

Summary
How are E3 ubiquitin ligases configured to match substrate quaternary structures? Here, by studying the yeast GID complex (mutation of which causes deficiency in glucose-induced degradation of gluconeogenic enzymes), we discover supramolecular chelate assembly as an E3 ligase strategy for targeting an oligomeric substrate. Cryoelectron microscopy (cryo-EM) structures show that, to bind the tetrameric substrate fructose-1,6-bisphosphatase (Fbp1), two minimally functional GID E3s assemble into the 20-protein Chelator-GIDSR4, which resembles an organometallic supramolecular chelate. The Chelator-GIDSR4 assembly avidly binds multiple Fbp1 degrons so that multiple Fbp1 protomers are simultaneously ubiquitylated at lysines near the allosteric and substrate binding sites. Importantly, key structural and biochemical features, including capacity for supramolecular assembly, are preserved in the human ortholog, the CTLH E3. Based on our integrative structural, biochemical, and cell biological data, we propose that higher-order E3 ligase assembly generally enables multipronged targeting, capable of simultaneously incapacitating multiple protomers and functionalities of oligomeric substrates.
Keywords: E3, ubiquitin, ubiquitin ligase, GID, CTLH, gluconeogenesis, RING, cryo-EM, supramolecular assembly, metabolic regulation
Graphical abstract
Highlights
-
•
GID E3 ligase assembly resembles a behemoth organometallic supramolecular chelate
-
•
Multipronged targeting of oligomeric structure and metabolic function of substrate
-
•
Yeast Gid7, human WDR26, and MKLN1 mediate supramolecular assembly of GID/CTLH E3s
-
•
Human CTLH E3 structural and mechanistic features parallel yeast GID E3 ligase
Structural, biochemical, and cellular data reveal supramolecular assembly of yeast GID E3 ligase. GID E3 forms a chelate-like structure tailored to target the oligomeric structure and metabolic function of the gluconeogenic enzyme substrate Fbp1. The orthologous human CTLH E3 contains a GID-like core assembly that forms a supramolecular chelate and utilizes a GID-like ubiquitin ligase mechanism.
Introduction
Cells rapidly adapt their metabolic pathways in response to nutrient availability (Tu and McKnight, 2006; Zaman et al., 2008; Zhu and Thompson, 2019). Shifts in metabolic enzyme activities are achieved by regulation at every conceivable level. Metabolite-responsive transcriptional programs activate pathways that maximally use available nutrients and repress those rendered unnecessary or counterproductive. For oligomeric enzymes, catalytic activities are subject to metabolite-mediated allosteric control (Koshland, 1963a, 1963b; Monod et al., 1963). In eukaryotes, undesired metabolic activities are often terminated by ubiquitin-mediated degradation (Nakatsukasa et al., 2015).
Degradation is typically controlled by recognition of proteins as substrates of E3 ubiquitin (Ub) ligases. However, little is known about whether or how E3 ligases are specifically tailored for oligomeric assemblies of metabolic enzymes. One of the first identified targets of nutrient-dependent degradation, budding yeast fructose-1,6-bisphosphatase (Fbp1), is an oligomer (Chiang and Schekman, 1991). Fbp1 is a gluconeogenic enzyme essential for yeast growth on non-fermentable carbon sources. A shift from gluconeogenic to glycolytic conditions renders gluconeogenesis superfluous. Accordingly, Fbp1 activity and expression are curtailed (Gancedo, 1971; Schork et al., 1994a, 1994b, 1995). The switch to glycolytic conditions induces Ub-mediated degradation of Fbp1 and other gluconeogenic enzymes, including malate dehydrogenase (Mdh2) and phosphoenolpyruvate carboxykinase (Pck1), mediated by the multiprotein E3 ligase termed "GID"; yeast mutants of Gid subunits are glucose-induced-degradation deficient (Braun et al., 2011; Chiang and Schekman, 1991; Hämmerle et al., 1998; Menssen et al., 2012; Regelmann et al., 2003; Santt et al., 2008; Schork et al., 1994b, 1995). Although the GID E3 is conserved across eukaryotes and regulates important physiology (Lampert et al., 2018; Liu et al., 2020; Liu and Pfirrmann, 2019; Salemi et al., 2017), its regulation and targets are best characterized in budding yeast.
Much like well-studied multiprotein E3 ligases, such as anaphase-promoting complex/cyclosome (APC/C) or cullin-RING ligases, GID is not a singular complex—a core catalytic and scaffolding assembly is modulated by other subunits (Barford, 2020; Karayel et al., 2020; Liu and Pfirrmann, 2019; Melnykov et al., 2019; Qiao et al., 2020; Rusnac and Zheng, 2020; Watson et al., 2019). The constituents of various GID assemblies and how they achieve regulation are beginning to emerge. Previous structural studies have elucidated the core assembly and recapitulated some GID regulation (Qiao et al., 2020). Briefly, a core inactive complex, GIDAnt, contains the heterodimeric E3 ligase RING and RING-like subunits (Gid2 and Gid9) and scaffold subunits (Gid1, Gid5, and Gid8). Coexpression of these subunits in insect cells enables purification of recombinant GIDAnt and systematic interrogation of GID functions. Within the GIDAnt scaffold, Gid5 can bind the structurally homologous, interchangeable substrate-binding receptors Gid4 and Gid10 (Karayel et al., 2020; Melnykov et al., 2019; Qiao et al., 2020). Of these, the molecular basis of substrate binding by Gid4 is well understood: glucose-induced incorporation of Gid4 into the GID E3 enables recognition of substrate “Pro/N-degron” motifs depending on an N-terminal proline (Chen et al., 2017; Dong et al., 2018; Hämmerle et al., 1998; Regelmann et al., 2003; Santt et al., 2008). Indeed, in vitro, adding Gid4 transforms GIDAnt into an active GIDSR4 complex that collaborates with the cognate E2, Ubc8 (also known as Gid3) to ubiquitylate Mdh2, as explained by a structure of GIDSR4 (Qiao et al., 2020). Mutations probing the GIDSR4 structure also showed that this assembly is required for glucose-induced Fbp1 degradation in vivo (Qiao et al., 2020).
Perplexingly, despite the crucial role of Fbp1 in regulating gluconeogenesis, its ubiquitylation has not been reconstituted in vitro using defined GID E3 ligase components. In vivo, Fbp1 degradation depends on another protein, Gid7, which associates with other Gid subunits (Menssen et al., 2012; Regelmann et al., 2003; Santt et al., 2008). Gid7 is evolutionarily conserved across eukaryotes. Mammals even have two orthologs, WDR26 and MKLN1, which are subunits of the “CTLH” complex that corresponds to the yeast GID E3 (Boldt et al., 2016; Francis et al., 2013; Kobayashi et al., 2007; Lampert et al., 2018; Liu and Pfirrmann, 2019; Salemi et al., 2017). The CTLH E3, named for the preponderance of CTLH domains (in Gid1, Gid2, Gid7, Gid8, and Gid9 and their orthologs), has intrinsic E3 ligase activity, although Pro/N-degron substrates have not yet been identified despite human Gid4 binding this motif (Cao et al., 2020; Dong et al., 2018; Lampert et al., 2018; Liu et al., 2020; Liu and Pfirrmann, 2019; Maitland et al., 2019; Zavortink et al., 2020).
Here we reconstitute a minimal GID E3 ligase active toward Fbp1 by combining GIDSR4 and Gid7. Cryoelectron microscopy (cryo-EM) reveals its structure as a 20-protein supramolecular chelate E3 ligase assembly specifically tailored for Fbp1’s quaternary structure. Structural and biochemical data suggest that the human Gid7 orthologs likewise transform a GIDSR4-like E3 ligase core into higher-order assemblies. Our data reveal supramolecular chelate assembly of a pre-existing, functionally competent E3 ligase complex as a structural and functional principle to achieve multipronged Ub targeting tailored to an oligomeric substrate.
Results
Reconstitution of Fbp1 ubiquitylation
Considering that the Gid7 protein, not visualized previously, is required for glucose-induced Fbp1 degradation in vivo (Regelmann et al., 2003), we tested its effect in vitro. Our assay setup probes modulation of the core recombinant GIDAnt assembly upon adding other purified components individually or in combination. First, adding Gid4 marginally stimulated Fbp1 ubiquitylation despite substantially potentiating ubiquitylation of Mdh2, another canonical Pro/N-degron substrate, and Pck1, whose recognition by the GID E3 remains elusive (Figure 1A). However, adding Gid7 together with Gid4 substantially increased Fbp1 ubiquitylation. Comparing reactions with wild-type (WT) Ub or a Ub version lacking lysines (K0Ub) that cannot form polyUb chains indicated that adding Gid7 increases substrate consumption, the number of modified Fbp1 sites, and the number of Ubs in polyUb chains (Figures 1A and 1B). Second, the remarkable activation upon addition of Gid7 was specific to Fbp1; effects on Pck1 were negligible, and effects on Mdh2 were nuanced in increasing polyUb chain length while attenuating the amount of Mdh2 molecules consumed in the assay (Figure 1A). Third, adding Gid7 actually suppressed intrinsic GID E3 ligase activity, as shown by effects on Ub transfer from a pre-formed Ubc8∼Ub intermediate to free lysine in solution (Figure S1A). Binding of Fbp1’s degron per se is insufficient to overcome this inhibition because Gid7 likewise subdued ubiquitylation of a model peptide substrate in which Fbp1’s degron sequence, PTLV, is connected to a lysine acceptor through an intervening flexible linker (Figure S1B).
Figure 1.

Fbp1 ubiquitylation and degradation require a distinct Gid7-containing GID E3 ligase
(A) Fluorescence scans of SDS-PAGE gels showing in vitro ubiquitylation assays. These assays test the roles of Gid4 and Gid7 in ubiquitylation of C-terminally fluorescently labeled Fbp1 (left), Mdh2 (center), and Pck1 (right). GIDAnt contains 2 protomers each of Gid1 and Gid8 and 1 of Gid2, Gid5, and Gid9. An asterisk indicates that substrates are fluorescently labeled.
(B) In vitro ubiquitylation assay as in (A) but performed with lysine-less Ub (K0Ub) to determine the number of Fbp1 ubiquitylation sites.
(C) Plots showing fraction of Fbp1 ubiquitylation as a function of concentration of GIDSR4 (left) or its complex with Gid7 (center and right). Km values were determined by fitting to the Michaelis-Menten equation. Error bars, SD (n = 2).
(D) Comparison of kcat between GIDSR4 and its complex with Gid7, determined from plots in Figure S1D. Error bars, SD (n = 2).
(E) Assessing in vivo ubiquitylation of Fbp1 (C-terminally 3×FLAG-tagged at the endogenous locus) under carbon starvation (ethanol) and after 2 h of carbon recovery in WT and ΔGid7 yeast strains. Following capture of ubiquitylated proteins with TUBEs (tandem ubiquitin binding entities), Fbp1-3×FLAG was visualized by anti-FLAG immunoblotting.
(F) Glucose-induced degradation in vivo of exogenously expressed substrates Fbp1 (left), Mdh2 (center), and Pck1 (right), quantified using the promoter reference technique. Substrate levels were quantified as the ratio of substrate detected relative to the level after switching from carbon starvation to carbon recovery conditions in WT, ΔGid4 (top panels), and ΔGid7 (bottom panels) strains. Points represent mean, and error bars represent SD (n ≥ 3).
See also Figure S1.
To gain mechanistic insights, we quantified effects of including Gid7 in a chromatographically purified version of the E3 by performing enzyme kinetics. Compared with GIDSR4, a version of the E3 complex fully incorporating Gid7 displayed a relatively 10-fold lower Michaelis-Menten constant, Km, for Fbp1 ubiquitylation and 10-fold increase in the reaction turnover number kcat (Figures 1C, 1D, S1C, and S1D). Adding purified Gid7 to GIDSR4 had similar effects (Figures 1C and S1C).
Consistent with the biochemical data, glucose-induced ubiquitylation of Fbp1 in vivo is impaired by Gid7 deletion (Figure 1E). To examine effects on degradation, we employed a promoter reference technique that monitors degradation of exogenously expressed proteins (here, C-terminally FLAG-tagged Fbp1, Mdh2, or Pck1) while normalizing for effects on expression (Chen et al., 2017; Oh et al., 2017). Our assay agreed with prior studies showing that glucose-induced degradation of Fbp1, Mdh2, and Pck1 depends on Gid4 (Chen et al., 2017; Qiao et al., 2020; Regelmann et al., 2003; Santt et al., 2008). However, Gid7 deletion substantially stabilized only Fbp1 (Figure 1F). This deficit in Fbp1 degradation upon Gid7 deletion was not rescued by Gid4 overexpression (Figure S1E). Also, quantitative mass spectrometry analyses of the yeast proteome confirmed that, of known gluconeogenic GID E3 substrates, Fbp1 was most affected by Gid7 deletion (Figure S1F).
A supramolecular Chelator-GIDSR4 E3 assembly encapsulates the tetrameric Fbp1 substrate
To understand the mechanism of Fbp1 recognition by the GID E3, we purified an Fbp1-active recombinant complex and analyzed its structure by cryo-EM (Figures S2A and S4; Table S1). A 13-Å-resolution map of the assembly even without the substrate showed a remarkable GID E3 structure: an exterior oval supporting several inward-pointing globular domains. Strikingly, the longest exterior dimension of 305 Å is roughly comparable with that of a singly capped 26S proteasome, 1.3 times that of the multiprotein Fanconi anemia E3 ligase complex and 1.5 times that of APC/C (Figure 2A) (Brown et al., 2016; Chen et al., 2016; Haselbach et al., 2017; Lander et al., 2012; Schweitzer et al., 2016; Shakeel et al., 2019; Wehmer et al., 2017). Unlike these compact assemblies, however, this GID complex displays a behemoth hollow center with interior edges of 270 and 130 Å in the longest and shortest dimensions, respectively—larger than a cullin-RING ligase ubiquitylating a substrate (Baek et al., 2020).
Figure 2.

Multidentate capture of the Fbp1 tetramer by the Chelator-GIDSR4 assembly
(A) Cryo-EM map of GID E3 active toward Fbp1 compared for scale with low-pass-filtered maps of the singly capped 26S proteasome (EMDB: EMD-3536; PDB: 5MPB), Fanconi anemia core complex (EMDB: EMD-10290; PDB: 6SRI), APC/C (EMDB: EMD-3433; PDB: 5L9T), and cullin-RING E3 ubiquitylation complex (EMDB: EMD-10585; PDB: 6TTU).
(B) Cryo-EM maps and molecular weights of recombinant GID assemblies. Structurally determined GIDSR4 (left, low-pass-filtered, dark gray, EMDB: EMD 10327; PDB: 6SWY) is a stoichiometric complex of Gid1, Gid8, Gid5, Gid4, Gid2, and Gid9. The purification conditions used here include an additional Gid1-Gid8 subcomplex (gray) bound to GIDSR4 (center, taken for the biochemical assays). The oval higher-order Chelator-GIDSR4 assembly includes Gid7 dimers (right, white).
(C) Coomassie-stained SDS-PAGE (left) and cryo-EM maps of endogenous yeast GIDAnt (center) and Chelator-GIDAnt (right) assemblies (prepared by anti-FLAG immunoprecipitation of lysates from yeast with Gid5 3×FLAG tagged and Gid7 hemagglutinin (HA) tagged at their endogenous loci and grown under conditions when Gid4 is not induced).
(D) Cryo-EM map of Chelator-GIDSR4 (gray) bound to the Fbp1 tetramer (brown). The close up shows 2 red Gid4 protomers (modeled from PDB: 6SWY) simultaneously contacting the docked Fbp1 crystal structure.
The organization of the oval GID assembly was gleaned from comparison with cryo-EM maps of subcomplexes (Figure 2B; Table S1). Two copies of the previously defined GIDSR4 structure (Qiao et al., 2020) fit in the large assembly. An additional Gid1-Gid8 subcomplex can be observed bound to GIDSR4. These duplicated Gid1 and Gid8 protomers are components of recombinant GIDAnt used for biochemical assays (Qiao et al., 2020) but are not visible upon map refinement to high resolution. We interpreted the remaining density in the large oval GID assembly as Gid7 dimers, one at each vertex, given size exclusion chromatography-multi angle light scattering (SEC-MALS) data indicating that purified Gid7 dimerizes (Figure S2B). The data reveal a 1.5-MDa eicosameric GID assembly composed of 4 Gid1: 2 Gid2: 2 Gid4: 2 Gid5: 4 Gid7: 4 Gid8: 2 Gid9 protomers.
We sought to determine whether this GID assembly might be formed in vivo. Prior studies did (Santt et al., 2008) or did not (Qiao et al., 2020) observe Gid7 cosedimenting with other GID proteins in density gradients. This raised the possibility that, like the equally giant 26S proteasome, some subunits or regulatory partners may be prone to dissociation; for example, based on lysis conditions (Leggett et al., 2002). Thus, we examined sedimentation of a core subunit, Gid8 tagged at the endogenous locus, as a marker for a GID assembly because it cosediments with all other GIDSR4 subunits even under relatively harsh lysis conditions (Qiao et al., 2020). Yeast lysates prepared by cryomilling were subjected to sucrose density gradient fractionation. Anti-FLAG immunoblotting showed Gid8 migrating at a lower molecular weight in a Gid7 deletion compared with the WT, irrespective of whether yeast was grown under carbon starvation or recovery in glucose, which induces GID E3 ligase activity (Figure S2C). Moreover, cryo-EM data of endogenous GID purified from yeast grown under carbon starvation yielded 3D reconstructions corresponding to the recombinant assemblies with and without Gid7 at 14.2- and 9.5-Å resolution, respectively (Figures 2C and S2D).
Why is the minimum E3 ligase for Fbp1 so gigantic and hollow? Given the substantial effect on Km in our enzyme kinetics analyses, we hypothesized that such an assembly would form to accommodate the substrate. To characterize the substrate, we determined the crystal structure of yeast Fbp1, which confirmed its tetrameric assembly (Figures 2D and S2B; Table S2). We next resolved a cryo-EM structure with Fbp1 bound to the GID E3, which led to several conclusions (Figure 2D; Table S1). First, Fbp1 was readily docked in the center of the large GID E3 oval. Second, two Fbp1 edges approach the substrate binding Gid4 subunits within each GIDSR4 on opposite sides of the oval. Third, the density attributed to Gid7 does not directly contact Fbp1 but connects two Fbp1-binding GIDSR4 complexes. Thus, Gid7 activates GID E3 activity toward Fbp1 indirectly by driving supramolecular assembly.
The resultant GID assembly resembles an organometallic supramolecular chelate in which multiple giant organic molecules capture a much smaller ligand through multiple discrete points of contact. Thus, we call the giant oval complex “Chelator-GIDSR4” based on its supramolecular assembly in which two GIDSR4 complexes simultaneously capture degrons displayed from two protomers in the tetrameric Fbp1 substrate.
High-resolution structures of modules in Chelator-GIDSR4
A series of focused refinements enabled building atomic models of the three functionally distinct modules comprising Chelator-GIDSR4 (Figures 3A, S2E, S3A, and S4; Table S1): (1) the substrate receptor scaffolding (SRS) module contained in GIDSR4, responsible for bridging the substrate receptor to the other E3 ligase subunits; (2) the catalytic (Cat) module, also present in GIDSR4, which binds and activates the Ubc8∼Ub intermediate; and (3) a previously undescribed supramolecular assembly (SA) module.
Figure 3.

High-resolution details of Chelator-GIDSR4 modular assembly
(A) Focused refined maps of the substrate receptor scaffolding (SRS), catalytic (Cat), and supramolecular assembly (SA) modules, colored according to subunit identity, fit in half of the overall map of Fbp1-bound Chelator-GIDSR4 (top center). The GIDSR4 structure (PDB: 6SWY) fits the SRS module (Gid1SRS, dark green; Gid8SRS, salmon; Gid5, purple; Gid4, red). A brown arrow points to Gid4’s substrate binding site (top right). The Cat module comprises Gid2 (sky blue) and Gid9 (navy). Zinc ions are shown as gray spheres. Ubc8~Ub was modeled by aligning Gid2 RING with an E2~Ub-bound RING structure (PDB: 5H7S). The SA module comprises Gid1SA (green), Gid8SA (pink) and 2 Gid7 protomers, Gid7to-Cat (yellow), and Gid7to-SRS (orange) facing the Cat or SRS module, respectively. Superscript text refers to a module for a given Gid1 or Gid8 protomer. Arrows point to connected modules.
(B) Cartoon of Chelator-GIDSR4 with close ups of intermodule CTLH-CRAN:CTLH-CRAN interactions fit into the map of Chelator-GIDSR4 (gray).
(C) Intramodule LisH-CRAC:LisH-CRAC (solid ribbon) interactions in Chelator-GIDSR4.
See also Figures S3 and S4 and Table S1.
A 3.4-Å map of the Chelator-GIDSR4 SRS module fit the prior coordinates for this region (PDB: 6SWY) (Figures 3A and S4B). As described previously, the globular substrate-binding domain of Gid4 fits snugly in a complementary concave surface of the scaffold subunit Gid5. This arrangement is supported by a base from Gid1SRS and Gid8SRS, which form an intricate heterodimer involving their LisH-CTLH-CRA domains.
Focused refinement over the Cat module yielded a 3.8-Å-resolution reconstruction (Figures 3A and S4C). The map quality permitted de novo building and refinement of atomic coordinates for the majority of Gid2 and Gid9 (Figure S3A). The catalytic function is mediated by a region of Gid2 that adopts an E3 ligase RING domain fold (albeit stabilized by a single zinc in the E2∼Ub binding site) together with a portion of Gid9 that adopts a unique RING-like (RING-L) structure (Figure S3B; Braun et al., 2011; Qiao et al., 2020; Regelmann et al., 2003). Folding of the Gid2 RING depends on its incorporation into the intricately configured Gid2-Gid9 heterodimer. The Gid2 RING is embedded in an unprecedented intermolecular heart-shaped domain, stabilized by Gid9 elements, including an intermolecular zinc-binding domain; a belt that encases roughly three quarters of the base of Gid2’s RING; the RING-L domain, which packs against the remaining side of Gid2’s RING; and the extreme C terminus, which contributes to Gid2’s RING in a manner analogous to canonical RING dimers (Budhidarmo et al., 2012). Gid2 and Gid9 are further intertwined by their N termini co-assembling in an ∼70-Å-long 4-helix coiled coil (Figures 3A and S3A).
Within Chelator-GIDSR4, the two Gid2-Gid9 E3 ligase domains face the two degron-binding Gid4 subunits. A model of the Gid2 RING-Ubc8∼Ub intermediate based on published isolated RING E3-E2∼Ub complexes shows the Gid2 RING domain recruiting Ubc8, whereas its linked Ub would be activated by Gid2 and Gid9 in the canonically activated conformation (Figures 3A and S3B; Dou et al., 2012; Plechanovová et al., 2012; Pruneda et al., 2012). The model explains the previously reported effects of Gid2 and Gid9 point mutations on Fbp1 degradation (Qiao et al., 2020).
A 3.6-Å resolution map of the SA module within Chelator-GIDSR4 enabled building of an atomic model (Figures 3A and S4D). The two Gid7 protomers form an asymmetric dimer on one side of the module. Gid1SA and Gid8SA form an interdigitated scaffold that connects the Gid7 dimer to the Cat module.
Each Gid7 protomer consists of an N-terminal LisH-CTLH-CRA motif and an atypical β-propeller. The LisH-CTLH-CRA motifs form elongated helical double-sided dimerization domains (Figure S3C). The LisH and CTLH helices initially progress in one direction. The distal end is capped by the first two CRA helices. The remaining CRA helices reverse and traverse the length of the domain, pack against CTLH helices along the way, and terminate adjacent to the LisH helices. We refer to one side of the LisH-CTLH-CRA structure as “LisH-CRAC” because it contains the LisH and C-terminal CRA helices. Accordingly, the other side is called “CTLH-CRAN.” The Gid7 LisH-CRAC motifs mediate homodimerization, much like LisH-CRAC motifs mediate heterodimerization between Gid1SRS and Gid8SRS and between Gid2 and Gid9 (Qiao et al., 2020).
β-Propellers are protein interaction domains formed by toroidally arranged β sheet “blades” (Chen et al., 2011a). The 7-bladed propellers from the two Gid7 protomers ensue from the LisH-CTLH-CRA motifs at different relative angles and interact with each other. The resultant asymmetric double-propeller domain binds part of Gid1SA. The SA module is further stabilized by distinctive interactions between the CTLH-CRAN domains from Gid1SA, a loop from Gid8SA, and the CTLH-CRAN domain from a Gid7 protomer we call Gid7to-Cat because it points toward the Cat module (Figure S3D). The remainder of the Gid1SA and Gid8SA subcomplex superimposes on corresponding regions of Gid1SRS and Gid8SRS. At the two edges of the SA module, the CTLH-CRAN domains from the SRS-facing Gid7 protomer (Gid7to-SRS) and Gid8SA connect to the SRS and Cat modules, respectively.
Supramolecular chelate assembly is supported by inter- and intramodule LisH-CTLH-CRA domain interactions
The relative arrangement of E3 ligase elements—the Gid4 substrate receptor and the Gid2-Gid9 RING-RING-L complex—in Chelator-GIDSR4 depends on the exterior oval band. The oval is established by two types of intersubunit interactions—within the modules and mediating intermodule connections—in a daisy chain-like arrangement of LisH-CTLH-CRA domains (Figures 3B and 3C).
In Chelator-GIDSR4, the modules are connected to each other by outward-facing heterotypic dimerization of CTLH-CRAN domains at the edges of each module (Figure 3B). The CTLH-CRAN domains connect modules in a side-by-side manner. In the GIDSR4 assembly, the SRS and Cat modules are adjoined by interactions between the CTLH-CRAN domains of Gid8SRS and Gid9. The Cat and SA modules are bridged by interactions between the CTLH-CRAN domains of Gid2 and Gid8SA. Notably, Gid2’s CTLH-CRAN domain also packs against Gid9’s RING-L domain, which may explain how formation of the Chelator-GIDSR4 assembly affects intrinsic Ub transferase activity (Figures 3A, S1A, and S3B). The oval structure also depends on adjoining the SRS and SA modules through interactions between the CTLH-CRAN domains of Gid1SRS and Gid7to-SRS. Despite the similarity of intermodule interactions at a secondary structural level, specificity is dictated by contacts between domains, ensuring formation of the Chelator-GIDSR4 assembly.
Chelator-GIDSR4 assembly mediates avid recruitment of the tetrameric substrate Fbp1
Comparing the major classes of Chelator-GIDSR4 alone or bound to Fbp1 showed relative repositioning of the SRS module toward the center of the oval to bind the substrate, resembling a Venus flytrap capturing its prey (Figure 4A). An individual Fbp1 Pro/N-degron was visualized bound to Gid4 in a locally refined map of SRS (Figures 4B and S4B). Fbp1’s N-terminal proline and two subsequent residues are recruited much like short peptides binding human Gid4 (Chen et al., 2017; Dong et al., 2018; Hämmerle et al., 1998). Comparing the substrate-bound Chelator-GIDSR4 structure with the substrate-free GIDSR4 (Qiao et al., 2020) shows remodeling of several Gid4 loops to embrace the N-terminal residues PTL of the Fbp1 substrate (Figure 4B).
Figure 4.

Chelator-GIDSR4 assembly specifies multivalent binding for the tetrameric Fbp1 substrate
(A) Superimposed maps of substrate-free (gray) and Fbp1-bound Chelator-GIDSR4 (brown) show relative inward movement of SRS modules (ribbon) upon substrate recruitment.
(B) Conformational differences between Gid4 in GIDSR4 (PDB: 6SWY, gray) and Fbp1-bound Chelator-GIDSR4 (red). The first three residues of Fbp1 (the Pro/N-degron) bound to Gid4 are shown as sticks.
(C) Crystal structure of the Fbp1 tetramer, with the N-terminal region (residues 2–19), including the degron not visible in the electron density, depicted as dotted lines. Fbp1 protomers are shown in various brown shades.
(D) Competitive in vitro ubiquitylation assays probing multivalent E3-substrate interactions. Chelator-GIDSR4 has two substrate binding sites and two catalytic centers, whereas two other E3 assemblies (GIDSR4 or GIDSR4 + Gid7MUT lacking the LisH-CTLH-CRA motif, Δ1–285) have only one substate binding site and one catalytic center. Substrates are oligomeric (tetrameric Fbp1) or monomeric (a peptide harboring a single acceptor Lys, Fbp1-pep) and fluorescently labeled at the C terminus (denoted by an asterisk). Competitors are oligomeric (tetrameric Fbp1tet K/R, with preferred target lysines mutated to arginines) or monomeric (lysine-less peptide, Fbp1pep K0).
Notably, the Pro/N-degrons and several subsequent residues are not visible in the Fbp1 crystal structure, suggesting that they are intrinsically disordered (Figure 4C). These elements could emanate from opposite sides of the disk-like Fbp1 catalytic domain. In the complex with Chelator-GIDSR4, degrons from both sides appear to simultaneously ensnare Gid4 substrate receptors. Such avid binding would rationalize the 10-fold lower Km in Fbp1 ubiquitylation assays (Figure 1C). To further test the possibility of avid substrate capture, we performed competitive qualitative ubiquitylation assays. Unlabeled monomeric and tetrameric Fbp1 competitors had a comparable inhibitory effect on ubiquitylation of fluorescent Fbp1 by GIDSR4 or GIDSR4 mixed with a Gid7 mutant that does not support supramolecular assembly (Figure 4D). However, compared with an unlabeled monomeric inhibitor, the unlabeled Fbp1 tetramer was strikingly more effective at impeding Chelator-GIDSR4 ubiquitylation of fluorescent Fbp1. The same inhibitory trends were observed for ubiquitylation of a fluorescent monomeric peptide substrate, confirming that the Fbp1 tetramer complements the Chelator assembly. The data are consistent with avid Fbp1 recruitment to Chelator-GIDSR4 depending on supramolecular assembly of the E3 ligase and its substrate.
Chelator-GIDSR4 assembly establishes dual site-specific Ub targeting
We next mapped regions of Fbp1 engaging the ubiquitylation active sites. Locating di-Gly sites by mass spectrometry identified Chelator-GIDSR4-mediated ubiquitylation of two pairs of neighboring lysines, K32/K35 and K280/K281, preferentially from 18 potential target lysines on the surface of Fbp1 (Figure S5). The importance of these lysines was confirmed mutationally (Figures 5A and 5B). Use of K0 Ub had shown modification of up to two sites in an Fbp1 protomer during the time course of the experiment (Figure 1B). Eliminating either lysine pair reduced this to monoubiquitylation, with a slightly greater effect on the K32/K35 mutant (Figure 5A). The results suggest that either region can be ubiquitylated independent of the other but that, for a given protomer, ubiquitylation is restricted to one lysine within a pair. Testing the effects of the mutations on Fbp1 degradation confirmed the importance of these lysines in vivo, with substantial stabilization even upon mutating only the K32/K35 lysine pair (Figure 5B).
Figure 5.

Chelator-GIDSR4 configures simultaneous targeting of specific lysine clusters in metabolic regulatory regions of the Fbp1 tetramer
(A) In vitro ubiquitylation of Fbp1-6xHis, detected by anti-His immunoblotting, with WT (top) or K0 (bottom) Ub, testing the effects of mutating the major Fbp1 Ub-targeted lysines identified by mass spectrometry.
(B) Glucose-induced degradation in vivo of exogenously expressed WT or lysine mutant versions of Fbp1. Substrate levels were quantified as the ratio of substrate detected relative to the level after switching from carbon starvation to carbon recovery conditions. Points represent mean, and error bars represent SD (n = 3).
(C) Structural model of Chelator-GIDSR4-mediated ubiquitylation of Fbp1. Ubc8~Ub was modeled by aligning a RING-E2~Ub structure (PDB: 5H7S) on Gid2 RING. Dotted lines indicate disordered Fbp1 N termini. Close ups show major Fbp1 ubiquitylation sites near substrate (Fructose-1,6-bisphosphate, F-1,6-BP) and allosteric AMP binding sides modeled from structures with human Fbp1 (PDB: 5ZWK and 5ET6).
(D) Structure-based cartoon of Fbp1 ubiquitylation as shown in (C). Stars and hexagons represent substrate-binding and allosteric sites in Fbp1, respectively.
(E) In vitro Fbpase activity of purified WT, polyubiquitylated, and mutant Fbp1 (K32A/K35A/K280A/K281A).
(F) Fbpase activity assay as in (E), testing the responses of purified WT, polyubiquitylated, and mutant Fbp1 (K32A/K35A/K280A/K281A) to the allosteric inhibitor AMP.
See also Figure S5.
To understand how the Chelator-GIDSR4 supramolecular assembly determines regulation, we generated a structural model of ubiquitylation (Figures 5C and 5D). Fbp1 was first anchored via two degrons, one from each side binding a Gid4. Ubc8∼Ub was modeled on the Gid2-Gid9 RING-RING-L domains based on homology to another RING-E2∼Ub assembly (Nayak and Sivaraman, 2018). Fbp1 was subjected to constrained rotation to localize the K32 and K35 region of one protomer adjacent to one active site. This led to two striking observations. First, the K32 and K35 regions of two pairs of protomers are adjacent to each other. Second, and unexpectedly, when a K32 and K35 region is aligned with one active site, the K280 and K281 region of a different Fbp1 protomer is simultaneously situated in the other Chelator-GIDSR4 active site. Thus, the Chelator-GIDSR4 supramolecular assembly complements the tetrameric structure of Fbp1 by enabling simultaneous capture of two Pro/N degrons and simultaneous ubiquitylation of multiple protomers within the Fbp1 tetramer.
Given that Fbp1 is allosterically regulated in response to metabolite binding (Ke et al., 1990a, 1990b), we inspected the structure for potential functional importance of the ubiquitylation sites (Figures 5C and 5D). Intriguingly, the K32 and K35 residues reside in a loop abutting the allosteric site that regulates Fbp1 activity by binding the non-competitive inhibitor AMP (Ke et al., 1990b). K280 and K281 are located adjacent to another interprotomer interface, relatively near the substrate binding site (Ke et al., 1990a). We thus examined the effects of Chelator-GIDSR4 ubiquitylation on Fbp1 activity. A K32A/K35A/K280A/K281A mutant and a ubiquitylated version of Fbp1 show Fbpase activity in our assay. However, allosteric modulation by AMP was substantially impaired in both cases (Figures 5E and 5F). Thus, Chelator-GIDSR4 targets sites related to Fbp1’s metabolic function.
Structural and mechanistic parallels in human CTLH E3
To determine whether structural principles governing activity of the yeast GID E3 are conserved in higher eukaryotes, we studied the human CTLH complex, whose subunits mirror those of Chelator-GIDSR4 (Figure 6A).
Figure 6.

Higher-order assemblies of human CTLH E3
(A) Color-coded guide to yeast GID subunits and their human orthologs in the CTLH complex (top). Two colors indicate multiple protomers of a subunit. Cartoon colored as in the top, representing CTLH oval assembly where the SA module is the WDR26-YPEL5 dimer (bottom).
(B) 3.2-Å-resolution segmented map of CTLH SRS module (RANBP9-TWA1-ARMC8-hGid4) obtained by focused refinement of CTLHSR4 (top) and its corresponding model (bottom).
(C) Cryo-EM maps of CTLH assemblies containing the Cat (RMND5A-MAEA), SRS (RANBP9-TWA1-ARMC8 alone or bound to hGid4), and/or supramolecular assembly (WDR26 with or without YPEL5) modules, as indicated. Subunits are colored according to the guide in (A). Top left: low-resolution map of WDR26-mediated SA of CTLH (RANBP9-TWA1-ARMC8-MAEA-RMND5A-WDR26). Right: 6.5-Å-resolution map of the human CTLH SRS module (RANBP9-TWA1-ARMC8-hGid4) subcomplex with an SA module comprising WDR26-YPEL5. Bottom panel: the yeast Gid2-Gid9 structure in the corresponding CTLH Cat module.
(D) 10.4-Å-resolution map of the human CTLH SRS module with MKLN1 as the SA module. The second copy of the SRS module in the subcomplex is transparent.
(E) Immunoblots of fractions from sucrose gradients of K562 cell lysates, probed with the indicated antibodies.
(F) Immunoblots probing for the core CTLH subunit (RANBP9) in fractions from sucrose gradients of lysates from parental K562 and WDR26−/−/MKLN1−/−, MKLN1−/−, WDR26−/−, and MAEA−/− knockout cells. Black boxes delineate high- and low-molecular weight (MW) peak fractions.
(G) As in (F) but probed as indicated with anti-MKLN1 or -WDR26 antibodies. ∗, WDR26 band.
See also Figures S6 and S7 and Table S1.
We first reconstituted a recombinant complex that we call “CTLHSR4,” which parallels yeast GIDSR4. A low-resolution cryo-EM envelope showed that the corresponding human subunits form SRS (hGid4-ARMC8-RANBP9-TWA1) and Cat (RMND5A-MAEA) modules (Figure S6A). As for yeast GIDSR4 (Qiao et al., 2020), the CTLHSR4 Cat module is relatively poorly resolved, but the coordinates for the yeast Gid2-Gid9 subcomplex derived from Chelator-GIDSR4 readily fit in the density. A 3.2-Å-resolution map obtained by focused refinement enabled building of atomic coordinates for the human SRS module, which superimposes on its yeast counterpart (Figures 6B, S6B and S7; Table S1).
We tested whether the structural conservation extended to the enzymatic mechanism. Because the Pro/N-end degron targets of the CTLH E3 remain unknown, we generated a model peptide substrate: an N-terminal PGLW sequence reported previously to optimally bind hGid4 (Dong et al., 2018, 2020), connected via a flexible linker to a C-terminal target lysine. With this peptide substrate, we tested the effects of structure-based point mutations on ubiquitylation. The hGid4 residues mediating its incorporation into CTLHSR4 and RMND5A and MAEA residues that activate UBE2H∼Ub are crucial for peptide substrate ubiquitylation (Figures S6C–S6H). Moreover, as with GIDSR4 (Qiao et al., 2020), only K48 of all Ub lysines was sufficient to support polyUb chain formation by CTLHSR4, albeit to a substantially lesser degree than WT Ub (Figure S6I). Thus, it seems that the human CTLH core module parallels that in yeast GID assemblies.
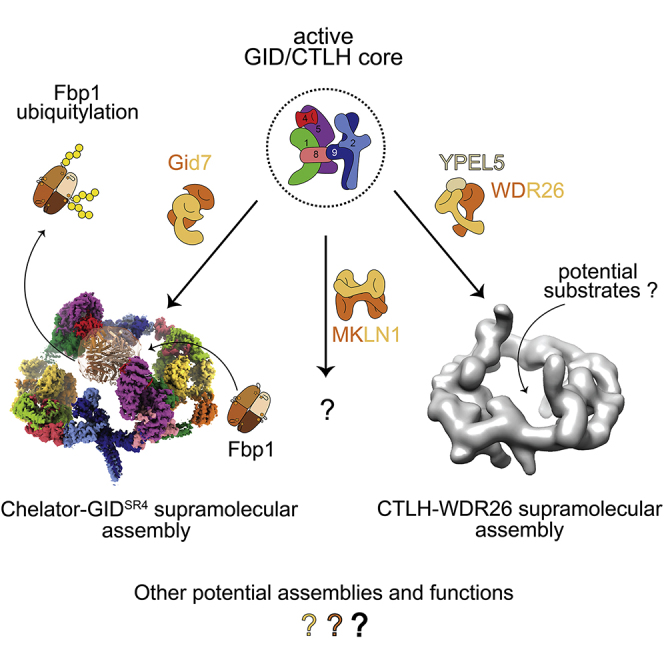
We examined by cryo-EM whether the human Gid7 orthologs WDR26 and MKLN1 have capacity for supramolecular assembly. We obtained reconstructions for two subcomplexes containing WDR26. Coexpressing WDR26 with scaffolding and catalytic subunits (ARMC8-RANBP9-TWA1-RMND5A-MAEA) yielded a complex broadly resembling Chelator-GIDSR4 in that it forms a hollow oval of similar dimensions (Figures 6A and 6C). Docking structures of human and yeast subcomplexes into the density showed that a WDR26 dimer is the SA module. However, WDR26 binds directly to RANBP9-TWA1 in the scaffold, without duplicates of these subunits corresponding to yeast Gid1SA-Gid8SA. The distinct WDR26-dependent supramolecular assembly places four—not two—ARMC8 subunits poised to each bind a hGid4 to capture substrate degrons in the CTLH oval.
The distinctive arrangement of SA and SRS modules was preserved in a 6-Å resolution map of WDR26, RANBP9, TWA1, ARMC8, hGid4, and the poorly understood CTLH subunit YPEL5 (Figure 6C; Table S1). Interestingly, YPEL5 binds at the junction of the two protomers in the WDR26 double-propeller domain.
A low-resolution map showed yet another SA for another human Gid7 ortholog, MKLN1, bound to the CTLH SRS module (Figure 6D; Table S1). Like WDR26, MKLN1 binds directly to RANBP9-TWA1 in the scaffold without intervening duplicates of these subunits. However, in accordance with previous studies (Delto et al., 2015; Kim et al., 2014), MKLN1 forms a tetramer. Four MKLN1 protomers bind between two CTLH SRS modules, demonstrating potential for even higher-order CTLH complex assemblies.
We confirmed roles of WDR26 and MKLN1 in human CTLH complex assembly by sedimentation analyses of lysates from K562 cells or lines in which the human Gid7 orthologs were deleted. Immunoblotting of fractions from sucrose density gradients of parental K562 cell lysates showed comigration of CTLH subunits, corresponding to a complex with a molecular weight greater than that predicted for a uniformly stoichiometric assembly (600–800 kDa according to standards) (Figure 6E). However, probing migration of the core subunit RANBP9 as a marker for the CTLH complex showed that the assembly changes markedly, toward fractions of 150–350 kDa, in CRISPR-Cas9 genome-edited lines lacking WDR26, MKLN1, or both or the Cat module subunit MAEA (Figures 6F and S6J). Interestingly, migration of WDR26 and MKLN1 in higher-molecular-weight fractions is not interdependent (Figure 6G), possibly indicating that each Gid7 ortholog can reside in distinct CTLH assemblies. Much of the total CTLH population shifted to lower-molecular-weight fractions upon deletion of WDR26, with a lesser effect of deleting MKLN1. This may suggest that a greater proportion of the CTLH complex in these cells depends on WDR26 for supramolecular assembly, perhaps because of a higher relative concentration of WDR26 or factors differentially regulating WDR26 or MKLN1 assembly into CTLH complexes.
Overall, the results suggest that CTLH E3 assemblies contain SRS, Cat, and SA modules with features resembling those of Chelator-GIDSR4. Moreover, differences in structural configuration of complexes containing MKLN1 or WDR26 offer prospects that CTLH may adopt a variety of supramolecular E3 assemblies that could impart distinct functionalities.
Discussion
Here we discovered multipronged substrate targeting by an E3 ligase chelate supramolecular assembly tailored to the oligomeric quaternary structure of its metabolic enzyme substrate. In the absence of chelate assembly, GIDSR4 is a competent E3 ligase that can bind a substrate degron, activate the intrinsic reactivity of its E2 partner (the Ubc8∼Ub intermediate), and promote Ub transfer from Ubc8 to a recruited substrate (Qiao et al., 2020). GIDSR4 is also competent in vivo insofar as Gid7 is not required for glucose- and GID-dependent degradation of several substrates (Figure 1). Instead of binding directly to its specified substrate Fbp1, Gid7 alters the GID assembly (Figures 2 and 3).
Although other E3s have been reported to self-assemble (Balaji and Hoppe, 2020), this is typically achieved by catalytic or substrate receptor subunits; for example, the dimeric RING domains of single-subunit E3s or dimeric F-box and BTB substrate receptors in multisubunit cullin-RING ligases (Dou et al., 2012; McMahon et al., 2006; Ogura et al., 2010; Plechanovová et al., 2012; Welcker et al., 2013; Zhuang et al., 2009). Substrate-bound multivalent E3s can undergo liquid-liquid phase-separation (Bouchard et al., 2018). However, the transformation into Chelator-GIDSR4 is a distinctive, extreme, and specific adjustment of E3 ligase architecture (Figures 2 and 3).
Resembling an organometallic chelate interacting with its central ligand, Chelator-GIDSR4’s multiple distinct points of contact with Fbp1 not only include the degron-binding sites from two opposing Gid4 substrate receptors but also the ubiquitylation active sites from Ubc8∼Ub intermediates activated by two opposing Gid2-Gid9 catalytic domains (Figures 4, 5, and 7). Relative to the monodentate GIDSR4, the Chelator-GIDSR4 assembly enables more molecules within the Fbp1 tetramer to be ubiquitylated simultaneously, increasing Ub density on a given Fbp1 tetramer (Figures 1A and 1B). Interestingly, there is not a 1:1 correspondence between the number of degron binding sites in Chelator-GIDSR4 and the number of degrons in Fbp1. The Fbp1 tetramer has four exposed potential degrons, two on each side, both seemingly poised to capture one central-facing Gid4 in Chelator-GIDSR4 (Figure 4C). An excess number of degrons is reminiscent of substrates recruited to the cullin-RING ligase receptor Cdc4, whose single binding site can continually and dynamically sample multiple degrons (Mittag et al., 2008). For Chelator-GIDSR4-bound Fbp1, we speculate that the arrangement of degrons allows their rapid interchange. This could potentially mediate switching between the protomers positioned adjacent to the active sites.
Figure 7.

Molecular logic of multipronged Ub targeting of Fbp1 by Chelator-GIDSR4
Supramolecular chelate assembly specifies oligomeric metabolic enzyme targeting. (1) Opposing Gid4 subunits avidly bind multiple degrons of tetrameric Fbp1. (2) Opposing RING-E2~Ub active sites simultaneously target specific lysine clusters. (3) Targeted lysines map to metabolically important regions of oligomeric substrate.
The human CTLH E3 complex displays striking parallels to Chelator-GIDSR4, albeit with interesting twists. In particular, the different Gid7 orthologs form distinct supramolecular assemblies (Figure 6). We speculate that the unique assemblies define distinct functions, as implied by varying phenotypic alterations upon their individual mutation (Bauer et al., 2018; Nassan et al., 2017; Skraban et al., 2017; Zhen et al., 2020).
Taken together with previous data (Lampert et al., 2018; Qiao et al., 2020), it is now clear that there is not a single yeast GID or human CTLH complex. Rather, GID and CTLH are examples of responsive systems of multiprotein assemblies with an active E3 core that can be elaborated by supramolecular assembly. Although the function of one such assembly is shown here, the variations revealed by human Gid7 orthologs suggest that they, and presumably other subunits, also co-configure substrate binding and ubiquitylation active sites in accordance with the molecular organization and quaternary structure of particular substrates. The Chelator model presented here demonstrates how GID (and presumably CTLH) utilizes an elegant molecular logic: the response to a signal such as glucose availability converges on numerous aspects of its substrate’s structure and function to achieve precise physiological regulation (Figure 7).
Limitations
Chelator-GIDSR4 is remarkably specific in ubiquitylating particular Fbp1 lysines in metabolic regulatory regions. However, the physiological roles of Fbp1 ubiquitylation impairing allosteric regulation and metabolic function are unknown. Future studies will be required to determine how metabolic flux is coupled with GID-dependent ubiquitylation during termination of gluconeogenesis.
Although Chelator-GIDSR4 is active toward Mdh2 and Pck1, it is unclear why these oligomeric substrates are less dependent than Fbp1 on Gid7-mediated supramolecular assembly. One speculative possibility could be that any potential advantage of avid binding is offset by accessibility of numerous ubiquitylation sites to GIDSR4. Future studies will be required to understand how Pck1 and other GID E3 substrates, including the Gid4 substrate receptor itself, are recognized and ubiquitylated (Hämmerle et al., 1998; Karayel et al., 2020; Menssen et al., 2018).
Finally, although discovery of the Chelator configuration provides a basis for understanding higher-order GID assembly, what other assemblies or sub-assemblies may form and their functions remain unknown. Clearly, other arrangements are observed for human CTLH complexes with WDR26. MKLN1 forms an even higher-order assembly with the human SRS module. Some yeast GID assemblies migrate in the void volume, as seen by size-exclusion chromatography (Figure S2A). Moreover, the mechanistic roles of additional subunits, including YPEL5 (Figure 6), or regulatory partners, such as Cdc48/p97, remain unknown (Barbin et al., 2010; Lampert et al., 2018). We await future studies revealing functions of other variations of GID and CTLH assemblies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2 antibody | Sigma Aldrich | Cat# F1804; RRID: AB_262044 |
| Anti-HA antibody produced in rabbit | Sigma Aldrich | Cat# H6908; RRID: AB_260070 |
| Goat anti-rabbit IgG Dylight488 conjugated | Invitrogen | Cat# 35552; RRID: AB_844398 |
| Goat anti-mouse IgG Dylight633 conjugated | Invitrogen | Cat# 35512; RRID: AB_1307538 |
| Anti-rabbit peroxidase antibody produced in goat | Sigma Aldrich | Cat# A9169; RRID: AB_258434 |
| Anti-mouse IgG Peroxidase antibody produced in goat | Sigma Aldrich | Cat# A4416; RRID: AB_258167 |
| Anti-His antibody produced in mouse | Cell Signaling Technology | Cat# 9991; RRID: AB_2797714 |
| Goat polyclonal anti-RMND5A antibody | Santa Cruz | Cat# sc-161202, RRID: AB_2181510 |
| Sheep polyclonal anti-MAEA antibody | R&D Systems | Cat# AF7288-SP, RRID: AB_10971438 |
| Rabbit polyclonal anti-RANBP9 antibody | Abnova | Cat# PAB16671; RRID: AB_10677213 |
| Rabbit polyclonal anti-TWA1 antibody | Novus | Cat# NBP1-32596; RRID: AB_2274921 |
| Mouse monoclonal anti-ARMC8 antibody | Santa Cruz | Cat# sc-365307; RRID: AB_10850172 |
| Mouse monoclonal anti-MKLN1 antibody | Santa Cruz | Cat# sc-398956; RRID: AB_2737249 |
| Rabbit polyclonal anti-WDR26 antibody | Bethyl Laboratories | Cat# A302-245A; RRID: AB_1730876 |
| Rabbit polyclonal anti-YPEL5 antibody | Thermo Fisher | Cat# PA5-26957; RRID: AB_2544457 |
| Sheep polyclonal anti-hGid4 | This study | N/A |
| HaloLink Resin | Promega | Cat# G1912 |
| ANTI-FLAG M2 affinity gel | Sigma Aldrich | Cat# A2220 |
| His-Select Nickel affinity gel | Sigma Aldrich | Cat# P6611 |
| Glutathione Sepharose 4B | GE Healthcare | Cat# 17075605 |
| StrepTactin Sepharose High Performance resin | cytiva | Cat# 28935599 |
| His-Halo UBAUBQLN1 | This study | N/A |
| Critical commercial assays | ||
| EnzChek Phosphate Assay Kit | ThermoFisher Scientific | Cat# E6646 |
| Bacterial and virus strains | ||
| E. coli BL21 RIL (DE3) | MPIB | N/A |
| E. coli DH5α | MPIB | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| complete EDTA free | Roche | Cat# 05056489001 |
| Aprotinin from bovine lung | Sigma | A1153-10MG |
| Leupeptin | Sigma | L2884-250MG |
| Benzamidine | Sigma | B6506-25G |
| GGGGGFYVK-FAM | MPIB | N/A |
| PTLVNGWPR | MPIB | N/A |
| PTLVNGPRRDSTEGFTGRGWSGRGWS KGGK-FAM |
MPIB | N/A |
| PGLWRSPRRDSTEGFTGRGWSGRG WSKGGK-FAM |
MPIB | N/A |
| 3xFLAG peptide | MPIB | N/A |
| Deposited data | ||
| Apo Chelator-GIDSR4 | This study | EMDB: EMD-12541 |
| Chelator-GIDSR4 + Fbp1 | This study | EMDB: EMD-12557 |
| GIDSR4 | This study | EMDB: EMD-12548 |
| SRS module | This study | EMDB: EMD-12559; PDB: 7NS3 |
| Cat module | This study | EMDB: EMD-12560; PDB: 7NS4 |
| SA module | This study | EMDB: EMD-12563; PDB: 7NSB |
| Endogenous GIDAnt | This study | EMDB: EMD-12538 |
| Endogenous Chelator-GIDAnt | This study | EMDB: EMD-12540 |
| CTLHSR4 | This study | EMDB: EMD-12537 |
| CTLHSR4 SRS module | This study | EMDB: EMD-12564; PDB: 7NSC |
| CTLH-WDR26 SA and SRS modules | This study | EMDB: EMD-12545 |
| CTLH-WDR26 supramolecular assembly | This study | EMDB: EMD-12542 |
| CTLH-MKLN1 SA and SRS modules | This study | EMDB: EMD-12547 |
| Fbp1 (crystal structure) | This study | PDB: 7NS5 |
| Proteomics data | This study | PXD024462 |
| Raw image data | This study | http://dx.doi.org/10.17632/rfpsg6939c.1 |
| Experimental models: Cell lines | ||
| Sf9 Insect cells | Thermo Fisher | Cat# 11496015 |
| High Five Insect cells | Thermo Fisher | Cat# B85502 |
| K562 human cells | ATCC | ATCC#CCL-243; RRID: CVCL_00004 |
| Experimental models: Organisms/strains | ||
| Saccharomyces cerevisiae: Strain S288C: BY4741; MATa his3Δ1leu2Δ0 met15Δ0 ura3Δ0 | Euroscarf | Cat# Y00000 |
| CRLY12; BY4741, Gid4::KANMX | This study | N/A |
| CRLY14; BY4741, Gid7::KANMX | This study | N/A |
| CRLY45; BY4741, Gid8::Gid8-3xFLAG-KANMX | Qiao et. al., 2020 | N/A |
| CRLY131; BY4741, Gid2::3xFLAG-Gid2 (K365A) | Qiao et. al., 2020 | N/A |
| CRLY241; BY4741, Gid7::KANMX, Gid8::Gid8-3xFLAG-HPHNT1 | This study | N/A |
| CRLY267; BY4741, Gid7::Gid7-3xHA-HPHNT1, Gid5::Gid5-3xFLAG-KANMX | This study | N/A |
| CRLY498; BY4741, Fbp1::Fbp1-3xFLAG-HPHNT1, Pdr5::NATNT2 | This study | N/A |
| CRLY504; BY4741, Fbp1::Fbp1-3xFLAG-HPHNT1, Pdr5::NATNT2, Gid7:KANMX | This study | N/A |
| Recombinant DNA | ||
| pCSJ95 | Chen et al., 2017 | N/A |
| pCSJ125 | Chen et al., 2017 | N/A |
| VBP6; pRS313-pGPD-Pck1-3xFLAG-CYC-pGPD-DHFR-HA-CYC | This study | N/A |
| DSJC1; pRS313-pGPD-Fbp1-K32R/K35R-3xFLAG-CYC-pGPD-DHFR-HA-CYC | This study | N/A |
| DSJC2; pRS313-pGPD-Fbp1-K280R/K281R-3xFLAG-CYC-pGPD-DHFR-HA-CYC | This study | N/A |
| DSJC3; pRS313-pGPD-Fbp1-K32R/K35R/K280R/K281R-3xFLAG-CYC-pGPD-DHFR-HA-CYC | This study | N/A |
| pRS415-pTEF-CYC | This study | N/A |
| pRS415-pTEF-GFP-ScGid4-CYC | This study | N/A |
| pLIB Gid1 | This study | N/A |
| pLIB Gid2 | This study | N/A |
| pLIB Gid4 | This study | N/A |
| pLIB Gid5 | This study | N/A |
| pLIB Gid7 | This study | N/A |
| pLIB Gid8-TEV-2xStrep | This study | N/A |
| pLIB Gid8 | This study | N/A |
| pLIB Gid9 | This study | N/A |
| pLIB RANBP9 | This study | N/A |
| pLIB RMND5A | This study | N/A |
| pLIB hGid4 | This study | N/A |
| pLIB ARMC8 | This study | N/A |
| pLIB 2xStrep-3C-ARMC8 | This study | N/A |
| pLIB MAEA | This study | N/A |
| pLIB WDR26 | This study | N/A |
| pLIB GST-TEV-WDR26 | This study | N/A |
| pLIB MKLN1 | This study | N/A |
| pLIB YPEL5 | This study | N/A |
| pLIB MAEA Y394A | This study | N/A |
| pLIB RMND5A R340A | This study | N/A |
| pLIB RMND5A I338A/L339A | This study | N/A |
| pLIB GST-TEV-Uba1 | This study | N/A |
| pBIG2 Gid1:Gid8-TEV-2xS:Gid5:Gid4:Gid2:Gid9 | This study | N/A |
| pBIG2 Gid1:Gid8-TEV-2xS:Gid5:Gid2:Gid9 | This study | N/A |
| pBIG2 Gid1:Gid8-TEV-2xS:Gid5:Gid2:Gid9:Gid7 | This study | N/A |
| pBIG2 Gid1:Gid8-TEV-2xS:Gid5:Gid4:Gid2:Gid9:Gid7 | This study | N/A |
| pBIG1 RANBP9:TWA1-TEV-2xS:ARMC8 | This study | N/A |
| pBIG1 RANBP9:TWA1:2xS-3C-ARMC8 | This study | N/A |
| pBIG2 RANBP9:TWA1-TEV-2xS:ARMC8:RMND5A:MAEA | This study | N/A |
| pBIG2 RANBP9:TWA1-TEV-2xS:ARMC8:RMND5A | This study | N/A |
| pBIG2 RANBP9:TWA1-TEV-2xS:ARMC8:MAEA | This study | N/A |
| pGEX GST-TEV-Gid4 (Δ1-116) | This study | N/A |
| pGEX GST-TEV-Gid7 | This study | N/A |
| pGEX GST-TEV-Gid7 (Δ1-285) | This study | N/A |
| pGEX GST-TEV-hGid4 (Δ1-99) | This study | N/A |
| pGEX GST-TEV-hGid4 (R189A) | This study | N/A |
| pGEX GST-TEV-hGid4 (Y154A) | This study | N/A |
| pGEX GST-TEV-hGid4 (F174A) | This study | N/A |
| pGEX GST-TEV-hGid4 (F239A) | This study | N/A |
| pGEX GST-TEV-hGid4 (C156D) | This study | N/A |
| pGEX GST-TEV-hGid4 (E298A) | This study | N/A |
| pGEX GST-TEV-hGid4 (H147D) | This study | N/A |
| pGEX GST-TEV-hGid4 (Y158A/F174A) | This study | N/A |
| pGEX GST-TEV-hGid4 (Y158A/F239A) | This study | N/A |
| pGEX GST-TEV-hGid4 (F174A/F239A) | This study | N/A |
| pGEX GST-TEV-hGid4 (Y297A/F229A) | This study | N/A |
| pGEX GST-TEV-hGid4 (Δ297-300) | This study | N/A |
| pRSF Ubc8-6xHis | This study | N/A |
| pRSF Fbp1-6xHis | This study | N/A |
| pRSF Fbp1 (K32R/K35R)-6xHis | This study | N/A |
| pRSF Fbp1 (K280R/K281R)-6xHis | This study | N/A |
| pRSF Fbp1 (K32R/K35R/K280R/K281R)-6xHis | This study | N/A |
| pRSF Fbp1-GGGGS-sortag-6xHis | This study | N/A |
| pRSF Mdh2-GGGGS-sortag-6xHis | This study | N/A |
| pRSF Pck1-GGGGS-sortag-6xHis | This study | N/A |
| pQlink Fbp1-TEV-V5-2xS | This study | N/A |
| pRSF Ube2H-6xHis | This study | N/A |
| pGEX GST-3C-Ub | This study | N/A |
| pGEX GST-3C-Ub K0 (all K > R) | This study | N/A |
| pGEX GST-3C-Ub K6 (all K > R; R6K) | This study | N/A |
| pGEX GST-3C-Ub K11 (all K > R; R11K) | This study | N/A |
| pGEX GST-3C-Ub K27 (all K > R; R27K) | This study | N/A |
| pGEX GST-3C-Ub K29 (all K > R; R29K) | This study | N/A |
| pGEX GST-3C-Ub K33 (all K > R; R33K) | This study | N/A |
| pGEX GST-3C-Ub K48 (all K > R; R48K) | This study | N/A |
| pGEX GST-3C-Ub K63 (all K > R; R63K) | This study | N/A |
| pET3b Ub | This study | N/A |
| pET29 sortase A | Chen et al., 2011b | N/A |
| Software and algorithms | ||
| FOCUS | Biyani et al., 2017 | https://focus.c-cina.unibas.ch/documentation.php |
| SerialEM | Mastronarde, 2003 | https://bio3d.colorado.edu/SerialEM/ |
| MOTIONCOR2 | Zheng et al., 2017 | https://emcore.ucsf.edu/ucsf-software |
| Gctf | Zhang, 2016 | https://www2.mrc-lmb.cam.ac.uk/download/gctf/ |
| Gautomatch | Kai Zhang | https://www2.mrc-lmb.cam.ac.uk/download/gautomatch-053/ |
| Relion3.0/3.1 | Fernandez-Leiro and Scheres, 2017; Scheres, 2012; Zivanov et al., 2018 | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| Phyre2 | Kelley et al., 2015 | http://www.sbg.bio.ic.ac.uk/∼phyre2/html/page.cgi?id=index |
| SWISS-MODEL | Waterhouse et al., 2018 | https://swissmodel.expasy.org |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX | Goddard et al., 2018 | https://www.rbvi.ucsf.edu/chimerax/ |
| PyMOL v2.1 | Schrödinger | https://pymol.org/2/ |
| CCP-EM | Burnley et al., 2017 | https://www.ccpem.ac.uk/download.php |
| Buccaneer | Cowtan, 2006 | http://www.ysbl.york.ac.uk/∼cowtan/buccaneer/buccaneer.html |
| Coot | Emsley and Cowtan, 2004; Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Adams et al., 2010; Afonine et al., 2018; DiMaio et al., 2013 | https://www.phenix-online.org/ |
| Molprobity | Chen et al., 2010 | http://molprobity.biochem.duke.edu/ |
| Image Studio | LI-COR Biosciences | https://www.licor.com/bio/image-studio/ |
| Fiji/ImageJ | Schindelin et al., 2012 | https://imagej.net/Welcome |
| GraphPad Prism version 8.0 | GraphPad Software | http://www.graphpad.com:443/ |
| ImageQuant TL Toolbox version 8.2 | Cytiva (formerly GE Healthcare) | https://www.cytivalifesciences.com |
| DeepEMhancer | Sanchez-Garcia et al., 2020 | http://www.biorxiv.org |
| Other | ||
| QUANTIFOIL® R1.2/1.3, 100 Holey Carbon Films, Grids: Cu 200 mesh | Quantifoil Micro Tools GmbH | https://www.quantifoil.com |
| IMEM | Thermo Fisher | Cat# 12440-053 |
Resource availability
Lead contact
Information and requests for resources and reagents should be directed to the Lead Contact, Prof. Dr. Brenda Schulman (schulman@biochem.mpg.de).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The accession codes for the PDB models and EM maps are available in RCSB and EMDB, respectively, as follows: Apo Chelator-GIDSR4, EMDB: EMD-12541; Chelator-GIDSR4 + Fbp1, EMDB: EMD-12557; GIDSR4, EMDB: EMD-12548; SRS module, EMDB: EMD-12559, PDB: 7NS3; Cat module, EMDB: EMD-12560; PDB: 7NS4; SA module, EMDB: EMD-12563; PDB: 7NSB; Endogenous GIDAnt, EMDB: EMD-12538; Endogenous Chelator-GIDAnt, EMDB: EMD-12540; CTLHSR4, EMDB: EMD-12537; CTLHSR4 SRS module, EMDB:EMD-12564; PDB: 7NSC; CTLH-WDR26 supramolecular assembly, EMDB: EMD-12542; CTLH-WDR26 SA and SRS modules, EMDB: EMD-12545; CTLH-MKLN1 SA and SRS modules, EMDB: EMD-12547; Fbp1 crystal structure, PDB: 7NS5.
All proteomics data have been deposited on ProteomeXchange with the dataset identifier PRIDE: PXD024462.
All the unprocessed image data have been deposited to Mendeley Data : http://dx.doi.org/10.17632/rfpsg6939c.1
Method details
Yeast strain construction and growth conditions
The yeast strains used in this study are specified in the Key Resources Table. They were constructed as derivatives of BY4741 using standard genetic techniques (Janke et al., 2004, Knop et al., 1999, Storici and Resnick, 2006) and were verified using PCR, DNA sequencing and immunoblotting (to confirm protein expression). Unless stated otherwise, yeast strains were grown to OD600 of 1.0 in synthetic dropout (SD-glucose; 0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% glucose, amino acid mix) or yeast peptone-based medium (YPD; 1% yeast extract, 2% peptone, 2% glucose) as indicated in the respective assays.
In vivo yeast substrate degradation assays
Degradation assays were performed to test the dependency of Fbp1, Mdh2, and Pck1 degradation on Gid4 and Gid7 (Figure 1F) using the promoter reference technique adapted from Oh et al. (2017). The respective strains were transformed with a plasmid harboring the open reading frame of either Fbp1-3xFLAG, Mdh2-3xFLAG or Pck1-3xFLAG and the control protein DHFR-HA, both expressed from identical promoters. Cells were grown in SD-glucose medium to OD600 of 1.0 before being starved in SE medium (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% ethanol, amino acid mix) for 19 hours. Subsequently, an equivalent of 1 OD600 was transferred to SD-glucose medium containing 0.5 mM tetracycline that inhibits translation of the respective substrate and DHFR by binding to specific RNA-regions within their ORFs. At the indicated time points, 1 mL or 1 OD600 of cells was harvested. Cell lysis was performed by resuspending the pellets in 800 μL 0.2 M NaOH and incubating them on ice for 20 minutes with subsequent centrifugation at 11,200xg for 1 minute at 4°C. The pellets were aspirated and resuspended in 50 μL HU buffer (8 M Urea, 5% SDS, 1 mM EDTA, 100 mM DTT, 200 mM Tris-HCl, pH 6.8, protease inhibitor, bromphenol blue), heated at 70°C for 10 minutes and then centrifuged again for 5 minutes at 11,200xg at 4°C. Protein levels of the substrates and a control protein DHFR were visualized by immunoblotting with anti-FLAG and anti-HA antibodies, respectively, and imaged using a Typhoon scanner (GE Healthcare). The bands were quantified using the ImageStudioLite software (LI-COR) and the substrate signal was normalized relative to the DHFR signal for every sample. At least three biological replicates were considered for all in vivo assays and the standard deviation was presented using error bars.
To validate the major ubiquitylation sites in Fbp1 in vivo (Figure 5B), the above-described PRT degradation assays were carried out in a similar manner with Fbp1-3xFLAG mutants, in which the lysines targeted by Chelator-GIDSR4 (K32, K35, K280 and K281) were mutated to arginine.
To test if overexpression of Gid4 affects degradation of Fbp1 in ΔGid7 yeast (Figure S1E), the GFP-Gid4 overexpression plasmid was transformed together with the Fbp1-3xFLAG PRT plasmid into different yeast strains (WT, ΔGid7 and a Gid2K365A catalytically inactive mutant). The cells were grown in SD medium lacking histidine and leucine, which served as selection markers for the Gid4 overexpression plasmid. After 8 h growth in SD-glucose media, samples of 1 OD600 were harvested and analyzed as described above.
Purification of endogenous yeast GID for cryo EM
To purify endogenous GID complex, 3 l of a yeast strain with Gid7 and Gid5 C-terminally tagged at their endogenous loci with an HA and 3xFLAG tag, respectively, were grown in YPD medium for 8 hours. Subsequently, the cells were washed and resuspended to OD600 of 1.0 in YPE medium (1% yeast extract, 2% peptone, 2% ethanol). Cells were harvested at OD600 of 18.0. The pellet was resuspended in the lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM CaCl2, 0.2 M sorbitol, complete protease inhibitor tablets) and frozen in liquid nitrogen in the form of small beads. For lysis, the frozen yeast pellets were subjected to cryogenic grinding using a cryo-mill (SPEX Sample Prep-6875 Freezer/Mill). The obtained yeast powder was thawed and centrifuged at 35,000 rpm for 10 minutes, and the resultant supernatant was incubated with ANTI-FLAG M2 affinity resin for an hour. After thorough washing, the protein was eluted using 3xFLAG peptide and visualized by Coomassie-stained SDS-PAGE. The eluted complex was concentrated to 1 mg/ml and analyzed by cryo EM.
Sucrose gradient fractionation of yeast lysates (Figure S2C)
Yeast strains with Gid8 C-terminally tagged at its endogenous locus with a 3xFLAG tag, with or without Gid7 deleted were grown in YPD media for 8 hours. Subsequently, they were switched to YPE medium and grown for 19 to 24 hours. One part of both cultures was harvested, while the other was switched to YPD medium for glucose recovery and harvested after 2 hours. The pellets were resuspended and lysed using a cryo-mill (as described above). To perform sucrose gradient fractionation of yeast lysates, roughly 300-500 mg of yeast powder was resuspended in the lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM CaCl2, 0.2 M sorbitol, complete protease inhibitor tablets). To aid in resolubilization, lysates were incubated for 15 minutes at 4°C with gentle agitation, and then pre-cleared by centrifugation at 17,000xg for 10 minutes. Protein concentration was normalized by Bradford assay, lysates were loaded onto a 5%–40% sucrose gradient, and centrifuged at 34,300 rpm for 16 hours at 4°C. Gradients were then fractionated into fourteen equal fractions and loaded onto a 12% SDS-PAGE gel. Proteins were visualized by immunoblotting and imaged with Amersham Typhoon imager (GE Healthcare).
In vivo Fbp1 ubiquitylation assay (Figure 1E)
Yeast strains with Fbp1 tagged at its endogenous locus with 3xFLAG were grown to OD600 of 1.0-1.5 in YPD, pelleted by centrifugation at 3,000 rpm for 3 min, washed with pre-warmed YPE, resuspended to an OD600 = 1 in fresh, pre-warmed YPE, and grown at 30°C for 18 hours. Cultures for the ethanol condition were then diluted to an OD = 1 in fresh, pre-warmed YPE containing 1% DMSO. For the recovery condition, cells were pelleted by centrifugation at 3,000 rpm for 3 minutes, and resuspended in fresh pre-warmed YPD containing 1% DMSO. After two hours of growth at 30°C, 50 ODs of cells were pelleted by centrifugation at 3,000 rpm for 3 minutes, and flash frozen in liquid nitrogen.
Samples were resuspended in 1 mL lysis buffer (50 mM Tris-HCl, pH7.5, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.1% SDS, 1% NP-40, 0.5% Na-deoxycholate, 1% glycerol, 20 mM NEM, and complete protease inhibitor tablets), and lysed by 3 rounds of 20 s in a FastPrep-24 instrument, resting 5 minutes on ice between each round. Lysates were then pre-cleared by centrifugation at 4,000xg for 10 minutes, and the supernatant was added to pre-equilibrated His-Halo-UBAUBQLN1-conjugated agarose beads, and incubated for 2 hours at 4°C with gentle rotation. Beads were separated by centrifugation at 800xg for 1 minute, washed once with lysis buffer and four times with wash buffer (50 mM Tris-HCl, pH7.5, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 0.1% SDS, 1% NP-40, 0.5% Na-deoxycholate, 1% glycerol). Proteins were eluted by addition of sample buffer, and heating at 95°C for 5 minutes. Samples were then loaded on a 12% SDS-PAGE gel and visualized by immunoblotting.
Plasmid preparation and Mutagenesis
All the genes encoding yeast GID subunits and the substrates Fbp1, Mdh2 and Pck1 were originally amplified from S. cerevisiae BY4741 genomic DNA. The genes coding for subunits of human CTLH were obtained from human cDNA library (Max Planck Institute of Biochemistry), except for hGid4, which was codon-optimized for bacterial expression system and synthesized by GeneArt gene synthesis service (Thermo Fisher Scientific). The sequences of all the CTLH genes correspond to the canonical UniProt sequences, besides ARMC8, for which isoform 2 (missing the residues 2-15 of the canonical sequence) was used based on the prior literature (Kobayashi et al., 2007).
The constructs for recombinant protein expression were generated by Gibson assembly method (Gibson et al., 2009), whereas the mutant versions of the genes were prepared by the QuickChange protocol (Stratagene). All the coding sequences used for protein expression were verified by DNA sequencing. To express GID/CTLH subunits from a single baculoviral expression vector, the genes were combined by the biGBac method (Weissmann et al., 2016). All the plasmids used in this study are listed in the Key resources table.
Insect cell expression and purification of GID/CTLH complexes
Both yeast GID and human CTLH complexes used for the biochemical assays and cryo EM were expressed in insect cells. For protein expression, Hi5 insect cells were transfected with recombinant baculovirus variants carrying the respective protein-coding sequences and grown for 60 to 72 hours in EX-CELL 420 Serum-Free Medium at 27°C. After harvesting, insect cell pellets were resuspended in a lysis buffer containing 50 mM HEPES pH 7.5, 200 mM NaCl, 5 mM DTT, 10 μg/ml leupeptin, 20 μg/ml aprotinin, 2 mM benzamidine, EDTA-free Complete protease inhibitor tablet (Roche, 1 tablet per 50 mL of buffer) and 1 mM PMSF.
All recombinant yeast GID complexes were purified from insect cell lysates by StrepTactin affinity chromatography by pulling on a twin-Strep tag fused at the Gid8 C terminus. Further purification was performed by anion exchange chromatography and size exclusion chromatography (SEC) in the final buffer containing 25 mM HEPES pH 7.5, 200 mM NaCl and 5 mM (Buffer A) or 1 mM DTT (Buffer B) for cryo EM and biochemical assays, respectively. To ensure a stoichiometric level of the substrate receptor Gid4 in all cryo EM samples, all GID complexes were expressed without Gid4, and a bacterially-expressed truncated version of Gid4 (Δ1-116) was added at a 2-fold molar excess to GidAnt (Gid1-Gid8-Gid2-Gid9-Gid5) before final SEC. To assemble Chelator-GIDSR4, both Gid4 (Δ1-116) and Gid7 were added to GidAnt at a 2-fold molar excess before final SEC. For the sample of Chelator-GIDSR4 with Fbp1 bound, 2-fold molar excess of the substrate was added to a purified and concentrated complex just before cryo EM grids preparation. A list of yeast GID complexes analyzed by cryo EM along with strategies for their expression and purification is shown below:
-
1.
Chelator-GIDSR4: Gid1, Gid2, Gid5, Gid8-2xS, Gid9 coexpressed in Hi5 insect cells; bacterially expressed Gid4 (Δ1-116) and Gid7 added before final SEC; purified by StrepTactin affinity, IEX and SEC
-
2.
Fbp1-bound Chelator-GIDSR4: Gid1, Gid2, Gid5, Gid8-2xS, Gid9 coexpressed in Hi5 insect cells; bacterially expressed Gid4 (Δ1-116) and Gid7 added before final SEC; purified by StrepTactin affinity, IEX and SEC; Fbp1-6xHis added directly before cryo EM grids preparation
-
3.
GIDSR4: Gid1, Gid2, Gid5, Gid8-2xS, Gid9 coexpressed in Hi5 insect cells; bacterially expressed Gid4 (Δ1-116) added before final SEC; purified by StrepTactin affinity, IEX and SEC
CTLHSR4 and CTLH-MKLN1 subcomplex comprising SA and SRS modules were purified from insect cell lysates by StrepTactin affinity chromatography by pulling on a twin-Strep tag fused at the TWA1 C terminus, whereas the CTLH-WDR26 subcomplex comprising SA and SRS modules was pulled on a twin-Strep tag at the ARMC8 N terminus. Further purification was performed by anion exchange chromatography and size exclusion chromatography in Buffer A or Buffer B. As for yeast GID, the CTLH subcomplexes used for cryo EM were saturated with hGid4 by mixing them with the bacterially-expressed truncated version of hGid4 (Δ1-99) and running SEC. CTLH-WDR26 supramolecular assembly was purified from lysates by a tandem affinity chromatography, by first pulling on TWA1-2xS and then GST-WDR26. The pull-down fractions were run on SEC in Buffer A. A list of human CTLH complexes analyzed by cryo EM along with strategies for their expression and purification is shown below:
-
1.
CTLH-WDR26 supramolecular assembly: RANBP9, TWA1-2xS, ARMC8, RMND5A, MAEA, GST-WDR26 coexpressed in Hi5 insect cells; purified by tandem StrepTactin and GST affinity and SEC
-
2.
CTLH-WDR26 SA and SRS modules: RANBP9, TWA1, 2xS-ARMC8, WDR26, YPEL5 coexpressed in Hi5 insect cells; bacterially expressed hGid4 (Δ1-99) added before final SEC; purified by StrepTactin affinity, IEX and SEC
-
3.
CTLH-MKLN1 SA and SRS modules: RANBP9, TWA1-2xS, ARMC8, MKLN1 coexpressed in Hi5 insect cells; bacterially expressed hGid4 (Δ1-99) added before final SEC; purified by StrepTactin affinity, IEX and SEC
-
4.
CTLHSR4: RANBP9, TWA1-2xS, ARMC8, RMND5A, MAEA coexpressed in Hi5 insect cells; bacterially expressed hGid4 (Δ1-99) added before final SEC; purified by StrepTactin affinity, IEX and SEC
Bacterial expression and purification
All bacterial expressions were performed in E. coli BL21 (DE3) RIL cells in a Terrific Broth (TB) medium overnight at 18°C.
All the mutant and WT versions of Gid4 (both yeast and human ortholog) and Gid7 were expressed as GST-TEV fusions. After harvesting, cell pellets were resuspended in the lysis buffer containing 50 mM HEPES pH 7.5, 200 mM NaCl, 5 mM DTT and 1 mM PMSF and purified from bacterial lysates by glutathione affinity chromatography, followed by overnight digestion at 4°C with tobacco etch virus (TEV) protease to liberate the GST tag. Further purification was carried out with size exclusion chromatography in Buffer B. Additionally, a pass-back over glutathione affinity resin was performed to get rid of the remaining uncleaved GST-fusion protein and free GST. Ubc8, Ube2H, Ub (for generating ubiquitylated Fbp1), Fbp1 (WT and mutants), Mdh2 and Pck1 were expressed as their C-terminally 6xHis-tagged versions. After harvesting, cell pellets were resuspended in the lysis buffer containing 50 mM HEPES pH 7.5, 200 mM NaCl, 5 mM β-mercaptoethanol, 10 mM imidazole and 1 mM PMSF, and purified from bacterial lysates by nickel affinity chromatography, followed by anion exchange and size exclusion chromatography in Buffer A or Buffer B (for structural studies and activity assays, respectively). Fbp1-V5-2xS (for Fbpase activity assays) was purified by StrepTactin affinity chromatography and SEC in Buffer B.
Untagged WT ubiquitin used for in vitro assays was purified via glacial acetic acid method (Kaiser et al., 2011), followed by gravity S column ion exchange chromatography and size exclusion chromatography in Buffer B. Different Ub variants as well as WT Ub used for the ubiquitin chain type determination assay were expressed as GST-3C fusions and purified by glutathione affinity chromatography, followed by incubation with HRV-3C protease for 3 hours at room temperature. Further purification was done with size exclusion chromatography in Buffer B.
Fluorescent tagging of the GID substrates Fbp1, Mdh2 and Pck1 used for all the biochemical assays was performed with a sortase A-mediated reaction, which catalyzed fusion of fluorescein to the C terminus of the substrate. The reaction mix contained 50 μM of the substrate, which was C-terminally tagged with a sortag (LPETGG) and a 6xHis tag, 250 μM of a fluorescent peptide (GGGGGFYVK-FAM) and 50 μM of sortase A (Chen et al., 2011b). The labeling reaction was carried out for 30 minutes at room temperature in a buffer comprising 50 mM Tris-HCl pH 8, 150 mM NaCl and 10 mM CaCl2. The reaction mixture was consecutively passed-back through the Ni-NTA Sepharose resin to get rid of unreacted Fbp1. Further purification was done with size exclusion chromatography in Buffer B.
All the labeled and unlabeled peptides used in the biochemical assays were synthesized in the MPIB Biochemistry Core Facility.
In vitro biochemical assays
All in vitro activity assays were performed at room temperature in a buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl, 5 mM ATP and 10 mM MgCl2. To ensure that all the reaction mixtures contained equal concentrations of WT and mutant versions of Gid4 and Gid7, these proteins were added exogenously for all assays besides kinetics. To analyze kinetics of Fbp1 ubiquitylation, the assays were performed with purified GIDSR4 and Chelator-GIDSR4 obtained by co-expressing all of their subunits, as well as GIDSR4 mixed with Gid7 before starting the reaction. All the reactions were quenched at indicated time points by mixing an aliquot of the total reaction mix with SDS-PAGE loading buffer. Ubiquitylation of fluorescent substrates was visualized by a fluorescent scan of SDS-PAGE gel using the Amersham Typhoon imager (GE Healthcare).
Biochemical assays with yeast GID
The influence of Gid7 and Gid4 on ubiquitylation of the full-length (Figures 1A and 1B) and peptide versions (Figure S1B) of Fbp1 was tested in a multiturnover assay format using 0.2 μM Uba1, 1 μM Ubc8-6xHis, 0.5 μM GIDAnt, 0 or 1 μM Gid4, 0 or 2 μM Gid7, 1 μM full-length Fbp1-FAM or a fluorescently labeled model peptide substrate and 20 μM Ub (WT or all K > R (K0) version). The model peptide substrate was designed with the N-terminal Fbp1 sequence (aa 2-16) and a single lysine placed at position 27 (to span the distance between the substrate receptor Gid4 and the catalytic center measured in the structure of Chelator-GIDSR4). Similarly, the influence of Gid7 and Gid4 on ubiquitylation of other gluconeogenic substrates, Mdh2 and Pck1, was tested in a multiturnover assay using their fluorescently labeled versions and carried out under identical conditions (Figure 1A). The same assay format and conditions were employed to qualitatively compare Fbp1 ubiquitylation activity of GIDSR4 exogenously mixed with Gid7 to that of the SEC-purified Chelator-GIDSR4 (containing co-expressed Gid7) (Figure S1C). All the assays were performed in at least duplicates and some of them were quantified using image analysis software ImageQuant (GE healthcare; version 8.2).
To test the influence of Gid7 on intrinsic activity of GID E3, a substrate-independent pulse-chase assay monitoring discharge of Ubc8∼Ub to free lysine in solution was employed (Figure S1A). In the pulse reaction, loading of Ubc8 was performed by mixing 0.5 μM Uba1, 10 μM Ubc8-6xHis, 30 μM Ub, 2.5 mM MgCl2 and 1 mM ATP. After 15 minutes at room temperature, Ubc8 loading was stopped by incubation of the pulse mixture with 50 mM EDTA on ice for 5 minutes. For the chase reaction, the quenched pulse mixture was mixed with an equal volume of the chase-initiating mixture containing 1 μM GIDSR4 complex, 0 or 2 μM Gid7 (WT or Δ1-285 mutant) and 25 mM lysine pH 8.0. The discharge was carried out at room temperature, quenched at different time points and visualized by non-reducing SDS-PAGE stained with Coomassie.
Avid binding of Fbp1 to Chelator-GIDSR4 was verified by performing a competition ubiquitylation assay in a multiturnover format (Figure 4D). The reactions were initiated by mixing 0.2 μM Uba1, 1 μM Ubc8-6xHis, 0.5 μM E3 GIDSR4, 0 or 2 μM Gid7 (WT or its Δ1-284 mutant), 0.5 μM of fluorescently labeled tetrameric Fbp1 or a monomeric model peptide substrate containing Fbp1 degron (as described above), 20 μM of an unlabeled competitor (full-length Fbp1-6xHis with major target lysines K32, K35, K280, K281 mutated to R or a lysine-less 9-residue peptide containing Fbp1 N-terminal sequence) and 20 μM Ub. Before starting the reaction, GIDSR4 was incubated with Gid7 for 3 minutes.
To validate the preferred ubiquitylation sites in Fbp1 determined by proteomics, multi-turnover ubiquitylation assays were performed using mutants of Fbp1, in which the pairs of major target lysines were mutated to arginine separately or together (Figure 5A). The reaction mixtures contained 0.2 μM Uba1, 1 μM Ubc8-6xHis, 0.5 μM GIDAnt, 1 μM Gid4, 2 μM Gid7, 1 μM Fbp1-6xHis (WT or target K > R mutants) and 20 μM Ub (WT or its all K > R (K0) version). Ubiquitylation of the substrates was visualized by immunoblotting with anti-His antibody.
Determination of kinetic parameters of Fbp1 ubiquitylation by GID E3
To examine the effect of Gid7 on the Michaelis-Menten constant Km for Fbp1 ubiquitylation by GID E3 (Figure 1C), multiturnover assays were performed by titrating the E3 concentration and with substrate levels that were below Km. Assays were performed with GIDSR4, GIDSR4 mixed with Gid7 as well as a purified Chelator-GIDSR4 (GIDSR4 coexpressed with Gid7). Reactions were quenched at time points such that the initial velocities of all reactions were well within the linear range (determined by running time courses for reactions that contained the highest E3 concentration from the titrations). Reactions with GIDSR4 comprised 0.2 μM Uba1, 1 μM Ubc8, 0.25-8 μM GIDSR4, 0.5 μM Fbp1-FAM and 20 μM Ub, and were quenched after 8 minutes. For GIDSR4 exogenously mixed with Gid7, reactions comprised 0.2 μM Uba1, 1 μM Ubc8, 0.025-0.8 μM GIDSR4 mixed with a 2-fold excess of Gid7, 0.1 μM Fbp1-FAM and 20 μM Ub, and were quenched after 3 minutes. In the case of Chelator-GIDSR4, the reaction mixes contained 0.2 μM Uba1, 1 μM Ubc8, 0.03-1 μM Chelator-GIDSR4, 0.1 μM Fbp1-FAM and 20 μM Ub, and the reactions were quenched after 2 minutes. Reaction substrate and products were resolved by SDS-PAGE and quantified using ImageQuant (GE healthcare; version 8.2). Fraction of Fbp1 that had been modified by one or more ubiquitins was then plotted as a function of E3 concentration in GraphPad Prism and fit to the Michaelis-Menten equation using non-linear curve fitting. All reactions were performed in duplicate.
Since the method described in the previous paragraph involved titration of E3 levels rather than that of the substrate, kcat was estimated using the following protocol. Initial velocities were measured for both GIDSR4 and Chelator-GIDSR4 by performing a time course where the ratios of both E3 to Km and substrate to Km were the same for each E3 complex (2.7 and 0.4, respectively). The fraction of ubiquitylated Fbp1 was plotted in GraphPad Prism as a function of time (Figure S1D) and the rate of the reaction was estimated by linear regression. Having calculated the rate, initial velocities V0 were calculated using the following equation: . Vmax was then estimated using a modified form of the Michaelis-Menten equation: , where because the substrate concentration was 2.5 times lower than Km. To obtain kcat values, Vmax was divided by the E3 concentration:.
Biochemical assays with human CTLHSR4
All in vitro ubiquitylation assays with CTLHSR4 were performed using a 30-residue fluorescent model peptide substrate harboring an N-terminal hGid4-interacting sequence PGLW and a single lysine placed at position 27, which is an optimal distance between the catalytic module and hGid4 based on the cryo EM structure.
To probe the residues of hGid4 that mediate its incorporation into CTLHSR4, structure-based hGid4 mutants (corresponding to homologous mutations in yeast Gid4 (Qiao et al., 2020) were tested in a binding test (Figure S6D) and ubiquitylation assays (Figure S6E). For the binding test, 10-fold molar excess of the purified WT and mutant hGid4 (Δ1-99) was mixed with 20 μg of RANBP9-TWA1-ARMC8-RMND5A-MAEA complex (tagged with a twin-Strep tag at TWA1 C terminus) in a buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl and 1 mM DTT. After incubating the proteins for 30 minutes on ice, 40 μL of the StrepTactin resin was added to the mixture and further incubated for 1 hour. As a control, RANBP9-TWA1-ARMC8-RMND5A-MAEA complex and hGid4 were mixed with StrepTactin alone. After throughout wash of the resin, elution fractions were collected and analyzed with SDS-PAGE stained with Coomasie. Ubiquitylation reactions were performed in a multiturnover format by mixing 0.2 μM Uba1, 2 μM Ube2H-6xHis, 1 μM RANBP9-TWA1-ARMC8-RMND5A-MAEA complex, 1 μM hGid4 (Δ1-99, WT or an indicated mutant), 0.5 μM fluorescent model peptide substrate and 20 μM Ub.
The catalytic mechanism of CTLH was examined by testing mutants of RMND5A and MAEA in substrate-independent discharge reactions (Figure S6G) and ubiquitylation assays (Figure S6H). The substrate-independent reactions monitored the discharge of Ube2H∼Ub to free lysine in solution in a pulse-chase format, applying the conditions as described for the assay with yeast GID. For the ubiquitylation multiturnover assays, the reactions contained 0.2 μM Uba1, 2 μM Ube2H-6xHis, 1 μM RANBP9-TWA1-ARMC8-RMND5A-MAEA complex (containing either WT or indicated mutants of RMND5A or MAEA), 1 μM hGid4 (Δ1-99), 0.5 μM fluorescent model peptide substrate and 20 μM Ub.
For characterizing the ubiquitin chain type formed by CTLHSR4 in conjunction with Ube2H, a multiturnover assay was performed (Figure S6I). The reaction mix contained 0.2 μM Uba1, 2 μM Ube2H-6xHis, 1 μM RANBP9-TWA1-ARMC8-RMND5A-MAEA complex, 1 μM hGid4 (Δ1-99), 0.5 μM fluorescent model peptide substrate and 20 μM Ub (WT, lysine-less (all K > R) or one of its single-lysine variants (with all but one lysine mutated to arginine)).
SEC for initial characterization of GID supramolecular assembly
For initial test of how Gid7 affects GID complex assembly (Figure S2A), 200 μL of 10 μM Gid7 and GIDSR4 alone or together (mixed in 1:1 ratio) were loaded onto a Superose 6 column (GE Healthcare) equilibrated with 25 mM HEPES 7.5, 150 mM NaCl and 5 mM DTT. SEC fractions were analyzed with Coomassie-stained SDS-PAGE.
SEC-MALS
To determine the oligomeric state of Fbp1 and Gid7 (Figure S2B), the proteins were subjected to SEC-MALS analysis. For each run, 100 μl of samples at 1 mg/mL were loaded onto Superdex 200 column equilibrated with a buffer containing 25 mM HEPES pH 7.5, 150 mM NaCl and 5 mM DTT. SEC-MALS was conducted in the MPIB Biochemistry Core Facility.
Fbp1 enzyme activity assay
To test the effect of Fbp1 ubiquitylation on its activity (Figure 5E) and sensitivity to allosteric regulation by AMP (Figure 5F), EnzChek Phosphate Assay Kit (ThermoFisher Scientific) was employed. This assay quantifies inorganic phosphate (Pi) released from fructose-1,6-bisphosphate by Fbp1 through enzymatic conversion of 2-amino-6-mercapto-7-methyl-purine riboside (MESG) to ribose 1-phosphate and 2-amino-6-mercapto-7-methylpurine by purine nucleoside phosphorylase (PNP). This leads to a shift in maximum absorbance from 330 nm for MESG to 360 nm for the final reaction product (2-amino-6-mercapto-7-methylpurine).
To obtain fully ubiquitylated Fbp1, it was subjected to an overnight multiturnover ubiquitylation reaction at room temperature consisting of 0.2 μM Uba1, 1 μM Ubc8, 0.5 μM GIDAnt, 1 μM Gid4, 2 μM Gid7, 10 μM Fbp1-V5-2xS and 100 μM 6xHis-3c-Ub. The reaction mix was run on SEC (using Superose 6 column) to separate different components of the assay. Fractions corresponding to the ubiquitylated Fbp1 were pooled and incubated with Ni-NTA resin for 30 minutes. After throughout wash, the bound protein was eluted and visualized by SDS-PAGE.
Fbp1 activity assays were performed according to the manufacturer’s instructions at room temperature. First, all the reagents provided in the assay kit (MESG, PNP and 20x reaction buffer), 0.5 mM fructose-1,6-bisphosphate substrate and 0.6 mM AMP (only for the Fbp1 inhibition assay) were pre-mixed and incubated for 5 min. Then, the reaction was initiated by addition of 53 nM of the WT, target lysine mutant (K32A/K35A/K280A/K281A) or fully ubiquitylated Fbp1, and the reaction progress was followed by measuring a time-course of absorbance at 360 nm (A360, absorbance of the final reaction product) using CLARIOStar Plus microplate reader (BMG LABTECH) in a UV-transparent 96-well plate. The values of A360 obtained for the buffer-only control were subtracted from all the experimental measurements, which were then plotted in GraphPad Prism.
Analysis of global proteome of WT versus ΔGid7 yeast (Karayel et al., 2020)
To test which proteins are dependent on Gid7 for their in vivo degradation, we compared the global proteome of WT with that of the ΔGid7 yeast (Figure S1F). Cells were grown in SD media to an OD of 1-1.5, pelleted by centrifugation, washed in pre-warmed SE media, and resuspended to an OD of 1 in fresh, pre-warmed SE media. Cultures were then allowed to grow at 30°C for 18 hours, after which cells were again pelleted by centrifugation, and resuspended in fresh, pre-warmed SD media to an OD of 1. Following growth at 30°C for 2 hours, 50 ODs of cells were pelleted by centrifugation, flash frozen and stored at −80°C until lysis. The frozen pellets were mixed with SDC lysis buffer (1% SDC and 100 mM Tris pH 8.5) and immediately heat-treated for 5 minutes at 95°C. Lysates were homogenized by sonication at 4°C using a Bioruptor and then diluted to achieve equal protein concentrations in a 96-well plate. Samples were next incubated for 5 minutes at 45°C with 40 mM CAA and 10 mM TCEP for reduction and alkylation and digested overnight at 37°C using trypsin (1:100 w/w, Sigma-Aldrich) and LysC (1/100 w/w, Wako). Next day, peptide material was desalted using SDB-RPS StageTips (Empore) (Kulak et al., 2014) and resuspended in buffer A (0.2% TFA/2% ACN). Peptide concentrations were measured by absorbance at 280 nm (Nanodrop 2000, Thermo Scientific) and equalized using buffer A∗. 300 ng peptides were subjected to LC-MS/MS analysis.
Samples were loaded onto a 20 cm reversed phase column (75 μm inner diameter, packed in house with ReproSil-Pur C18-AQ 1.9 μm resin (Dr. Maisch GmbH)). The column temperature was maintained at 60°C using a homemade column oven. A binary buffer system, consisting of buffer I (0.1% formic acid (FA) and buffer II (80% ACN plus 0.1% FA), was used for peptides separation, at a flow rate of 450 nl/min. An EASY-nLC 1200 system (Thermo Fisher Scientific), directly coupled online with the mass spectrometer (Q Exactive HF-X, Thermo Fisher Scientific) via a nano-electrospray source, was employed for nano-flow liquid chromatography. We used a gradient starting at 5% buffer B, increased to 35% in 18 and a half minute, 95% in a minute and stayed at 95% for three and a half min. The mass spectrometer was operated in DIA mode. Full MS resolution was set to 120,000 with a full scan range of 300-1650 m/z, a maximum fill time of 60 ms and an automatic gain control (AGC) target of 3e6. One full scan was followed by 12 windows with a resolution of 30,000 in profile mode. Precursor ions were fragmented by stepped higher-energy collisional dissociation (HCD) (NCE 25.5, 27,30%).
Spectronaut version 13 (Biognosys) was used to analyze DIA raw files using the yeast FASTA file (Swissprot, 2018) and the proteome library previously published (Karayel et al., 2020) with default settings and enabled cross run normalization. The Perseus software package version 1.6.0.7 was used for the data analysis (Tyanova et al., 2016). Protein intensities were log2-transformed and filtered to make sure that identified proteins showed expression in all biological triplicates of at least one condition. The missing values were subsequently replaced by random numbers that were drawn from a normal distribution (width = 0.3 and down shift = 1.8). For volcano plots, we used permutation-based FDR, which was set to 0.05 in conjunction with an S0-parameter of 0.1 to determine the significance.
Determination of preferentially targeted lysines in Fbp1 by LC-MS/MS (Figure S5)
To determine the preferentially targeted lysines in Fbp1, it was ubiquitylated by Chelator-GIDSR4 and subjected to proteomic analysis. To capture the initial ubiquitylation events, the assay was performed in a single-turnover pulse-chase format, wherein the concentration of the substrate was significantly exceeding that of E2∼Ub. In the pulse, 10 μM Ubc8 was loaded with 30 μM lysine-less ubiquitin mutant (all K > R) and 0.5 μM Uba1 for 15 minutes at room temperature and quenched with 50 mM EDTA. To start the chase, the pulse reaction was mixed with an equal volume of the chase-initiating mixture containing 1 μM GIDAnt, 2 μM Gid7, 2 μM Gid4 and 4 μM Fbp1-6xHis and incubated at room temperature. After 1 minute, the reaction was quenched by adding 10 mM DTT, which was then removed by desalting before proteomic analysis.
Proteins were digested and prepared for LC-MS/MS measurements as previously described (Qiao et al., 2020). Briefly, samples were diluted in digestion buffer (1 M urea in 50 mM ammonium bicarbonate, pH 8.0), followed by addition of TCEP and CAA to a final concentration of 10 mM and 40 mM, respectively. After reduction and alkylation for 5 minutes at 45°C, samples were enzymatically digested using either trypsin (1:20 w/w, Sigma-Aldrich) alone, trypsin (1:40 w/w)/GluC (1:40 w/w, BioLab) or trypsin (1:40 w/w)/AspN (1:40 w/w, Promega) at 37°C overnight. Thereafter, protease activity was quenched and peptides were loaded and cleaned on SDP-RPS StageTips. Peptides were subsequently eluted with 1.25% ammonium hydroxide/80% ACN, dried using a SpeedVac centrifuge (Eppendorf, Concentrator plus) and resuspended in buffer A (2% ACN/0.1% TFA) for LC/MS-MS analysis.
Peptide concentration was estimated by UV spectrometry and approximately 200 ng were loaded on a 50 cm reversed phase column (75 μm inner diameter, packed in-house with ReproSil-Pur C18-AQ 1.9 μm resin (Dr. Maisch GmbH)). Column temperature was maintained at 60°C using a homemade column oven. Peptides were separated with a binary buffer system of buffer A (0.1% formic acid (FA)) and buffer B (80% acetonitrile plus 0.1% FA), at a flow rate of 300 nl/min. We used an EASY-nLC 1200 system (Thermo Fisher Scientific), which was directly coupled online with the mass spectrometer (Q Excative HF-X, Thermo Fisher Scientific) via a nano-electrospray source. Peptides were eluted with a gradient starting at 3% buffer B and stepwise increased to 8% in 8 min, 36% in 32 min, 45% in 4 minutes and 95% in 4 min. The mass spectrometer was operated in Top12 data-dependent mode (DDA) with a full scan range of 250-1350 m/z at 60,000 resolution with an automatic gain control (AGC) target of 3e6 and a maximum fill time of 20 ms. Precursor ions were isolated with a width of 1.4 m/z and fragmented by higher-energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 28%. Fragment scans were performed at a resolution of 30,000, an AGC of 1e5 and a maximum injection time of 110 ms. Dynamic exclusion was enabled and set to 15 s.
Raw MS data were searched against UniProt Yeast FASTA using MaxQuant (version 1.6.2.10) with a 1% FDR at peptide and protein level. Cysteine carbamidomethylation was set as fixed, protein N-terminal acetylation, methionine oxidation and lysine diGly as variable modifications. The minimum peptide length was set to 7 amino acids, enzyme specificity was set to trypsin and two missed cleavages were allowed, permitting a maximum of 5 modifications per peptide. MS/MS spectra identifying ubiquitylated peptides of interest were obtained and exported using MaxQuant Viewer.
Cell culture and generation of CRISPR-Cas9 knock out cell lines
K562 erythroleukemia cell line was obtained from ATCC (CCL-243™) and cultured in IMDM completed with 10% (v/v) FBS (GIBCO) and antibiotics (100 units/ml penicillin, 0.1 mg/ml streptomycin, GIBCO). Cell densities were kept between 0.1-1 × 106 cells/mL, and cultures were regularly checked for the absence of mycoplasma contamination. For CRISPR-Cas9-(D10A) nickase-mediated functional knockouts of MAEA, MKLN1 and WDR26, paired sense and antisense guide RNAs (gRNA) were designed to target MAEA in exon 2, exon 5 in MKLN1 and exon 1 in WDR26 genetic locus. Sense and antisense gRNA were cloned into pBABED-U6-Puromycin plasmid (gift from Thomas Macartney, University of Dundee, UK) and pX335-Cas9(D10A) (Addgene) (Cong et al., 2013), respectively. K562 cells were co-transfected with vectors encoding the pair of gRNAs using Lipofectamine LTX reagent (Invitrogen) following manufacturer’s instructions. Twenty-four hours after transfection, cells were selected in puromycin (2 μg/ml) for 2 days, followed by expansion, and single-cell dilution to obtain cell clones. Successful knockout clones were confirmed by immunoblotting and genomic sequencing of targeted loci (Figure S6J).
Human cell lysate fractionation by sucrose density gradient
1 × 107 cells were harvested by centrifugation at 360 x g, washed once with ice-cold PBS, and resuspended in lysis buffer (40 mM HEPES pH 7.5, 120 mM NaCl, 1 mM EGTA, 0.5% NP40, 1 mM DTT, and Complete protease inhibitor mix (Roche)). Cells were homogenized by pushing them 10 times through a 23G syringe. The obtained lysate was cleared by centrifugation at 23,000 x g for 30 minutes at 4°C. 3 mg of total protein were loaded on top of a 5%–40% sucrose gradient (weight/volume, in lysis buffer) and centrifuged in a SW60 rotor at 34,300 rpm for 16 hours at 4°C. Fractions were collected from top of the gradient and separated by SDS-PAGE, followed by immunoblotting using the following antibodies: RMND5A (Santa Cruz), MAEA (R&D systems), RANBP9 (Novus Biologicals), TWA1 (Thermo Fisher), ARMC8 (Santa Cruz), WDR26 (Bethyl Laboratories), MKLN1 (Santa Cruz) and YPEL5 (Thermo Fisher). Antibodies that recognize hGid4 were generated by immunizing sheep with bacterially expressed GST-hGid4 (Δ1-99). Western blots were developed using Clarity Western ECL Substrate (BioRad) and imaged using Amersham Imager 600 (GE Lifesciences).
Cryo EM sample preparation and Imaging
Cryo EM grids were prepared using Vitrobot Mark IV (Thermo Fisher Scientific) operated at 4°C and 100% humidity. 3.5 μl of freshly purified proteins at 0.3-0.5 mg/ml were applied to glow-discharged Quantifoil holey carbon grids (R1.2/1.3 200 mesh). Grids were immediately blotted with Whatman no. 1 filter paper (blot time: 3 s, blot force: 3) and vitrified by plunging into liquid ethane.
Cryo EM data were first screened and collected on a Talos Arctica or Glacios transmission electron microscope (Thermo Fisher Scientific) operated at 200 kV, equipped with a Falcon III (Thermo Fisher Scientific) or K2 (Gatan) direct electron detector, respectively. Automated data collection was carried out using EPU software (Thermo Fisher Scientific) or SerialEM (Mastronarde, 2003). High-resolution datasets were collected on a Titan Krios (Thermo Fisher Scientific) microscope operated at 300 kV, equipped with a post-column GIF and a K3 Summit direct electron detector (Gatan) operating in a counting mode. SerialEM was used to automate data collection (Mastronarde, 2003). Details of cryo EM data collection and map refinement are listed in Table S1.
Cryo EM data processing
Frames were motion-corrected with dose weighting using MotionCorr2 (Zheng et al., 2017) and subjected to estimation of contrast transfer function parameters with Gctf (Zhang, 2016). Auto-picking of particles was performed with Gautomatch (https://www.mrc-lmb.cam.ac.uk/kzhang/) and for most datasets, it was aided by provision of a template obtained from previous low-resolution datasets. For Titan Krios datasets, movies were being pre-processed on-the-fly during data collection with Focus (Biyani et al., 2017), which also automatically discarded poor quality images. All the subsequent stages of data processing were carried out with Relion (Fernandez-Leiro and Scheres, 2017; Scheres, 2012; Zivanov et al., 2018). To clean up the data, extracted particles were subjected to either several rounds of 2D classification, followed by a 3D classification or submitted directly to a masked 3D classification. The chosen subset of particles was subjected to auto-refinement without and with a mask. To improve the quality of maps obtained after consensus refinement, a 3D classification without particle alignment was performed and a class having the most complete features was selected.
High-resolution maps of yeast substrate receptor scaffolding (SRS), catalytic (Cat) and supramolecular assembly (SA) modules were obtained from the Chelator-GIDSR4 dataset with its substrate Fbp1 bound. For the SRS module, a more resolved half of the Chelator-GIDSR4 was first auto-refined and a focused 3D classification without particle alignment was performed with a mask over GIDSR4. Then, focused refinement was performed, wherein the Cat module was masked out. For Cat and SA modules, the number of particles used for alignment was doubled by taking advantage of the Chelator-GIDSR4 having C2 symmetry. First, the map of the entire complex was auto-refined with C2 symmetry imposed and masks were created for each of its halves. Then, the signal for each half was separately subtracted and the resulting semi-elliptical particles were aligned by auto-refinement. After masking out the SRS module, a focused 3D classification without particle alignment was performed separately for Cat and SA modules. After one more round of 3D classification without particle alignment with a higher T-value, the particles were subjected to CTF refinement and final auto-refinement.
For high-resolution CTLHSR4 dataset, the density corresponding to the catalytic module (RMND5A-MAEA) was masked out due to its mobility relative to the substrate receptor scaffolding module (RANBP9-TWA1-ARMC8-hGid4). For visualization of less resolved parts of the map, such as RANBP9LisH-CRAC-TWA1 as well as the N- and C-termini of ARMC8, subsequent rounds of focused 3D classifications with masks over these regions were carried out. The final auto-refinement was preceded by a CTF refinement.
All maps were post-processed by automatic B-factor weighting and high-resolution noise substitution in Relion. In addition, to aid in building atomic models, the refined maps of the Chelator-GIDSR4 SA module and CTLHSR4 SRS module were post-processed with DeepEMhancer (Sanchez-Garcia et al., 2020) and are deposited as additional maps in EMDB. The estimated resolutions of all reconstructions are based on the gold-standard Fourier Shell Correlation (FSC) at 0.143 criterion. Simplified schematic of processing for both Titan Krios datasets are presented in Figures S4 and S7 (for Chelator-GIDSR4 and CTLHSR4, respectively).
Model building and refinement
Manual building of all models was performed with Coot (Emsley and Cowtan, 2004; Emsley et al., 2010), whereas structure visualization and analysis was carried out with Chimera (Pettersen et al., 2004), ChimeraX (Goddard et al., 2018) and Pymol-v2.1 (https://pymol.org/2/).
The atomic model of CTLH substrate receptor scaffolding module was prepared as follows. Most of ARMC8 was built automatically with Buccaneer (Cowtan, 2006) and refined manually with Coot. The model of the substrate receptor hGid4 was generated by docking its crystal structure (PDB: 6CDC) into the EM map and manual building of its N- and C-termini. The crystal structure of RANBP9 SPRY domain (PDB: 5JI7) was fitted into the electron density map and served as a starting point for manual building of its downstream region. Manual building of TWA1 was guided by fitting parts of its homology model into the map (generated by SWISS-MODEL (Waterhouse et al., 2018), based on the structure of yeast Gid8 in GIDSR4, PDB: 6SWY) and secondary structure prediction obtained from Phyre2 server (Kelley et al., 2015).
The structure of the SRS module in Chelator-GIDSR4 was generated by fitting the atomic coordinates of the corresponding part of GIDSR4 (PDB: 6SWY) and manual refinement. The loops of Gid4 surrounding its substrate-binding cavity as well as Fbp1 degron were built manually. Coordinates of most of Gid8 and Gid1 in the SA module were fitted from the structure of GIDSR4 and the missing or differing parts, such as Gid1 CTLH-CRAN, were built manually. The LisH-CRAC as well as CTLH-CRAN domains of Gid7 were built manually, guided by secondary structure predictions. Manual building of Gid7 β-propellers was aided by their homology model from Phyre2 (Kelley et al., 2015). All of the Cat module was built manually and the geometry of the zinc binding site was constrained to account for tetrahedral coordination of zinc ions.
All the models were subjected to iterative rounds of manual building with Coot and real space refinement in PHENIX (Adams et al., 2010; Afonine et al., 2018; DiMaio et al., 2013) until a satisfactory model quality, in terms of its geometry and agreement with the map, was obtained.
Fbp1 crystallization and data processing
Crystallization trials of Fbp1-6xHis were performed in the MPIB Crystallization Facility. Before setting up the crystallization trays, the purified Fbp1 was concentrated to 12 mg/mL and combined with 0.5 mM of its allosteric inhibitor AMP and the substrate fructose-1,6-bisphosphate. Crystals used for X-ray data collection were obtained at 4°C in the buffer containing 16% PEG 3350, 0.2 M MgCl2 and 0.1 M Bis-Tris pH 6 using a vapor diffusion method performed in a sitting-drop format. Crystals were cryoprotected using 20% ethylene glycol and stored by flash freezing in liquid nitrogen until data collection.
Diffraction dataset was recorded at X10SA beam line, Swiss Light Source (SLS) in Villingen, Switzerland. Data were recorded at 0.5 degree rotation intervals using Dectris Pilatus 2M-F detector. Data were indexed, integrated, and scaled using XDS package to a resolution limit of 1.95 Å. Phasing was performed through molecular replacement using a structure of human Fbp1 (PDB: 1FTA) with PHASER integrated into the PHENIX software suite (Adams et al., 2010; Afonine et al., 2018; DiMaio et al., 2013). Model building was done using Coot (Emsley and Cowtan, 2004; Emsley et al., 2010), whereas refinement was carried out with phenix.refine. Details of X-ray diffraction data collection and refinement statistics are listed in Table S2.
Quantification and statistical analysis
For the assays described in the section “In vivo yeast substrate degradation assays,” protein bands visualized by western blots were quantified using ImageStudioLite software (Li-Cor). For statistical analysis, at least three biological replicates were considered and the standard deviation of the replicates was presented using error bars.
Fluorescently labeled proteins in in vitro ubiquitylation reactions were quantified in ImageQuant (GE Healthcare) and the calculated fractions of ubiquitylated substrates were plotted in GraphPad Prism. All in vitro assays were performed in at least duplicates and the standard deviation represented by error bars are shown wherever necessary. For determination of Km for Fbp1 ubiquitylation by GID E3 with kinetics, the fraction of ubiquitylated Fbp1 was fit to the Michaelis-Menten equation in GraphPad Prism. kcat was calculated based on a slope of a linear phase of Fbp1 ubiquitylation reaction fitted in GraphPad Prism.
Acknowledgments
We thank A. Varshavsky for promoter reference plasmids; S. Übel and S. Pettera for peptide synthesis; D. Bollschweiler, T. Schäfer, J. Basquin, K. Valer-Saldana, and S. Pleyer for assistance with cryo-EM and crystallography; Swiss Light Source, Villigen, Switzerland for Fbp1 crystal data collection; M. Strauss for assistance with initial negative-stain data collection on the yeast Chelator-GID complex; M. Yamaguchi for Gid7 constructs; G. Kleiger for guidance regarding kinetics; I. Paron for technical assistance with mass spectrometry; and K. Baek, B. Bräuning, and the Schulman lab for advice and support. The B.A.S. lab is funded by the Leibniz Prize from Deutsche Forschungsgemeinschaft (SCHU 3196/1-1). B.A.S. and M.M. are supported by the Max Planck Society.
Author contributions
Initial conceptualization, D.S., J.C., and B.A.S.; methodology, D.S., J.C., J.R.P., C.R.L., O.K., A.F.A., and B.A.S.; investigation, D.S., J.C., S.Q., C.R.L., L.A.H., K.V.G., F.M.H., O.K., J.R.P., and A.F.A.; resources, D.S., J.C., K.V.G., C.R.L., S.v.G., and A.F.A.; writing – original draft, D.S., J.C., and B.A.S.; writing – review & editing, D.S., J.C., C.R.L., A.F.A., and B.A.S.; supervision, M.M., A.F.A., and B.A.S.; funding acquisition, M.M. and B.A.S.
Declaration of interests
B.A.S. is an honorary professor at Technical University of Munich, Germany and adjunct faculty at St. Jude Children’s Research Hospital, Memphis, TN, USA and is on the scientific advisory board of Interline Therapeutics.
Published: April 26, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.03.025.
Supplemental information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Klaholz B.P., Moriarty N.W., Poon B.K., Sobolev O.V., Terwilliger T.C., Adams P.D., Urzhumtsev A. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 2018;74:814–840. doi: 10.1107/S2059798318009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K., Krist D.T., Prabu J.R., Hill S., Klügel M., Neumaier L.M., von Gronau S., Kleiger G., Schulman B.A. NEDD8 nucleates a multivalent cullin-RING-UBE2D ubiquitin ligation assembly. Nature. 2020;578:461–466. doi: 10.1038/s41586-020-2000-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaji V., Hoppe T. Regulation of E3 ubiquitin ligases by homotypic and heterotypic assembly. F1000Res. 2020;9 doi: 10.12688/f1000research.21253.1. F1000 Faculty Rev-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbin L., Eisele F., Santt O., Wolf D.H. The Cdc48-Ufd1-Npl4 complex is central in ubiquitin-proteasome triggered catabolite degradation of fructose-1,6-bisphosphatase. Biochem. Biophys. Res. Commun. 2010;394:335–341. doi: 10.1016/j.bbrc.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Barford D. Structural interconversions of the anaphase-promoting complex/cyclosome (APC/C) regulate cell cycle transitions. Curr. Opin. Struct. Biol. 2020;61:86–97. doi: 10.1016/j.sbi.2019.11.010. [DOI] [PubMed] [Google Scholar]
- Bauer A., Jagannathan V., Högler S., Richter B., McEwan N.A., Thomas A., Cadieu E., André C., Hytönen M.K., Lohi H. MKLN1 splicing defect in dogs with lethal acrodermatitis. PLoS Genet. 2018;14:e1007264. doi: 10.1371/journal.pgen.1007264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biyani N., Righetto R.D., McLeod R., Caujolle-Bert D., Castano-Diez D., Goldie K.N., Stahlberg H. Focus: The interface between data collection and data processing in cryo-EM. J. Struct. Biol. 2017;198:124–133. doi: 10.1016/j.jsb.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Boldt K., van Reeuwijk J., Lu Q., Koutroumpas K., Nguyen T.M., Texier Y., van Beersum S.E., Horn N., Willer J.R., Mans D.A., UK10K Rare Diseases Group An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 2016;7:11491. doi: 10.1038/ncomms11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard J.J., Otero J.H., Scott D.C., Szulc E., Martin E.W., Sabri N., Granata D., Marzahn M.R., Lindorff-Larsen K., Salvatella X. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol. Cell. 2018;72:19–36.e8. doi: 10.1016/j.molcel.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun B., Pfirrmann T., Menssen R., Hofmann K., Scheel H., Wolf D.H. Gid9, a second RING finger protein contributes to the ubiquitin ligase activity of the Gid complex required for catabolite degradation. FEBS Lett. 2011;585:3856–3861. doi: 10.1016/j.febslet.2011.10.038. [DOI] [PubMed] [Google Scholar]
- Brown N.G., VanderLinden R., Watson E.R., Weissmann F., Ordureau A., Wu K.P., Zhang W., Yu S., Mercredi P.Y., Harrison J.S. Dual RING E3 Architectures Regulate Multiubiquitination and Ubiquitin Chain Elongation by APC/C. Cell. 2016;165:1440–1453. doi: 10.1016/j.cell.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhidarmo R., Nakatani Y., Day C.L. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012;37:58–65. doi: 10.1016/j.tibs.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Burnley T., Palmer C.M., Winn M. Recent developments in the CCP-EM software suite. Acta Crystallogr. D Struct. Biol. 2017;73:469–477. doi: 10.1107/S2059798317007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W.X., Kabelitz S., Gupta M., Yeung E., Lin S., Rammelt C., Ihling C., Pekovic F., Low T.C.H., Siddiqui N.U. Precise Temporal Regulation of Post-transcriptional Repressors Is Required for an Orderly Drosophila Maternal-to-Zygotic Transition. Cell Rep. 2020;31:107783. doi: 10.1016/j.celrep.2020.107783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.K., Chan N.L., Wang A.H. The many blades of the β-propeller proteins: conserved but versatile. Trends Biochem. Sci. 2011;36:553–561. doi: 10.1016/j.tibs.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Chen I., Dorr B.M., Liu D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA. 2011;108:11399–11404. doi: 10.1073/pnas.1101046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Wu J., Lu Y., Ma Y.B., Lee B.H., Yu Z., Ouyang Q., Finley D.J., Kirschner M.W., Mao Y. Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. USA. 2016;113:12991–12996. doi: 10.1073/pnas.1614614113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.J., Wu X., Wadas B., Oh J.H., Varshavsky A. An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science. 2017;355:eaal3655. doi: 10.1126/science.aal3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang H.L., Schekman R. Regulated import and degradation of a cytosolic protein in the yeast vacuole. Nature. 1991;350:313–318. doi: 10.1038/350313a0. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Delto C.F., Heisler F.F., Kuper J., Sander B., Kneussel M., Schindelin H. The LisH motif of muskelin is crucial for oligomerization and governs intracellular localization. Structure. 2015;23:364–373. doi: 10.1016/j.str.2014.11.016. [DOI] [PubMed] [Google Scholar]
- DiMaio F., Echols N., Headd J.J., Terwilliger T.C., Adams P.D., Baker D. Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods. 2013;10:1102–1104. doi: 10.1038/nmeth.2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Zhang H., Li L., Tempel W., Loppnau P., Min J. Molecular basis of GID4-mediated recognition of degrons for the Pro/N-end rule pathway. Nat. Chem. Biol. 2018;14:466–473. doi: 10.1038/s41589-018-0036-1. [DOI] [PubMed] [Google Scholar]
- Dong C., Chen S.J., Melnykov A., Weirich S., Sun K., Jeltsch A., Varshavsky A., Min J. Recognition of nonproline N-terminal residues by the Pro/N-degron pathway. Proc. Natl. Acad. Sci. USA. 2020;117:14158–14167. doi: 10.1073/pnas.2007085117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H., Buetow L., Sibbet G.J., Cameron K., Huang D.T. BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 2012;19:876–883. doi: 10.1038/nsmb.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Leiro R., Scheres S.H.W. A pipeline approach to single-particle processing in RELION. Acta Crystallogr. D Struct. Biol. 2017;73:496–502. doi: 10.1107/S2059798316019276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis O., Han F., Adams J.C. Molecular phylogeny of a RING E3 ubiquitin ligase, conserved in eukaryotic cells and dominated by homologous components, the muskelin/RanBPM/CTLH complex. PLoS ONE. 2013;8:e75217. doi: 10.1371/journal.pone.0075217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gancedo C. Inactivation of fructose-1,6-diphosphatase by glucose in yeast. J. Bacteriol. 1971;107:401–405. doi: 10.1128/jb.107.2.401-405.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A., 3rd, Smith H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämmerle M., Bauer J., Rose M., Szallies A., Thumm M., Düsterhus S., Mecke D., Entian K.D., Wolf D.H. Proteins of newly isolated mutants and the amino-terminal proline are essential for ubiquitin-proteasome-catalyzed catabolite degradation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. J. Biol. Chem. 1998;273:25000–25005. doi: 10.1074/jbc.273.39.25000. [DOI] [PubMed] [Google Scholar]
- Haselbach D., Schrader J., Lambrecht F., Henneberg F., Chari A., Stark H. Long-range allosteric regulation of the human 26S proteasome by 20S proteasome-targeting cancer drugs. Nat. Commun. 2017;8:15578. doi: 10.1038/ncomms15578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C., Magiera M.M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., Knop M. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;Aug;21(11) doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kaiser S.E., Riley B.E., Shaler T.A., Trevino R.S., Becker C.H., Schulman H., Kopito R.R. Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods. 2011;8:691–696. doi: 10.1038/nmeth.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayel O., Michaelis A.C., Mann M., Schulman B.A., Langlois C.R. DIA-based systems biology approach unveils E3 ubiquitin ligase-dependent responses to a metabolic shift. Proc. Natl. Acad. Sci. USA. 2020;117:32806–32815. doi: 10.1073/pnas.2020197117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke H.M., Thorpe C.M., Seaton Ba., Lipscomb W.N., Marcus F. Structure refinement of fructose-1,6-bisphosphatase and its fructose 2,6-bisphosphate complex at 2.8 A resolution. J. Mol. Biol. 1990;212:513–539. doi: 10.1016/0022-2836(90)90329-k. [DOI] [PubMed] [Google Scholar]
- Ke H.M., Zhang Y.P., Lipscomb W.N. Crystal structure of fructose-1,6-bisphosphatase complexed with fructose 6-phosphate, AMP, and magnesium. Proc. Natl. Acad. Sci. USA. 1990;87:5243–5247. doi: 10.1073/pnas.87.14.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.H., Hong S.K., Hwang K.Y., Kim E.E. Structure of mouse muskelin discoidin domain and biochemical characterization of its self-association. Acta Crystallogr. D Biol. Crystallogr. 2014;70:2863–2874. doi: 10.1107/S139900471401894X. [DOI] [PubMed] [Google Scholar]
- Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;Jul;15(10B) doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kobayashi N., Yang J., Ueda A., Suzuki T., Tomaru K., Takeno M., Okuda K., Ishigatsubo Y. RanBPM, Muskelin, p48EMLP, p44CTLH, and the armadillo-repeat proteins ARMC8alpha and ARMC8beta are components of the CTLH complex. Gene. 2007;396:236–247. doi: 10.1016/j.gene.2007.02.032. [DOI] [PubMed] [Google Scholar]
- Koshland D.E., Jr. Correlation of Structure and Function in Enzyme Action. Science. 1963;142:1533–1541. doi: 10.1126/science.142.3599.1533. [DOI] [PubMed] [Google Scholar]
- Koshland D.E., Jr. Properties of the active site of enzymes. Ann. N Y Acad. Sci. 1963;103:630–642. doi: 10.1111/j.1749-6632.1963.tb53723.x. [DOI] [PubMed] [Google Scholar]
- Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Lampert F., Stafa D., Goga A., Soste M.V., Gilberto S., Olieric N., Picotti P., Stoffel M., Peter M. The multi-subunit GID/CTLH E3 ubiquitin ligase promotes cell proliferation and targets the transcription factor Hbp1 for degradation. eLife. 2018;7:e35528. doi: 10.7554/eLife.35528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander G.C., Estrin E., Matyskiela M.E., Bashore C., Nogales E., Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett D.S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R.T., Walz T., Ploegh H., Finley D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- Liu H., Pfirrmann T. The Gid-complex: an emerging player in the ubiquitin ligase league. Biol. Chem. 2019;400:1429–1441. doi: 10.1515/hsz-2019-0139. [DOI] [PubMed] [Google Scholar]
- Liu H., Ding J., Köhnlein K., Urban N., Ori A., Villavicencio-Lorini P., Walentek P., Klotz L.O., Hollemann T., Pfirrmann T. The GID ubiquitin ligase complex is a regulator of AMPK activity and organismal lifespan. Autophagy. 2020;16:1618–1634. doi: 10.1080/15548627.2019.1695399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitland M.E.R., Onea G., Chiasson C.A., Wang X., Ma J., Moor S.E., Barber K.R., Lajoie G.A., Shaw G.S., Schild-Poulter C. The mammalian CTLH complex is an E3 ubiquitin ligase that targets its subunit muskelin for degradation. Sci. Rep. 2019;9:9864. doi: 10.1038/s41598-019-46279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde D. SerialEM: A Program for Automated Tilt Series Acquisition on Tecnai Microscopes Using Prediction of Specimen Position. Microsc. Microanal. 2003;9:1182–1183. [Google Scholar]
- McMahon M., Thomas N., Itoh K., Yamamoto M., Hayes J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006;281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- Melnykov A., Chen S.J., Varshavsky A. Gid10 as an alternative N-recognin of the Pro/N-degron pathway. Proc. Natl. Acad. Sci. USA. 2019;116:15914–15923. doi: 10.1073/pnas.1908304116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen R., Schweiggert J., Schreiner J., Kusevic D., Reuther J., Braun B., Wolf D.H. Exploring the topology of the Gid complex, the E3 ubiquitin ligase involved in catabolite-induced degradation of gluconeogenic enzymes. J. Biol. Chem. 2012;287:25602–25614. doi: 10.1074/jbc.M112.363762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen R., Bui K., Wolf D.H. Regulation of the Gid ubiquitin ligase recognition subunit Gid4. FEBS Lett. 2018;592:3286–3294. doi: 10.1002/1873-3468.13229. [DOI] [PubMed] [Google Scholar]
- Mittag T., Orlicky S., Choy W.Y., Tang X., Lin H., Sicheri F., Kay L.E., Tyers M., Forman-Kay J.D. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. USA. 2008;105:17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J., Changeux J.P., Jacob F. Allosteric proteins and cellular control systems. J. Mol. Biol. 1963;6:306–329. doi: 10.1016/s0022-2836(63)80091-1. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K., Okumura F., Kamura T. Proteolytic regulation of metabolic enzymes by E3 ubiquitin ligase complexes: lessons from yeast. Crit. Rev. Biochem. Mol. Biol. 2015;50:489–502. doi: 10.3109/10409238.2015.1081869. [DOI] [PubMed] [Google Scholar]
- Nassan M., Li Q., Croarkin P.E., Chen W., Colby C.L., Veldic M., McElroy S.L., Jenkins G.D., Ryu E., Cunningham J.M. A genome wide association study suggests the association of muskelin with early onset bipolar disorder: Implications for a GABAergic epileptogenic neurogenesis model. J. Affect. Disord. 2017;208:120–129. doi: 10.1016/j.jad.2016.09.049. [DOI] [PubMed] [Google Scholar]
- Nayak D., Sivaraman J. Structure of LNX1:Ubc13∼Ubiquitin Complex Reveals the Role of Additional Motifs for the E3 Ligase Activity of LNX1. J. Mol. Biol. 2018;430:1173–1188. doi: 10.1016/j.jmb.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Ogura T., Tong K.I., Mio K., Maruyama Y., Kurokawa H., Sato C., Yamamoto M. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc. Natl. Acad. Sci. USA. 2010;107:2842–2847. doi: 10.1073/pnas.0914036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.H., Chen S.J., Varshavsky A. A reference-based protein degradation assay without global translation inhibitors. J. Biol. Chem. 2017;292:21457–21465. doi: 10.1074/jbc.M117.814236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Plechanovová A., Jaffray E.G., Tatham M.H., Naismith J.H., Hay R.T. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature. 2012;489:115–120. doi: 10.1038/nature11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda J.N., Littlefield P.J., Soss S.E., Nordquist K.A., Chazin W.J., Brzovic P.S., Klevit R.E. Structure of an E3:E2∼Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao S., Langlois C.R., Chrustowicz J., Sherpa D., Karayel O., Hansen F.M., Beier V., von Gronau S., Bollschweiler D., Schäfer T. Interconversion between Anticipatory and Active GID E3 Ubiquitin Ligase Conformations via Metabolically Driven Substrate Receptor Assembly. Mol. Cell. 2020;77:150–163.e9. doi: 10.1016/j.molcel.2019.10.009. [DOI] [PubMed] [Google Scholar]
- Regelmann J., Schüle T., Josupeit F.S., Horak J., Rose M., Entian K.D., Thumm M., Wolf D.H. Catabolite degradation of fructose-1,6-bisphosphatase in the yeast Saccharomyces cerevisiae: a genome-wide screen identifies eight novel GID genes and indicates the existence of two degradation pathways. Mol. Biol. Cell. 2003;14:1652–1663. doi: 10.1091/mbc.E02-08-0456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnac D.V., Zheng N. Structural Biology of CRL Ubiquitin Ligases. Adv. Exp. Med. Biol. 2020;1217:9–31. doi: 10.1007/978-981-15-1025-0_2. [DOI] [PubMed] [Google Scholar]
- Salemi L.M., Maitland M.E.R., McTavish C.J., Schild-Poulter C. Cell signalling pathway regulation by RanBPM: molecular insights and disease implications. Open Biol. 2017;7:170081. doi: 10.1098/rsob.170081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia R., Gomez-Blanco J., Cuervo A., Carazo J., Sorzano C., Vargas J. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. bioRxiv. 2020 doi: 10.1101/2020.06.12.148296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santt O., Pfirrmann T., Braun B., Juretschke J., Kimmig P., Scheel H., Hofmann K., Thumm M., Wolf D.H. The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol. Biol. Cell. 2008;19:3323–3333. doi: 10.1091/mbc.E08-03-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schork S.M., Bee G., Thumm M., Wolf D.H. Catabolite inactivation of fructose-1,6-bisphosphatase in yeast is mediated by the proteasome. FEBS Lett. 1994;349:270–274. doi: 10.1016/0014-5793(94)00668-7. [DOI] [PubMed] [Google Scholar]
- Schork S.M., Bee G., Thumm M., Wolf D.H. Site of catabolite inactivation. Nature. 1994;369:283–284. doi: 10.1038/369283a0. [DOI] [PubMed] [Google Scholar]
- Schork S.M., Thumm M., Wolf D.H. Catabolite inactivation of fructose-1,6-bisphosphatase of Saccharomyces cerevisiae. Degradation occurs via the ubiquitin pathway. J. Biol. Chem. 1995;270:26446–26450. doi: 10.1074/jbc.270.44.26446. [DOI] [PubMed] [Google Scholar]
- Schweitzer A., Aufderheide A., Rudack T., Beck F., Pfeifer G., Plitzko J.M., Sakata E., Schulten K., Förster F., Baumeister W. Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc. Natl. Acad. Sci. USA. 2016;113:7816–7821. doi: 10.1073/pnas.1608050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeel S., Rajendra E., Alcón P., O’Reilly F., Chorev D.S., Maslen S., Degliesposti G., Russo C.J., He S., Hill C.H. Structure of the Fanconi anaemia monoubiquitin ligase complex. Nature. 2019;575:234–237. doi: 10.1038/s41586-019-1703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skraban C.M., Wells C.F., Markose P., Cho M.T., Nesbitt A.I., Au P.Y.B., Begtrup A., Bernat J.A., Bird L.M., Cao K. WDR26 Haploinsufficiency Causes a Recognizable Syndrome of Intellectual Disability, Seizures, Abnormal Gait, and Distinctive Facial Features. Am. J. Hum. Genet. 2017;101:139–148. doi: 10.1016/j.ajhg.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F., Resnick M.A. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymology. 2006;409 doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- Tu B.P., McKnight S.L. Metabolic cycles as an underlying basis of biological oscillations. Nat. Rev. Mol. Cell Biol. 2006;7:696–701. doi: 10.1038/nrm1980. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson E.R., Brown N.G., Peters J.M., Stark H., Schulman B.A. Posing the APC/C E3 Ubiquitin Ligase to Orchestrate Cell Division. Trends Cell Biol. 2019;29:117–134. doi: 10.1016/j.tcb.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehmer M., Rudack T., Beck F., Aufderheide A., Pfeifer G., Plitzko J.M., Förster F., Schulten K., Baumeister W., Sakata E. Structural insights into the functional cycle of the ATPase module of the 26S proteasome. Proc. Natl. Acad. Sci. USA. 2017;114:1305–1310. doi: 10.1073/pnas.1621129114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann F., Petzold G., VanderLinden R., Huis In ’t Veld P.J., Brown N.G., Lampert F., Westermann S., Stark H., Schulman B.A., Peters J.M. biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl. Acad. Sci. USA. 2016;113:E2564–E2569. doi: 10.1073/pnas.1604935113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M., Larimore E.A., Swanger J., Bengoechea-Alonso M.T., Grim J.E., Ericsson J., Zheng N., Clurman B.E. Fbw7 dimerization determines the specificity and robustness of substrate degradation. Genes Dev. 2013;27:2531–2536. doi: 10.1101/gad.229195.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman S., Lippman S.I., Zhao X., Broach J.R. How Saccharomyces responds to nutrients. Annu. Rev. Genet. 2008;42:27–81. doi: 10.1146/annurev.genet.41.110306.130206. [DOI] [PubMed] [Google Scholar]
- Zavortink M., Rutt L.N., Dzitoyeva S., Henriksen J.C., Barrington C., Bilodeau D.Y., Wang M., Chen X.X.L., Rissland O.S. The E2 Marie Kondo and the CTLH E3 ligase clear deposited RNA binding proteins during the maternal-to-zygotic transition. eLife. 2020;9:e53889. doi: 10.7554/eLife.53889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen R., Moo C., Zhao Z., Chen M., Feng H., Zheng X., Zhang L., Shi J., Chen C. Wdr26 regulates nuclear condensation in developing erythroblasts. Blood. 2020;135:208–219. doi: 10.1182/blood.2019002165. [DOI] [PubMed] [Google Scholar]
- Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Thompson C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019;20:436–450. doi: 10.1038/s41580-019-0123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang M., Calabrese M.F., Liu J., Waddell M.B., Nourse A., Hammel M., Miller D.J., Walden H., Duda D.M., Seyedin S.N. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol. Cell. 2009;36:39–50. doi: 10.1016/j.molcel.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife. 2018;7:e42166. doi: 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession codes for the PDB models and EM maps are available in RCSB and EMDB, respectively, as follows: Apo Chelator-GIDSR4, EMDB: EMD-12541; Chelator-GIDSR4 + Fbp1, EMDB: EMD-12557; GIDSR4, EMDB: EMD-12548; SRS module, EMDB: EMD-12559, PDB: 7NS3; Cat module, EMDB: EMD-12560; PDB: 7NS4; SA module, EMDB: EMD-12563; PDB: 7NSB; Endogenous GIDAnt, EMDB: EMD-12538; Endogenous Chelator-GIDAnt, EMDB: EMD-12540; CTLHSR4, EMDB: EMD-12537; CTLHSR4 SRS module, EMDB:EMD-12564; PDB: 7NSC; CTLH-WDR26 supramolecular assembly, EMDB: EMD-12542; CTLH-WDR26 SA and SRS modules, EMDB: EMD-12545; CTLH-MKLN1 SA and SRS modules, EMDB: EMD-12547; Fbp1 crystal structure, PDB: 7NS5.
All proteomics data have been deposited on ProteomeXchange with the dataset identifier PRIDE: PXD024462.
All the unprocessed image data have been deposited to Mendeley Data : http://dx.doi.org/10.17632/rfpsg6939c.1