

The stabilized R-spine of oncogenic BRAF mutants impairs RAF inhibitor association and confers drug resistance.
Abstract
Although targeting BRAF mutants with RAF inhibitors has achieved promising outcomes in cancer therapy, drug resistance remains a remarkable challenge, and underlying molecular mechanisms are not fully understood. Here, we characterized a previously unknown group of oncogenic BRAF mutants with in-frame insertions (LLRins506 or VLRins506) of αC-β4 loop. Using structure modeling and molecular dynamics simulation, we found that these insertions formed a large hydrophobic network that stabilizes R-spine and thus triggers the catalytic activity of BRAF. Furthermore, these insertions disrupted BRAF dimer interface and impaired dimerization. Unlike BRAF(V600E), these BRAF mutants with low dimer affinity were strongly resistant to all RAF inhibitors in clinic or clinical trials, which arises from their stabilized R-spines. As predicted by molecular docking, the stabilized R-spines in other BRAF mutants also conferred drug resistance. Together, our data indicated that the stability of R-spine but not dimer affinity determines the RAF inhibitor resistance of oncogenic BRAF mutants.
INTRODUCTION
The Ser/Thr protein kinase, RAF, is a key component of RAS/RAF/mitogen-activated protein kinase kinase (MEK)/extracellular signal–regulated kinase (ERK) signaling that consists of three isoforms: CRAF (or RAF1), BRAF, and ARAF in mammalian cells (1). RAF plays a central role in cell biology and its aberrant activation induces developmental disorders and cancers (2). Under physiological conditions, RAF is recruited to plasma membrane by active RAS, where it forms homo/heterodimers and releases its catalytic activity toward MEK. A number of studies have identified dimerization as a key event in signal transduction mediated by RAF (3–13). The dimerization of RAF facilitates assembly of R-spine, a typical structure of active kinases, and triggers its catalytic activity (9, 14–18). On the other hand, it helps the recruitment of MEK and subsequent MEK dimerization, both of which are indispensable for the phosphorylation of MEK by RAF (10, 11). The different propensity of dimerization among RAF isoforms leads to their distinct ability to turn on ERK signaling with an order that BRAF > CRAF > ARAF (9, 10). In addition to active RAS and isoform propensity, RAF dimerization can be regulated on other layers such as genetic alterations, inhibitor association, and ERK-mediated feedback phosphorylation (19, 20).
In cancer genomes, genetic alterations that activate RAF mainly occur upstream (receptor tyrosine kinases and RAS), or on RAF itself, particularly BRAF probably by virtue of its high basal activity (19, 21). Although BRAF mutation/alteration exists in only ~7% cases of all cancers, it is highly prevalent in some cancer types such as melanoma, thyroid cancers, and histiocytosis (22, 23). Biochemical studies have suggested that cancer-related BRAF mutants might be classified as three groups: (i) highly constitutively active mutants (i.e., V600E) that achieve active conformation independent of dimerization and turn on downstream signaling by phosphorylation, (ii) kinase-dead or kinase-impaired mutants (i.e., V471F) that stimulate together with active RAS downstream signaling through dimerizing with and triggering the catalytic activity of wild-type paralogs, and (iii) intermediate active mutants (i.e., G469A) that are activated through enhanced dimerization and directly turn on downstream signaling (24). However, there are still some unique RAF mutants that cannot fall into any of these categories. For example, BRAF mutants with in-frame deletions of β3-αC loop (i.e., ΔNVTAP) have a high activity and a high dimer affinity (10, 25, 26). Moreover, even in the same group, different BRAF mutants might be activated through distinct mechanisms. As reported before, BRAF(V600E) achieves its active conformation through forming the K507-E600 salt bridge (27), whereas BRAF(L485F) and BRAF(L505H) do this through stabilizing their R-spines (18), although all of them have a high activity independent of dimerization. These findings indicate that BRAF mutants have diverse characters and complicated functional modes, whose understanding would improve precision therapies against cancers harboring BRAF mutations.
To target oncogenic BRAF mutants, RAF inhibitors such as vemurafenib and dabrafenib have been developed and applied to clinic treatment of cancers (28, 29). Although these inhibitors achieve a promising outcome on BRAF(V600E)–harboring cancers, a majority of cancers with BRAF mutation, at initial treatment phase, unfortunately, their efficacy is abrogated by quick-rising resistance (30). Mechanistic studies have shown that RAF inhibitor resistance arises from enhanced RAF dimerization by active RAS or by alternative RAF splicing at mRNA level that delete its N terminus (31–34). At present, how elevated RAF dimer affinity causes drug resistance remains unclear. To resolve this problem, the second-generation RAF inhibitors, PLX8394 and LY3009120, have been developed and are undergoing clinical trials, which either impairs dimerization upon association with BRAF mutants or blocks the activity of both protomers in BRAF dimers (35, 36). However, whether all these inhibitors in clinic or clinical trials would cover BRAF mutants other than V600E in cancer genomes needs to be further determined.
RESULTS
LLRins506 and VLRins506 define a previously unknown group of oncogenic BRAF mutants
Recently, a previously unknown group of BRAF mutants with in-frame insertion of three residues on αC-β4 loop was identified by cancer genomic sequencings (37, 38). These insertions arose from altered mRNA splicing (LLRins506) or from genomic DNA duplication (VLRins506) of BRAF (Fig. 1A) and existed in Langerhans cell histiocytosis or astrocytoma. According to the three-dimensional (3D) structure of BRAF kinase domain, the αC-β4 loop constitutes a large portion of dimer interface and positions αC-helix together with the β3-αC loop (Fig. 1B), both of which are essential for the catalytic activity of BRAF (10, 39). The insertion of three residues (LLR or VLR) in this loop would generate some new interactions that may alter local conformation and activity of BRAF. Thus, we measured the activity of LLRins506 and VLRins506 in 293T transfectants by using anti–phospho-ERK1/2 immunoblot. As shown in Fig. 1C, these mutants had much higher activity in contrast to wild-type counterpart, which is comparable with those of BRAF(V600E) and BRAF(ΔNVTAP), two well-defined constitutively active BRAF mutants (10, 22, 25, 26). To ensure that LLRins506 and VLRins506 are constitutively active mutants that activate ERK signaling independent of endogenous RAF molecules, we expressed these mutants in BRAF−/− or CRAF−/− fibroblasts and found that they could activate downstream pathway regardless of BRAF or CRAF deficiency (Fig. 1D). This finding was also supported by our short hairpin RNA (shRNA)–mediated knockdowns in which down-regulation of CRAF, ARAF, or KSR1 in LLRins506-reconstituted BRAF−/− fibroblasts has no effect on ERK signaling (fig. S1). Furthermore, LLRins506 and VLRins506 had strong ability to induce foci formation in both wild-type and RAF-deficient fibroblasts (Fig. 1E). Together, these data indicated that in-frame insertions of LLR or VLR on the αC-β4 loop of BRAF create constitutively active kinases that transform cells.
Fig. 1. LLRins506 and VLRins506 activate BRAF and transform cells independent of endogenous RAFs.
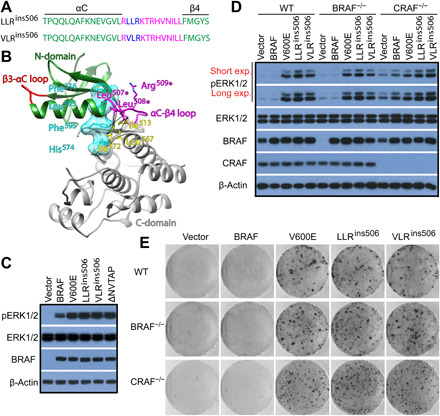
(A and B) LLRins506 and VLRins506 occur in the αC-β4 loop of BRAF. (A) LLRins506 and VLRins506 in the primary sequence of αC-β4 loop of BRAF. (B) 3D structural model of LLRins506. The N- and C-lobes of BRAF kinase domain were depicted as ribbons and colored with green or gray. The β3-αC and the αC-β4 loops, which play a critical role in positioning the αC-helix, were shown in red or magenta color. The position of R-spine residues within the structure were shown as cyan translucent surface representation. The inserted residues of LLRins506 (magenta color), R-spine residues (cyan color), and some of neighboring hydrophobic residues (yellow color) were shown as stick representation. Potential hydrophobic contacts that may be formed between LLR residues and R-spine residues as well as neighboring residues were indicated with dotted lines. (C) LLRins506 and VLRins506 mutants strongly activated the ERK signaling as BRAF(V600E) and BRAF(ΔVNTAP) did when expressed in 293T cells. The activity of ERK1/2 in 293T transfectants that express BRAF mutants was measured by anti–phospho-ERK1/2 immunoblotting. (D) LLRins506 and VLRins506 mutants activated the ERK signaling as BRAF(V600E) did in wild-type (WT), BRAF−/−, and CRAF−/− fibroblasts. The activity of ERK1/2 in fibroblast stable cell lines that express BRAF mutants was determined as that in (C). (E) LLRins506 and VLRins506 mutants induced foci formation as BRAF(V600E) did when expressed in fibroblasts. The foci formation assay was carried out as described in Materials and Methods. All images are representative of at least three independent experiments.
LLRins506 and VLRins506 activate BRAF by assembling a large hydrophobic network that involves in R-spine
To understand how LLRins506 or VLRins506 triggers the catalytic activity of BRAF, we mutated these three amino acids into Ala with different combinations. As shown in Fig. 2A, AAAins506 or AARins506 mutants did not exhibit any activity toward downstream MEK-ERK signaling when expressed in 293T cells in contrast to both wild-type BRAF and LLRins506 or VLRins506 mutants, suggesting that the hydrophobicity, but not the length of αC-β4 loop, is responsible for elevating the catalytic activity of BRAF. Furthermore, we found that the activity of ALRins506 was comparable to those of LLRins506 and VLRins506, significantly higher than the activity of LARins506 and wild-type BRAF, when expressed in 293T cells (Fig. 2B), suggesting that the second Leu (Leu508*), occupied by Thr508 in wild-type counterpart, plays a major role in activating LLRins506 and VLRins506 mutants although the first Leu or Val may have minor effect. To explore the structural and mechanistic basis of how this additional hydrophobic residue enhances the catalytic activity of BRAF, we carried out structural modeling of LLRins506 mutant on the basis of the structure of wild-type BRAF in dimeric state. In the model of LLRins506 mutant, hydrophilic Lys507 and Thr508 residues were replaced by two hydrophobic Leu residues, while Arg509 residue was retained (Fig. 2C and fig. S2). Moreover, Leu508* of LLRins506 mutant was optimally positioned to form hydrophobic interactions with neighboring residues such as Ile513, Leu567, Ile572, and in close proximity (within 5.5 Å) of Phe595 in R-spine (RS2), which leads to assembly of a large hydrophobic network. To further refine our structural model of LLRins506 mutant, we carried out unrestrained equilibrium molecular dynamics (MD) simulations of dimeric wild-type BRAF and LLRins506 mutant systems. Both systems were simulated for 500 ns in their apo-states for a total of 1 μs. We found that most fluctuations in both systems were centered around αC-helix, αC-β4 loop, activation loop region (near residues 600 to 650), and terminal regions (figs. S3 and S4). Analysis of MD-generated trajectories of dimeric wild-type BRAF and LLRins506 mutant further strengthened our speculation regarding the role of Leu508*. In LLRins506 mutant, Leu508* on average formed ~5 times more hydrophobic contacts with its neighboring residues including Leu507*, Ile513, Tyr566, Leu567, Ile572, and Phe595 compared to Thr508 in wild-type BRAF (Fig. 2D, top). In particular, Leu508* had significantly greater number of hydrophobic contacts with Phe595 of the R-spine than Thr508 in wild-type BRAF (Fig. 2D, bottom). This finding from MD simulations clearly established the central organizing role of Leu508* in LLRins506 mutant, which facilitates assembly of a large hydrophobic network including R-spine and thereby shifts the conformational equilibrium of BRAF to favor catalysis. Next, we validated our computational observations by using mutagenic assays that could potentially weaken the hydrophobic network formed by Leu508* in LLR/VLRins506 mutants. As shown in Fig. 2 (E to G), mutating Ile513, Leu567, or Ile572 into Ala significantly restricted the ability of ALRins506 to activate ERK signaling when expressed in 293T cells (Fig. 2, E to G). However, when identical mutations were introduced into BRAF(V600E) whose active conformation is stabilized by the salt bridge between K507 and E600 (27), we did not observe any remarkable effect. Similarly, this type of mutations only partially impaired the activity of BRAF(ΔNVTAP) whose active conformation is stabilized by enhanced dimerization (fig. S5A) (10, 25). Consistent with these findings, ALRins506 was more sensitive to F595 alteration than BRAF(V600E) and BRAF(ΔNVTAP), and F595V mutation abolished its ability to activate ERK signaling but not those of BRAF(V600E) and BRAF(ΔNVTAP) when expressed in 293T cells (Fig. 2H). To further confirm that LLRins506 or VLRins506 stabilizes active conformation of BRAF through facilitating assembly of a large hydrophobic network that involves in R-spine, we determined whether these mutants were resistant to R-spine disturbance and found that although mutating the R-spine residue #4 (RS4), Phe516 into Leu, completely blocked the activity of wild-type BRAF, it had no effect on ALRins506 mutant as well as BRAF(V600E) and BRAF(ΔNVTAP) (Fig. 2, I and J, and fig. S5B). Collectively, our data indicate that LLRins506 or VLRins506 activates BRAF by assembling a large hydrophobic network that includes and stabilizes R-spine.
Fig. 2. LLRins506 and VLRins506 trigger the catalytic activity of BRAF through assembling a large hydrophobic network that involves in R-spine.
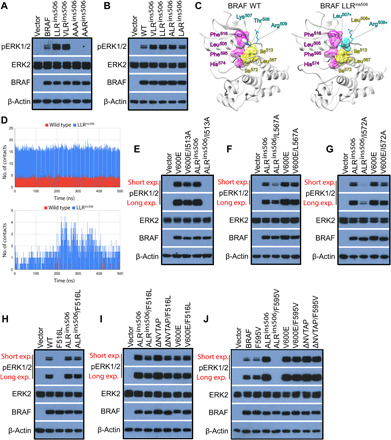
(A and B) The Leu508* plays a determinant role in activation of BRAF by LLRins506 and VLRins506. BRAF and its mutants were expressed in 293T cells and their activity was measured as phospho-ERK1/2 by immunoblotting. (C) Configuration of R-spine and αC-β4 loop of wild-type BRAF (left) and LLRins506 mutant (right) structural models. Residues of αC-β4 loop, R-spine, and neighboring hydrophobic residues were shown in cyan, pink, or yellow colors, respectively, while the rest of protein as gray ribbon. (D) Total number of hydrophobic contacts formed by Thr508 in wild-type BRAF (red) or by Leu508* in LLRins506 mutant (blue) with neighboring residues (top) and number of Thr508-Phe595 contacts in wild-type BRAF (red) or Leu508*-Phe595 contacts (blue) in LLRins506 mutant (bottom) observed during simulation were presented as histograms. The number of contacts presented in the plot was averaged across both protomers in a given dimer. (E to G) Altering the hydrophobic network formed by Leu508 through mutating either Ile513, Leu567, or Ile572 abolished the activity of BRAF(ALRins506). (H to J) BRAF(ALRins506) is resistant to the alteration of R-spine albeit sensitive to that of Phe595. The activity of BRAF and its mutants in (E) to (J) was determined as that in (A). All images are representative of at least three independent experiments.
LLRins506 and VLRins506 impair BRAF dimerization by altering dimer interface
Because the residues from αC-β4 loop constitute a large portion of dimer interface of RAF kinases (8), we next investigated whether LLR insertion on this loop altered RAF dimerization besides stabilizing R-spine. We found that the dimer interface of LLRins506 mutant was significantly rearranged compared to that of wild-type BRAF upon MD simulations, which lastly breaks the LLRins506 dimer over the course of simulation, although there was no major structural changes observed within individual protomers (Fig. 3A). We further assessed the relative changes of interprotomer contacts of wild-type and LLRins506 dimers before (t = 0 ns) and after (t = 500 ns) simulations by measuring the interprotomer pairwise minimum distances of dimer interface residues and visualized as a heatmap (Fig. 3B). The breaking of LLRins506 dimer exposed previously buried residues to the solvent, resulting in a remarkable reduction of dimer interface area from 1360.4 to 439.0 Å2. In contrast, the dimer interface area of wild-type BRAF was changed only marginally from 1158.4 to 1138.1 Å2 over simulation. Correspondingly, the solvent accessible surface area of dimer interface residues of LLRins506 mutant increased significantly compared to that of wild-type BRAF (Fig. 3C and tables S1 and S2). We thought that the extension of the αC-β4 loop in LLRins506 mutant displaced hydrophilic Thr508, Arg509, and His510 further into the interface region and thus potentially disintegrated the dimer interface of LLRins506 mutant. To validate our observations from simulations, we directly measured the relative dimer affinity of LLRins506 and VLRins506 mutants by using complimentary split luciferase assay (10, 40, 41), a living-cell protein interaction method similar to bioluminescence resonance energy transfer (42). Although LLRins506- or VLRins506-fused split luciferase pairs generated a stronger luciferase signal than that derived from monomeric BRAF mutant (R509H/AAE) (10), it was much weaker than those from both wild-type BRAF and BRAF(V600E)–fused split luciferase pairs (Fig. 3D), suggesting that LLRins506 and VLRins506 mutants have much less dimer affinity than wild-type BRAF and BRAF(V600E). Moreover, the weak luciferase signal from LLRins506-fused split luciferase pair could be elevated by different extents with KRAS(G12D) coexpression or high-dose dabrafenib treatment (fig. S6), indicating that the weak dimerization of LLRins506 could be enhanced upon RAS or RAF inhibitor engagement. Alternatively, we further confirmed these findings by using coimmunoprecipitation assay. When coexpressed in 293T cells, hemagglutinin (HA)–tagged wild-type BRAF and BRAF(V600E) were pulled down by FLAG-tagged counterparts, whereas it was barely reproduced with either LLRins506 or VLRins506 mutants (Fig. 3E), indicating that LLRins506 and VLRins506 mutants form much weaker homodimers in vivo. Consistently, we also found that this type of mutants barely formed heterodimers with wild-type BRAF, CRAF, ARAF, or KSR1 when coexpressed in 293T cells although their interaction with MEK is not altered (fig. S7).
Fig. 3. LLRins506 or VLRins506 disrupts the dimerization of BRAF.
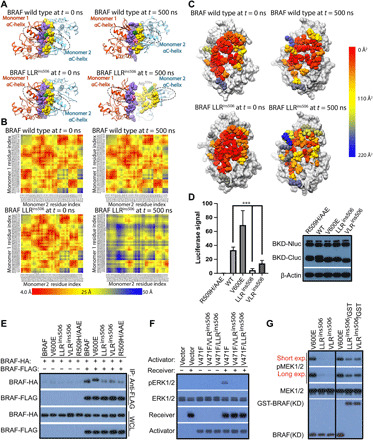
(A to C) Molecular simulation shows that LLRins506 impairs BRAF homodimerization. (A) The relative position of two protomers in wild-type BRAF versus LLRins506 dimers. All protomers were depicted as ribbons and dimer interface residues as spheres. Purple and pink were used to label dimer interface and Arg509 in one protomer (orange), while yellow and green for these elements in the other (light blue). (B) Visualization of interprotomer distances for interface residue pairs of wild-type BRAF versus LLRins506 mutant. (C) The solvent accessible surface areas of dimer interface residues in wild-type BRAF versus LLRins506 mutant. (D and E) LLRins506 mutant has much less dimer affinity than wild-type BRAF and V600E. The dimer affinity of wild-type BRAF and mutants was determined by complimentary split luciferase assay (D) and coimmunoprecipitation (E) as described in Materials and Methods. ***P < 0.001. (F) Kinase-dead LLRins506 and VLRins506 mutants are not able to transactivate CRAF. BRAF mutants were respectively coexpressed with CRAF receiver in 293T cells, and ERK signaling was detected by anti–phospho-ERK1/2 immunoblotting. (G) LLRins506 and VLRins506 mutants lose their catalytic activity in vitro, which is rescued by glutathione S-transferase (GST) fusion. BRAF mutants were purified from 293T transfectants by immunoprecipitation, and their catalytic activity was measured by in vitro kinase assay. All images are representative of at least three independent experiments.
Because the dimerization of RAFs is critical for their activation and catalytic activity toward MEK, kinase-dead RAF mutants with low dimer affinity hardly transactivate catalysis-competent RAF-binding partners and constitutively active RAF mutants with low dimer affinity frequently lose their catalytic activity toward MEK in vitro by virtue of dimer dissociation (10, 40, 41). Thus, we checked whether these phenomena occurred on LLRins506 or VLRins506 mutants given their low dimer affinity. As shown in Fig. 3F, catalytic spine-fused LLRins506 or VLRins506 mutant (V471F/LLRins506 and V471F/VLRins506) was not able to trigger the activity of CRAF in RAF coactivation assay (9, 18, 41, 43). Furthermore, purified LLRins506 and VLRins506 mutants from 293T transfectants barely phosphorylated MEK in vitro, whose activity can be restored by glutathione S-transferase (GST) fusion–enhanced dimerization (Fig. 3G). Together, these data demonstrated that LLR/VLR insertion on αC-β4 loop disorders BRAF dimer interface and remarkably impairs its dimerization.
LLRins506 and VLRins506 mutants are resistant to all clinical and preclinical RAF inhibitors albeit sensitive to MEK inhibitor
To target oncogenic BRAF mutants, the first-generation RAF inhibitors (vemurafenib and dabrafenib) had been developed and applied to clinic cancer treatment, and the second-generation inhibitors that have less paradoxical effect (PLX8394) or target both protomers of RAF dimer (LY3009120) were undergoing clinical trials (28, 29). We wondered whether these inhibitors could be used to target LLRins506 and VLRins506 mutants in cancer genomes. To test this notion, we examined the effects of these RAF inhibitors on 293T transfectants that express LLRins506 or VLRins506 mutants as well as BRAF(V600E) and BRAF(ΔNVTAP). As reported before, all these RAF inhibitors effectively blocked the activity of BRAF(V600E) (Fig. 4A), and only LY3009120 strongly inhibited the activity of BRAF(ΔNVTAP) (Fig. 4B). However, these inhibitors had little to no effects on LLRins506 and VLRins506 mutants (Fig. 4, C and D) and their median inhibitory concentrations (IC50s) against LLRins506 or VLRins506 mutants versus BRAF(V600E) had at least >10-fold difference (Fig. 4E). We further determined whether sorafenib, a nonspecific RAF inhibitor, could effectively inhibit the activity of LLRins506 or VLRins506, but unfortunately, it also failed as RAF-specific inhibitors did (fig. S8A). To construct an effective therapeutic approach against LLRins506- or VLRins506-harboring cancers, we next determined whether clinical MEK inhibitors such as trametinib, binimetinib, AZD6244, and cobimetinib were able to block MEK-ERK signaling activated by LLRins506 or VLRins506 mutants. Although all of them were effective compared with RAF inhibitors, trametinib exhibited the best ability to inhibit ERK signaling triggered by LLRins506 (fig. S8B). Unexpectedly, we found that ERK signaling driven by LLRins506 or VLRins506 mutants were slightly more sensitive to trametinib than those triggered by BRAF(V600E) or BRAF(ΔNVTAP) (Fig. 4, E and F), suggesting that trametinib might service as a good therapeutic approach against cancers harboring LLRins506 or VLRins506 mutants.
Fig. 4. LLRins506 and VLRins506 mutants are resistant to all RAF inhibitors in clinic or clinical trials albeit sensitive to MEK inhibitor.
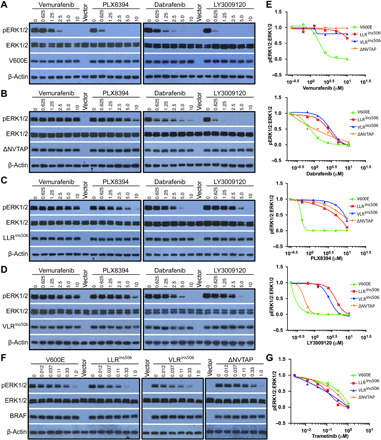
(A to E) Vemurafenib, dabrafenib, PLX8394, and LY3009120 are not able to effectively inhibit the activity of LLRins506 and VLRins506 mutants as they do on BRAF(V600E) or BRAF(ΔNVTAP). 293T transfectants that express individual BRAF mutants were treated with different drugs at indicated concentration for 3 hours, and their ERK signaling was measured by anti–phospho-ERK1/2 immunoblotting (A to D) and quantified to generate graphs by using GraphPad Prism 6 (E). (F and G) MEK inhibitor, trametinib, effectively inhibits the ERK signaling evoked by LLRins506 and VLRins506 mutants. The drug response of 293T transfectants that express individual BRAF mutants was determined as that in (A) to (E). All images are representative of at least three independent experiments.
The stabilized R-spine of LLRins506 or VLRins506 mutants impairs RAF inhibitor association
Acquired resistance to RAF inhibitors is a major challenge in current targeted therapy against BRAF-mutated cancers (19, 30). Molecular mechanisms underlying this phenomenon are complicated and not fully understood. Although an elevated dimer affinity had been shown responsible for RAF inhibitor resistance of BRAF(V600E) splicing variants (34) or BRAF(ΔNVTAP) (10, 25), it was not applicable to LLRins506 and VLRins506 mutants, given their low dimer affinity. To understand why LLRins506 and VLRins506 mutants are resistant to RAF inhibitors, here we first examined the structures of BRAF/inhibitor complexes that are available in the Protein Data Bank (PDB) database and found that RAF inhibitors locked BRAF in configurations with distorted or broken R-spine (fig. S9). This raised a possibility that the stabilized R-spine in LLRins506 and VLRins506 mutants impairs association of RAF inhibitors. To justify this notion, we docked dabrafenib, vemurafenib, LY3009120, and PLX8394, respectively, to representative structures of BRAF in either R-spineintact or R-spinebroken configurations and calculated their docking scores. Comparing the docking poses of these inhibitors in R-spinebroken (yellow) and R-spineintact (purple) configurations, we found that the disengagement between Phe595 and Leu505 in R-spinebroken configuration compared to that in R-spineintact configuration provided access to the hydrophobic “back pocket” for inhibitors, which allows them to bind deeper and achieves better docking scores (Fig. 5A and table S3). To validate this computational analysis, we directly measured the affinity of LLRins506 mutant with these RAF inhibitors by using microscale thermophoresis (MST) method and found that it had at least nine-fold higher IC50 for all tested inhibitors than BRAF(V600E) in vitro (Fig. 5B), indicating that this type of BRAF mutants can barely associate with RAF inhibitors. To further confirm that the stabilized R-spine impairs RAF inhibitor docking and results in drug resistance, we next determined whether other oncogenic BRAF mutants with a stabilized R-spine were resistant to RAF inhibitors as LLR/VLRins506 mutants did. In our previous studies, we had shown that L505H and L485F mutations stabilized R-spine of BRAF through distinct manners by different extends and thus elevated its kinase activity (18). As expected, these two BRAF mutants exhibited a strong resistance to all tested inhibitors with an exception for L485F when treated with LY3009120 (Fig. 5, C to E). Together, these data consistently demonstrated that the stability of R-spine in BRAF mutants is responsible for their drug resistance.
Fig. 5. The stabilized R-spine of LLRins506 or VLRins506 mutants impairs RAF inhibitor association, which also occurs in other BRAF mutants with stabilized R-spine.
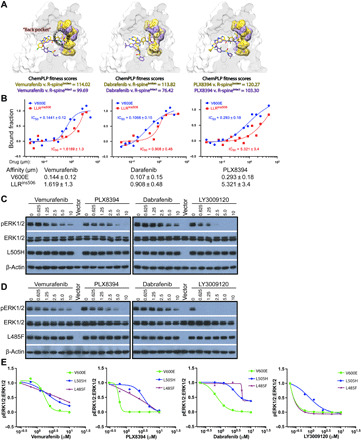
(A) Docking poses of vemurafenib, dabrafenib, and PLX8394 on BRAF with an intact (yellow) or broken (purple) R-spine. Configurations of intact and broken R-spine were shown in stick representation with translucent surfaces, while the ligands only in stick representation. (B) RAF inhibitors have much less affinity with LLRins506 mutant than BRAF(V600E). The apparent dissociation constant values were measured by using MST method in buffer with 1 mM adenosine 5′-triphosphate. (C to E) BRAF(L505H) and BRAF(L485F) that have enhanced R-spine exhibited similar resistance to RAF inhibitors. The drug response of 293T transfectants that express individual BRAF mutants was determined as in Fig. 4 (A to E). All images are representative of at least three independent experiments.
LLRins506 and VLRins506 induce in vivo tumors that are resistant to RAF inhibitor treatment
Because LLRins506 and VLRins506 mutants constitutively activated oncogenic ERK signaling and transformed immortalized fibroblast in vitro, we next evaluated their oncogenicity in vivo by using xenografted tumor mouse model. As shown in Fig. 6 (A to C), immortalized fibroblasts that express LLRins506 or VLRins506 mutant formed fibroblastomas upon subcutaneous injection into nonobese diabetic (NOD)–severe combined immunodeficient (SCID) mice as those that express BRAF(V600E) or BRAF(ΔNVTAP) did, indicating that LLRins506 and VLRins506 mutants are truly cancer drivers. Because these fibroblast cell lines were generated by reconstituting BRAF−/− fibroblasts with BRAF (or mutant)–internal ribosomal entry site (IRES)–green fluorescent (GFP) cassettes, and BRAF mutants were restricted at a comparable level of endogenous BRAF by cell sorting (fig. S10), this finding would not be an artifact arising from protein overexpression. To construct therapeutic strategies for treating cancers harboring LLRins506 or VLRins506 mutation, we next determined the efficacies of RAF inhibitors (vemurafenib, dabrafenib, and PLX8394) and MEK inhibitor (trametinib) against fibroblastomas induced by LLRins506 or VLRins506 mutant. Although both RAF inhibitors and MEK inhibitor impaired the growth of BRAF(V600E)–induced fibroblastomas by different extends (trametinib > dabrafenib > vemurafenib > PLX8394), only MEK inhibitor exhibited a strong inhibitory effect on that of LLRins506/VLRins506 mutant–induced fibroblastomas (Fig. 6, D to F). Consistent with this finding, our immunohistochemical staining revealed that administrating with MEK inhibitor, trametinib, but not with RAF inhibitors significantly decreased the level of phospho-ERK1/2 and Ki67 in LLRins506/VLRins506 mutant–induced fibroblastomas (Fig. 6G). Together, these data demonstrated that LLRins506 and VLRins506 are real driver mutations in cancer genomes and cancers harboring this type of mutations could be treated effectively with MEK inhibitor, trametinib.
Fig. 6. LLRins506 and VLRins506 mutants induce tumor formation in vivo, which is resistant to RAF inhibitors albeit sensitive to MEK inhibitor.
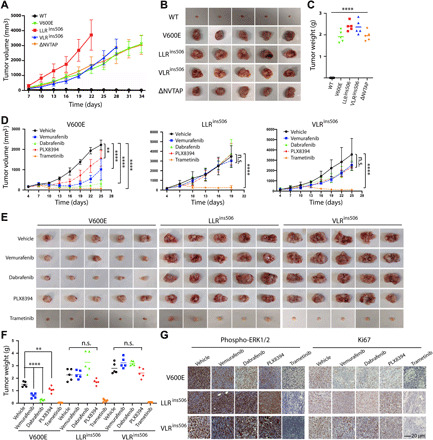
(A to C) LLRins506 and VLRins506 mutants function as a cancer driver and induce fibroblastomas as BRAF(V600E) and BRAF(ΔNVTAP) in vivo. BRAF−/− fibroblasts reconstituted with wild-type BRAF or its mutants were subcutaneously injected into NOD-SCID mice, and tumor growth was tracked as described in Materials and Methods. At the experimental end point, tumors were harvested and weighted (n = 5 per group; ****P < 0.0001). (D to F) Xenograft tumors induced by LLRins506 and VLRins506 mutants are resistant to vemurafenib, dabrafenib and PLX8394 albeit sensitive to trametinib. The experiments were carried out as that in (A) to (C), except that NOD-SCID mice were administered orally and daily with vehicle or RAF/MEK inhibitors (vemurafenib, 120 mg/kg; dabrafenib, 200 mg/kg; PLX8394, 150 mg/kg; trametinib, 3 mg/kg; n = 5 per group; **P < 0.01, and ****P < 0.0001). (G) Immunohistochemistry staining analysis of xenografted tumors from (E). The activity of ERK1/2 and cell proliferation were assessed by staining for phospho-ERK1/2 and Ki67, respectively. All images are representative of at least three independent experiments. n.s., not significant.
The in-frame insertion of hydrophobic residues in αC-β4 loop also activates epidermal growth factor receptor in cancer genomes by stabilizing its active conformation
The discovery of LLRins506 and VLRins506 mutants led us to identify other oncogenic kinase mutants that are activated through a same principle. By surveying cancer genomic databases, we found that there were a notable number of epidermal growth factor receptor (EGFR) mutants that have in-frame insertions in αC-β4 loop (fig. S11A). Although previous studies had shown that these EGFR mutants were constitutively active (44, 45), how the inserted residues trigger the activity of EGFR was not clear. To address this question, we first measured the activity of representative EGFR mutants with in-frame insertion in αC-β4 loop (ASVins769 and SVDins770) and found that they activated ERK signaling when expressed in 293T cells (fig. S11B). A single Val insertion at the same location also triggered the activity of EGFR as those representative insertions did, suggesting that a hydrophobic residue at position 772* plays a major role in activating EGFR. Next, we carried out structural modeling of ASVins769 and SVDins770 mutants by using wild-type EGFR structure as template (46) and found that the additional Val772* from ASVins769 or SVDins770 formed hydrophobic interactions with neighboring Leu1017 (fig. S11C), while the additional Ser771* either compensated for the loss of Asp770-Arg776 interaction in ASVins769 mutant or provided an additional interacting partner for Arg776 in SVDins770 mutant. Because the Asp770-Arg776 salt-bridge connects the αC-β4 loop with Leu777 (RS4) of the R-spine via Arg776 and is a hallmark of the active conformation in wild-type EGFR, the additional hydrophobic Val772* and polar Ser771* would strengthen a network of polar and hydrophobic interactions adjacent to R-spine and thus shift the conformational equilibrium of EGFR toward active form similar to that in LLRins506 and VLRins506 mutants of BRAF albeit through a different, indirect mode of action. Like LLRins506 and VLRins506 mutants of BRAF, these EGFR mutants did not need dimerization for maintaining their active conformation and a dimer interface mutation (V948R) did not inhibit their activity (fig. S11D). Together, our data implied that in-frame insertions in αC-β4 loop activate EGFR by stabilizing its active conformation.
DISCUSSION
Genetic mutations/alterations activate BRAF through diverse mechanisms, whose understanding is indispensable for developing precise targeted therapy. Recently, a previously unknown group of BRAF mutants with in-frame insertion of LLRins506 or VLRins506 in αC-β4 loop were discovered in Langerhans cell histiocytosis and astrocytoma (37, 38). However, whether LLRins506 and VLRins506 are driver mutations and how these mutations turn on the catalytic activity of BRAF as well as whether they can be targeted by clinical or preclinical RAF inhibitors are not clear. In this study, we have characterized LLRins506 and VLRins506 mutants and shown that these highly active BRAF mutants turn on downstream signaling and transform cells independent of wild-type paralogs. Mechanistically, we have demonstrated that LLRins506 or VLRins506 triggers the catalytic activity of BRAF by assembling a large hydrophobic network that includes and stabilizes R-spine. We argue that such a R-spine stabilizing network of interactions could shift the conformational equilibrium of BRAF favoring the active form. Because the αC-β4 loop of BRAF constitutes a large portion of dimer interface and its alteration potentially impairs or enhances BRAF dimerization (8), we have further determined the effect of LLR/VLRins506 on the dimerization of BRAF and found that LLR/VLRins506 disrupts dimer interface, which markedly weakens BRAF dimerization. The enhanced dimerization of oncogenic BRAF mutants has been linked to drug resistance, and BRAF mutants with a high dimer affinity such as BRAF(ΔVNTAP) and BRAF(V600E) splicing variants without N terminus (V600E/ΔNT) have been shown to be strongly resistant to the first-generation RAF inhibitors vemurafenib and dabrafenib as well as the second-generation RAF inhibitor PLX8394 (10, 25, 34). Unexpectedly, LLRins506 and VLRins506 mutants are also resistant to these inhibitors, and even to LY3009120, a pan-RAF inhibitor that blocks the activity of BRAF mutants with or without elevated dimer affinity (36), although they have an even lower dimer affinity than wild-type BRAF. This finding indicates that dimer affinity does not directly correlates with drug resistance of BRAF mutants. To understand molecular basis underlying RAF inhibitor resistance, we examined the structural conformations of BRAF complexes associated with all these four inhibitors and found that an engagement of these inhibitors disrupted the R-spine of BRAF in all cases to varying degrees, and an intact R-spine of BRAF would impair their binding, suggesting that a highly stabilized R-spine is a key factor that determines drug resistance of BRAF mutants. To strengthen our conclusion, we further examined the drug sensitivity of BRAF(L505H) and BRAF(L485F), two other BRAF mutants with stabilized R-spines, and found that they exhibited similar drug resistance as LLRins506 and VLRins506 mutants. Together, our study has revealed the root of RAF inhibitor resistance and clearly demonstrated that R-spine stability but not dimer affinity confers the drug resistance of BRAF mutants, which has important implications in precise targeted therapies for cancers harboring BRAF mutations.
Stabilizing R-spine not only triggers the catalytic activity of RAF but also dampens its drug response. A number of studies have suggested that diverse oncogenic alterations can stabilize R-spine of RAF by different extends through distinct mechanisms and thus create constitutively active kinases that are resistant to RAF inhibitors (9, 10, 18, 25, 34). The first type of alterations includes variations that improve dimerization, such as BRAF(V600E/ΔNT) and BRAF(ΔNVTAP). R-spine of these BRAF mutants is enhanced through a coordinative effect caused by high-affinity dimerization. The activity of these mutants cannot be blocked by vemurafenib, dabrafenib, and PLX8394 but by LY3009120 because its methyl-(methylamino)pyridopyrimindyl group can replace F595 (RS2) residue and assemble a much more stable hydrophobic core than R-spine itself (36). The second type of alterations occurs on the residues of R-spine, such as L505H and L505M, which directly strengthen R-spine (18). These BRAF mutants likely have a more stable R-spine than the first type of BRAF mutants and thus exhibit a partial resistance to LY3009120. The third type of alterations stabilizes R-spine of BRAF through an assembly of a large hydrophobic network that associates with R-spine. L485F and LLR/VLRins506 mutants belong to this group. As reported before, F485, F498, and I527 form a hydrophobic network that linked to R-spine in L485F mutant (18), and here we have shown that Leu508*, I513, L567, and I572 constitute an even larger hydrophobic network that involves in R-spine in LLR/VLRins506 mutants. Dependent of the stability of R-spine, these mutants might have a strong resistance (i.e., LLR/VLRins506 mutants) or little to no resistance (i.e., L485F) to LY3009120, albeit highly resistant to vemurafenib, dabrafenib, and PLX8394. Unfortunately, for all types of BRAF mutants, we are not able to quantify their R-spine stability by virtue of no reliable approaches available. This denies our effort to correlate the R-spine stability of BRAF mutants directly with their drug binding affinity or resistance.
To understand molecular mechanism that underlies RAF inhibitor resistance resulted from R-spine stabilization, we have carried out molecular docking analysis, which clearly shows that different RAF inhibitors have their own preferring R-spine configurations although all of them distort R-spine upon association. Vemurafenib and dabrafenib have priority for the DFG-in configuration, while LY3009120 for the DFG-out configuration. Although the structure of BRAF/PLX8394 complex has not been resolved, our data suggests that this inhibitor prefers a binding mode similar to vemurafenib and dabrafenib (type 1½ inhibitor) rather than that of LY3009120 (type 2 inhibitor). Nevertheless, our docking studies support that both types of 1½ and 2 inhibitors bind poorly to BRAF when the R-spine is assembled (R-spineintact) and could putatively explain how and why R-spine-stabilizing mutations such as LLRins506/VLRins506 confer resistance to these different types of inhibitors. This conclusion can be further strengthened by the docking scores of RAF inhibitors on different configurations of BRAF (table S3). Although RAF inhibitors had significantly higher best docking scores for R-spinebroken configurations, they achieved either comparable (dabrafenib and LY3009120) or slightly lower (vemurafenib and PLX8394) best docking scores for R-spineintact configurations of LLRins506 mutant comparing to those of wild-type BRAF. These data suggest that the drug resistance of BRAF mutations such as LLRins506/VLRins506 is more dominantly prompted by a higher population of R-spineintact state in the LLRins506/VLRins506 mutants’ conformational ensemble than that in the wild-type BRAF’s, as a consequence of a shift in the conformational equilibrium of BRAF induced by these R-spine–stabilizing mutations. In contrast, direct effects from these mutations may be minor and limited to specific drugs. In addition, our study also demonstrates that an integrated approach that combines protein structure modeling, drug docking, and experimental testing can be very effective to identify and understand the drug resistance of various BRAF mutants, given the fact that a large number of non-V600E BRAF mutants exist in cancer genomes and lack high-resolution experimental structures.
Although resistant to all RAF inhibitors, LLRins506 and VLRins506 mutants are sensitive to MEK inhibitor, trametinib, both in vitro and in vivo. Therefore, we will next examine the therapeutic efficacy of trametinib against LLR/VLRins506-harboring cancers through clinical trials. However, given the toxicity of MEK inhibitors, we will develop novel RAF inhibitors that can effectively target LLRins506 and VLRins506 mutants as well as other RAF mutants with stabilized R-spine, which requires us to resolve their high-resolution structures in future study. Nevertheless, these works would propel the precise therapy of cancers driven by BRAF mutations.
MATERIALS AND METHODS
Antibodies, biochemicals, plasmids, and cell lines
Antibodies used in this study include anti–phospho-ERK1/2 (no. 4370), anti–phospho-MEK1/2 (no. 9154), anti-MEK1/2 (no. 9124), anti-ARAF (no. 4432), and anti-HA (no. 3724) (Cell Signaling Technology); anti-FLAG (F3165), anti-BRAF (SAB5300503), anti-CRAF (SAB5300393), and anti–β-actin (A2228) (Sigma-Aldrich); anti-KSR1 (no. PA5-75208, Invitrogen); anti-ERK1/2 (A0229, AB clonal); anti-Ki67 (ab16667, Abcam); and horseradish peroxidase–labeled secondary antibodies (the Jackson laboratory). All antibodies were diluted according to the manufacturers’ recommended protocols.
Vemurafenib, dabrafenib, PLX8394, LY3009120, and trametinib were purchased from Selleckchem. All other chemicals were obtained from Sigma-Aldrich.
Plasmids that encode BRAF, CRAF, MEK1, or their mutants were generated by polymerase chain reaction and Gibson assembly. pCDNA3.1(+) vector (Invitrogen) was used for transient expression, pMSCV-MCS-IRES-GFP retroviral vector (derived from pMSCV vector, Clontech) for stable expression, and pET-28a vector (Novagen) for bacterial expression. The shRNAs that target murine CRAF, ARAF, and KSR1 were designed by using a website software (http://katahdin.cshl.org/siRNA/RNAi.cgi?type=shRNA) and were inserted into pLL3.7 lentiviral vector (Addgene) by using traditional molecular cloning methods. The targeting sequences were listed as follows:
ARAF shRNA no. 1: 5′-GGCTCATCAAAGGAAGAAA-3′
ARAF shRNA no. 2: 5′-GGAAGGCATGAGTGTCTAT-3′
ARAF shRNA no. 3: 5′-GGTCTACAGGTCATCAAA-3′
CRAF shRNA no. 1: 5′-GGAATGGAATGAGCTTACA-3′
CRAF shRNA no. 2: 5′-GGAGATGTTGCAGTAAAGA-3′
CRAF shRNA no. 3: 5′-GGAATGAGCTTACATGACT-3′
KSR1 shRNA no. 1: 5′-GTGCCAGAAGAGCATGATTTT-3′
KSR1 shRNA no. 2: 5′-CATGGGTTATCTTCATGCAAA-3′
Wild-type, BRAF−/−, and CRAF−/− fibroblasts were gifts from M. Baccarini at the University of Vienna (47, 48). Human embryonic kidney (HEK) 293T cell line was obtained from American Type Culture Collection.
Cell culture, transfection, and transduction
All cell lines were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (Hyclone). Cell transfection was carried out by using the polyethylenimine transfection reagent. To generate stable cell lines that express wild-type BRAF or its mutants, viruses were prepared and applied to infect target cells according to our previous studies (10, 18). Infected cells were sorted by using flow cytometry.
Protein expression and purification
6xHis-tagged MEK1(K97A) was expressed in BL21(DE3) strains and purified by using a nickel column (Qiagen) as described before (10). FLAG-tagged BRAF(V600E) and BRAF(LLRins506) were respectively expressed in HEK-293T cells and purified by using anti-FLAG affinity gel and 3xFLAG peptide (Sigma-Aldrich) and following the manufacturer’s protocol.
Immunoprecipitation, in vitro kinase assay, and immunoblotting
Immunoprecipitations were performed as described previously (9, 18, 43). Briefly, whole-cell lysates from 293T transfectants that express wild-type BRAF or its mutants were mixed with anti-FLAG beads (A2220) (Sigma-Aldrich), rotated in cold room for 60 min, and washed three times with radioimmunoprecipitation buffer. For in vitro kinase assays of BRAF mutants, the immunoprecipitants were washed once with kinase reaction buffer (25 mM Hepes, 10 mM MgCl2, 0.5 mM Na3VO4, 0.5 mM dithiothreitol, pH 7.4) and then incubated with 20-μl kinase reaction mixture [2-μg His-tagged MEK1(K97A) and 100 μM adenosine 5′-triphosphate in 20-μl kinase reaction buffer] per sample at room temperature for 10 min. Kinase reaction was stopped by adding 5 μl per sample of 5× Laemmli sample buffer and determined by immunoblotting. Otherwise, the immunoprecipitants were directly mixed with 2× Laemmli sample buffer before detection. The immunoblotting was carried out as described before (41).
Foci formation assay
The foci formation assay was performed as described before (43). Immortalized mouse fibroblasts infected with retroviruses encoding wild-type BRAF or its mutants were plated at 5 × 103 cells per 60-mm dish and fed every other day. Twelve days later, cells were fixed with 2% formaldehyde and stained with Giemsa solution (Sigma-Aldrich).
Complementary split luciferase assay
293T transfectants that express different pairs of Nluc- and Cluc-fused BRAF proteins were plated in 24-well Krystal black image plates at a seeding density of 2 × 105 per well. Twenty-four hours later, d-luciferin (0.2 mg/ml) was added to the culture, and the incubation was allowed for 30 min before the luciferase signals were measured by using Promega GloMax-Multi Detection System.
BRAF and EGFR structural modeling
Standard structural modeling
For modeling all missing regions (except for the αC-β4 loop) in the kinase domain (amino acids 455 to 723) of BRAF, canonical sequence was extracted from UniProt database (AC: P15056) and added to the crystal structure of BRAF resolved in its dimeric state (PDB ID: 4XV1) (35) by using MODELLER v9.13 (49). A total of 500 models were generated for every single query-template alignment and the best model was selected with DOPE score (50).
Advanced structural modeling
The standard structural modeling method was adjusted, when we modeled the αC-β4 loop in dimeric wild-type BRAF and LLRins506 mutant. The interprotomer hydrogen bonds formed by Arg at position 509 with the corresponding residues in the other protomer (Phe516, Thr508 in wild type, and Leu508 in LLRins506) were retained by the imposition of distance restraints on the donor and acceptor atoms (51, 52). In addition, we adopted a sliding-window alignment scheme to model the αC-β4 loop region in LLRins506 mutant with greater accuracy: Ten distinct query-template sequence alignments were generated with each alignment using a different three-residue region in the template (fig. S2). For wild-type BRAF, a total of 500 models were generated with a single alignment, while for LLRins506 mutant, a total of 5000 models were generated with these 10 alignments. For modeling two EGFR mutants ASVins769 and SVDins770, the canonical sequence of human EGFR was obtained from UniProt database (AC: P00533). A similar advanced structural modeling method was applied respectively to two mutants by using the wild-type EGFR structure (PDB ID: 2GS6) (46) as the template. In both mutants, the distinctive turn-like conformation adopted by Asn771, Pro772, and His773 in the αC-β4 loop was retained using distance restraints (51, 52). The sliding-window alignment scheme was used to generate 10 alignments within the αC-β4 region, which yields a total of 5000 models for each mutant. For BRAF LLRins506 and the two EGFR mutants, the best models were always selected with Discrete Optimized Protein Energy (DOPE) score (provided in the Supplementary Materials).
MD simulations
MD simulations of dimeric wild-type BRAF and LLRins506 mutants in apo state were performed with GROMACS 2018 suite of programs (53). The N (─NH2) and C terminus (─COOH) of proteins were rendered neutral. The systems were described using parameters derived from the OPLS-AA/L all-atom force field. Proteins were first solvated with single-point charge water in a cubic box with a solute-box distance of 15 Å, followed by addition of neutralizing counterions (Na+ and Cl−). Short-range nonbonded interactions were accounted for with a 14 Å cutoff distance, while long-range interactions were estimated using the particle mesh Ewald method. Periodic boundary conditions were applied in the X, Y, and Z directions. Then, systems were energy-minimized using steepest descent method, followed by equilibration for 100 ps each under NVT and NPT conditions for stabilizing temperature and pressure around 310 K and 1.0 bar, respectively. Furthermore, equilibration was performed with position restraints applied on all heavy atoms of the protein. Following equilibration, production MD runs were carried out for 500 ns without any restraints, using Nosé-Hoover thermostat and Parrinello-Rahman barostat for maintaining temperature and pressure.
Analysis of crystal structures and MD trajectories
Analysis of the crystal structures and MD-generated trajectories was carried out using a combination of programs available within the GROMACS 2018 suite and in-house Perl scripts. Hydrogen bonds and salt-bridge interactions in modeled structures and MD trajectories were defined using geometric parameters used by Mishra et al. (54), while hydrophobic contacts were characterized on the basis of parameters reported by Pierri and co-workers (55).
Molecular docking
For ligand-bound wild-type BRAF structures considered in our docking study, the protocol described above was used to model missing regions in the structures of BRAF loading with vemurafenib, dabrafenib, LY3900120, and adenosine 5′-diphosphate analog (PDB IDs: 5ITA, 4XV2, 5C9C, and 6PP9), respectively. For active-like BRAF LLRins506 mutant, we used PDB ID: 6PP9 as template and used the sliding window scheme to generate and select the best model. Docking studies were carried out using GOLD 2020.1 (56) and docked poses were assessed on the basis of empirical ChemPLP fitness score (a higher ChemPLP score represents for more favorable protein-ligand interaction). The 3D coordinates of ligands used for docking, including vemurafenib (CID: 42611257), dabrafenib (CID: 44462760), PLX8394 (CID: 90116675), and LY3009120 (CID: 71721540), were obtained from PubChem. The structures of BRAF kinase domain used as receptors for docking and the top-ranked docked poses were provided in the Supplementary Materials.
MST analysis
The affinity of BRAF(V600E) and LLRins506 mutants with different RAF inhibitors (vemurafenib, dabrafenib, and PLX8394) was measured using the Monolith NT.115 from NanoTemper Technologies. The MST analysis was carried out as described before (11). Briefly, BRAF mutants purified from 293T transfectants were labeled with a fluorescent dye NT-647 (Cysteine Reactive) according to the manufacturer’s protocol. Then, a series of protein solutions with different concentrations were prepared by consecutive twofold dilutions in buffer containing 25 mM Hepes (pH 7.5), 150 mM NaCl, 0.2% IGEPAL, and 0.1 mM Tris(2-carboxyethyl)Phosphine (TCEP). The labeled proteins were mixed with unlabeled drugs at a volume ratio of 1:1 and loaded into silica capillaries after a short incubation at room temperature. The measurements were performed at 25°C by using 20% light-emitting diode power and 40% MST power. The laser-on and laser-off intervals were 30 and 5 s, respectively. NanoTemper Analysis (x86) software was used to fit the data and to determine the apparent dissociation constant values.
Animal studies
For xenograft experiments, female NOD-SCID mice (6 to 8 weeks) were subcutaneously injected with 5 × 106 cells per mice in 1:1 Matrigel (Corning). Tumor volumes were monitored by digital calipers twice a week and calculated using the formula: volume = (width)2 × length/2. Vemurafenib (120 mg/kg), dabrafenib (200 mg/kg), PLX8394 (150 mg/kg), and trametinib (3 mg/kg) were administered orally and daily when tumors reached an average volume of ~100 mm3 according to the previous studies (57, 58). At the experiment endpoint, mice were euthanized, and tumors were harvested for ex vivo analysis and subsequent histology. All operations were approved by the Animal Ethics Committee of National Cancer Centre Singapore (NCCS) with Institutional Animal Care and Use Committee (IACUC)–approved animal protocol.
Immunohistochemistry staining
Tumors were fixed in 10% buffered formalin overnight and embedded according to the standard procedures. Tumor sections were cut to 4-μm thickness, mounted on glass slides, and air-dried at room temperature. After antigen retrieval, tumor sections were stained with antibodies and then with hematoxylin. Images of tumor sections were taken with a bright light microscope at ×10.
Statistical analysis
All statistical analysis in this study was performed using GraphPad InStat (GraphPad Software, CA, USA). Statistical significance was determined by two-tailed Student’s t test in animal studies and error bars represent SD to show variance between samples in each group, or by one-sample t test in other experiments and error bars represent SD to show variance between independent experiments.
Acknowledgments
We thank M. Baccarini at the University of Vienna for the gifts of BRAF−/− fibroblasts and CRAF−RA fibroblasts, and K. Sabapathy, K. Man Hui, and J. Yuan from the National Cancer Centre Singapore and D. Virshup and M. Wang from the Duke-NUS Medical School for help in experimental approaches and comments on this manuscript. The MD simulations were performed on resources of the National Supercomputing Centre, Singapore (www.nscc.sg). Funding: This study was funded by Asia Fund for Cancer Research (AFCR-2017/2019-JH), SingHealth Foundation (AM/TP011/2018), NMRC (OFIRG18nov-0078), and Duke-NUS Khoo Bridge Funding Award (Duke-NUS-KBrFA/2020/0036) (to J.H.); and by the Biomedical Research Council of A*STAR (to R.N.V.K.D. and H.F.). Author contributions: J.Y., R.N.V.K.D., H.F., and J.H. designed the study; J.Y. and J.H. searched databases/literatures for BRAF and EGFR mutations in cancer genomes; J.Y., Z.T., W.H.N., K.C.G., M.P.M., A.F., Z.H.T., Y.R.M.S., U.D., and J.H. carried out molecular biology, biochemistry, and cell biology experiments; J.Y. and K.C.G. constructed mouse xenograft models and performed immunohistology analysis; P.L., Z.C., and J.H. supervised all wet experiments and interpreted experimental data; R.N.V.K.D. and H.F. carried out all in silico studies and interpreted computational data; J.Y., R.N.V.K.D., H.F., and J.H. wrote the manuscript; H.F. and J.H. revised manuscript; and all authors commented and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data required for supporting the conclusion in the paper are present in the main text and/or the Supplementary Materials and Methods. The plasmids used in this study can be provided by J.H. pending scientific review and a completed material transfer agreement. Requests for the plasmids should be submitted to hu.jiancheng@nccs.com.sg.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg0390/DC1
REFERENCES AND NOTES
- 1.Lavoie H., Therrien M., Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 16, 281–298 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Maurer G., Tarkowski B., Baccarini M., Raf kinases in cancer–roles and therapeutic opportunities. Oncogene 30, 3477–3488 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Farrar M. A., Alberola-lla J., Perlmutter R. M., Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature 383, 178–181 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Luo Z., Tzivion G., Belshaw P. J., Vavvas D., Marshall M., Avruch J., Oligomerization activates c-Raf-1 through a Ras-dependent mechanism. Nature 383, 181–185 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Weber C. K., Slupsky J. R., Kalmes H. A., Rapp U. R., Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 61, 3595 (2001). [PubMed] [Google Scholar]
- 6.Wan P. T. C., Garnett M. J., Roe S. M., Lee S., Niculescu-Duvaz D., Good V. M., Project C. G., Jones C. M., Marshall C. J., Springer C. J., Barford D., Marais R., Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Garnett M. J., Rana S., Paterson H., Barford D., Marais R., Wild-type and mutant B-RAF Activate C-RAF through distinct mechanisms involving heterodimerization. Mol. Cell 20, 963–969 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Rajakulendran T., Sahmi M., Lefrançois M., Sicheri F., Therrien M., A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Hu J., Stites E. C., Yu H., Germino E. A., Meharena H. S., Stork P. J. S., Kornev A. P., Taylor S. S., Shaw A. S., Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154, 1036–1046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan J., Ng W. H., Lam P. Y. P., Wang Y., Xia H., Yap J., Guan S. P., Lee A. S. G., Wang M., Baccarini M., Hu J., The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene 37, 5719–5734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan J., Ng W. H., Tian Z., Yap J., Baccarini M., Chen Z., Hu J., Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci. Signal. 11, eaar6795 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Santos E., Crespo P., The RAS-ERK pathway: A route for couples. Sci. Signal. 11, eaav0917 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Park E., Rawson S., Li K., Kim B.-W., Ficarro S. B., Pino G. G.-D., Sharif H., Marto J. A., Jeon H., Eck M. J., Architecture of autoinhibited and active BRAF–MEK1–14-3-3 complexes. Nature 575, 545–550 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornev A. P., Taylor S. S., Ten Eyck L. F., A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 14377–14382 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor S. S., Kornev A. P., Protein kinases: Evolution of dynamic regulatory proteins. Trends Biochem. Sci. 36, 65–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor S. S., Shaw A., Hu J., Meharena H. S., Kornev A., Pseudokinases from a structural perspective. Biochem. Soc. Trans. 41, 981–986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw A. S., Kornev A. P., Hu J., Ahuja L. G., Taylor S. S., Kinases and pseudokinases: Lessons from RAF. Mol. Cell. Biol. 34, 1538–1546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu J., Ahuja L. G., Meharena H. S., Kannan N., Kornev A. P., Taylor S. S., Shaw A. S., Kinase regulation by hydrophobic spine assembly in cancer. Mol. Cell. Biol. 35, 264–276 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degirmenci U., Wang M., Hu J., Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cell 9, 198 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brummer T., McInnes C., RAF kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene 39, 4155–4169 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Roberts P. J., Der C. J., Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W. C., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A., Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Holderfield M., Deuker M. M., McCormick F., McMahon M., Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dankner M., Rose A. A. N., Rajkumar S., Siegel P. M., Watson I. R., Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 37, 3183–3199 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Chen S.-H., Zhang Y., Horn R. D. V., Yin T., Buchanan S., Yadav V., Mochalkin I., Wong S. S., Yue Y. G., Huber L., Conti I., Henry J. R., Starling J. J., Plowman G. D., Peng S.-B., Oncogenic BRAF deletions that function as homodimers and are sensitive to inhibition by RAF dimer inhibitor LY3009120. Cancer Discov. 6, 300–315 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Foster S. A., Whalen D. M., Özen A., Wongchenko M. J., Yin J., Yen I., Schaefer G., Mayfield J. D., Chmielecki J., Stephens P. J., Albacker L. A., Yan Y., Song K., Hatzivassiliou G., Eigenbrot C., Yu C., Shaw A. S., Manning G., Skelton N. J., Hymowitz S. G., Malek S., Activation mechanism of oncogenic deletion mutations in BRAF, EGFR, and HER2. Cancer Cell 29, 477–493 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Haling J. R., Sudhamsu J., Yen I., Sideris S., Sandoval W., Phung W., Bravo B. J., Giannetti A. M., Peck A., Masselot A., Morales T., Smith D., Brandhuber B. J., Hymowitz S. G., Malek S., Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer Cell 26, 402–413 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Yuan J., Dong X., Yap J., Hu J., The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 13, 113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yaeger R., Corcoran R. B., Targeting alterations in the RAF–MEK pathway. Cancer Discov. 9, 329–341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lito P., Rosen N., Solit D. B., Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Heidorn S. J., Milagre C., Whittaker S., Nourry A., Niculescu-Duvas I., Dhomen N., Hussain J., Reis-Filho J. S., Springer C. J., Pritchard C., Marais R., Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatzivassiliou G., Song K., Yen I., Brandhuber B. J., Anderson D. J., Alvarado R., Ludlam M. J. C., Stokoe D., Gloor S. L., Vigers G., Morales T., Aliagas I., Liu B., Sideris S., Hoeflich K. P., Jaiswal B. S., Seshagiri S., Koeppen H., Belvin M., Friedman L. S., Malek S., RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Poulikakos P. I., Zhang C., Bollag G., Shokat K. M., Rosen N., RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poulikakos P. I., Persaud Y., Janakiraman M., Kong X., Ng C., Moriceau G., Shi H., Atefi M., Titz B., Gabay M. T., Salton M., Dahlman K. B., Tadi M., Wargo J. A., Flaherty K. T., Kelley M. C., Misteli T., Chapman P. B., Sosman J. A., Graeber T. G., Ribas A., Lo R. S., Rosen N., Solit D. B., RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang C., Spevak W., Zhang Y., Burton E. A., Ma Y., Habets G., Zhang J., Lin J., Ewing T., Matusow B., Tsang G., Marimuthu A., Cho H., Wu G., Wang W., Fong D., Nguyen H., Shi S., Womack P., Nespi M., Shellooe R., Carias H., Powell B., Light E., Sanftner L., Walters J., Tsai J., West B. L., Visor G., Rezaei H., Lin P. S., Nolop K., Ibrahim P. N., Hirth P., Bollag G., RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Peng S.-B., Henry J. R., Kaufman M. D., Lu W.-P., Smith B. D., Vogeti S., Rutkoski T. J., Wise S., Chun L., Zhang Y., Van Horn R. D., Yin T., Zhang X., Yadav V., Chen S.-H., Gong X., Ma X., Webster Y., Buchanan S., Mochalkin I., Huber L., Kays L., Donoho G. P., Walgren J., McCann D., Patel P., Conti I., Plowman G. D., Starling J. J., Flynn D. L., Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell 28, 384–398 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Héritier S., Hélias-Rodzewicz Z., Chakraborty R., Sengal A. G., Bellanné-Chantelot C., Thomas C., Moreau A., Fraitag S., Allen C. E., Donadieu J., Emile J.-F., New somatic BRAF splicing mutation in Langerhans cell histiocytosis. Mol. Cancer 16, 115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khater F., Langlois S., Cassart P., Roy A.-M., Lajoie M., Healy J., Richer C., St-Onge P., Piché N., Perreault S., Cellot S., Marzouki M., Jabado N., Sinnett D., Recurrent somatic BRAF insertion (p.V504_R506dup): A tumor marker and a potential therapeutic target in pilocytic astrocytoma. Oncogene 38, 2994–3002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor S. S., Shaw A. S., Kannan N., Kornev A. P., Integration of signaling in the kinome: Architecture and regulation of the αC Helix. Biochim. Biophys. Acta 1854, 1567–1574 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan J., Ng W. H., Yap J., Chia B., Huang X., Wang M., Hu J., The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho–Ser-621 in the C terminus of CRAF. J. Biol. Chem. 293, 14276–14284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yap J. A., Yuan J. A., Tee Z. H. A., Huang X. A., Ng W. H. A., Hu J. A., Characterize disease-related mutants of RAF family kinases by using a set of practical and feasible methods. JoVE, e59795 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Lavoie H., Thevakumaran N., Gavory G., Li J. J., Padeganeh A., Guiral S., Duchaine J., Mao D. Y. L., Bouvier M., Sicheri F., Therrien M., Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat. Chem. Biol. 9, 428–436 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu J., Yu H., Kornev A. P., Zhao J., Filbert E. L., Taylor S. S., Shaw A. S., Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc. Natl. Acad. Sci. U.S.A. 108, 6067–6072 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasuda H., Park E., Yun C.-H., Sng N. J., Lucena-Araujo A. R., Yeo W.-L., Huberman M. S., Cohen D. W., Nakayama S., Ishioka K., Yamaguchi N., Hanna M., Oxnard G. R., Lathan C. S., Moran T., Sequist L. V., Chaft J. E., Riely G. J., Arcila M. E., Soo R. A., Meyerson M., Eck M. J., Kobayashi S. S., Costa D. B., Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) Exon 20 insertion mutations in lung cancer. Sci. Transl. Med. 5, 216ra177 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruan Z., Kannan N., Altered conformational landscape and dimerization dependency underpins the activation of EGFR by αC–β4 loop insertion mutations. Proc Natl. Acad. Sci. U.S.A. 115, E8162–E8171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J., An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Galabova-Kovacs G., Matzen D., Piazzolla D., Meissl K., Plyushch T., Chen A. P., Silva A., Baccarini M., Essential role of B-Raf in ERK activation during extraembryonic development. Proc. Natl. Acad. Sci. U.S.A. 103, 1325–1330 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jesenberger V., Procyk K. J., Rüth J., Schreiber M., Theussl H.-C., Wagner E. F., Baccarini M., Protective role of Raf-1 in Salmonella-induced macrophage apoptosis. J. Exp. Med. 193, 353–364 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Šali A., Blundell T. L., Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Shen M., Sali A., Statistical potential for assessment and prediction of protein structures. Protein Sci. 15, 2507–2524 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deepak R. N. V. K., Sankararamakrishnan R., Unconventional N-H…N hydrogen bonds involving proline backbone nitrogen in protein structures. Biophys. J. 110, 1967–1979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deepak R. N. V. K., Sankararamakrishnan R., N–H···N hydrogen bonds involving histidine imidazole nitrogen atoms: A new structural role for histidine residues in proteins. Biochemistry 55, 3774–3783 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 54.Mishra N. K., Deepak R. N. V. K., Sankararamakrishnan R., Verma S., Controlling in vitro insulin amyloidosis with stable peptide conjugates: A combined experimental and computational study. J. Phys. Chem. B 119, 15395–15406 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Onofrio A., Parisi G., Punzi G., Todisco S., Noia M. A. D., Bossis F., Turi A., Grassi A. D., Pierri C. L., Distance-dependent hydrophobic–hydrophobic contacts in protein folding simulations. Phys. Chem. Chem. Phys. 16, 18907–18917 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Jones G., Willett P., Glen R. C., Leach A. R., Taylor R., Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Hu-Lieskovan S., Mok S., Homet Moreno B., Tsoi J., Robert L., Goedert L., Pinheiro E. M., Koya R. C., Graeber T. G., Comin-Anduix B., Ribas A., Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 7, 279ra41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okimoto R. A., Lin L., Olivas V., Chan E., Markegard E., Rymar A., Neel D., Chen X., Hemmati G., Bollag G., Bivona T. G., Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 13456–13461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg0390/DC1