

Abstract
This article presents a mechanistic study of the photomediated growth of silver nanoprisms. The data show that the photochemical process is driven by silver redox cycles involving reduction of silver cations by citrate on the silver particle surface and oxidative dissolution of small silver particles by O2. Bis(p-sulfonatophenyl)phenylphosphine increases the solubility of the Ag+ by complexing it and acts as a buffer to keep the concentration of Ag+ at 20 μM. The silver particles serve as photocatalysts and, under plasmon excitation, facilitate Ag+ reduction by citrate. Higher Ag+ concentrations favor a competitive thermal process, which results in increased prism thickness.
Introduction
The light-induced transformation of spherical silver nanoparticles into triangular prisms is both synthetically useful and mechanistically intriguing.1–4 Under the proper conditions, this reaction is highly controllable, can be carried out on a bulk scale, and is remarkably efficient, with transformation yields approaching 100% (yield is defined by the number of particles with a triangular shape, regardless of mild truncation). Additionally, in certain cases the excitation wavelength can be used to control prism edge length.2,4 Although there have been some hypotheses proposed to explain the formation of the silver nanoprisms, including crystal-twinning theory5–9 and face-selective ligand passivation,10,11 to date, there has not been a systematic study that has resulted in a mechanism that is consistent with all of the experimental observations made regarding this unusual transformation. Herein, we describe the results of a study aimed at identifying the chemical role of each reagent used in this photomediated process. Furthermore, we probe the role of plasmon excitation in reaction initiation and particle shape transformation. Finally, we examine the silver nanocrystal growth process in terms of both photochemical and thermal control.
It is important to consider that two different photochemical strategies have been reported for the preparation of silver nanoprisms, both of which originated from our laboratories. The first involves dual-beam excitation,2 while the other involves only single-beam excitation.4 In the dual-beam case, one of the excitation beams suppresses prism fusion, and the other beam controls the prism edge length. Under these conditions, a single beam will result in bimodal distributions of prisms (large and small, in terms of edge length). However, it has been found that the cluster fusion process also can be regulated by solution pH, which allows one to control nanoprism edge length using only a single beam.4 Above pH 11, cluster fusion is not efficient due to electrostatic repulsion of the prisms. This article will focus primarily on describing the mechanism of nanoprism growth under single-beam excitation conditions. However, we will also comment on the implications of this work for the dual-beam preparation route.
Experimental Section
Chemicals and Materials.
Silver nitrate (99.998%), trisodium citrate dihydrate (99.9%), sodium borohydride (99%), and deuterium oxide (99.96 atom % D) were purchased from Aldrich and used as received. Bis(p-sulfonatophenyl)phenylphosphine dihydrate dipotassium (BSPP) was purchased from Strem Chemicals, Inc. All H2O was purified by a Barnstead Nanopure water purification system (resistance = 18.1 MΩ).
Instrumentation.
UV-visible spectra were recorded on a Jasco V-530 UV/vis spectrometer. Transmission electron microscopy (TEM) imaging was performed on a Hitachi H8100 transmission electron microscope (200 kV). Nuclear magnetic resonance (NMR) spectra were collected at 25 °C on a Varian Mercury-300 instrument. For 31P NMR spectroscopy, the spectra were referenced to H3PO4 (external standard). All of the irradiation experiments were performed with a halogen lamp (Dolan-Jenner, MI-150) as the light source. An optical bandpass filter (25 mm diameter, Intor Inc.) centered at 550 ± 20 nm was employed to control the irradiation wavelength. The solution pH value was measured with a pH meter (Oakton, pH 510).
Preparation of Silver Nanoparticles.
Silver nanoparticles were prepared according to literature methods.4 In a typical synthesis, Nanopure water (95 mL), AgNO3 (0.5 mL, 20 mM), and sodium citrate (1 mL, 30 mM) were combined in a 250 mL three-neck flask. The flask was immersed in an ice bath, and the solution was bubbled with N2 under vigorous stirring for ~20 min. One milliliter of aqueous NaBH4 (50 mM, freshly prepared with ice-cold Nanopure water prior to injection) was rapidly injected into the solution. Over the next 15 min, 5 drops of NaBH4 solution were added every 2 min into the solution. Then 1 mL of BSPP (5 mM) and 1 mL of NaBH4 solutions were added dropwise to the reaction mixture. The resulting Ag colloid was gently stirred for 5 h in the ice bath and allowed to age overnight at ~4 °C in the dark. Immediately after synthesis of the silver nanoparticles, the average size of the silver nanoparticles was 3.0 ± 0.5 nm. After aging overnight at 4 °C, the nanoparticle size increased to 5.0 ± 0.9 nm. This silver colloid exhibits an extinction band with λmax at 395 nm.
Photomediated Silver Nanoprism Growth.
In a typical experiment, 20 mL of the silver nanoparticle solution at pH 9.6 was irradiated with a halogen lamp (150 W) coupled with an optical bandpass filter centered at 550 ± 20 nm (beam irradiance of ~200 000 lx at full power of the halogen lamp). The photoreaction was monitored by UV-vis spectroscopy (Figure 1a). After irradiation for 5 h, the silver colloid exhibited two major extinction bands at 670 and 920 nm, respectively. Consistently, TEM analysis of the final nanoprisms showed a bimodal size distribution (Figure 1b). The larger prisms have an edge length (~130 nm) that is approximately twice the edge length of the small structures (~65 nm). The average thickness of these nanoprisms is 9.1 ± 0.5 nm.
Figure 1.
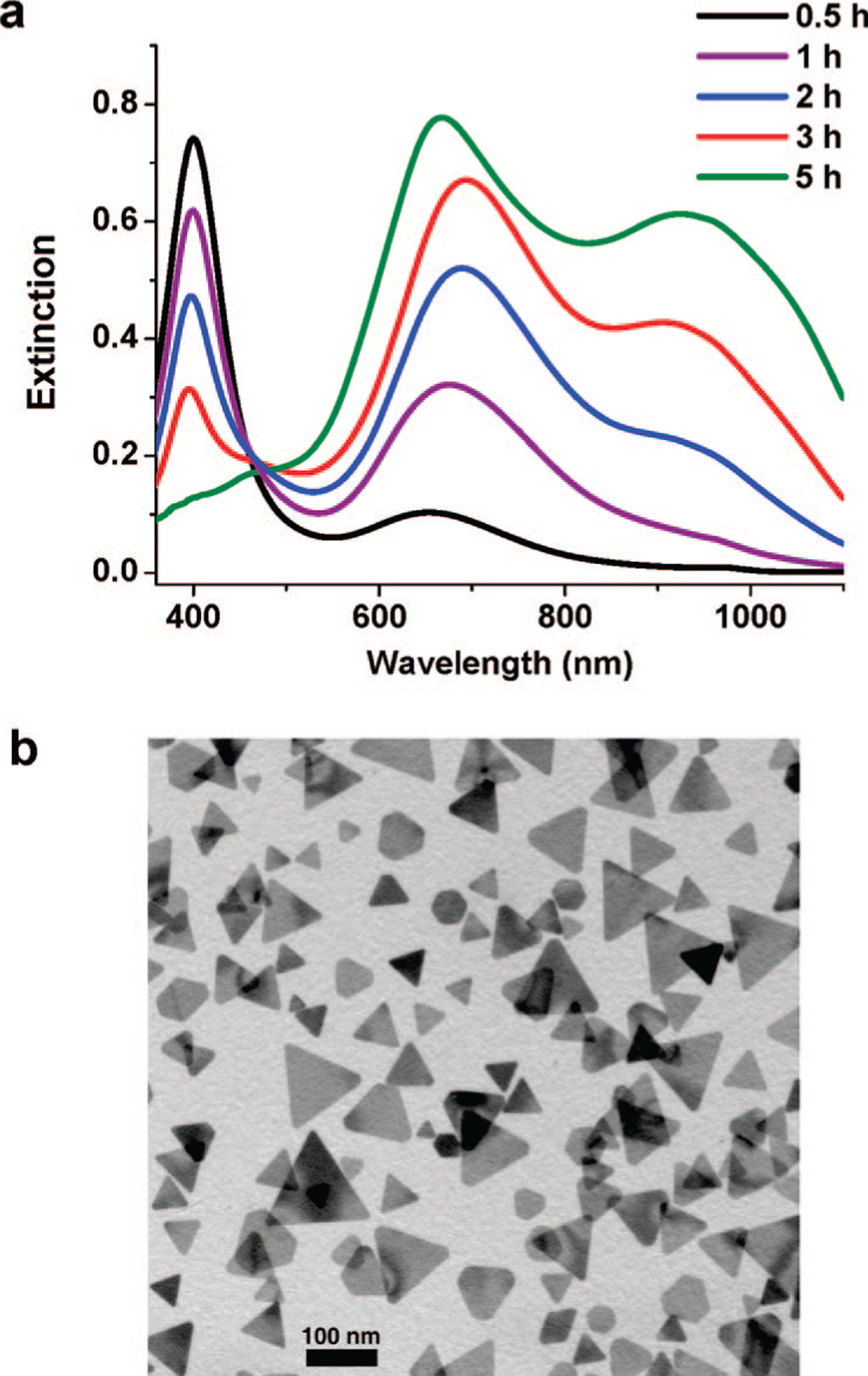
(a) Time-resolved extinction spectra of the nanoparticle solution with an irradiation time of 0.5–5 h. (b) Representative TEM image of the product after irradiation for 5 h. The scale bar is 100 nm.
Results and Discussion
To develop an understanding of the mechanism behind silver nanoprism formation, the chemical fate of each reagent in the photomediated process has been considered. This systematic analysis is used to determine the chemistry that is taking place during the synthesis of the Ag nanoprisms, thereby establishing a platform from which we can build an understanding of the various processes effecting nanoprism formation. After establishing the chemical factors at play, the nanoprism reaction is viewed from the perspective of nanocrystal growth, which we consider in three phases. The interplay of redox chemistry and crystal growth in the context of plasmon photoexcitation is then used to construct a mechanism from which we can understand the photomediated evolution of Ag nanoprisms in solution.
Chemical Role of Trisodium Citrate.
Citrate has been widely used as a thermal reducing agent in the preparation of colloidal silver nanoparticles.12,13 In the thermal reduction of silver nitrate, citrate is degraded into acetoacetate and formate.13 To determine the chemical roles of citrate in the photoreaction involving prism formation, we followed the reaction and fate of the citrate (1, Scheme 1) by 1H NMR spectroscopy. Aliquots of the reaction mixture (0.8 mL) were withdrawn after 0, 1, 3, and 5 h of photoexcitation and mixed with D2O (0.2 mL) for NMR analysis. A standard presaturation method was applied to suppress the signal of HOD at δ 4.80.14 The 1H NMR spectrum of the initial nanoparticle solution revealed that citrate 1 did not change in the presence of silver without irradiation (Figure 2). The initial citrate concentration is approximately 0.3 mM. After irradiation for 1 h, a new resonance appeared at δ 3.49, which is assigned to 1, 3-acetonedicarboxylate (ADE, 2),13 the initial product of citrate oxidation (Figure 2 and Scheme 1). Compound 2 is not stable and undergoes further decomposition to form acetoacetate (3), which exhibits resonances at δ 3.41 and 2.25 (Figure 2 and Scheme 1). After 5 h of irradiation, the concentration of ADE is estimated to be ~0.05 mM based upon the stoichiometry of the reaction. In this photomediated process, citrate, in addition to being a reducing agent, also seems to significantly influence the shape of the final particle product. Nanoprisms are not observed if citrate is absent or replaced by other carboxylate-containing compounds, such as tricarballylate, citramalate, and aconitate. Indeed, the only other carboxylate-containing molecule that we have studied that seems to effect prism formation is isocitrate. However, even in the case of isocitrate, although prisms form, other irregularly shaped large particles also are produced.
Scheme 1.
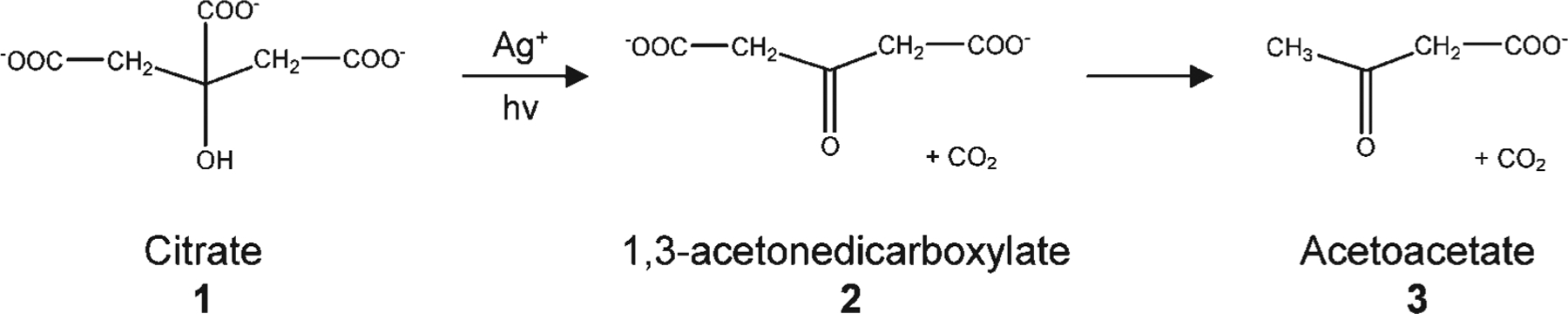
Degradation Pathway for Citrate during the Photoreaction
Figure 2.
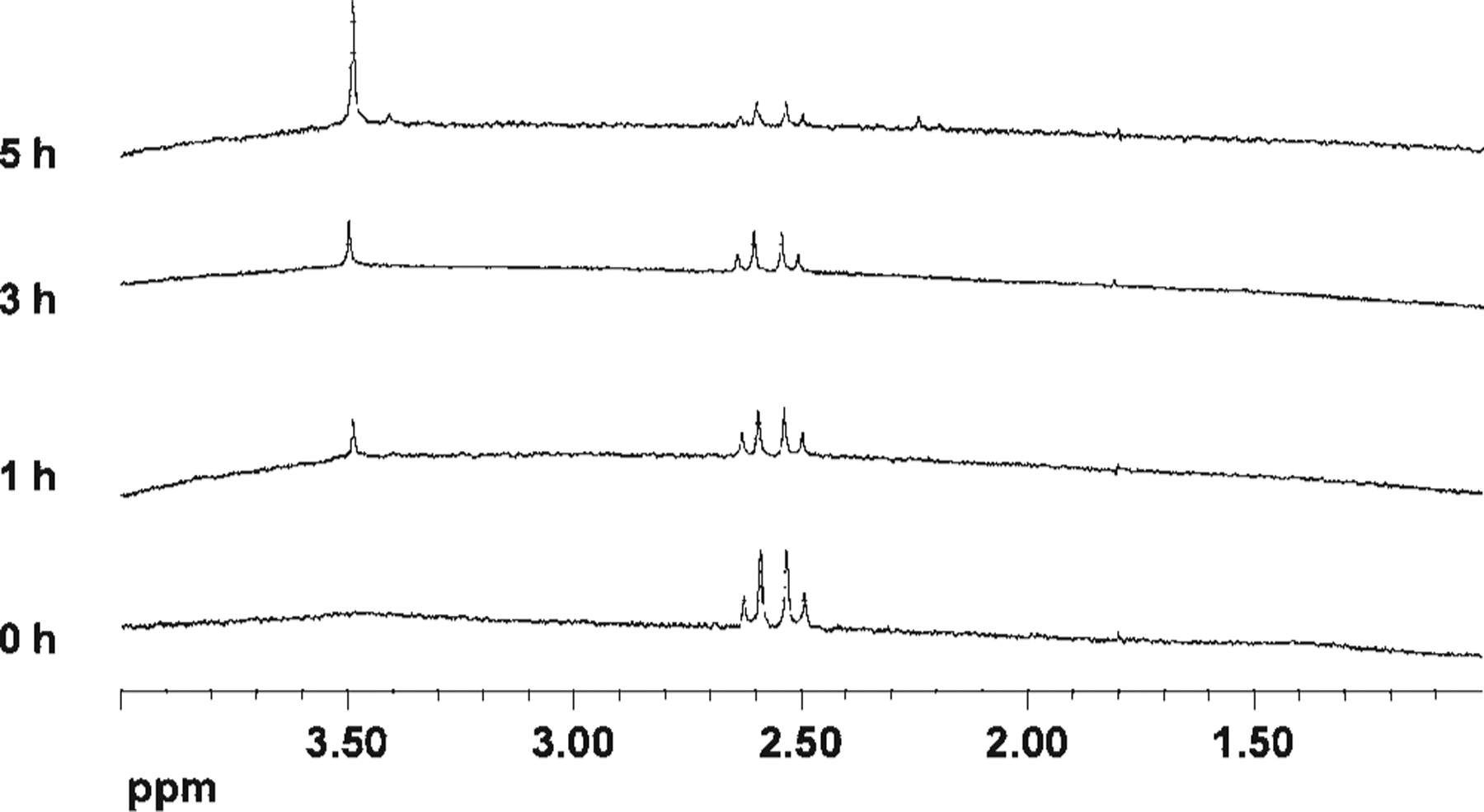
1H NMR spectroscopy mapping the fate of citrate during the photochemical prism growth process.
Chemical Role of BSPP.
BSPP is a water-soluble derivative of triphenylphosphine that has been commonly used as a stabilizing ligand in the synthesis of metal nanoparticles in organic media.15 An aqueous solution of BSPP exhibits a strong absorption band at 268 nm (Figure 3a). Over time, this band collapses, with the concomitant growth of a new band centered at 229 nm, indicating that BSPP has been oxidized to a compound that is analogous to triphenylphosphine oxide (BSPP=O).16 This transformation was confirmed by 31P NMR spectroscopy (Figure 3b), which shows a decrease in a resonance at δ −6.49, assigned to BSPP, and a concomitant increase in a new resonance at δ 36.4, assigned to BSPP=O.
Figure 3.
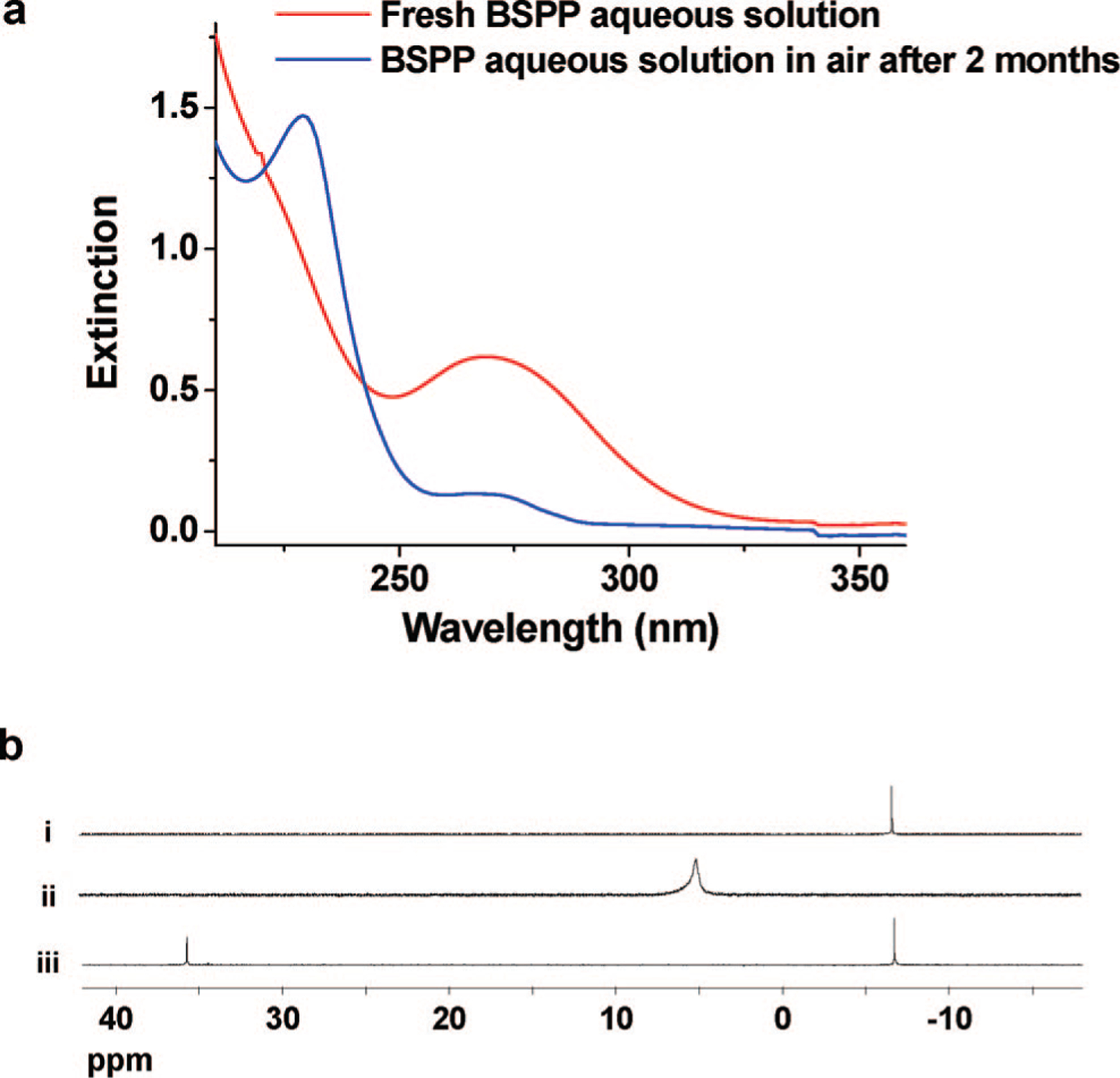
(a) UV-vis spectrum of a 0.05 mM BSPP solution at time 0 and after 2 months in air. (b) 31P NMR spectra of (i) BSPP, δ −6.49 ppm; (ii) BSPP:Ag+ = 5:1, δ 5.05 ppm; and (iii) BSPP and BSPPO, δ −6.49, 36.40 ppm.
BSPP will coordinate with Ag+ to form a coordination complex, while BSPP=O, under comparable conditions, does not form a complex with Ag+. This complexation reaction also can be followed by 31P NMR spectroscopy, where one sees the resonance at δ −6.49 disappear and the formation of a new resonance at δ 5.05 assigned to the BSPP-Ag+ complex (Figure 3b, trace ii). The peak broadening observed in the NMR spectrum is attributed to the fast exchange between the free and complexed BSPP.17,18
Interestingly, photoconversion of silver nanoparticles still occurs in the absence of BSPP, albeit at a retarded rate. In addition to resulting in slower rates of transformation, these conditions lead to many large, irregularly shaped particles as well as nanoprisms. This experiment demonstrates that BSPP is not essential for prism formation but does substantially affect the rate at which prisms are formed and the overall yield. The primary role of the BSPP seems to be to solubilize the Ag+ ions that have been formed by oxidation of small Ag nanoparticles (vide infra). Inductively coupled plasma mass spectrometry (ICP-MS) analysis reveals that, in the presence of BSPP (0.05 mM in a standard case), the elemental concentration of silver cations in the solution stays constant at 20 ± 2 μM over the entire photoreaction; however, if BSPP is absent during irradiation (using the same time and power), the solution concentration of Ag+ is 4 ± 1 μM, 5 times lower than the Ag+ concentration in the presence of BSPP. Note that Ag+ concentration includes the BSPP-Ag+ complex.
Chemical Role of Oxygen.
Oxygen is required for the transformation of the spherical seeds into Ag triangular prisms. When a colloid containing the isotropic seed particles and BSPP is irradiated under nitrogen, no prisms form. When oxygen is added to a solution containing the isotropic Ag seed particles and BSPP (0.05 mM), the particles dissolve, as indicated by a decrease in the intensity of their surface plasmon resonance (SPR), and the Ag+ concentration in solution increases as described above. Therefore, the major role of oxygen is to chemically convert the silver particles into Ag+, which is subsequently complexed by BSPP and used as the Ag+ source for the photoreaction to form prisms. The reaction can be terminated at any time if the oxygen is removed from the solution by evacuation or nitrogen bubbling, presumably because the supply of silver cations is decreased in the absence of oxygen. In contrast, the addition of excess BSPP (0.5 mM) dramatically accelerates the dissolution process. Under these conditions, the silver nanoparticles completely dissolve in 5 min. Finally, this dissolution process is accompanied by an increase in solution pH from 10.02 to 10.25, since the oxidation of Ag0 in H2O generates OH− (eq 1).
| (1) |
It is interesting to note that the results described thus far seem to suggest that the dissolution of Ag0 is not necessary as long as there is a source of Ag+. Indeed, one can prepare prisms by irradiating a mixed sample of silver seeds, citrate, and additional AgNO3.3 However, the quality of prisms is relatively low and the reaction is more difficult to control, because the concentration of Ag+ is constantly changing. In contrast, the spherical seeds and the BSPP in the photoreaction act like a buffer to help keep the concentration of Ag+ constant (~20 μM, see above) throughout the bulk of the chemical transformation.
Chemical Role of Silver Nanoparticles.
The silver nanoparticles used in the photoreaction range in size from 2 to 6 nm in diameter and have two different functions in the photoreaction. First, we have observed that particles larger than 10 nm do not work well in the photoconversion process.2 The small silver nanoparticles are more susceptible to oxidation than their larger counterparts due to their lower redox potentials.19,20 These particles work as a feedstock of silver cations for the photoreaction. In contrast, the larger Ag particles serve as plasmonic seeds and photocatalysts for Ag+ reduction by citrate and photoexcitation (vide infra).
Growth Step I: Plasmon Excitation-Initiated Photoreaction.
The initial colloid of silver nanoparticles exhibits an extinction maximum at 395 nm. However, magnifying the spectrum (Figure 4a) reveals that silver colloid absorbance spans the entire visible range. However, the chemical reagents present, such as AgNO3, citrate, and BSPP, show no significant absorbance in the visible range (<0.001 O.D./mL; beyond the detection limit of the spectrometer, Figure 4b). Therefore, the extinction of the colloid in the visible range must derive, in part, from excitation of the SPR of the silver nanoparticles, which means that these nanoparticles can be excited by visible light to varying degrees, depending upon wavelength.
Figure 4.
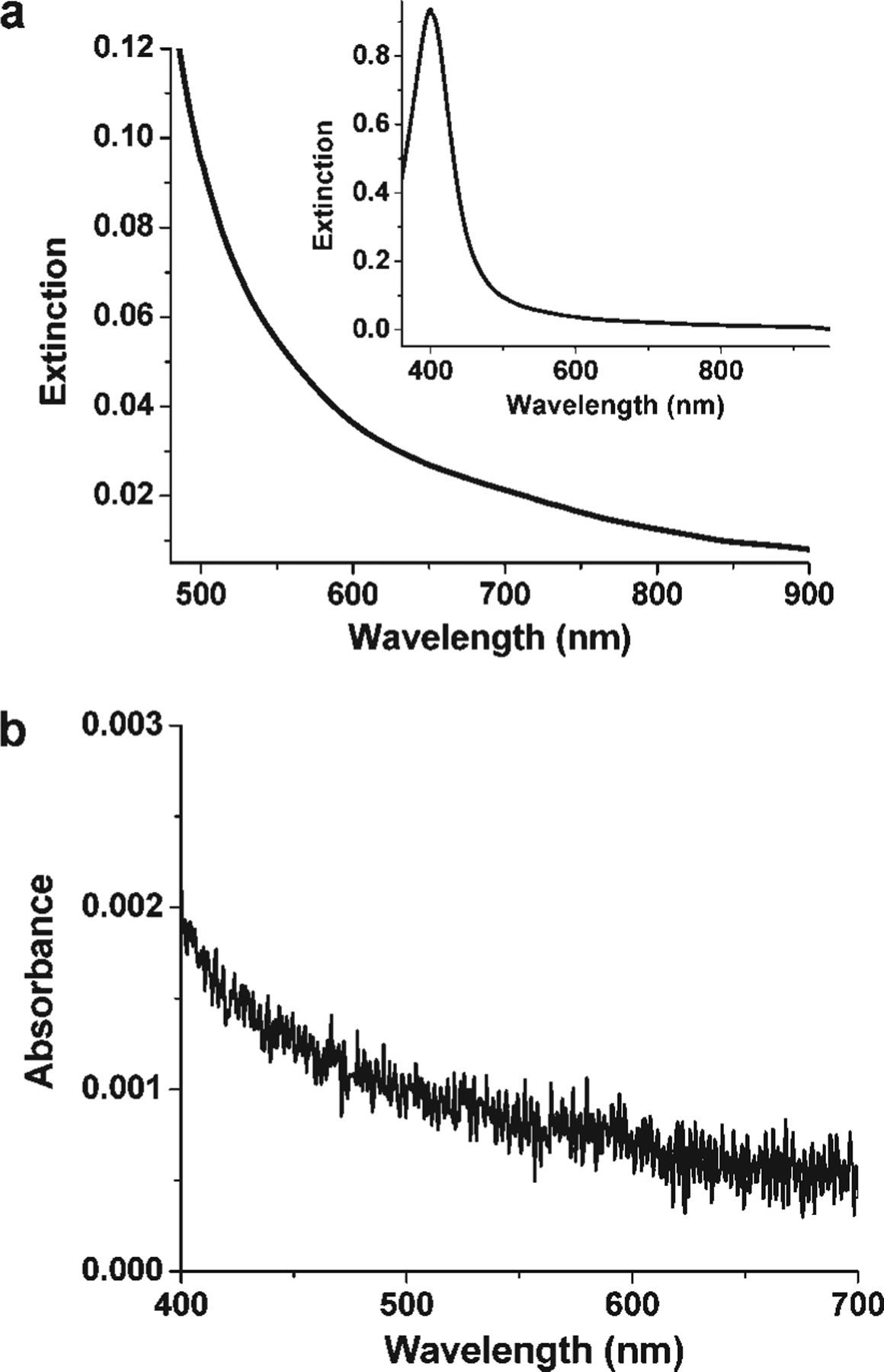
(a) Expanded extinction spectrum of a silver nanoparticle solution prior to irradiation. The inset is the full spectrum. (b) UV-vis spectrum of a mixture of AgNO3, citrate, and BSPP. Each reagent is 1 mM.
The significance of plasmon excitation in the photomediated nanoprism growth has been recently demonstrated by using gold nanoparticles as plasmonic seeds and experimental labels.21 This strategy takes advantage of the distinct optical signatures and image contrast between gold and silver, and enables one to track a reaction by both spectroscopic and electron microscopy techniques. After a mixture of gold (11 nm) and silver nanoparticles (5 nm) was irradiated with 550 nm light for 30 min, all of the gold nanoparticles were coated with silver shells, and the SPR band of the gold particles had blue-shifted. Importantly, gold seeds with different plasmon frequencies required different excitation wavelengths (550 nm for 11 nm spherical gold seeds, and 1064 nm for 120 nm edge length triangular gold prism seeds) to effect the silver shell formation.21 This work, combined with the observations made herein, unambiguously shows that the plasmon excitation of the gold or silver nanoparticles induces silver ion reduction onto their surfaces.
The photochemical reaction induced by plasmon excitation has been proposed by several groups as a charge transfer between adsorbates and the hot electrons (holes) that are likely produced by plasmon decay.22–24 In the presence of oxygen, there is always an equilibrium between the Ag particle and Ag+ in solution, and as stated previously, the small particles (<2 nm) are more susceptible to oxidation than the larger particles due to their low redox potentials.25,26 Indeed, ICP analysis of a solution after removal of all of the silver particles by filtration shows that the Ag+ concentration is ~20 μM. Therefore, when a silver colloid is excited by incident light, the hot electrons generated on the surfaces of the nanoparticles can be transferred to the unoccupied orbitals of the adsorbed silver cations. The hot holes are then filled with electrons from the oxidization of adsorbed citrate (decarboxylation of citrate) to ADE and CO2 (Scheme 2).3
Scheme 2.
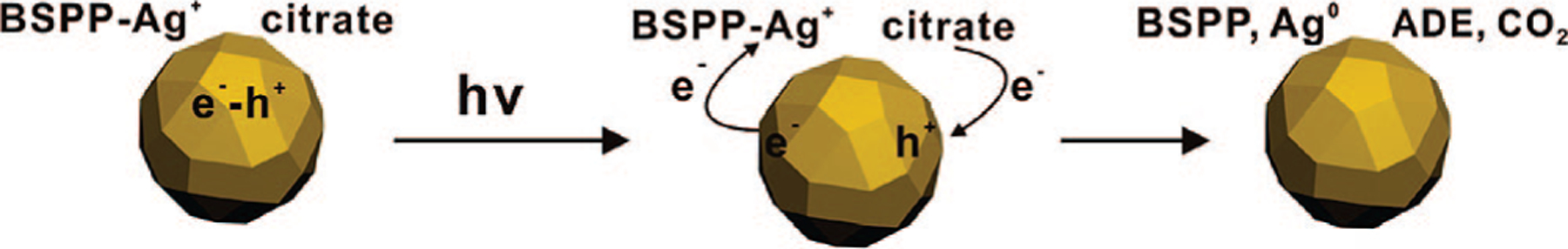
Proposed Redox Reaction Occurring on the Silver Nanoparticle Surface upon Plasmon Excitation
The reduction of Ag+ by citrate is thermodynamically allowed based upon their redox potentials (EAg+/Ag = 0.7996 V vs NHE; and at pH above 8).27 The reaction proceeds at elevated temperatures (refluxing aqueous conditions)13 but is extremely slow at room temperature. Indeed, when stored in the dark at room temperature, a mixture of AgNO3 and citrate is quite stable, showing little evidence of reaction for several days. This implies that the required activation energy for the reaction of citrate and Ag+ is relatively high, so that at room temperature, the electrons of citrate are not able to efficiently overcome the energy barrier for conversion of Ag+ to Ag0 (Figure 5). This makes the reaction quite slow (unobservable in the course of our experiments) at room temperature. Note that neither Ag+ nor citrate absorbs light in the visible range; however, irradiation at visible wavelengths can initiate this reaction, presumably because of a trace concentration of silver seed particles that form via the slow thermal process. It is also worth pointing out that the transformation of isotropic silver seeds into anisotropic silver nanoprisms is slow at the beginning but accelerates once the silver particles grow to the point where they are absorbing a significant amount of light due to plasmon excitation (vide infra). These observations imply that silver nanoparticles serve as photocatalysts that facilitate the reduction of silver ions onto their crystalline faces. Without light, even in the presence of metal nanoparticles (Au or Ag), no significant reaction is observable between Ag+ and citrate.
Figure 5.
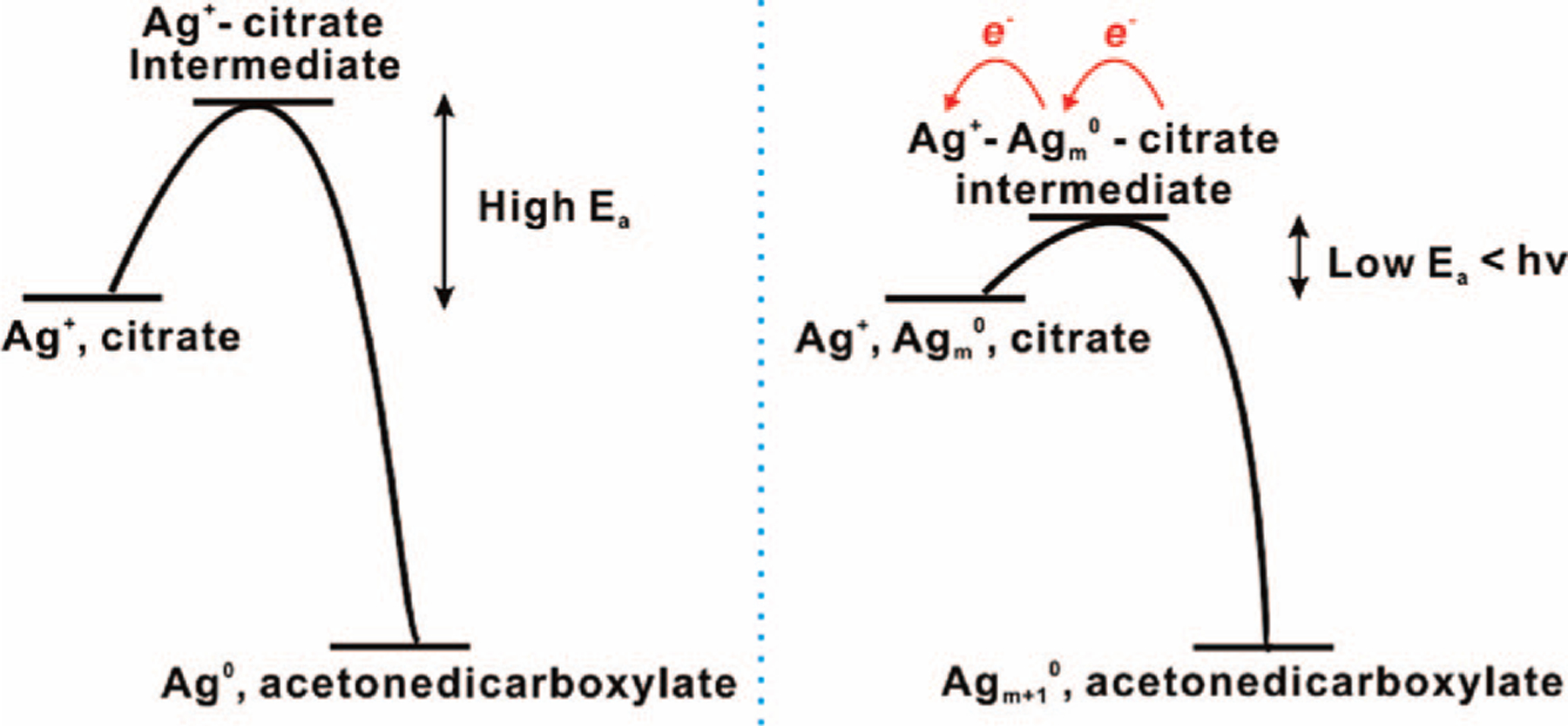
Scheme showing the activation energy for the reaction between Ag+ and citrate, and the proposed effect of silver nanoparticles upon photoexcitation.
For silver deposition to occur on the particle surface, both ligand dissociation and a silver ion source are necessary. Ligand dissociation can be initiated through the coupling of photoexcited electrons and metal-ligand bond vibrations.28 Photothermal effects also can induce localized ligand dissociation,28–31 and such effects can be significantly enhanced under plasmon resonance excitation.30 It has been reported that metal particles might be broken up under strong pulsed laser excitation due to the photothermal heat accumulation.32,33 However, in the experiments described herein, the light intensity is much weaker as compared with these laser conditions (1015-1018 W/m2 for the pulsed laser; 102-103 W/m2 for the halogen lamp), and we have not observed any evidence of splintering of larger silver nanoparticles into smaller ones. Instead, we propose that the source of silver cations comes from the oxidative dissolution of the small silver seed particles, per the evidence described above.
Growth Step II: Shape Transformation of Spherical Silver Nanoparticles to Small Nanoprisms.
Our observations suggest that as the SPR of the isotropic silver particle seed is excited, silver cations are continuously deposited on the nanoparticle surface and the particle gradually becomes anisotropic and takes on the form of a triangular nanoprism. There are several key observations that provide important clues regarding the mechanism responsible for this shape-change process. First, during the intermediate phases of nanoprism growth (after 30 min of irradiation), small triangular prisms can be visualized by TEM (Figure 6a). Since dipole plasmon excitation induces ultrafast charge separation on the nanoparticle surface,34 this likely leads to face-selective silver cation reduction, thereby causing anisotropic crystal growth. Alternatively, one could also envision Ag+ ions selectively adsorbing on different faces, which leads to subsequent selective photodeposition of Ag on those faces. Although this process cannot be completely ruled out, if this were the case, one would expect triangular prisms from thermal growth processes with similar reagents. However, such reactions, unless one uses strongly binding ligands, typically lead to isotropic pseudospherical particles. The idea of dipole excitation causing an anisotropic distribution of charge and reducing equivalents on the particle surface is consistent with our recent observations using gold particles as plasmon reaction labels, as well as the observations of others,3,21 which show inhomogeneous silver shell growth at early stages of the growth process. These results indicate that the exact shape of the seed particle is not the dominant factor in controlling the photoinduced silver prism growth process other than, in part, contributing to the excitation wavelength required for prism growth, which correlates well with the dipole plasmon resonance of the seed particle. In other words, any shaped seed can be transformed into a triangular prism through dipole plasmon excitation, and it is the randomness of the initial deposition that then begins anisotropic growth. The implications of this process as it applies to the formation of plate-like silver structures were first described in detail by Brus et al., who related the proportion of Ag+ deposition to the near-field enhancement caused by dipole excitation, and consequently the initiation of anisotropic particle growth.3
Figure 6.

(a) TEM image of the intermediate product, including ellipsoidal particles and a small triangular plate, in the silver colloid after irradiation for 30 min. (b) High-resolution TEM image showing stacking faults in the prisms during growth.
In addition to the photochemical process, many groups have suggested that the generation of [111] twin planes and stacking faults are key factors influencing particle shape. This work also was carried out in the context of prismatic plate-like structures.5–9,35 The researchers suggested that the twin-created re-entrant grooves enable preferential attachment of adatoms with lower nucleation energy to form a new atomic layer. Therefore, particle growth is accelerated parallel to the twin planes, extends the lowest energy crystal facet ([111]), and drives the particle toward a well-defined platelet structure. These [111] stacking faults have been observed by us (Figure 6b) as well as others,6,8 and all of our data collected thus far are consistent with this hypothesis.
Theoretical studies have suggested that irradiation of Ag nanoparticles in the red region of the dipole resonance causes dipole excitation.34,36 Previous studies have demonstrated that the effective wavelength range that can induce nanoprism formation is between 450 and 750 nm.2 However, if the photoreaction is carried out with λex = 400 nm (the SPR band of the 5 nm silver nanoparticle seed), the plasmon resonance signature of the resulting colloid shifts to only 420 nm. TEM analysis of the resulting colloid shows little evidence of prism formation but rather shows small oblate particles (Figure 7). At this wavelength, both dipole and multipole SPRs of the particle are excited, which does not favor selective prism growth.
Figure 7.

TEM image of a silver colloid after irradiation with 400 nm light for 3 h.
It also has been proposed that slow reduction of Ag+ assists twin generation during crystal growth9,10 and hence results in the formation of silver nanoprisms. Conventional silver nanoparticle growth usually involves rapid reduction of silver ion precursors through a thermal process, which leads to spherical or pseudospherical particles such as cuboctahedrons and icosahedrons.9,10 In the photoreaction described herein, the concentration of silver cations is maintained at 20 μM throughout the reaction (vide supra), and the kinetic stability of the citrate allows the Ag+ deposition on particle surfaces to occur preferentially under photochemical control. However, in the absence of BSPP, the photoreaction goes relatively slowly since the Ag+ concentration is significantly lower (4 μM), and we obtain a considerable amount of irregular-shaped particles in the final product. This observation indicates that, if the photochemical growth rate is slow and approaches the growth rate of the thermal growth process, isotropic growth will be competitive with anisotropic growth, and a broad distribution of product shapes will be observed. Therefore, if triangular prisms are the desired product, one wants to enhance the photochemical process without accelerating the rate of the thermal process. Consistent with this hypothesis, reducing irradiation light intensity (to 20% of full power of the halogen lamp) decreases the photoreaction rate (Figure 8a) and the yield of silver prisms in the product (Figures 1b and 8b).
Figure 8.
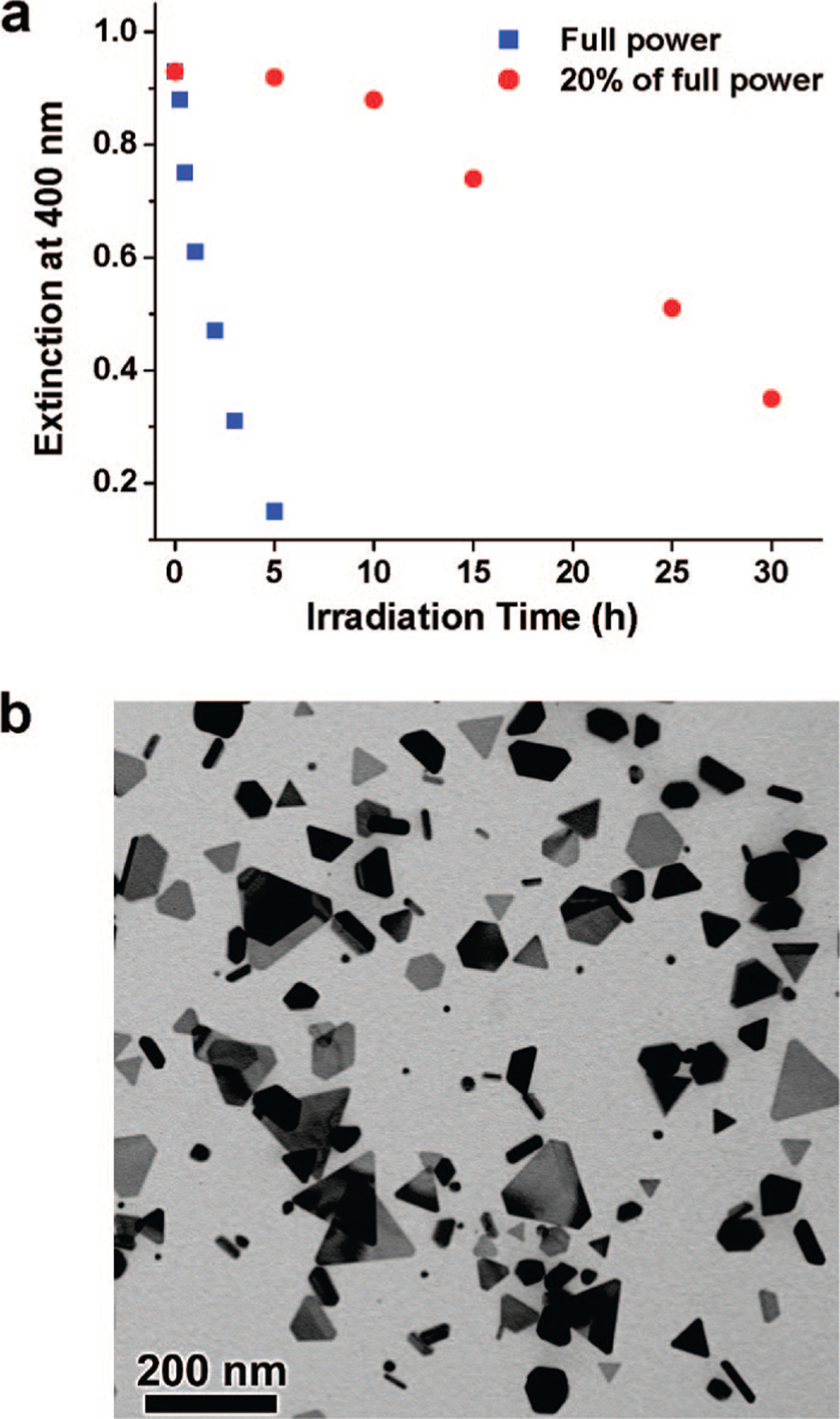
(a) Extinction of silver colloid at 400 nm as a function of irradiation time at full irradiation power (blue squares) and 20% of the full power (red circles). Note that, at 20% full power, the rate of consumption of the silver particle seeds is initially slow but accelerates after larger particles are formed with plasmon resonances that can more efficiently absorb the excitation light. (b) TEM image of the silver colloid after irradiation with 20% of the full power for 30 h.
Growth Step III: Growth from Small Nanoprism Seeds to Large Prisms.
Once the silver colloid exhibits an extinction band at 670 nm (30 min after irradiation with a band-pass filter centered at 550 nm), this band continues to increase in intensity over the next 4–5 h. During this period, concurrent with the shape transformation of spherical particles, edge-length expansion of small nanoprisms occurs rapidly, where re-entrant grooves on the edge planes provide fast-growing sites upon plasmon excitation (Scheme 3).
Scheme 3.
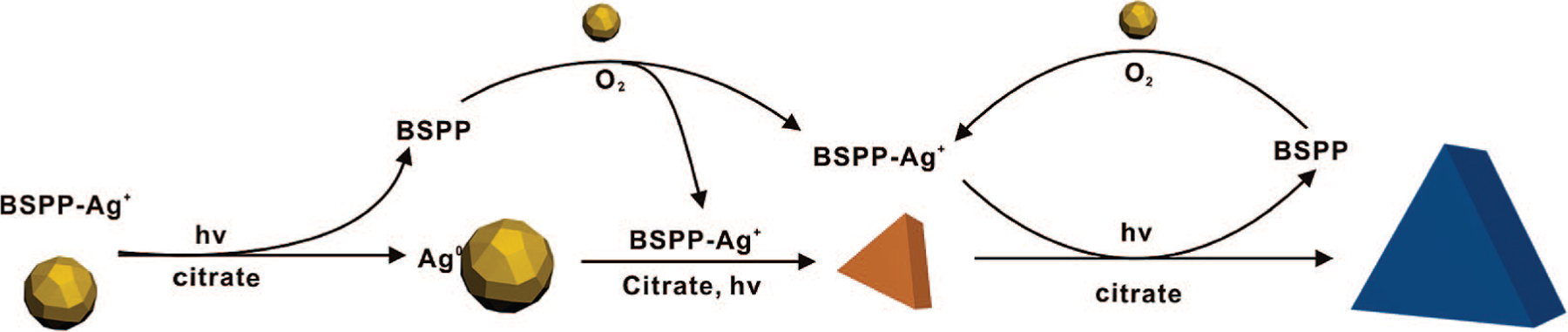
Proposed Photomediated Growth Pathway of Silver Nanoprisms from Spherical Nanoparticles
It is well known that the facets comprising silver crystals have different surface free energies (σ111 < σ100 < σ110).37 These surface energy differences are responsible for the different bonding energies and chemical reactivity of the crystal faces. Thus, the [111] face has the lowest growth rate, and prism edge lengths grow preferentially over thickness. As the prism grows, this preferential growth is also driven by stoichiometry reasons because it takes more silver to increase thickness over edge length due to the relatively large ratio of face surface area to edge surface area (Table 1). Note that nanoprism thickness is almost independent of excitation wavelengths (from 450 to 750 nm), and the final prism thickness is ~10 nm, regardless of edge length.2
Table 1.
Ratio of Face Surface Area to Edge Surface Area for a Triangular Silver Nanoprism with Different Edge Lengths at a Fixed Thickness of 10 nm
| edge length of triangular silver nanoprisms (nm) | ratio of face surface area to edge surface area |
|---|---|
| 10 | 0.29 |
| 20 | 0.57 |
| 30 | 0.86 |
| 40 | 1.15 |
| 60 | 1.72 |
| 80 | 2.29 |
| 100 | 2.87 |
Theoretical studies indicate that the strongest electromagnetic fields generated by the dipole excitation of triangular nanoprisms are localized on the prism tips.34,36 Therefore, the prism tips exhibit higher growth rates than the edges. This observation explains why the reaction leads to structures with sharp triangle tips during prism edge length expansion (Scheme 4). As the edge lengths of the nanoprisms increase, the dipole plasmon bands of the prisms red-shift with respect to the excitation wavelength. Thus, when the prism growth is close to complete, the irradiation wavelength sits in the blue region of the prism dipole plasmon resonance so that dipole excitation is weak. During this stage, the in-plane quadrupole plasmon resonance of triangular nanoprisms may begin to be excited.34,36 As a result, the prism edges are preferentially excited, which induces a higher growth rate of the edges than the tips, leading to the generation of nanoprisms with highly faceted tip truncation (appearing as hexagonal prismatic plates, Scheme 4; see Supporting Information for details). This conclusion is in agreement with our observation that there is always a population of truncated nanoprisms in addition to the sharp-tip triangular prism product.
Scheme 4.

Triangular Prisms with Sharp Tips Form from Dipole Excitation because Tip Growth Is Faster than Edge Growth; Truncated Prisms Form from Quadrupole Excitation because under These Conditions, Edge Growth Is Faster than Tip Growth
When the small silver nanoparticle seeds are completely consumed, the prism growth stops, even if some prisms can still experience weak excitation. If additional silver cations are introduced into the nanoprism colloid (see Supporting Information for details), prism growth resumes upon plasmon excitation. However, these additional silver ions lead to an increase in prism thickness in addition to some change in the edge length (Figure 9a). When the concentration of Ag+ in the prism growth solution is increased to 0.4 mM through the addition of AgNO3, the average prism thickness can be increased to 25.7 ± 2.8 nm (more than twice the thickness of the original nanoprisms). This prism thickness increase is attributed to the high concentration of additional Ag+, which accelerates the thermal silver ion reduction process that is not edge-selective like the photochemical process. Therefore, both the edge length and the thickness of the nanoprisms grow. Since the SPR bands of silver nanoprisms are more sensitive to changes in thickness than edge lengths,34 the prism dipole resonance shows a blue shift over the first hour after Ag+ addition (Figure 9b), corresponding to an increase in prism thickness (note that increasing the edge length only causes a red shift the SPRs). Once the Ag+ concentration is depleted from the thermal deposition of silver on the [111] faces, the photochemically controlled prism edge length growth becomes competitive again, resulting in a red shift of the prism dipole plasmon resonance.
Figure 9.
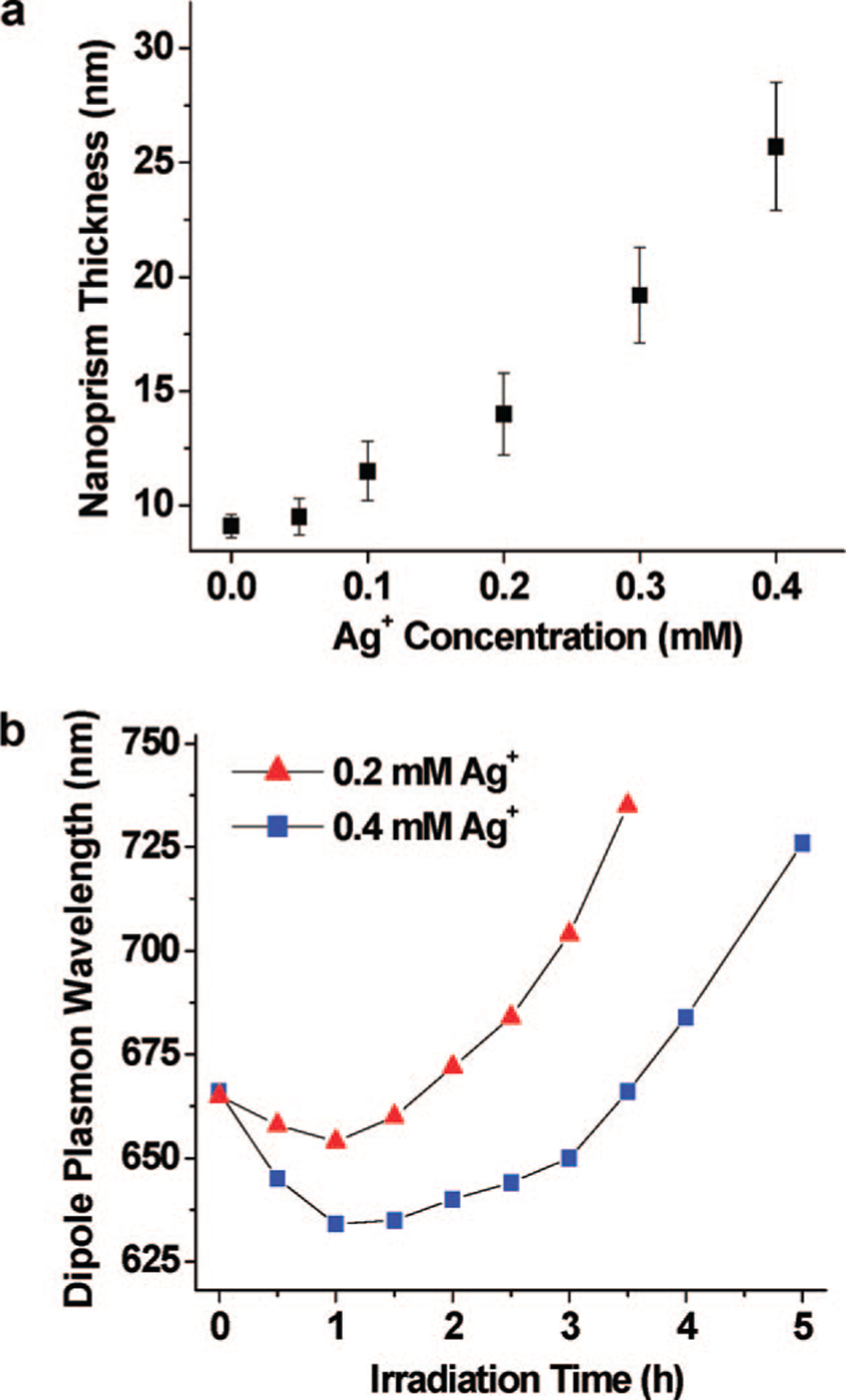
(a) Plot of nanoprism thickness as a function of Ag+ concentration. [0.0] represents the point where no Ag+ has been added to the solution. The other points are where the Ag+ concentration has been raised through the addition of AgNO3. After irradiation with 550 nm light, the final nanoprisms have an average thickness between 9.1 ± 0.5 and 25.7 ± 2.8 nm, depending upon Ag+ concentration. (b) Dipole plamon wavelength of the nanoprism colloid as a function of irradiation time and Ag+ concentration.
Bimodal and Unimodal Growth Control.
In the procedure used herein to synthesize the prisms, a bimodal distribution of sizes is observed. This bimodal distribution was also observed in the original photochemical preparatory procedure for prisms.2 The distribution can be observed by electron microscopy, and there are characteristic spectroscopic features that signal this distribution. Notably, as a population of prisms is grown with an average edge length of 65 nm, an extinction band at 670 nm associated with the dipole plasmon resonance of these structures is observed. This band steadily grows until a critical concentration is reached, at which point cluster fusion begins to take place, resulting in the formation of larger prisms with an average edge length of 130 nm and a characteristic dipole plasmon resonance at ~920 nm (Figure 1). Consistent with the hypothesis of the cluster fusion process, (1) a critical concentration of small prisms is required to form the larger structures, (2) a bimodal distribution is observed by spectroscopy and electron microscopy studies, (3) the larger structures have an edge length approximately twice the edge length of the small structures, and (4) we do not see a gradual shift in plasmon resonance as the small prisms are converted into the larger ones. In addition, in separate work, we have shown that this process can be inhibited by controlling the charge on the smaller prisms.4 Since the prism surfaces are passivated with negatively charged ligands, such as BSPP and citrate, the nanoprisms must overcome electrostatic repulsion in order to participate in the fusion process. Consistent with these hypotheses, we have recently reported that one can achieve unimodal growth of silver nanoprisms by adjusting solution pH.4 At high pH, such as 11.2, the interprism repulsion is significantly enhanced so that the prisms cannot approach each other, thereby prohibiting the prism fusion process.
Our previous studies showed that a secondary excitation of the out-of-plane quadrupole plasmon resonance of silver nanoprisms can also be used to suppress prism fusion.2 Though the function of this secondary excitation is not yet fully understood, we suggest that it likely increases the charge on the prism edge planes, thereby increasing interprism repulsion. Note that the out-of-plane quadrupole plasmon resonance is perpendicular to the triangular faces and induces excitation focused on the prism edges.34
Conclusions
This work provides a number of important observations and conclusions for the photomediated growth of silver nanoprisms from spherical silver nanoparticles. First, building off our early work1 and that of Brus,3 this photochemical process is driven by silver redox cycles involving reduction of silver cations by citrate on the silver particle surface and oxidative dissolution of small silver particles facilitated by BSPP (Scheme 3). Importantly, the small silver seeds and BSPP in the photoreaction act like a buffer to keep the Ag+ concentration constant (~20 μM), which facilitates the Ag+ deposition on the seed particle surface under photochemical control. By increasing irradiation light intensity, one can significantly enhance the photochemical process as compared with the thermal process, which increases the photoreaction rate and the yield of silver nanoprisms. High silver concentrations begin to favor the thermal process, which leads to less selectivity for the transformation of isotropic silver seeds into anisotropic triangular prisms.
Second, dipole and quadrupole plasmon excitation play important roles in the shape control process. The silver particles serve as photocatalysts and mediate the reduction of Ag+ by citrate on their surfaces under plasmon excitation. Dipole plasmon excitation favors sharp-tip silver prism growth by localizing energy at the tips, while in-plane quadrupole excitation favors truncated prism growth by localizing energy and facilitating silver deposition on the edges.
Third, nanoprism thickness can be increased by intentionally adding an additional source of Ag+ to the prism solution. This occurs because, at high Ag+ concentration, the thermal process is favored over the photochemical process, and one gets significant silver deposition on the [111] faces of the prisms. The increase of prism thickness correlates well with the Ag+ concentration added into the solution. This observation provides a straightforward way to tailor both the architectural parameters and spectroscopic features of the silver nanoprisms. Taken together, this work complements the growing body of work on the fundamental processes underlying triangular nanoprism growth.3,8,22,38–40
Supplementary Material
Acknowledgment.
Support for this work was provided by the AFOSR, ONR, and MRSEC program of the National Science Foundation (DMR-0076097) at the Material Research Center of Northwestern University. C.A.M. is also grateful for the NIH Director’s Pioneer Award. J.E.M. is grateful for a Northwestern University Presidential Fellowship.
Footnotes
Supporting Information Available: Plots of the dipole plasmon band wavelength of silver nanoprisms and the extinction band intensity of silver nanoprism colloid as a function of irradiation time; TEM images of silver nanoprisms after 2 h irradiation by 550 nm light and then 470 nm irradiation; and a table summarizing nanoprism edge length, thickness, and aspect ratio at different Ag+ concentration. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Jin RC; Cao YW; Mirkin CA; Kelly KL; Schatz GC; Zheng JG Science 2001, 294, 1901. [DOI] [PubMed] [Google Scholar]
- (2).Jin RC; Cao YC; Hao EC; Metraux GS; Schatz GC; Mirkin CA Nature 2003, 425, 487. [DOI] [PubMed] [Google Scholar]
- (3).Maillard M; Huang PR; Brus L Nano Lett 2003, 3, 1611. [Google Scholar]
- (4).Xue C; Mirkin CA Angew. Chem., Int. Ed 2007, 46, 2036. [DOI] [PubMed] [Google Scholar]
- (5).Elechiguerra JL; Reyes-Gasga J; Yacaman MJ J. Mater. Chem 2006, 16, 3906. [Google Scholar]
- (6).Germain V; Li J; Ingert D; Wang ZL; Pileni MP J. Phys. Chem. B 2003, 107, 8717. [Google Scholar]
- (7).Goessens C; Schryvers D; Van Landuyt J Microsc. Res. Techniq 1998, 42, 85. [DOI] [PubMed] [Google Scholar]
- (8).Salzemann C; Urban J; Lisiecki I; Pileni MP Adv. Funct. Mater 2005, 15, 1277. [Google Scholar]
- (9).Lofton C; Sigmund W Adv. Funct. Mater 2005, 15, 1197. [Google Scholar]
- (10).Xiong Y; Washio I; Chen J; Sadilek M; Xia Y Angew. Chem., Int. Ed 2007, 46, 4917. [DOI] [PubMed] [Google Scholar]
- (11).Sun YA; Xia YN Adv. Mater 2003, 15, 695. [Google Scholar]
- (12).Lee PC; Meisel DJ Phys. Chem 1982, 86, 3391. [Google Scholar]
- (13).Munro CH; Smith WE; Garner M; Clarkson J; White PC Langmuir 1995, 11, 3712. [Google Scholar]
- (14).Mitchell TN; Costisella B NMR–From Spectra to Structures: An Experimental Approach; Springer: Berlin, 2007; p 14. [Google Scholar]
- (15).Weare WW; Reed SM; Warner MG; Hutchison JE J. Am. Chem. Soc 2000, 122, 12890. [Google Scholar]
- (16).Jaffe HH; Freedman LD J. Am. Chem. Soc 1952, 74, 1069. [Google Scholar]
- (17).Petroski J; Chou MH; Creutz C Inorg. Chem 2004, 43, 1597. [DOI] [PubMed] [Google Scholar]
- (18).Woehrle GH; Brown LO; Hutchison JE J. Am. Chem. Soc 2005, 127, 2172. [DOI] [PubMed] [Google Scholar]
- (19).Henglein AJ Phys. Chem 1993, 97, 5457. [Google Scholar]
- (20).Jiang ZJ; Liu CY; Li YJ Chem. Lett 2004, 33, 498. [Google Scholar]
- (21).Xue C; Millstone JE; Li S; Mirkin CA Angew. Chem., Int. Ed 2007, 46, 8436. [DOI] [PubMed] [Google Scholar]
- (22).Redmond PL; Walter EC; Brus LE J. Phys. Chem. B 2006, 110, 25158. [DOI] [PubMed] [Google Scholar]
- (23).Lindstrom CD; Zhu XY Chem. Rev 2006, 106, 4281. [DOI] [PubMed] [Google Scholar]
- (24).Watanabe K; Menzel D; Nilius N; Freund HJ Chem. Rev 2006, 106, 4301. [DOI] [PubMed] [Google Scholar]
- (25).Chaki NK; Sharma J; Mandle AB; Mulla IS; Pasricha R; Vijayamohanan K Phys. Chem. Chem. Phys 2004, 6, 1304. [Google Scholar]
- (26).Jana NR; Gearheart L; Murphy CJ Chem. Commun 2001, 617. [Google Scholar]
- (27).Trettenhahn G; Koberl A Electrochim. Acta 2007, 52, 2716. [Google Scholar]
- (28).Jain PK; Qian W; El-Sayed MA J. Am. Chem. Soc 2006, 128, 2426. [DOI] [PubMed] [Google Scholar]
- (29).Richardson HH; Hickman ZN; Govorov AO; Thomas AC; Zhang W; Kordesch ME Nano Lett 2006, 6, 783. [DOI] [PubMed] [Google Scholar]
- (30).Govorov AO; Richardson HH Nano Today 2007, 2, 30. [Google Scholar]
- (31).Link S; Ei-Sayed MA Annu. Rev. Phys. Chem 2003, 54, 331. [DOI] [PubMed] [Google Scholar]
- (32).Link S; Burda C; Mohamed MB; Nikoobakht B; El-Sayed MA J. Phys. Chem. A 1999, 103, 1165. [Google Scholar]
- (33).Kamat PV; Flumiani M; Hartland GV J. Phys. Chem. B 1998, 102, 3123. [Google Scholar]
- (34).Kelly KL; Coronado E; Zhao LL; Schatz GC J. Phys. Chem. B 2003, 107, 668. [Google Scholar]
- (35).Rocha TCR; Zanchet DJ Phys. Chem. C 2007, 111, 6989. [Google Scholar]
- (36).Hao E; Schatz GC J. Chem. Phys 2004, 120, 357. [DOI] [PubMed] [Google Scholar]
- (37).Wang ZL J. Phys. Chem. B 2000, 104, 1153. [Google Scholar]
- (38).Millstone JE; Park S; Shuford KL; Qin L; Schatz GC; Mirkin CA J. Am. Chem. Soc 2005, 127, 5312. [DOI] [PubMed] [Google Scholar]
- (39).Millstone JE; Métraux GS; Mirkin CA Adv. Funct. Mater 2006, 16, 1209. [Google Scholar]
- (40).Métraux GS; Mirkin CA Adv. Mater 2005, 17, 412. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.