhibitor binding channel are required to maintain inhibitor occupancy while counteracting the SF3B transition to a closed state that is required for stable U2 snRNP interactions with the intron.
Keywords: SF3B, pre-mRNA splicing, SF3B inhibitors, herboxidiene analogs
Graphical Abstract
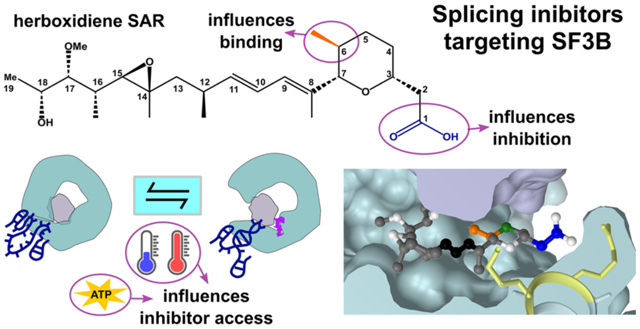
INTRODUCTION
The spliceosome is responsible for removing introns from gene transcripts in eukaryotes to generate messenger RNAs (mRNA). It assembles on each intron to be removed from a transcript from five U-rich small nuclear RNAs complexed in ribonucleoproteins (snRNPs), which join with dozens of additional proteins through a complicated series of interactions and structural rearrangements. In early spliceosome assembly, U2 snRNP, which consists of U2 snRNA, core proteins and the SF3A and SF3B multi-protein complexes, joins the intron to form the A-complex spliceosome. U2 snRNA base pairs with the neighboring nucleotides of the branch point sequence (BPS) to specify an adenosine residue near the end of intron as the branch point1,2. Because the 3’ splice site is typically the first AG dinucleotide within a certain downstream distance from the branch point, selection of the BPS also determines the end of introns3–7. Proteins involved in BPS selection, including SF3B components, are frequently mutated in some cancers and dysplasia, which suggests a role for intron definition in carcinogenesis8–11. Specific SF3B1 cancer mutations are linked to use of an aberrant branch point and 3’ splice site in some transcripts, indicating that SF3B1 normally helps ensure fidelity of BPS selection12–14.
Recent cryo-EM structural models of the spliceosome captured at the earliest stage of assembly provide some clues for how SF3B1 participates in BPS recognition1,15–17. The N- and C-terminal regions of the central C-shaped HEAT-repeat domain of SF3B1 come together and appear to clamp onto the branch helix formed by U2 snRNA and the intron. This conformation sequesters the bulged branch point adenosine into a pocket formed between PH5FA and SF3B1 HEAT repeats 15 and 16. Interestingly, in structures of U2 snRNP and isolated SF3B protein complex, the same domain exhibits a more open conformation that hinges at the same two HEAT repeats18,19. Together, these structures imply that closing the SF3B clamp is a critical step in BPS selection.
Pladienolide B (PB), spliceostatin A (SSA) and herboxidiene, first identified as potent inhibitors of tumor cell growth, are natural products that target SF3B and interfere with spliceosome assembly as U2 snRNP joins the intron to form the spliceosome A-complex20–25. Structures of SF3B bound to PB revealed the inhibitor cradled in a channel between PHF5A and SF3B1 HEAT repeats 15 and 16 in the open conformation and the inhibitor is proposed to block SF3B closing26,27. In structures where SF3B1 is closed over the branch helix, several residues that contact the inhibitor are rearranged to form the pocket for the branch point adenosine, which effectively eliminates the inhibitor channel.
We previously reported that splicing inhibition by PB, SSA and herboxidiene can be masked by addition of an excess of some inactive analogs regardless of the structural family28. We hypothesized that the inactive analogs compete for the inhibitor channel but lack a chemical feature that is required for either more stable binding or interfering with SF3B function. We test the model in this study with a systematic series of analogs and conclude that SF3B1 interactions with the C1 carboxylic acid of herboxidiene are essential for the inhibitor to maintain binding in the context of SF3B closure over the branch helix. We also find that all SF3B inhibitors exchange very slowly relative, which enabled us to investigate factors that influence inhibitor interactions via competition studies. Our evidence points to SF3B conformation controlling access to the inhibitor binding channel, which is further regulated by an ATP-dependent event.
By differentiating chemical features of herboxidiene that contribute to binding and inhibitory activity, our results will support its therapeutic development. Already, derivatives of PB and SSA show chemotherapeutic promise by decreasing the volume of human tumors implanted in mice and repressing expression and oncogenic splicing variant that confers chemotherapy resistance29–31. However, none have passed clinical trials29,32. Improvements based on structure activity relationship (SAR) studies are hampered by the structural complexity of these natural products. Our results here demonstrate that tuning the activity of herboxidiene, which has a somewhat simpler structure relative to SSA and PB, is more feasible and enables a deeper investigation into SF3B mechanism needed to realize the promise of splicing inhibitors for human health
RESULTS AND DISCUSSION
Order of addition affects competition between SF3B inhibitors and inactive analogs
We previously showed that when 1 μM herboxidiene, PB or SSA alone is included in an in vitro splicing reaction, splicing products are not detected. However, when 100 μM of an inactive analog of herboxidiene (iHB) is added simultaneously, splicing products are produced at levels nearly equivalent to control reactions with no added inhibitor. We tested whether the order of addition affects competition between SF3B inhibitors and iHB competitor. We reasoned that if inhibitor interactions exchange rapidly with SF3B, then incubating the inhibitor in nuclear extract prior to adding excess inactive competitor would have no effect on the competitor’s ability to rescue in vitro splicing from inhibition. Alternatively, if the inhibitor exchanges slowly or interferes irreversibly with SF3B function, then excess inactive competitor added after the inhibitor would not be able to replace it and result in low splicing efficiency. To distinguish between these two possibilities, we incubated 1 μM PB in nuclear extract for 10 minutes (binding phase), and then added 100 μM iHB for an additional 10 minutes (competition phase) (Figure 1a). We then tested the nuclear extracts for in vitro splicing efficiency (splicing phase). In control experiments, nuclear extract treated with PB or SSA followed by DMSO yielded no splicing products (Figure1b lanes 3 and 6, and 1c). An excess of inactive competitor iHB added before inhibitor PB or SSA rescues splicing efficiency to the level of DMSO controls, consistent with previous results with simultaneous addition (Figure 1b lanes 5 and 8, and 1c)28. In contrast, when PB or SSA is incubated with nuclear extracts before addition of excess iHB competitor, splicing efficiency is greatly reduced, although not completely lost relative to the inhibitor alone (Figure 1b, lanes 3–5 and 6–8, and 1B). We also tested less potent inactive competitor analogs of PB and SSA in the same manner with similar outcome (Supplemental Figure 2). Ordered competition with herboxidiene does yield a strong difference in splicing inhibition, which is consistent with its lower potency relative to SSA and PB (Supplemental Figure 2). Although our assay does not directly measure binding, these results are consistent with a very slow off-rate for PB and SSA, which opened the door to using the competition assay to assess factors that affect the interaction between SF3B and its inhibitors.
Figure 1. Order of addition affects competition between SF3B inhibitors and an inactive herboxidiene analog.
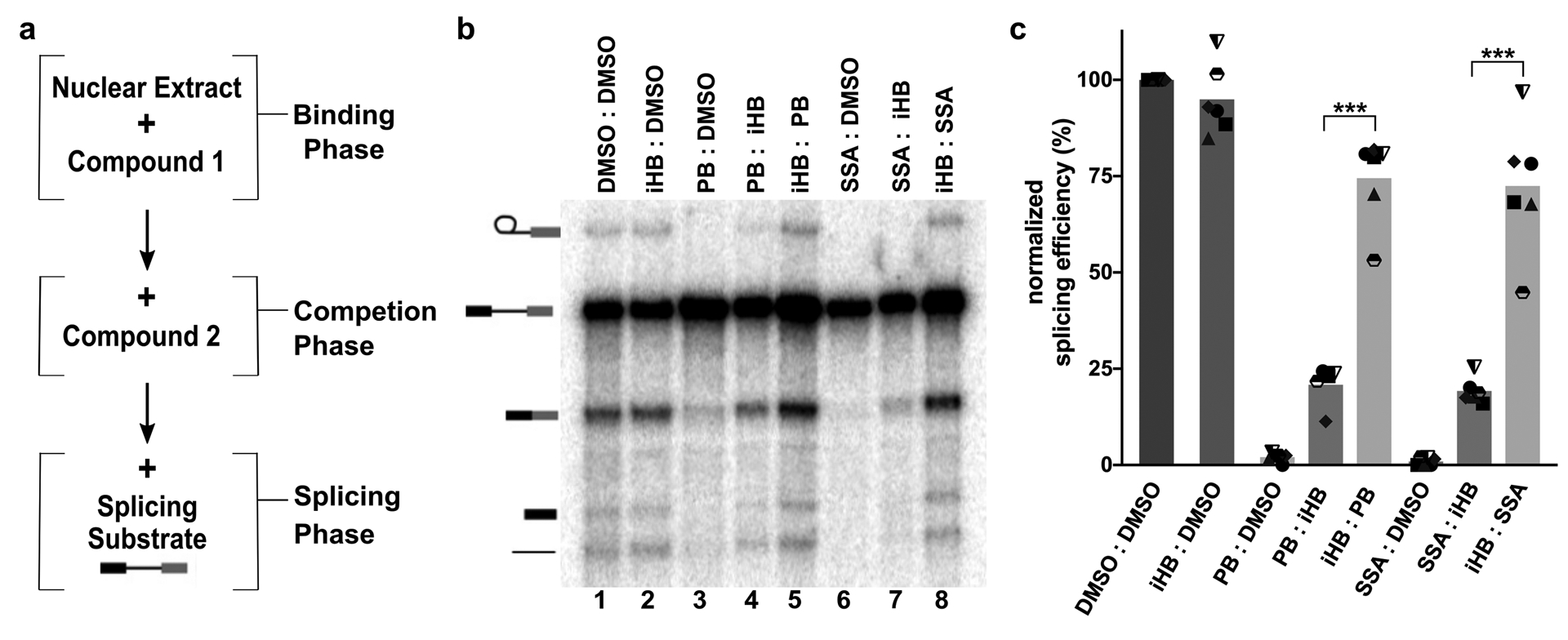
(a) Schematic of order of addition assay. (b) Representative denaturing gel analysis of in vitro splicing. Binding and competitions phases were 10 minutes at 4°C with DMSO, iHB (100 μM), PB (1 μM) or SSA (1 μM) and denoted Compound 1: Compound 2. Splicing phase was 30 minutes at 30°C. Band identities are illustrated on the left as (top to bottom): lariat intron product, pre-mRNA substrate, mRNA product, 5’ exon intermediate and linear intron product. Chemical structures of compounds are illustrated in Supplemental Figure 1. (c) Splicing efficiency quantified from denaturing gels plotted relative to DMSO control. Mean normalized splicing efficiency is displayed as bars, with experimental replicates values represented by different shape. *** p<0.001.
Inhibitor interactions with SF3B are modulated by temperature
Because structural models imply that an open SF3B1 conformation is required for inhibitor binding1,27, we reasoned that temperature may modulate SF3B conformational dynamics and affect accessibility of the inhibitor channel. We repeated the same order of addition assay and compared the effect of incubating inhibitors during the binding and competition phases at 4°C vs. 30°C, while the splicing phases remained at 30°C (Figure 1a). In the absence of competition, incubation temperature has no significant effect on splicing or splicing inhibition (Figure 2b, c). The result is different in the context of competition. When SSA, PB or herboxidiene is first incubated in nuclear extract at 30°C before addition of excess iHB competitor, the extract’s splicing efficiency is lower compared to when they are first incubated at 4°C (Figure 2a lanes 6 vs 7, 8 vs 9 and 10 vs 11). The effect of a higher temperature incubation providing inhibitors a stronger competitive advantage relative to lower temperature incubation also holds for less potent inactive competitor analogs of PB and SSA (Supplemental Figure 3). The difference is consistent with differential accessibility of the inhibitor binding channel in SF3B depending on its conformation. We speculate that higher temperature favors the SF3B1 open conformation to give the inhibitor more opportunity to interact with SF3B before the competitor is added, which enables the inhibitor to prevent splicing when substrate is introduced into the extract.
Figure 2. Temperature affects competition between SF3B inhibitors and inactive analogs.
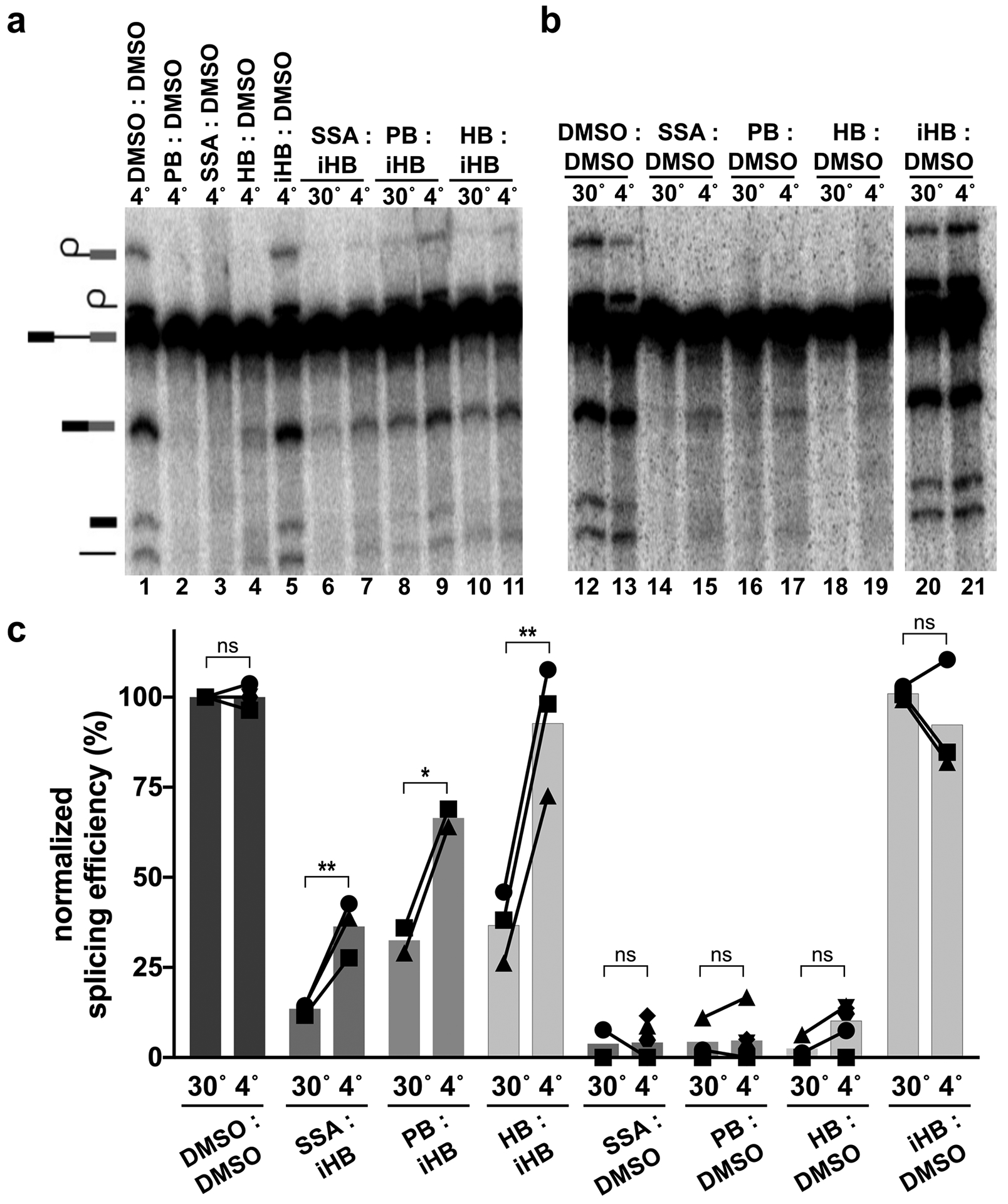
(a & b) Representative denaturing gels as described in Figure 1. Binding and competitions phases were 10 minutes at 4 or 30 °C with DMSO, HB (1 μM), PB (1 μM), SSA (1 μM) or iHB (100 μM). (c) Splicing efficiency quantified from denaturing gels as described in Figure 1. n.s. = p≥0.033, * p<0.033, ** p<0.002.
Temperature-dependent interactions with SF3B are not modulated by ATP
The mechanisms controlling SF3B conformation are not fully understood. A recent cryo-EM structure of the U2 snRNP revealed SF3B in the open conformation with the inhibitor binding channel accessible19. Notably, the RNA-dependent ATPase DDX46 (human ortholog of yeast Prp5) contacts SF3B1. In the competition assays described above, we included ATP during inhibitor incubation in the nuclear extracts. To test the hypothesis that the temperature dependent effects on inhibitor interactions with SF3B were mediated by an ATP-dependent event, we repeated the splicing assay but excluded ATP during the competition phase of the experiment. Surprisingly the absence of ATP during the competition phase did not abolish the effect of temperature on splicing inhibition. ATP-depleted extracts incubated with inhibitor at 30°C followed by excess competitor still exhibited lower splicing efficiency relative to competition at 4°C (Figure 3a lanes 4 vs. 5, 6 vs. 7, and 3b). We conclude that SF3B1’s ability to transition from a closed conformation that is refractory to inhibitor binding, to an open conformation that is available for inhibitor binding is influenced by temperature. Nevertheless, the presence of ATP does favor inhibitor interactions 30°C (Figure 3a lanes 4 vs. 6, and 3b), meaning that ATP promotes inhibitor access to SF3B independent of temperature, possibly through the activity of DDX46.
Figure 3. Temperature-dependent modulation of SF3B inhibition is independent of ATP.
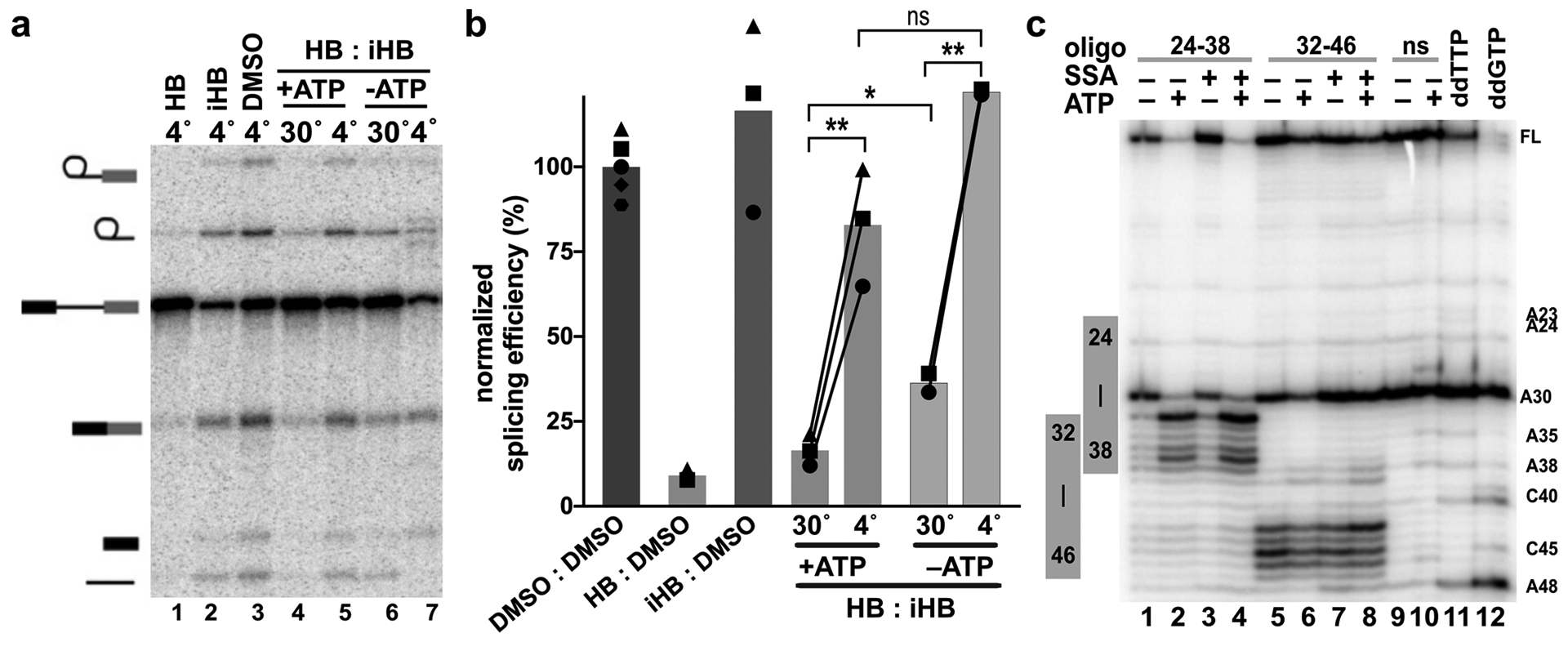
(a) Representative denaturing gel as described in Figure 1. Binding and competitions phases were 10 minutes with 1 μM HB or 100 μM iHB. (b) Splicing efficiency quantified from denaturing gels as described in Figure 1. (c) Primer extension analysis of targeted DNA-oligo mediated RNase H digestion of U2 snRNA in nuclear extracts. Extracts were incubated +/− ATP and 1 μM SSA for 10 minutes at 30°C prior to addition of oligos complementary to the indicated nucleotides. ns = non-specific oligo, ddTTP and ddGTP = sequencing lanes.
ATP-dependent changes in U2 snRNA BBS accessibility are not influenced by SF3B1 inhibitors
Stable incorporation of U2 snRNP in A-complex to establish the branch helix requires at least one ATP-dependent step33–35. ATP is also required for U2 snRNP association with pre-mRNA in the presence of both SSA and PB to form an unstable A-like complex in which the branch helix is not fully formed22,23,36. U2 snRNP conformation is also regulated by ATP24,37,38. Folco et al reported that the PB analog E7107 inhibits U2 snRNPs ability to engage an oligo containing the branch sequence, but that inhibition is bypassed by an ATP-dependent rearrangement of the snRNP24. We hypothesized that the small improvement of inhibitor competition conferred by the presence of ATP at 30°C is due to the same ATP-dependent rearrangement. To test this idea, we examined U2 snRNA accessibility in the presence and absence of ATP and SF3B inhibitor.
Upon addition of DNA oligos that are complementary to regions accessible for base pairing, endogenous RNase H in nuclear extract cleaves RNA in any RNA/DNA hybrids that form. We tested DNA oligos targeting U2 snRNA nucleotides between Stem I and Stem IIa, and mapped cleavage sites using primer extension. As predicted by previous studies, cleavage efficiency changes in some regions when extracts are first incubated with ATP38. The most predominate change is an increase in cleavage at nucleotides 33 through 38 with an oligo complementary to nucleotides 24–38 (Figure 3c lanes 1–4). Notably, these nucleotides are the GUAGUA nucleotides of the branch binding sequence that forms base pairs with an intron. They also sit in the loop region of the branchpoint-interacting stem loop (BSL) as recently visualized in the U2 snRNP structure19. Because ATP increases RNA cleavage in this region, an unwinding of the BSL, potentially by DDX46, is a possible explanation for the increased accessibility. Alternatively, release of DDX46 from the snRNP may allow access to the DNA oligo probe. Surprisingly, the SF3B inhibitor SSA does not interfere with the ATP-dependent changes in cleavage (Figure 3c). It is therefore possible that exposing the branch binding sequence of U2 snRNA mediates the ATP-requirement for U2 snRNP associates with an intron in the presence of inhibitor. The result also suggests that this event is independent of a transition from open to closed SF3B conformation, although we cannot rule out that an open SF3B is required. Notably, the previously described ATP-dependent conformational change in U2 snRNP affected by SF3B inhibitors remains to be characterized24.
Inactive analogs of SF3B inhibitors only lose some their competitive advantage over time
The idea that splicing inhibitors preventing SF3B closure raises an interesting question: How does iHB block SF3B inhibitor action, presumably by competing for the SF3B channel, while at the same time not affect splicing? One possible explanation is that the inactive compound exchanges rapidly from the binding site in comparison with the apparent slow off-rate for active compounds suggested by our order of addition experiments. If true, when inactive compound is incubated in extract before adding inhibitor, then competition will not show the same temperature dependence, because the inactive compound be quickly replaced by an active inhibitor with the final shift to 30°C for the splicing reaction. Furthermore, the ratio of active to inactive compound should affect both the extent and rate of competition. To test these predictions, we repeated the order of addition experiment at both 4° and 30°C, but added 100μM, 10μM or 1μM iHB first for 10 minutes, and followed with 1 μM SSA for another 10 minutes. As predicted, splicing is rescued to the same level at both temperatures when an excess of inactive competitor is added first (Figure 4a lanes 3 vs. 4, 6 vs.7,9 vs. 10 and 4b), and holds for competitions with inactive analogs of PB and SSA followed by herboxidiene, PB or SSA (Supplemental Figure 4a, b, c). When at a 1:1 ratio splicing was largely inhibited, but the small amount of splicing rescue was slightly higher for competition at 4°C relative to 30°C. This result is consistent with SSA having increased access to SF3B at the higher temperature as the inactive competitor leaves.
Figure 4. SF3B inhibitors exchange more slowly than inactive analogs.
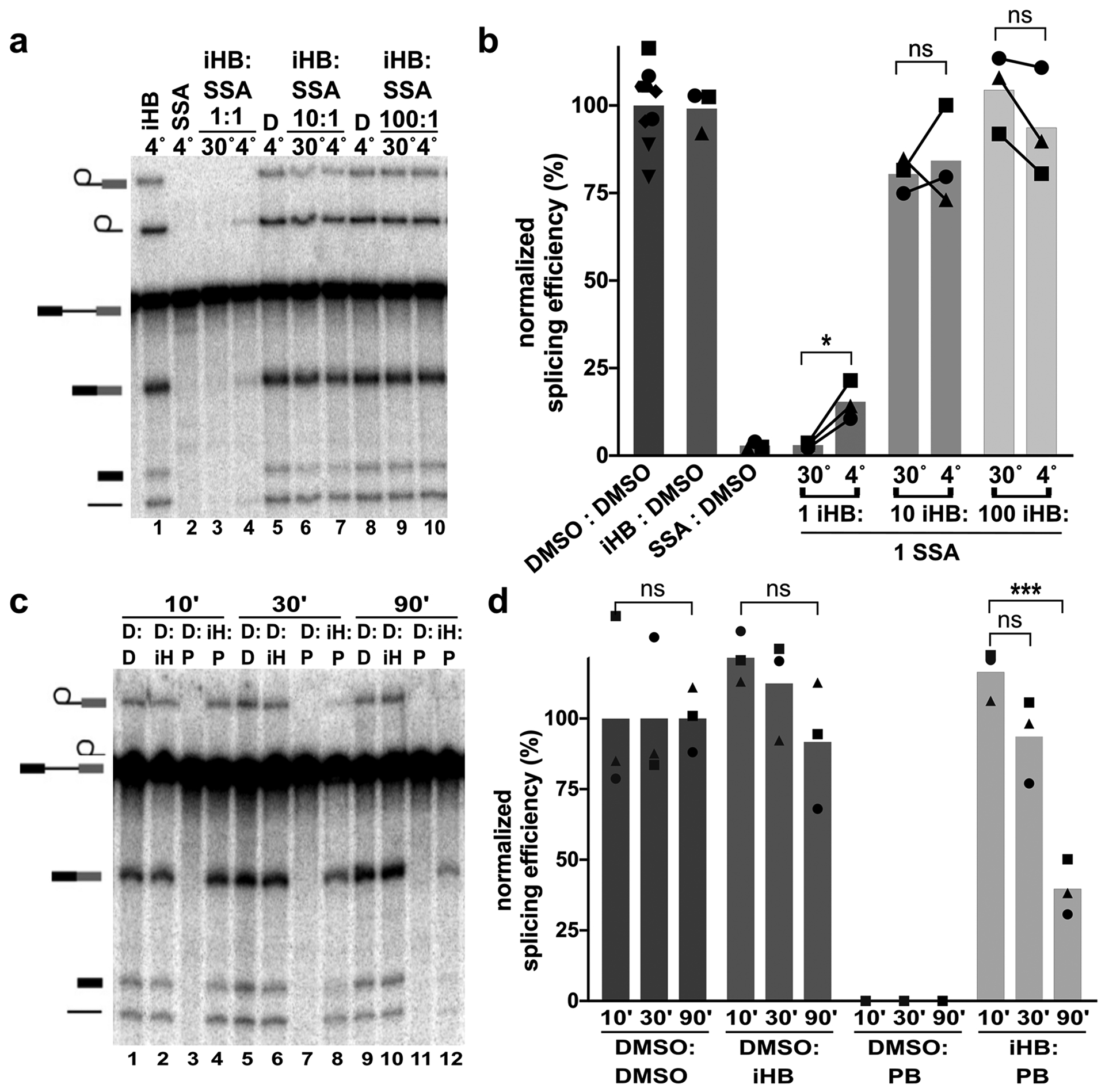
(a) Representative denaturing gel as described in Figure 1. Binding and competitions phases were 10 minutes with 1, 10 or 100 μM iHB and 1 μM SSA. (b) Splicing efficiency quantified from denaturing gels as described in Figure 1. (c) Representative denaturing gel with 10 minute binding phase and varying length competitions phases at 30°C. D= DMSO, iH = iHB, P=PB. (d) Splicing efficiency quantified for (c).
Differences in exchange rates for inactive competitors vs. SF3B inhibitors also predict that extending competition for a longer period of time should result in decreased splicing rescue when inactive competitor is followed by an inhibitor that has much slower exchange rate. We tested this expectation by incubating iHB in nuclear extract for 10 minutes at 30°C and then adding PB to compete for 10, 30 or 90 minutes (Figure 4c lanes 1–4, 5–8, 9–12 respectively and 4d). To further bias competition toward the active inhibitor, we used a ratio of 10:1 iHB to PB. Although splicing efficiency decreases somewhat with longer competition, iHB’s competitive advantage still remains after 90 minutes with only a 50% reduction relative to shorter competitions of 10 or 30 minutes (Figure 4c, d). Coupled with its ability to also rescue splicing at relatively low concentrations (1:1 and 1:10), it appears that even though iHB’s exchange rate from SF3B is higher than active inhibitors, it not rapid enough to explain why iHB has no effect on splicing.
Chemical features of herboxidiene differentially impact splicing inhibition and competition
To further explore the chemical features of herboxidiene (1) important for splicing inhibition and competition we tested a series of analogs in which we varied the substituents at C1 and C6 positions (see Figure 5 for compound modifications numbered (1)-(20). The inactive herboxidiene analog iHB (14) that we used for our competition assays differs from the active parent compound herboxidiene at two positions: an additional hydroxyl group at C5 of the tetrahydropyran ring, and conversion of the C1 carboxylic acid to its methyl ester. The C1 methyl ester is likely responsible for the lack of splicing inhibition, because the presence of the C5 hydroxyl group alone (2) has a minimal effect on compound activity39. The parent compound, herboxidiene (1), has an IC50 value for in vitro splicing of 0.3 μM39. Removing the methyl group at C6 (3) or replacement with a methylene group (4) had essentially no effect on compound activity, as their IC50 values remain sub-micromolar and comparable to herboxidiene (1). Flipping the chirality of the methyl group (7), creating a di-methyl (6) or cyclopropane at C6 (8) each resulted in >4-fold decreased inhibitory activity, while addition of a methyl ether (9) dropped activity >10-fold. Inclusion of an epoxy group (17 and 18) or a dichlorocyclopropane functionality (15) resulted over 500-fold decreased activity with an IC50 value greater than 100 μM, which we classify as inactive. From these results, we conclude that the presence and orientation of the methyl group at C6 in herboxidiene is not crucial for the inhibitor’s activity. However, increasing the size of the group at that position does have a deleterious effect. We also tested some of alterations at C6 (dimethyl, cyclopropane, methylether, dichlorocyclopropane) in combination with a methyl ester at C1. In this context, all compounds showed no activity at 100 μM, further supporting a critical role for the carboxylic acid functionality at C1. Notably, substitution of the carboxylic acid at C1 with carboxamide (10) along with an additional hydroxyl at C5 (12) strongly reduced splicing inhibition (>10–100-fold), but did not completely inactivate the compound in our assay system. Converting the C1 carboxylic acid to its hydroxymethyl derivative (11), in the context of the normally benign C5 hydroxyl addition, also strongly affected activity. We next asked at what level inactive herboxidiene analogs are able to compete with an inhibitor. Addition of most inactive analogs at 100 μM rescued >95% of splicing activity normally completely inhibited by 1 μM PB, although rescue was slightly reduced with two compounds that had both a methyl ester group at C1 and bulky adduct at C6. At a 10:1 ratio, compounds with larger adducts at C6 (compounds 15–20) again yielded lower splicing rescue.
Figure 5. Structure activity relationships for herboxidiene activity and SF3B interaction.
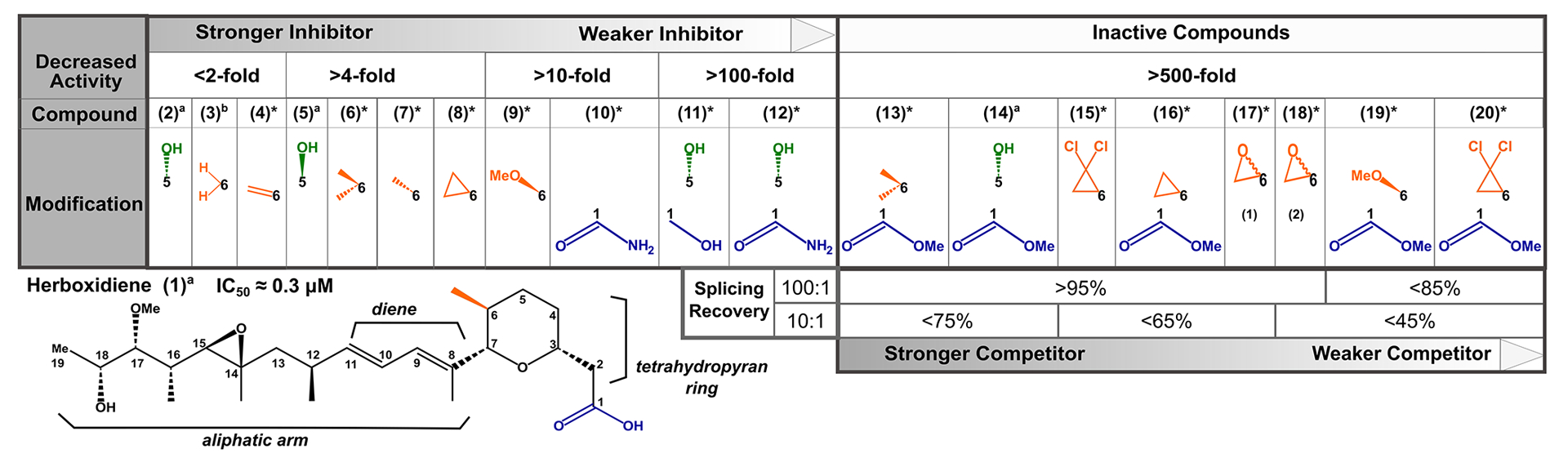
Summary of SAR results relative to the chemical structure of herboxidiene with modifications colored by their location and grouped by the magnitude of effect on inhibitor activity relative to the parent compound. Compounds with >500-fold decrease in inhibitory activity were tested for their ability to rescue splicing activity with 1 μM active splicing inhibitor at 100-fold and 10-fold excess. * indicates compounds new to this study, a39, b40.
Modeling herboxidiene interactions with SF3B
We modeled herboxidiene into the PB binding site of the SF3B crystal structure by overlaying the aliphatic arm and diene, and placing the carboxyl group over the ester of PB’s macrolide ring similar to Cretu et al.27. The model predicts that groups at C1 and C6 positions interact with residues from the U2 snRNP proteins PHF5A and SF3B1 (Figure 6a). The methyl group at C6 is close to a surface of PHF5A that remains relatively static in the context of SF3B1 closing. Compounds with larger adducts at this position (for example, 15) would sterically clash, which explains both their decreased potency as splicing inhibitors and decreased ability to compete with active inhibitors. The carboxyl at group at C1 in interacts with residues in HEAT repeat 15 of SF3B1, which undergo a large structural shift when SF3B1 closes to engage the branchpoint adenosine of an intron. Modifications at C1 decrease herboxidiene’s activity, showing that the position is important for inhibition, but have less impact on competition. Extended competitions in which inactive competitor is added first are consistent with iHB (14) having a higher exchange rate from the inhibitor binding channel relative to strong inhibitors. Still, why the inactive compounds are able to block SF3B inhibitors for extended periods of time, but do not block engagement of the intron at the same level is puzzling. It may be that our changes at C1, which removes potential hydrogen bonding partners, do not strongly affect affinity for the inhibitor binding channel present in an open SF3B1. Instead, a carboxylic acid at C1 may be important for stabilizing the position of nearby positively charged side chains and prevent their normal rearrangement to contact the intron during SF3B1 closing. Consistent with this idea, SF3B1 mutants have been identified as resistant to PB and herboxidiene41,42, and these mutations map to the positively charged amino acids (K1070, K1071, R1074, R1075) that are situated adjacent to C1 in our model (Figure 6b). We speculate that without the carboxylic acid functionality to compete with the SF3B1 conformational change required for intron engagement, inactive competitors are effectively ejected from SF3B upon closing. We aim to test this model in the future with inactive herboxidiene analogs that are engineered with a cross-linking group to stabilize their occupancy in the channel.
Figure 6. Model of early spliceosome assembly and SF3B inhibition.
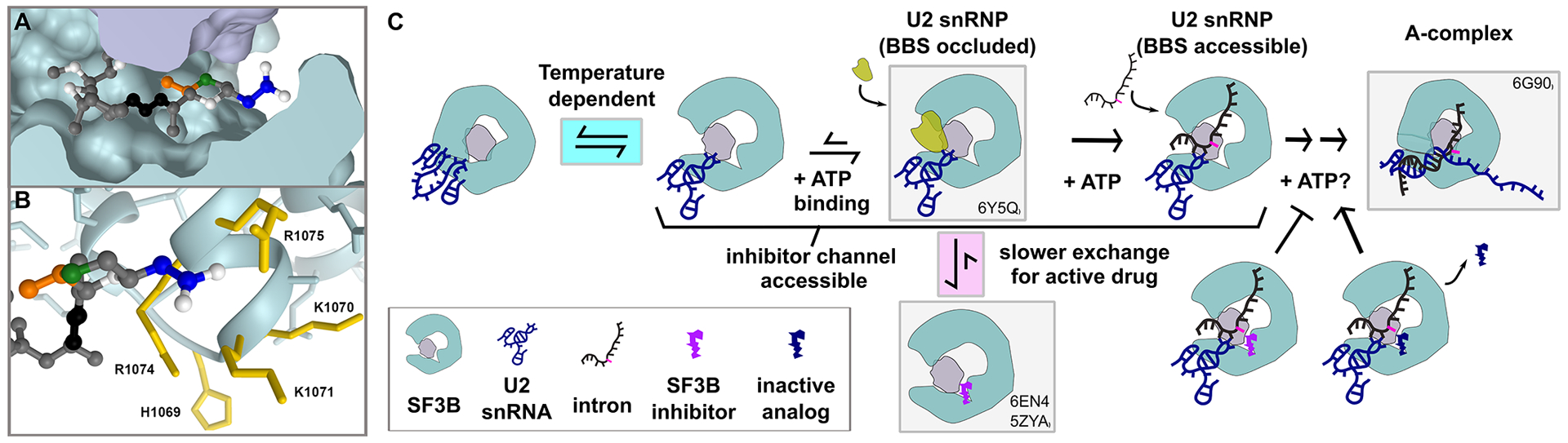
(a) Model of herboxidiene bound to SF3B inhibitor channel between SF3B (teal) and PHF5A (purple). Positions are colored as follows: C1 blue, C5 green, C6 orange, diene black. (b) Sidechains located near the modeled position of herboxidiene C1. Mutations of labeled residues in yellow confer resistance to Pladienolide B or herboxidiene. (c) Model of early spliceosome assembly steps and SF3B inhibitor action. Complexes boxed in gray are supported by indicated structure models1,19,26,27
Conclusion
The ability of iHB (14) to compete with inhibitors provides a unique way to study the parameters that affect inhibitor activity, and by extension their molecular target. Through order of addition experiments, we found that inhibitor interactions with SF3B are long lived and that access to the inhibitor channel changes in response to temperature and an ATP-dependent event. We also found that SF3B inhibitors do not interfere with an ATP-dependent rearrangement that exposes U2 snRNA. These results combined with previous studies lead us to expand the model of U2 snRNP addition to the spliceosome (Figure 6c). Although SF3B inhibitors do not prevent U2 snRNP from joining an intron in an A-like splicing complex22–24, the complex is less stable relative to the real A-complex, and likely has not yet completely formed the branch helix23,28. Notably even in the presence of inhibitor, ATP is required for U2 snRNP’s interaction with an intron. We hypothesize that this event may be related to the rearrangement that leads to increased accessibility of the U2 snRNA branch binding sequence. A recent cryo-EM structure revealed a population of U2 snRNPs in which the protein HTATSF1 is situated between the open “jaws” of SF3B1, while the branch binding sequence of U2 snRNA is folded in the BSL and surrounded by both HTATSF and DDX4619. It may be that DDX46 uses ATP to promotes these interactions to help stabilize the SF3B1 open conformation necessary for inhibitor occupancy of the binding channel. We hypothesize that an ATP-dependent restructuring of U2 snRNP, potentially involving the removal of HTATSF1 and/or release of DDX46 would allow an intron initial access to U2 snRNA to form the A-like complex stalled by SF3B inhibitors. We also suspect additional ATP-dependent rearrangements in U2 snRNP are required for branch helix formation given the extensive rearrangements of RNA/RNA and RNA protein interactions that must take place for U2 snRNP to take on the structure observed in the cryo-EM model of A-complex1. Nearly irreversible inhibitor interaction with SF3B prevents the transition to a closed SF3B1 conformation that is required to form or stabilize the branch helix. Nailing down the players and order of these interactions is the next challenge for splicing researchers, and SF3B inhibitors will likely continue to be important tools for helping to probe the process.
MATERIALS AND METHODS
Synthesis of SF3B1 inhibitors and analogs
Synthesis of SF3B1 inhibitors and herboxidiene analogs (2), (3), (5) and (14), are published39,40,43–45. Herboxidiene analogs (4), (6–3) and (15–20) were synthesized and purities were assessed by HPLC (>90% pure). The details of synthesis are described in a separate publication46.
In vitro splicing analysis
Pre-mRNA substrate was derived from the adenovirus major late (AdML) transcript (sequence shown in Supplemental Figure 5). 32P-UTP body-labeled G(5’)ppp(5’)G-capped substrate was generated by T7 run-off transcription followed by gel purification. Nuclear extract was prepared as previously described47 from HeLa cells grown in DMEM/F-12 1:1 and 5% (v/v) newborn calf serum. Order of addition experiments started with 60 mM potassium glutamate, 2 mM magnesium acetate, 2 mM ATP, 5 mM creatine phosphate, 0.05 mg/ml tRNA, 50% (v/v) HeLa nuclear extract and first compound and incubated at 4°C or 30 °C. Next the second compound was added, and the mixture was incubated for 10, 30 or 90 minutes at 4°C or 30°C. Then, radiolabeled pre-mRNA was added to a final concentration of 10 nM and the mixture incubated at 30°C for 30 or 60 minutes. The RNA was isolated from the reactions and separated on a 15% (v/v) denaturing polyacrylamide gel. 32P-labeled RNA species were visualized by phosphorimaging and quantified with ImageQuant software (Molecular Dynamics). Splicing efficiency was quantified as the amount of mRNA relative to total RNA and normalized to a dimethyl sulfoxide (DMSO) control reaction. Results were analyzed using unpaired t-test (two-tailed) using GraphPad Prism version 9.0.1 for Mac, GraphPad Software, San Diego, California USA.
For experiments testing ATP-dependency, HeLa nuclear extract was first depleted of ATP by incubation at 30°C for 15 minutes. Following inhibitor competitions with and without added ATP (2 mM), additional ATP (2 mM) was included with pre-mRNA substrate to allow for splicing.
RNase H cleavage analysis
A mixture of 40% HeLa nuclear extract (depleted of ATP by incubating for 10 minutes at 30°C) 60 mM potassium glutamate, 2 mM magnesium acetate, 0.1 mg /mL tRNA with and without 2 mM ATP and 1μM SSA or DMSO was incubated for 10 minutes at 30°C. DNA oligos complementary to U2 snRNA 24–38 nt or 32–46 nt were added at 5 μM and incubated for another 15 minutes at 30°C to allow for cleavage by endogenous RNase H. The oligos were degraded with addition of 1 μL RQ1 DNase for 10 minutes at 30°C. RNA was isolated and used as template for reverse-transcription primer extension reactions by first annealing with a 32P-radiolabeled primer complementary to U2 snRNA 97–117 nt. Reverse transcription reactions contained 50 mM Tris pH 7.9, 75 mM potassium chloride, 7 mM DTT, 3 mM magnesium chloride, 1 mM dNTPs, and 0.5 μg reverse transcriptase (MMLV variant). After a 30 minute incubation at 53°C, DNA was isolated and separated on a 10% (v/v) denaturing polyacrylamide gel that was dried and visualized as described previously.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award R01GM72649 to M.S.J and A.K.G. NIGMS award R25GM058903 and the National Science Foundation Graduate Research Fellowship under grant DGE-1842400 were used to support A.G.L
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
References:
- 1.Plaschka C, Lin PC, Charenton C & Nagai K Prespliceosome structure provides insights into spliceosome assembly and regulation. Nature 559, 419–422 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker R, Siliciano PG & Guthrie C Recognition of the TACTAAC box during mRNA splicing in yeast involves base pairing to the U2-like snRNA. Cell 49, 229–239 (1987). [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Anderson K & Moore MJ Evidence for a linear search in bimolecular 3’ splice site AG selection. Proc Natl Acad Sci U S A 97, 593–598 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taggart AJ, DeSimone AM, Shih JS, Filloux ME & Fairbrother WG Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19, 719–721 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith CW, Porro EB, Patton JG & Nadal-Ginard B Scanning from an independently specified branch point defines the 3’ splice site of mammalian introns. Nature 342, 243–247 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Smith CW, Chu TT & Nadal-Ginard B Scanning and competition between AGs are involved in 3’ splice site selection in mammalian introns. Mol Cell Biol 13, 4939–4952 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed R The organization of 3’ splice-site sequences in mammalian introns. Genes Dev 3, 2113–2123 (1989). [DOI] [PubMed] [Google Scholar]
- 8.Hubert CG et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes Dev 27, 1032–1045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quesada V et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44, 47–52 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Papaemmanuil E et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365, 1384–1395 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang AD et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11, 1438(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darman RB et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep 13, 1033–1045 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Alsafadi S et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7, 10615(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maji D, Grossfield A & Kielkopf CL Structures of SF3b1 reveal a dynamic Achilles heel of spliceosome assembly: Implications for cancer-associated abnormalities and drug discovery. Biochim Biophys Acta Gene Regul Mech 1862, 194440(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plaschka C, Lin PC & Nagai K Structure of a pre-catalytic spliceosome. Nature 546, 617–621 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai R, Wan R, Yan C, Lei J & Shi Y Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science 360, 1423–1429 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Cretu C et al. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol Cell 64, 307–319 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Zhang Z et al. Molecular architecture of the human 17S U2 snRNP. Nature 583, 310–313 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Mizui Y et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 57, 188–196 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Kotake Y et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol 3, 570–575 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Roybal GA & Jurica MS Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res 38, 6664–6672 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corrionero A, Minana B & Valcarcel J Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25, 445–459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Folco EG, Coil KE & Reed R The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev 25, 440–444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Effenberger KA et al. Coherence between cellular responses and in vitro splicing inhibition for the anti-tumor drug pladienolide B and its analogs. J Biol Chem 289, 1938–1947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finci LI et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev 32, 309–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cretu C et al. Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol Cell 70, 265–273 e8 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Effenberger KA, Urabe VK, Prichard BE, Ghosh AK & Jurica MS Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 22, 350–359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong DS et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 32, 436–444 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Yoshikawa Y et al. Design and Synthesis of 1,2-Deoxy-pyranose Derivatives of Spliceostatin A toward Prostate Cancer Treatment. ACS Med Chem Lett 11, 1310–1315 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seiler M et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 24, 497–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eskens FA et al. Phase I, Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin Cancer Res 19, 6296–6304 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Zhuang Y & Weiner AM A compensatory base change in human U2 snRNA can suppress a branch site mutation. Genes Dev 3, 1545–1552 (1989). [DOI] [PubMed] [Google Scholar]
- 34.Wu J & Manley JL Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev 3, 1553–1561 (1989). [DOI] [PubMed] [Google Scholar]
- 35.Konarska MM & Sharp PA Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49, 763–774 (1987). [DOI] [PubMed] [Google Scholar]
- 36.Effenberger KA, Urabe VK & Jurica MS Modulating splicing with small molecular inhibitors of the spliceosome. WIREs RNA 8, e1381(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abu Dayyeh BK, Quan TK, Castro M & Ruby SW Probing interactions between the U2 small nuclear ribonucleoprotein and the DEAD-box protein, Prp5. J Biol Chem 277, 20221–20233 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Black DL, Chabot B & Steitz JA U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell 42, 737–750 (1985). [DOI] [PubMed] [Google Scholar]
- 39.Ghosh AK, Ma N, Effenberger KA & Jurica MS Total Synthesis of GEX1Q1, Assignment of C-5 Stereoconfiguration and Evaluation of Spliceosome Inhibitory Activity. Org Lett 16, 3154–3157 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghosh AK et al. Design, synthesis and in vitro splicing inhibition of desmethyl and carba-derivatives of herboxidiene. Org Biomol Chem 14, 5263–5271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yokoi A et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J 278, 4870–4880 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Butt H et al. CRISPR directed evolution of the spliceosome for resistance to splicing inhibitors. Genome Biol 20, 73(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh AK & Li J A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org Lett 13, 66–69 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh AK & Anderson DD Enantioselective total synthesis of pladienolide B: a potent spliceosome inhibitor. Org Lett 14, 4730–4733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghosh AK & Chen ZH Enantioselective Syntheses of FR901464 and Spliceostatin A: Potent Inhibitors of Spliceosome. Org Lett 15, 5088–5091 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh AK et al. Design and synthesis of herboxidiene derivatives that potently inhibit in vitro splicing. Org Biomol Chem Advance Article, 10.1039/D0OB02532A (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dignam JD, Lebovitz RM & Roeder RD Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.