

Abstract
Motivation
In recent years, nanopore sequencing technology has enabled inexpensive long-read sequencing, which promises reads longer than a few thousand bases. Such long-read sequences contribute to the precise detection of structural variations and accurate haplotype phasing. However, deciphering precise DNA sequences from noisy and complicated nanopore raw signals remains a crucial demand for downstream analyses based on higher-quality nanopore sequencing, although various basecallers have been introduced to date.
Results
To address this need, we developed a novel basecaller, Halcyon, that incorporates neural-network techniques frequently used in the field of machine translation. Our model employs monotonic-attention mechanisms to learn semantic correspondences between nucleotides and signal levels without any pre-segmentation against input signals. We evaluated performance with a human whole-genome sequencing dataset and demonstrated that Halcyon outperformed existing third-party basecallers and achieved competitive performance against the latest Oxford Nanopore Technologies’ basecallers.
Availabilityand implementation
The source code (halcyon) can be found at https://github.com/relastle/halcyon.
1 Introduction
Recently, long-read single-molecule sequencing (lengths up to 2.4 Mbp) has been realized by Oxford Nanopore Technologies (ONT) with the introduction of MinION devices (Payne et al., 2019). Nanopore sequencing has been utilized in various applications such as in the detection of structural variation and cytosine methylation, along with metagenome de novo assembly (Cretu Stancu et al., 2017; De Coster et al., 2019; Gong et al., 2018; Jain et al., 2018; Simpson et al., 2017). Basecalling, i.e. translation from complex nanopore raw signals into nucleotide sequences, is first performed in nanopore sequencing pipelines. Error-prone basecalling adversely affects the entirety of downstream analyses incorporating nanopore sequencing, and therefore, the development of more accurate basecallers is critical. Although ONT has officially developed several basecallers, the details of their model specifications are not public. Thus, various third-party basecallers based on deep learning have been developed based on different approaches (Boža et al., 2017; Stoiber and Brown, 2017; Teng et al., 2018; Wang et al., 2018). However, the accuracy achieved by these basecallers at the individual read resolution is insufficient [approximately (Wick et al., 2019)]. Considering the significance of recent studies driven by ONT’s sequencing platform, there is high demand for the development of a more sophisticated basecaller. In turn, sequence data obtained from more accurate basecalling will enable more accurate detection of structural variations and cytosine methylation.
Almost all neural-network-based basecallers proposed to date are dependent to some extent on the recurrent neural network (RNN) model. The RNN is well-recognized to handle inputs with variable lengths and interpret complicated timestep dependencies of input sequences. Introducing such a technique in basecalling tasks would be reasonable because nanopore raw signals are produced by multiple nucleotides passing through a pore and interpreting such dependencies from complicated raw signals is essential.
However, a single sequence of RNN cells cannot handle a variable-length output from a given input. In the case of nanopore basecalling, the length of an output nucleotide sequence cannot be determined exactly from the length of the input raw signals. DeepNano (Boža et al., 2017) tackled this problem by dividing input raw signals into ‘events’ such that a single event corresponds to a single nucleotide. Although such an approach can ensure the training of neural networks is simple and intuitively resolve the problem of variable output dimension, the basecalling performance suffers from a bottleneck in the heuristic segmentation of signals to events, which is not exact.
Alternatively, another neural network technique with the potential to handle variable output dimension is the connectionist temporal classification (CTC) decoder (Graves et al., 2006); it has been used in processing speech signals. This technique was incorporated in the novel third-party basecaller, Chiron (Teng et al., 2018). However, although this technique can resolve the variable output dimension problem and can enable end-to-end learning from input raw signals into nucleotides, the CTC-decoder itself is not a technique proposed in current schemes; this implies that a more state-of-the-art technique would likely be required to boost the basecalling performance.
In addition, the encoder–decoder model has been frequently used in machine translation (Sutskever et al., 2014). This model has two RNNs, one of which, the encoder, can encode a variable-length input, whereas the other ‘decoder’ RNN can decode a variable-length output from the fixed dimensional encoded features. This model can be trained using matched input and output sequences, without any corresponding semantic information between the local parts of the inputs and the outputs.
Another essential technique commonly used in sequence-to-sequence learning is an attention mechanism (Bahdanau et al., 2015; Luong et al., 2015). Prior to the emergence of attention mechanisms, an encoder was used to represent the whole input sequence as a fixed-dimensional vector and a decoder started decoding from the vector. This manner of encoding was dependent largely on the end-part of an input sequence, with the decoder being unable to use sufficient information at the beginning of the input, especially when longer input sequences were used. The attention mechanism resolved this problem by representing a variable-length input sequence as a fixed-dimension context vector in each decoding timestep. Each context vector is obtained by weighting all timestep outputs of an encoder, wherein weights are calculated by a simple feed-forward network given all outputs and a current decoder cell state. An encoder–decoder model with attention mechanisms can learn appropriate attention in a backpropagation scheme. Notably, recent sequence-to-sequence models using this mechanism have achieved remarkable performance (Chen et al., 2018; Chiu et al., 2018). Moreover, recent studies have shown that the attention mechanism is superior to a conventional CTC decoder-based model even in speech recognition (Chorowski et al., 2015; Zeyer et al., 2018).
Thus, we decided to develop an improved basecaller, Halcyon, by utilizing an encoder–decoder model incorporating an attention mechanism. Halcyon incorporates a ‘monotonic’ attention mechanism, which enables the decoder to attend from an earlier part to a later part along an input sequence. Although this technique was originally introduced to accelerate decoding speed in inference at the expense of a small decrease in inference speed (Raffel et al., 2017), we incorporated this technique to stabilize the transition of attention, thereby improving basecalling precision.
2 Materials and methods
2.1 Deep neural network architecture
Halcyon combines a novel CNN module and RNN-based encoder and decoder. Whereas the CNN module is based on architectures commonly utilized in the field of image recognition, encoder and decoder modules are based on those used in the field of neural machine translation. The entire network was implemented using TensorFlow (Abadi et al., 2016).
2.1.1. Preliminaries
This study aimed to construct neural networks that directly translate raw input signals measured by a pore into corresponding nucleotide sequences that passed through the pore. Here, an input with a T-timestep signal is denoted by , and an N-base nucleotide sequence is denoted by , where is a 4-D vector indicating the probabilities of four nucleotides (A, T, G and C) at position k.
2.1.2. Inception-block-based CNN module
Input raw signals are first fed into a CNN module. This module incorporates inception blocks, which are state-of-the-art architectures in the field of computer vision. A single inception block has branches. Each branch has 1 × 1 convolution to prevent the expansion of channel dimensionality and a convolution layer with different widths of filters. Finally, output vectors from these branches are concatenated in a channel axis and fed into the next layer.
The motivation behind using this module is the need to extract local features of input raw signal and reduce the dimension of the input timestep axis. As the time-complexity of RNN is severely influenced by the timestep dimension, the reduction contributes to high throughput inference.
Each convolution block consists of a single layer convolution layer with a rectified linear unit (ReLU) activation function, followed by a batch-normalization layer.
The ReLU activation function is defined as
Batch normalization constitutes a technique to accelerate the learning of neural networks by normalizing each layer’s input within a training minibatch (Ioffe and Szegedy, 2015). Given a minibatch output of a single unit (where n is a minibatch size), the batch normalization layer calculates the mean value and variance value within the minibatch as
and the normalized output as
Then, it returns output instead of returning x, where . Here, ϵ, γ and β are parameters specific to the unit and are optimized in a backpropagation scheme. In the test, μ and are set to the average values over those used in training minibatches.
2.1.3. Encoder module
An RNN-based encoder plays an important role in capturing long-time dependencies in the timestep dimension and dealing with the variable lengths of inputs. LSTM is used in Halcyon as an RNN-based architecture. An LSTM layer is characterized by an LSTM cell and its recursive computation.
The function of a single cell at the timestep of t can be formulated as follows:
where denotes the Hadamard product between two vectors and , and denotes their vector concatenation. denotes the input from the previous network at t-timestep; in this case, the output of stacked inception blocks. and denote the synaptic-weight matrices and and denote bias vectors, all of which are shared among LSTM over all timesteps. σ denotes a sigmoid activation function . Such calculation is conducted recursively along the timestep axis.
To capture local dependencies in both the forward and backward directions along the timestep axis, bidirectional recurrent neural networks are incorporated (Schuster and Paliwal, 1997) in Halcyon; these networks conduct the same recursive calculation in the backward direction of the timestep axis. In each timestep, an output vector of a forward RNN cell and that of a backward RNN cell are concatenated, and the result is yielded to the next layer.
2.1.4. Decoder module using attention mechanisms
Given encoded features , the goal is to estimate the target nucleotide probabilities ; i.e. to model the conditional probability . The basic idea of modeling the probability using an LSTM layer can be formulated as
where is the fixed-dimensional representation of X given by the last hidden state of the encoder LSTM (theoretically, it has all information over an input sequence). We note that an output sequence length m cannot be defined by an input sequence length n as the number of electrical signal values measured per nucleotide exhibits some variation. We need to introduce an end-of-sequence symbol <EOS> to model output nucleotide sequences with all possible lengths. Here, each conditional probability is represented by the output of the decoder LSTM at t-timestep, a single fully connected layer, and a softmax function. Given the output of the LSTM at the timestep of t and the weight matrix of the fully connected layer W, the conditional probability for each nucleotide base is
where l denotes the number of output tokens including an end token, and gi denotes the ith element of the fully connected output vector .
However, such a model has a problem whereby the fixed-dimensional v contains little information for the beginning of an input sequence, with the problem becoming more serious when input sequences are longer. To handle this issue, we introduced attention mechanisms. Each probability of the elements of joint probability is formulated using attention mechanisms as
where is a hidden state of decoder LSTM at timestep i. The context vector with attention is dependent on the previous decoder hidden state and all hidden states of encoder LSTM cells X. The context vector is defined as the weighted sum of encoder hidden states as
where the weight αij for each hidden state is calculated by the softmax function to scored values as
where
A score function can be formulated as a simple trainable feed forward network. Among some variations of such score functions, we adopted Luong attention (Luong et al., 2015), in which the score function is defined by where is a synaptic weight matrix for the score function and it is shared over all timesteps. The score function calculates the importance of input features when predicting the output in timestep i, which enables the decoder to retrieve essential information from all encoded features selectively.
Further, we adopted a monotonic attention mechanism (Raffel et al., 2017). Monotonic attention is an attention mechanism that restricts the transition of attention in a left-to-right manner, which is suitable for the task of basecalling nanopore sequences. In general, monotonic attention is used to reduce the complexity in decoding; it was incorporated in Halcyon to decode more accurately. A ‘soft’ monotonic attention mechanism was used in both training and inference time.
2.1.5. Training and inference decoder
In a training phase, each decoder cell outputs likelihoods of nucleotides in each timestep, and then, the cell state is passed to the next decoder cell. In this timing, even if the decoder cell infers a wrong nucleotide, a correct nucleotide from a ground truth sequence will be passed to the next cell. Alternatively, in the inference for test data, a decoder cell cannot use the output token of the previous decoder cell, unlike a training decoder. Therefore, an inference decoder cell infers the likely nucleotide given the previous cell state, attended encoder’s features, and the token emitted by the previous cell. However, searching for an optimal nucleotide sequence Y that maximizes the conditional probability is too computationally expensive because the complexity grows exponentially with the number of nucleotide bases in the inferred sequence. To tackle this problem, a beam search strategy is commonly used, which retains the best k decoded paths with the highest probabilities at each timestep; k is termed the beam search width. Halcyon incorporated this strategy in the inference, with the beam search width set to 20 in all experiments except for the performance assessment of using different beam widths.
2.1.6. Scheduled sampling
Although each inference decoder cell can only use the previously decoded token, the training decoder cell always uses the token from the ground truth. Such discrepancy is known to produce rapidly accumulated errors in the decoding of inference. To resolve this issue, ‘schedule sampling’ was introduced (Bengio et al., 2015). Scheduled sampling is a technique used in a training phase, and it randomly samples the previously inferred token instead of sampling from the ground truth. Halcyon used this technique in the training phase against a longer input signal (3000 values long) with a sampling ratio of 0.3.
2.2 Data preparation
2.2.1. ONT MinION and Illumina sequencing
Genomic DNA (#NA18943) used in the HapMap project was purchased from the Coriell Institute (Camden, NJ). For MinION sequencing, a sequencing library was prepared from 1.5 µg of the DNA using Ligation Sequencing Kit 1D (SQK-LSK108; ONT, Oxford, UK) and Library Loading Bead Kit (EXP-LLB001; ONT) according to the manufacturer’s instructions. The library was loaded onto the R9.4 flow cell of the MinION sequencing device (ONT) and sequenced for 48 h. A total of 11 runs of MinION sequencing were conducted. For Illumina sequencing, a sequencing library was prepared from 200 ng of the DNA using the TruSeq Nano DNA Library Prep Kit (Illumina, San Diego, CA). Sequencing was performed with paired-end reads of 101 bp on a HiSeq 2500 platform according to the manufacturer’s instructions (Illumina).
2.2.2. Labeling of raw signals
Taiyaki (v5.1.0), the training models for basecalling Oxford Nanopore reads, was used to obtain labeled sequences. By using Taiyaki, nanopore raw signals are divided into segments, each of which corresponds to one nucleotide. By using such labeled reads, the arbitrary length of signals with a matched nucleotide sequence is easily obtained. We generated labeled signals with a length of 1000 and those with a length of 3000.
2.2.3. Training and Validation dataset
Halcyon was trained and evaluated in a hold-out validation scheme. Unlike other general machine learning problems such as image recognition, it is inappropriate to divide the dataset into a training dataset and a test dataset. Among the obtained nanopore raw reads, some reads are from the same region of a human whole genome sequence. If such sequences exist in both the training and test datasets, correctly evaluating the generalization performance of nanopore basecalling would be impossible because trivial overfitting to patterns of consecutive nucleotide sequences of a human genome would also contribute to an accurate basecalling. Therefore, a training dataset was defined as paired signals and nucleotide sequences aligned to even-numbered chromosomes (i.e. chr2, chr4,…, chr22), and the test dataset as those aligned to odd-numbered chromosomes (i.e. chr1, chr3, …, chr21).
2.3 Transfer learning against different input lengths
Halcyon was trained against different lengths of signals in a transfer-learning scheme to train the model against longer inputs effectively because starting with longer inputs might render attention-based training difficult. Therefore, Halcyon was trained against 1000-value-long signals and then against 3000-value-long signals. Such transfer learning was possible because (i) RNN-based encoders and decoders are applicable to inputs and outputs with different lengths, which are attributed to recurrent RNN cells, and (ii) parameters of CNN are fully dependent on the convolution kernels, the parameters of which are shared along the timestep axis.
2.4 Inference
In basecalling test data with arbitrary lengths, each set of input current signals was segmented into 3000-value-long signals with 800-value-long overlaps. These segmented reads were basecalled independently and merged into a single nucleotide sequence. In merging neighbor reads, pairwise local alignment against sequences supposed to be overlapped was conducted. A match score of +4, a mismatch penalty of -4.5, and gap/extend penalties of -5/-3 were used in the pairwise alignment.
2.5 Evaluation
The performance of Halcyon was compared with that of other existing basecallers with two viewpoints (i) ‘Individual read accuracy’: how accurately can each model basecall an individual sequence, and (ii) ‘SNV detection rate’: how accurately can SNVs be detected using whole basecalled sequences obtained from each model.
We selected Guppy [v3.6.0], Bonito[v0.1.5], Chiron [v.0.5.1] (Teng et al., 2018) and DeepNano [latest version from https://bitbucket.org/vboza/deepnano] (Boža et al., 2017) as basecallers for comparison. Guppy and Bonito were selected as basecallers developed by ONT officially, and the others were selected as third-party basecallers.
2.5.1. Read accuracy
The performance of basecalling for an individual example of an input current signal can be measured by calculating similarity between a basecalled sequence and the corresponding ground truth sequence. We defined the similarity according to the following criteria that can be calculated after conducting pairwise alignment between the two sequences; (i) the ratio of the number of nucleotide bases accurately basecalled calculated as , (ii) the ratio of the number of inserted nucleotide bases calculated as and (iii) the ratio of number of deleted bases calculated as . These metrics were calculated by aligning basecalled reads from each basecaller back to the reference sequence using minimap2 (Li, 2018).
2.5.2. SNV detection
SNV detection performance was measured by comparing the SNVs detected using whole nanopore basecalled reads with those detected using whole short read sequences. As short read sequences are highly accurate, we used the results as ground truths. SNVs were detected using basecalled nanopore reads obtained from our basecaller and the other baselines, each of which was compared with the ground truth SNVs.
Short-read sequences were aligned to the reference sequence using BWA MEM and then processed by Strelka2 (Kim et al., 2018), a fast and accurate variant caller. Resultant SNVs were then filtered to extract only SNVs with high quality (QUAL > 500). Nanopore basecalled reads were aligned using minimap2 (Edge and Bansal, 2019) and SNVs were detected by LongShot (Edge and Bansal, 2019). SNV detection recall and precision are calculated by using hap.py (v0.3.8) (Krusche et al., 2019). True positive rates given SNV positions for each depth (depth 6 20) are also calculated using the tool for each basecaller.
2.5.3. Basecalling speed
Basecalling speed of five basecallers are measured (i) using only 1-threaded CPU only and (ii) using 5-threaded CPU and 1 core of GPU in Ubuntu 18.04.2 LTS x86 64 bit 257606MiB RAM with CPU: Intel Xeon Gold 6130 @ 3.700 GHz, and GPU: NVIDIA Quadro GV100.
3 Results
Whole genome sequencing was conducted using ONT’s MinION device against one human sample, with these reads then being used to train the neural network-based basecaller and evaluate the basecalling performance. To obtain matched raw signals and the corresponding nucleotide sequences for training, we used Taiyaki, ONT’s training model. The statistics of resulting reads by Taiyaki are shown in Table 1. These labeled raw reads were then divided into training/test datasets according to the chromosomes.
Table 1.
Metrics of all 11 runs of MinION sequencing
| MinION run | Number of reads | Signals length | Nucleotide length |
|---|---|---|---|
| RUN 1 | 198 318 | 50 408 ± 37 154 | 4762 ± 3577 |
| RUN 2 | 90 619 | 53 587 ± 42 081 | 4700 ± 4220 |
| RUN 3 | 720 885 | 65 579 ± 41 308 | 6724 ± 4345 |
| RUN 4 | 605 642 | 72 292 ± 74 283 | 7040 ± 7380 |
| RUN 5 | 541 783 | 76 314 ± 76 775 | 7123 ± 7347 |
| RUN 6 | 255 240 | 75 450 ± 79 599 | 6795 ± 7372 |
| RUN 7 | 665 879 | 82 656 ± 82 728 | 7503 ± 7715 |
| RUN 8 | 1 016 413 | 72 082 ± 42 833 | 6413 ± 3905 |
| RUN 9 | 946 914 | 72 807 ± 44 096 | 6299 ± 3929 |
| RUN 10 | 569 715 | 72 186 ± 43 109 | 6316 ± 3866 |
| RUN 11 | 220 199 | 70 420 ± 46 432 | 5825 ± 3905 |
Note: The number of reads obtained in each run, a mean and a standard deviation of lengths of raw signals and the lengths of nucleotides basecalled by Guppy (exploited by Taiyaki) observed in each run are also shown.
The performance of Halcyon, was measured by comparing it with the performance of Guppy, Bonito, Chiron and DeepNano. The accuracy of basecallers in an individual read resolution was measured by basecalling all raw signals in a test dataset and aligning these reads to the reference sequence by minimap2 (Table 2). Figure 1a–c show the distribution of read identity, insertion error rate and deletion error rate of reads from the five evaluated basecallers. These metrics collectively constitute a heuristic measurement for read precision. The results showed that Halcyon achieved competitive performance against ONT’s cutting edge basecallers and outperformed the other third-party basecallers. Among baseline basecallers, Guppy achieved maximal performance, which is in agreement with recently reported results (Wick et al., 2019).
Table 2.
Read metrics for reads basecalled by five different basecallers
| Basecaller | Total reads | Total yield (Gb) | Read length | Read identity | Insertion rate | Deletion rate |
|---|---|---|---|---|---|---|
| Halcyon | 3 225 205 | 20.5 | 6359 ± 5702 | 0.894 ± 0.084 | 0.028 ± 0.023 | 0.041 ± 0.043 |
| Guppy | 3 150 600 | 20.5 | 6519 ± 5748 | 0.905 ± 0.080 | 0.021 ± 0.018 | 0.041 ± 0.044 |
| Bonito | 3 160 225 | 20.3 | 6410 ± 5664 | 0.902 ± 0.080 | 0.020 ± 0.016 | 0.045 ± 0.050 |
| Chiron | 2 129 764 | 17.4 | 8161 ± 5384 | 0.800 ± 0.061 | 0.047 ± 0.019 | 0.072 ± 0.033 |
| Deepnano | 2 783 926 | 18.4 | 6606 ± 5616 | 0.805 ± 0.055 | 0.042 ± 0.014 | 0.075 ± 0.030 |
Note: Except for total reads and total yield, the mean and standard deviation of each measurement is described. Read identity, insertion rate, deletion rate are obtained by aligning basecalled reads to reference by minimap2.
Fig. 1.
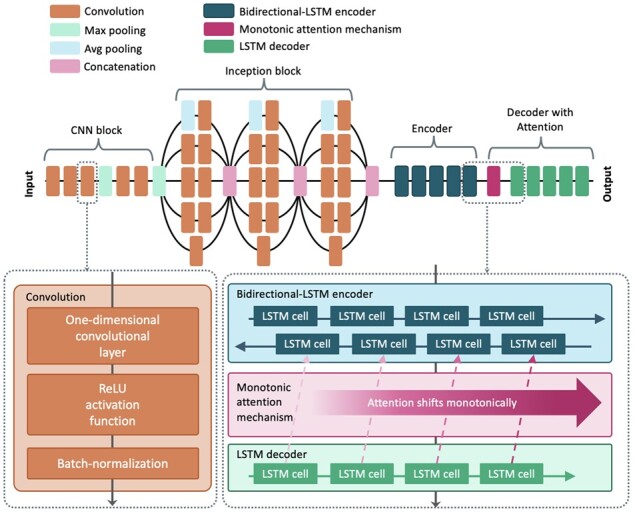
Overview of the network architecture of Halcyon from the input (nanopore raw signals) to the output (nucleotide sequence). Each convolution component is composed of a one-dimensional convolution layer with a rectified linear unit (ReLU) activation function followed by a batch normalization layer. A semantic relationship between the last layer among five stacked bidirectional LSTM encoding layers and the first layer among five stacked LSTM decoding layers is comprehended by monotonic attention
Although these results are obtained using the test dataset, they did not conclusively display Halcyon’s superiority in nanopore sequencing for the following reasons: (i) the basecalled reads in the test data were aligned to the reference sequences, which did not consider individual genome variation such as SNVs, and (ii) accuracy in a individual read resolution did not necessarily imply consensus accuracy, which is more valuable in practice, as actual sequencing analyses involve aggregating multiple-coverage sequences to obtain a consensus result. Therefore, we assessed SNV detection performance by utilizing short read sequence data. Whole genome sequencing against the same sample was performed using Illumina HiSeq. The obtained reads were aligned to the reference sequences using the Burrows-Wheeler aligner [BWA (Li and Durbin, 2010)], and then SNVs were detected using Strelka2 (Kim et al., 2018). For nanopore sequences, SNVs were detected by using LongShot (Edge and Bansal, 2019).
The SNV detection performance was measured in recall and precision obtained by hap.py, haplotype comparison tools by Illumina (Krusche et al., 2019). The evaluation pipeline is shown in Figure 2b. The resulting SNV detection recall and precision are illustrated in Figure 3f. In addition to the performance in individual read resolution, Halcyon achieved competitive performance against ONT’s basecallers. These results demonstrated that the performance of Halcyon was not overfitted against the utilized reference sequences, and the model would be the most useful in practical nanopore sequencing analyses.
Fig. 2.

(a) Overview of preparation of training datasets using ONT’s retraining model, Taiyaki. Labeled reads obtained by Taiyaki are then split into fixed-length raw signals and corresponding nucleotide sequences. (b) Overview of evaluation of different basecallers in terms of SNV-detection performance assuming short-read sequencing as the ground truth
Fig. 3.

Individual read statistics obtained by aligning basecalled reads to the reference sequence with minimap2. Distributions of (a) read identities, (b) insertion error rates and (c) deletion error rates calculated over all basecalled reads are illustrated using letter-value plots. The SNV detection rate measured by comparing SNVs detected by LongShot to those detected by Strelka2 using short-read sequences. (d) SNV detection rate overall each chromosome, and (e) true positive rate of SNV-detection for each read depth (6 20). Basecalling speed measured in terms of the number of nucleotide basecalled in a second. Speed of basecalling (f) measured using CPU with a single thread and (g) that measured using a single GPU and CPU with five threads
Further we investigated SNV detection performance for each read depth. Such investigation is important because (i) in the actual clinical application of nanopore reads, it might be necessary to create an important decision relying on limited coverage data, and (ii) observing the saturation of SNV detection rate along with read depth may aid in the determination of nanopore sequencing strategy. The result is shown in Figure 3g. Halcyon consistently performed similarly to ONT’s basecaller, with results similar to those obtained in Figure 3f. As basecalling speed is an important aspect, we measured the number of nucleotides basecalled in a second. Figure 3f and g showed the result using CPU and GPU respectively. The basecalling speed of Halcyon is slower than other basecaller except for Chiron in the CPU, and the slowest in the GPU.
One advantage of incorporating the attention mechanism is that one can understand semantic correspondence between raw signals and basecalled sequences. As shown in Figure 4, an attention matrix obtained in an inference phase represents the information, where you can understand which part of the raw signals is referred by Halcyon to infer a certain nucleotide. Retaining this information will be helpful when investigating a single nucleotide in detail, such as for the detection of cytosine methylation.
Fig. 4.
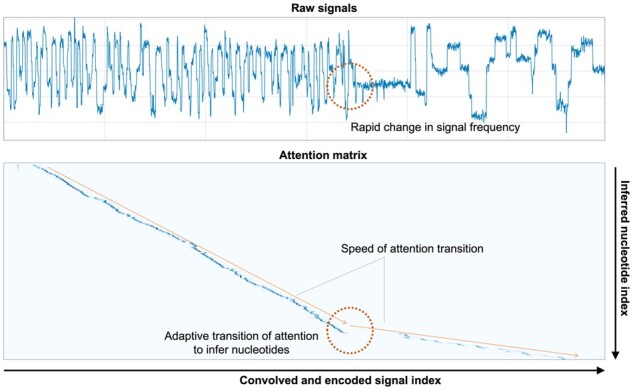
Actual row signal input (top) and an attention matrix obtained in the basecalling phase to infer nucleotides from the given signals (bottom). The number of signal values measured during single nucleotide passage through a pore changes rapidly at a certain point (indicated by a circle in the figure). In the corresponding part of the attention matrix, the gradient of attention transition speed also changes rapidly
4 Conclusion
We developed a novel basecaller incorporating state-of-the-art neural network techniques commonly utilized for sequence-to-sequence learning. Our proposed basecaller, Halcyon, achieved high performance for individual read resolution and the detection of SNVs using multiple reads. Given the recent advances in downstream analyses using long read sequences such as the detection of cytosine methylation and structural variations, obtaining accurate reads with semantic correspondence between raw signals and the reads using Halcyon would accelerate such applications and lead to biologically significant findings. Furthermore, as models of nanopore basecallers officially developed by ONT are not public, providing the neural network specification of a well-working basecaller will facilitate the development of a more sophisticated basecaller in the future.
Financial Support: none declared.
Conflict of Interest: none declared.
Contributor Information
Hiroki Konishi, Health Intelligence Center.
Rui Yamaguchi, Human Genome Center.
Kiyoshi Yamaguchi, Advanced Clinical Research Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan.
Yoichi Furukawa, Advanced Clinical Research Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan.
Seiya Imoto, Health Intelligence Center; Human Genome Center.
References
- Abadi M. et al. (2016). Tensorflow: A system for large-scale machine learning. In: 12th USENIX Symposium on OSDI, pp. 265–283.
- Bahdanau D. et al. (2015) Neural machine translation by jointly learning to align and translate. In: ICLR, 2015.
- Bengio S. et al. (2015) Scheduled sampling for sequence prediction with recurrent neural networks. In: NeurIPS, Vol. 28, pp. 1171–1179. [Google Scholar]
- Boža V. et al. (2017) DeepNano: deep recurrent neural networks for base calling in MinION Nanopore reads. PLoS One, 12, e0178751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.X. et al. (2018) The best of both worlds: combining recent advances in neural machine translation. arXiv preprint arXiv 1804.09849.
- Chiu C. et al. (2018) State-of-the-art speech recognition with sequence-to-sequence models. In: 2018 ICASSP, pp. 4774–4778.
- Chorowski J.K. et al. (2015) Attention-based models for speech recognition. In: NeurIPS, Vol. 28, pp. 577–585. [Google Scholar]
- Cretu Stancu M. et al. (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun., 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Coster W. et al. (2019) Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res., 29, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge P., Bansal V. (2019) Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun., 10, 4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong L. et al. (2018) Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat. Methods, 15, 455–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves A. et al. (2006) Connectionist temporal classification: labelling unsegmented sequence data with recurrent neural networks. In: ICML ’06, pp. 369–376.
- Ioffe S., Szegedy C. (2015) Batch normalization: accelerating deep network training by reducing internal covariate shift. In: PMLR, Vol. 37, pp. 448–456. [Google Scholar]
- Jain M. et al. (2018) Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol., 36, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. et al. (2018) Strelka2: fast and accurate variant calling for clinical sequencing applications. Nat. Methods, 15, 591–594. [DOI] [PubMed]
- Krusche P. et al. ; for Genomics, t. G. A. and Team, H. B. (2019) Best practices for benchmarking germline small-variant calls in human genomes. Nat. Biotechnol., 37, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics, 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luong M.-T. et al. (2015) Effective approaches to attention-based neural machine translation. In: Proceedings of the 2015 Conference on Empirical Methods in Natural Language Processing, pp. 1412–1421.
- Payne A. et al. (2019) BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics, 35, 2193–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffel C. et al. (2017) Online and linear-time attention by enforcing monotonic alignments. In: ICML ’17, Vol. 70, pp. 2837–2846. [Google Scholar]
- Schuster M., Paliwal K.K. (1997) Bidirectional recurrent neural networks. IEEE Trans. Signal Process., 45, 2673–2681. [Google Scholar]
- Simpson J.T. et al. (2017) Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods, 14, 407–410. [DOI] [PubMed] [Google Scholar]
- Stoiber M., Brown J. (2017) BasecRAWller: streaming nanopore basecalling directly from raw signal. bioRxiv, 133058.
- Sutskever I. et al. (2014) Sequence to sequence learning with neural networks. In: NeurIPS, Vol. 27, pp. 3104–3112. [Google Scholar]
- Teng H. et al. (2018) Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. GigaScience, 7, giy037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. (2018) Wavenano: a signal-level nanopore base-caller via simultaneous prediction of nucleotide labels and move labels through bi-directional wavenets. Quant. Biol., 6, 359–368. [Google Scholar]
- Wick R.R. et al. (2019) Performance of neural network basecalling tools for oxford nanopore sequencing. Genome Biol., 20, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyer A. et al. (2018) Improved training of end-to-end attention models for speech recognition. In: Proceedings of Interspeech, pp. 7–11.