

Abstract
The Warburg effect, representing enhanced glycolysis and lactate production in adequately oxygenated cancer cells, has been widely regarded to cause increased extracellular acidification. Converting pyruvate to lactate by lactate dehydrogenase A (LDHA) is the last step of glycolysis. Here, we report an interesting counterintuitive observation that inhibition of LDHA resulted in enhanced glycolysis in MDA-MB-231 breast cancer cells. The cells were treated with FX11 (7-benzyl-2,3-dihydroxy-6-methyl-4-propylnaphthalene-1-carboxylic acid), a specific LDHA inhibitor. Seahorse assay reported dose-dependent increases in both oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Independent biochemical measurements also confirmed the increase of lactate production under FX11 treatment. The reasons and mechanism of these observations of elevated ECAR and lactate production in the MDA-MB-231 breast cancer cells under FX11 treatment remain to be investigated.
Keywords: Lactate dehydrogenase A, FX11, OCR, ECAR, Lactate production, Triple-negative breast cancer
26.1. Introduction
Cancers are a large family of diseases that involve the abnormal growth of cells and the potential to invade and spread to other parts of the body. To understand the underlying mechanisms involved in cancer, many studies on cancer metabolism have been performed. One of the first such observations, as described by Otto Warburg, shows that the metabolic activities in cancer cells rely heavily on glycolysis, even in the presence of oxygen [1]. Although it was hypothesized that the glycolytic phenotype in cancer is due to a permanent impairment of cancer mitochondrial oxidative phosphorylation (OXPHOS), many studies have challenged this view to show that cancer cells have normal mitochondrial OXPHOS [2]. The Warburg effect or enhanced glycolysis under aerobic condition has been demonstrated in many types of cancers, including breast cancer [3].
The very last step of glycolysis is catalyzed by lactate dehydrogenase A (LDHA), which converts pyruvate to lactate coupled by NADH and NAD+. Although various LDH isoforms have been detected in breast cancer cells [4], studies have shown that LDHA is critical to cancer cells exhibiting the highly glycolytic Warburg phenotype and is overexpressed in many cancer types including breast cancer [5–7], indicating LDHA as a promising target for cancer prevention and treatment. In the current study, we explore the bioenergetics and metabolic effects of LDHA inhibition by FX11, which competes with NADH for LDHA binding, on the triple-negative breast cancer (TNBC) cell line, MDA-MB-231. We hypothesized that FX11-treated cells will show a decrease in lactate efflux to extracellular space as pyruvate cannot be converted to lactate effectively. Furthermore, we expected cells treated with FX11 to show higher reliance on OXPHOS and suppressed aerobic glycolysis since more pyruvate may be diverted to mitochondrial metabolism. However, counterintuitively, we observed that enhanced glycolysis and lactate generation were induced by FX11 treatment.
26.2. Methods
MDA-MB-231 cells were cultured in phenol-red-containing RPMI 1640 (Invitrogen, Cat no. 11875–085) supplemented with 10% fetal bovine serum (FBS). Cells were maintained at 37 °C with 5% CO2 and passaged at approximately 80% confluency using 0.25% trypsin-EDTA.
The Seahorse XFe96 Extracellular Flux Analyzer was used to perform the metabolic analysis. Briefly, MDA-MB-231 cells were seeded on a microplate at 20,000 cells per well and incubated at 37 °C with 5% CO2 overnight. The following day, the culture medium was replaced with Seahorse XF RPMI Medium (Agilent, Cat no. 103576–100) supplemented with 11 mM glucose and 2 mM L-glutamine. The plate was placed in a non-CO2 incubator about 1 hour before the start of the assay. For treatment, chemical agents at various concentrations were preloaded into the injection ports. After instrument calibration, three basal oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements (3-0-3, mix-wait-measure cycle) were performed, followed by control and FX11 injections (blank control with Seahorse medium; 0.1% DMSO; 1 μM, 3 μM, 5 μM, and 10 μM FX11). FX11 concentrations were prepared using serial dilutions, with 10 μM FX11 having the highest percentage of DMSO (0.1%). The effect of FX11 was monitored for a total of 15 cycles, approximately 100 minutes. There were seven wells as technical replicates for each concentration of FX11 and six biological replicates in total.
The amount of protein in each well was determined using the bicinchoninic acid (BCA) assay (Pierce Biotechnology, Cat no. G7570). Briefly, medium was removed from the wells, which were then rinsed with PBS with Ca2+ and Mg2+. Albumin concentrations (0, 0.125, 0.25, 0.5, and 1 mg/mL) were loaded into cell-free wells as standards. Cells were lysed using RIPA buffer with 0.5% PMSF protease inhibitor. Working reagent was added into all wells, and the plate was wrapped in aluminum foil and incubated for 30 minutes at 37 °C. Absorbance at 562 nm was measured using a plate reader (Enspire Multimode Plate Reader, model: 2300, Perkin Elmer). A linear regression of the reference standard absorbance and their concentrations was performed, and the results were used to quantify the protein concentrations in wells with cells.
The Seahorse metabolic flux assay data was exported from Wave software (v.2.6.0, Agilent Technologies) and analyzed using Microsoft Excel. Data from each well were first normalized to its protein amount. To combine all biological replicates, OCR and ECAR measurements were normalized to the third basal measurement to account for minor differences in basal OCR and ECAR across replicates. Data values greater than 1.5 times the interquartile ranges below the first quartile or above the third quartile were classified as outliers and removed from the analysis. The OCR and ECAR of individual wells were averaged across technical replicates first and then biological replicates.
Extracellular lactate and glucose concentrations were measured as with follows: MDA-MB-231 cells were seeded in a six-well plate at 0.5 million cells per well with 2 mL culture medium. After cells had attached overnight, the medium was removed, and the wells were rinsed with PBS with Ca2+ and Mg2+, followed by the addition of 1 mL FBS-free RPMI with phenol red. Treatments (0.1% DMSO and 5 μM FX11) were added to wells, and the plate was then incubated at 37 °C and 5% CO2. Cells treated with FX11 had equal amounts of DMSO as in the control. Samples of the medium (30 μL) were collected at 2, 6, and 24 hours post treatment and measured for extracellular glucose and lactate concentrations using the YSI 2300 STAT Plus Glucose and Lactate Analyzer (YSI Inc.) calibrated with YSI 2747 dual standards. Glucose and lactate measurements were also analyzed using Microsoft Excel.
To assess statistical significance, unpaired heteroscedastic two-tailed t-tests were performed. A modified alpha = 0.005 was used to mitigate type 1 error (Bonferroni correction). All p-values below this alpha indicate statistical significance and are reported as significant results.
26.3. Results
FX11 induced metabolic responses in the TNBC cell line, MDA-MB-231. After the initial baseline measurements and FX11 injection, we observed a dose-dependent increase in OCR (Fig. 26.1a). In particular, we observed a significant difference (p < 0.005) between DMSO and 3 μM FX11 at the first and last time points and a significant difference between DMSO and 5 μM at the first time point after FX11 injection (Fig. 26.1c). These results were in line with our hypothesis, where LDHA inhibition by FX11 leads to an increase in OCR, an indicator of increased OXPHOS.
Fig. 26.1.
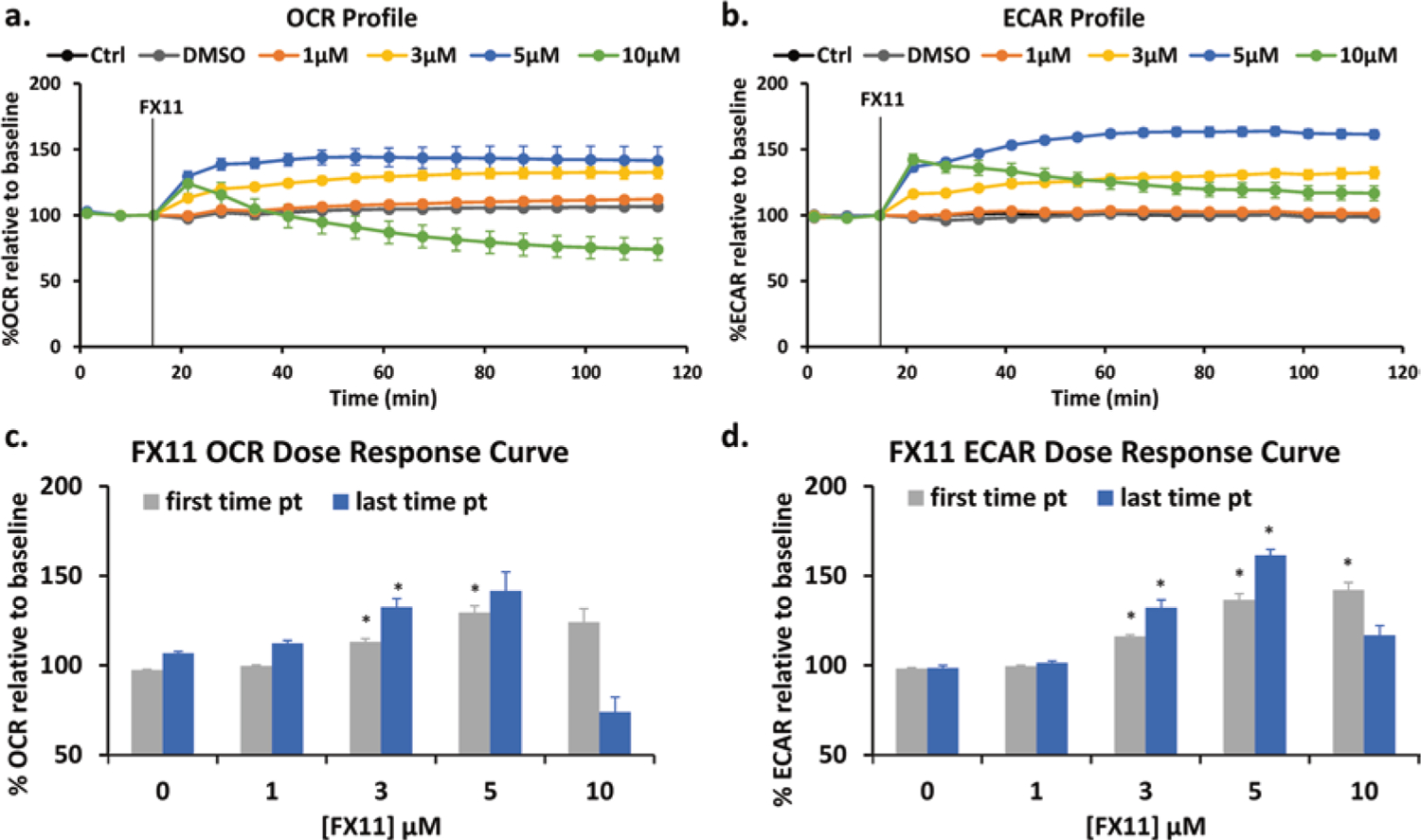
Effect of FX11 on the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in MDA-MB-231 cells. (a) OCR profile and (b) ECAR profile for control (Ctrl and DMSO) and FX11 treatments at various concentrations. Ctrl indicates blank control with medium. Results are reported as %OCR or %ECAR relative to the third time point (pt) baseline measurement ± SEM. The FX11 dose-dependent response was also plotted for (c) OCR and (d) ECAR based on first and last time points during the treatment period. Zero μM FX11 indicates DMSO control. The first time point indicates approximately 6 minutes post treatment, and the last time point indicates approximately 100 minutes post treatment. n = 6, *p < 0.005 with DMSO control
For the ECAR, we also observed a concentration-dependent increase induced by FX11 (Fig. 26.1b). In particular, we observed significant differences between DMSO and 3 μM and 5 μM at their first and last time points and between DMSO and 10 μM at the first time point after FX11 injection (Fig. 26.1d). These observations were not as expected, as we had hypothesized that inhibition of the last step of glycolysis by FX11 would decrease glycolysis and ECAR due to ineffective pyruvate to lactate conversion.
In both the OCR and ECAR responses, the 10 μM FX11 treatment showed a decrease over time. In fact, its OCR 2 hour post treatment was lower than its initial baseline measurement. This might reflect cytotoxic effects of high FX11 concentrations.
To further validate our ECAR observations, we performed extracellular lactate and glucose measurements on cells treated with DMSO and 5 μM FX11. Our results showed significantly higher extracellular lactate levels in extracellular media at 2, 6, and 24 hours post 5 μM FX11 treatment compared to that of the DMSO-treated group (Fig. 26.2a). Two additional independent experiments performed with differing seeding densities and medium volumes also showed significantly higher extracellular lactate levels in the medium of cells treated with 5 μM FX11 compared to that of the DMSO control (data not shown). This lactate enhancement phenomenon was further confirmed by an experiment using newly purchase MDA-MB-231 cells from ATCC. Extracellular glucose measurements showed lower levels of glucose in the medium of the 5 μM FX11-treated group at all treatment times but with the 24-hour treatment group being significantly different from the DMSO control (Fig. 26.2b), indicating an increased glucose consumption associated with increased ECAR and lactate production.
Fig. 26.2.
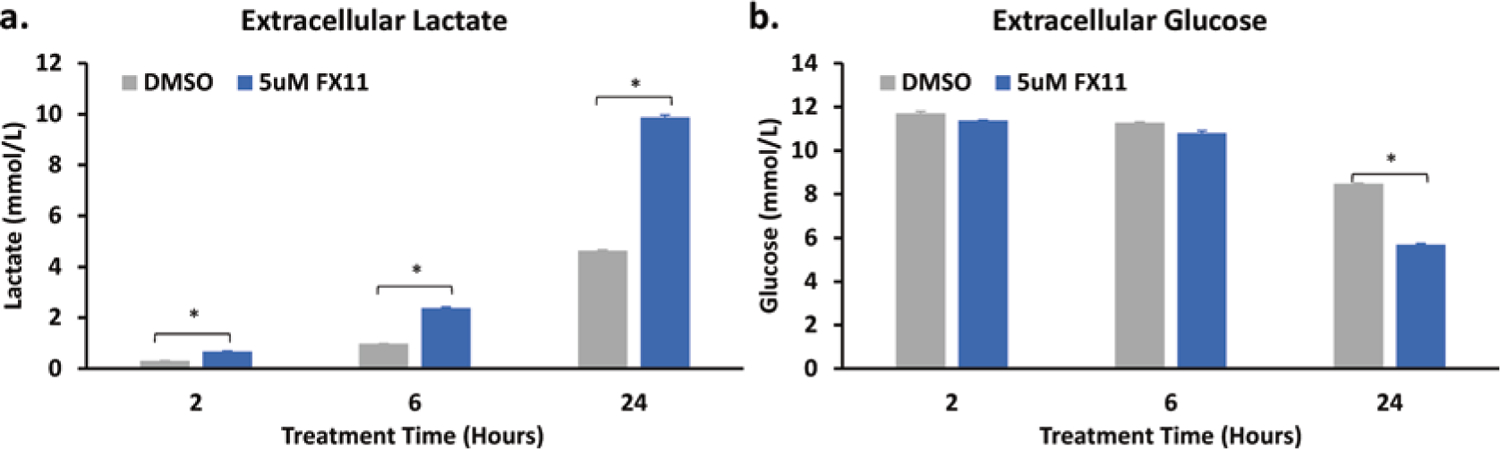
Effect of FX11 on extracellular lactate and glucose levels. (a) Lactate and (b) glucose concentration in the extracellular medium at 2, 6, and 24 hours post treatment. Lactate and glucose levels are reported as mean + SEM. n = 3, *p < 0.005
26.4. Discussion and Conclusion
Our results showed that FX11 increased the oxygen consumption rate in MDA-MB-231, as expected by our hypothesis, because reduced LDHA activity would favor the entry of pyruvate into the mitochondria for OXPHOS. This observation has also been reported in another human B-cell lymphoma cell line P493, where Le et al. observed increased oxygen consumption with the reduction of LDHA by siRNA and FX11 treatment [8]. With the halt in the conversion between pyruvate and lactate, we could assume that the aerobic glycolysis activity would decrease, which has been demonstrated in neuroblastoma cells treated with FX11 showing a decrease in lactate production [9] and in osteosarcoma cells also showing a decrease in lactate production and ECAR by genetic knockdown of LDHA [10].
Contrary to these observations and our hypothesis, our results showed an increased extracellular acidification rate, lactate production, and glucose consumption in breast cancer MDA-MB-231 cells, indicating an enhanced glycolytic activity. One possible explanation for this observation is to consider the role of the other major lactate dehydrogenase isoforms such as LDHB. It has been reported that neither LDHA nor LDHB genetic knockouts strongly reduced lactate efflux and that double disruption of LDHA and LDHB was required to ablate the lactate production in tumor cells [11]. It has also been reported that highly glycolytic TNBC cells exhibited high LDHB expression [4, 12] and that LDHB was used as a metabolic marker for neoadjuvant chemotherapy response in breast cancers [13]. These reports suggest that LDHB also likely plays an important role in regulating breast cancer metabolism. For future studies, we can investigate LDHB activity and whether LDHB contributes to the enhancement of ECAR and lactate production in MDA-MB-231 cells as observed here. We can also investigate whether FX11 may cause other effects on MDA-MB-231 cell metabolism, e.g., changing intracellular pH and extracellular glucose uptake rate. Although the glucose level used in cell culture is much higher than the extracellular glucose level in solid tumors [14, 15] and thus the results may not be applicable to clinical tumors, this study presents an interesting phenomenon worthy of further investigation.
Acknowledgments
This work was supported by US NIH Grant R01CA191207 (L. Z. Li). We thank David Nelson for his assistance in the operation of the YSI instrument.
References
- 1.Hsu PP, Sabatini DM (2008) Cancer cell metabolism: Warburg and beyond. Cell 134:703–707 [DOI] [PubMed] [Google Scholar]
- 2.Zheng J (2012) Energy metabolism of cancer: glycolysis versus oxidative phosphorylation (review). Oncol Lett 4:1151–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robey IF, Lien AD, Welsh SJ et al. (2005) Hypoxia-inducible factor-1alpha and the glycolytic phenotype in tumors. Neoplasia 7:324–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hussien R, Brooks GA (2011) Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol Genomics 43:255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman RD, Kaplan NO, Hall TC (1964) Lactic dehydrogenase in human neoplastic tissues. Cancer Res 24:389–399 [PubMed] [Google Scholar]
- 6.Koukourakis MI, Kontomanolis E, Giatromanolaki A et al. (2009) Serum and tissue LDH levels in patients with breast/gynaecological cancer and benign diseases. Gynecol Obstet Investig 67:162–168 [DOI] [PubMed] [Google Scholar]
- 7.Huang X, Li X, Xie X et al. (2016) High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast 30:39–46 [DOI] [PubMed] [Google Scholar]
- 8.Le A, Cooper CR, Gouw AM et al. (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107:2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rellinger EJ, Craig BT, Alvarez AL et al. (2017) FX11 inhibits aerobic glycolysis and growth of neuroblastoma cells. Surgery 161:747–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao S, Tu DN, Li H et al. (2016) Pharmacological or genetic inhibition of LDHA reverses tumor progression of pediatric osteosarcoma. Biomed Pharmacother 81:388–393 [DOI] [PubMed] [Google Scholar]
- 11.Zdralevic M, Brand A, Di Ianni L et al. (2018) Double genetic disruption of lactate dehydrogenases A and B is required to ablate the “Warburg effect” restricting tumor growth to oxidative metabolism. J Biol Chem 293:15947–15961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCleland ML, Adler AS, Shang Y et al. (2012) An integrated genomic screen identifies LDHB as an essential gene for triple-negative breast cancer. Cancer Res 72:5812–5823 [DOI] [PubMed] [Google Scholar]
- 13.Dennison JB, Molina JR, Mitra S et al. (2013) Lactate dehydrogenase B: a metabolic marker of response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res 19:3703–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gullino PM, Clark SH, Grantham FH (1964) The interstitial fluid of solid tumors. Cancer Res 24:780–794 [PubMed] [Google Scholar]
- 15.Vaupel P (2004) Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol 14:198–206 [DOI] [PubMed] [Google Scholar]