

The combination of directed migration and immunosuppressive shielding enables effective T cell therapy in solid tumors.
Abstract
CAR T cell therapy remains ineffective in solid tumors, due largely to poor infiltration and T cell suppression at the tumor site. T regulatory (Treg) cells suppress the immune response via inhibitory factors such as transforming growth factor–β (TGF-β). Treg cells expressing the C-C chemokine receptor 8 (CCR8) have been associated with poor prognosis in solid tumors. We postulated that CCR8 could be exploited to redirect effector T cells to the tumor site while a dominant-negative TGF-β receptor 2 (DNR) can simultaneously shield them from TGF-β. We identified that CCL1 from activated T cells potentiates a feedback loop for CCR8+ T cell recruitment to the tumor site. This sustained and improved infiltration of engineered T cells synergized with TGF-β shielding for improved therapeutic efficacy. Our results demonstrate that addition of CCR8 and DNR into CAR T cells can render them effective in solid tumors.
INTRODUCTION
The implementation of immune-checkpoint blockade into the standard of care for a growing number of malignant diseases has established the role of T cells in therapeutically inducible antitumor immunity (1, 2). Likewise, the therapeutic use of T cells, also termed adoptive T cell therapy (ACT), has been developed. This includes the ex vivo expansion and reinfusion of tumor-infiltrating lymphocytes or engineering patient T cells to express a cancer antigen–specific T cell receptor (TCR) bearing improved tumor-targeting capacities (3). Alternatively, patient T cells can be engineered to express a chimeric antigen receptor (CAR), which combines the specificity of an antibody with T cell–activating and costimulatory domains that acts as a fully synthetic receptor triggering a full-blown T cell response (4). CAR T cells have shown unparalleled efficacy in refractory patients with hematological malignancies such as acute lymphatic leukemia or diffuse large B cell lymphoma (5, 6). Nevertheless, a large proportion of patients acquire resistance during the course of their therapy due to target loss or mutation (7). In the more frequent solid cancer indications, efficacy of CAR T cells remains to be demonstrated, despite the strong proof-of-concept studies in xenograft models (8). These immunocompromised models should be complemented with immune-competent syngeneic models that can demonstrate the immune system’s impact on CAR T cells (9, 10). However, we and others have repeatedly observed that tumor control by TCR-specific T cells and also CAR T cells, despite appropriate target selection, is limited or absent in syngeneic models (11–13). This reflects the growing body of clinical observations with CAR T cells (7) and demonstrates the need for alternative methods to improve the activity of CAR T cells against solid tumors.
Major unaddressed obstacles to CAR T cell efficacy in solid tumors are poor infiltration at the tumor site and T cell suppression (14). Clinically relevant CAR T cell efficacy is unlikely to occur unless both aspects are tackled simultaneously. T regulatory (Treg) cells are key drivers of immunosuppression in solid tumors (15) largely via release of transforming growth factor–β (TGF-β) (16). More recently, another mechanism of immunosuppression by Treg cells has been identified and involves the ligand CCL1 binding to its receptor C-C chemokine receptor 8 (CCR8) (16). Accumulation of these CCR8+ Treg cells is associated with poor prognosis in breast cancer (15, 16). So far, this chemokine axis has not been exploited therapeutically. Since tumors of different origins actively recruit Treg cells through the CCL1-CCR8 axis, we reasoned that this axis could be therapeutically exploited by arming effector T (Teff) cells with CCR8 to improve recruitment to tumor sites. However, recruitment of CCR8-engineered T cells to the same sites as Treg cells will directly expose them to Treg cell–mediated suppression. To overcome this immunosuppression, we sought to use the well-characterized truncated variant of the TGF-β receptor 2 (17, 18). Combining a previously unused tumor recruitment mechanism with a well-established mechanism of immunosuppression prevention would generate the first T cell product that can address the two major limitations of ACT in solid tumors. We probed this hypothesis in murine and human models in vitro and in vivo and show that CAR T cell therapy can be rendered effective in various tumor models with this novel combinatorial approach.
RESULTS
CCL1 recruits CCR8+ T cells to tumors in a murine pancreatic cancer model
To further understand the determinants of successful infiltration of adoptively transferred antigen-specific T cells, we transferred ovalbumin (OVA)–specific, in vitro expanded T cells (OT-I T cells) transduced with green fluorescent protein (GFP) to mice bearing Panc02-OVA tumors (Fig. 1A). A week after transfer, we isolated the transferred T cells from spleen and tumor and performed reverse transcription polymerase chain reaction (RT-PCR)–based microarray analysis on predefined genes associated with T cell exhaustion and homing. As expected, markers of activation and exhaustion such as PD-1 (Programmed cell death protein 1) were up-regulated in tumor-infiltrating T cells (Fig. 1B). Of all the chemokine receptors, the only target that was significantly up-regulated was CCR8 (Fig. 1B), implying its role in facilitating T cell accumulation at the tumor site. Homing of CCR8+ cells is mainly driven by CCL1 (16, 19), which could be detected in ex vivo Panc02 tumors at both the RNA (Fig. 1C) and protein level (Fig. 1, D and E), and was detected in the tumor microenvironment at considerably higher levels than other organs (Fig. 1D). Activated T cells have previously been reported to produce CCL1 (20). Consistent with this, we found that tumor-infiltrating T cells produced high amounts of CCL1 upon stimulation (Fig. 1F). Upon antigen recognition, T cells produced more CCL1 with increasing effector-to-target ratios (Fig. 1G), highlighting a positive feedback loop, whereby local activation of T cells in the tumor induces their expression of CCL1, which, in turn, will further amplify the recruitment of CCR8-expressing immune cells. In contrast, the Panc02 tumor cells themselves did not release CCL1 (Fig. 1G).
Fig. 1. CCR8 is used by a subset of transferred antigen-specific T cells to traffic to a murine pancreatic cancer in a CCL1-dependent manner.
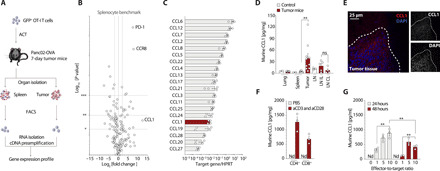
(A) Schematic experimental layout to determine the gene expression profile of OT-I T cells after adoptive transfer. (B) Volcano plot for gene expression profile of tumor-infiltrating OT-I T cell compared to a splenocyte benchmark. Gene expression is depicted on the x axis versus the P value depicted on the y axis. (n = 3 mice). (C) RT-PCR gene analysis of explanted Panc02-OVA tumors (n = 4). HPRT, hypoxanthine-guanine phosphoribosyltransferase. (D) Enzyme-linked immunosorbent assay (ELISA) for murine CCL1 on organ lysates of control animals (n = 3) or Panc02-OVA tumor-bearing animals (n = 10). LNIL, lymph node ipsilateral to the tumor; LNCL, lymph node contralateral to the tumor. (E) Fourteen-day Panc02-OVA tumor explants, embedded in OCT medium and frozen for cryosectioning and stained with CCL1 and 4′,6-diamidino-2-phenylindole (DAPI) (representative of n = 3 mice). (F) ELISA for murine CCL1 on CD4+ or CD8+ magnetic-activated cell-sorted T cells after 24-hour stimulation with anti-CD3 and anti-CD28 antibodies or vehicle solutions only (representative of n = 3 independent experiments). (G) ELISA for murine CCL1 of supernatants generated upon 24- or 48-hour culture of different effector to target ratios, namely, 0, 1, 5, or 10 OT-I T cells to 1 tumor target cell (a constant number of 20.000 Panc02-OVA tumor cells was used) (representative of n = 3 independent experiments). nd stands for nondetectable. ns stands for nonsignificant P > 0.05. Experiments show mean values ± SEM of at least triplicates and are representative of at least three independent experiments. P values are based on two-sided unpaired t test. Data shown in (D) are pooled from three independent experiments.
CCR8-transduced T cells improve ACT efficacy through CCL1-dependent tumor trafficking
The presence of CCR8 on tumor-infiltrating T cells implies that the CCL1-CCR8 axis recruits T cells to the tumor site. CCR8 is a chemokine receptor predominantly present on effector Treg cells. There, its expression is substantially higher compared to central Treg or conventional Teff cells (Fig. 2A) and may contribute to their infiltration into CCL1-expressing tumors. Thus, by ectopically introducing CCR8 into tumor-specific Teff cells, these cells may be able to mirror Treg cell migration patterns. However, CCR8 is not the only chemokine receptor found in Treg cells. CCL22 is the ligand of CCR4, which, similar to CCR8, is expressed on Treg cells, fostering an immunosuppressive environment in ovarian carcinoma and predicting poor survival (21). We therefore compared the relative expression of these two axes across several human tissues. We found substantial expression of CCL22 in numerous healthy tissues including the central nervous system, gastrointestinal tract, lung, and skin. In contrast, CCL1 was expressed at lower levels and restricted to the spleen, lung, testes, and brain (fig. S1A). These data suggest a favorable specificity profile with less off-target accumulation of CCR8-CAR T cells and increased specificity of the CCL1-CCR8 axis to tumors.
Fig. 2. CCR8 transduction in OT-I T cells improves ACT efficacy through CCL1-dependent tumor trafficking.
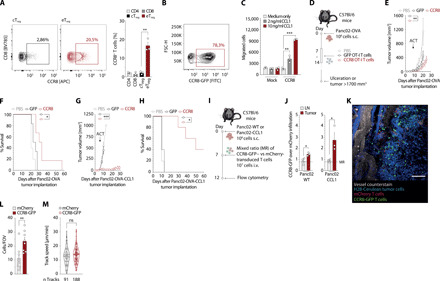
(A) CCR8 staining of live, CD45+CD3+CD4+FoxP3+ CD44high CD62L− eTreg and CD44low CD62L+ cTreg Panc02-OVA–infiltrating cells. CCR8 expression on CD4+, CD8+, eTreg, and cTreg T cells. (n = 3 mice). APC, Allophycocyanin. (B) CCR8-GFP transduction efficiency of murine T cells. FITC, fluorescein isothiocyanate. (C) In vitro migration of murine T cells to CCL1. (D) Experimental layout for (E) to (H). (E) Tumor growth curves of mice treated with a single intravenous (i.v.) injection of phosphate-buffered saline (PBS) or 107 GFP-transduced or CCR8-transduced OT-I T cells (n = 5 mice per group). (F) Tumor survival curves of (E). (G) Panc02-OVA-CCL1 tumor growth curves of mice treated with a single intravenous injection of PBS or 107 GFP-transduced or CCR8-transduced OT-I T cells (n = 5 mice per group). (H) Tumor survival curves of (G). (I) ACT tracking experiments in Panc02 or Panc02-CCL1 tumors by flow cytometry. s.c., subcutaneous. (J) Live, CD45.1+ tumor-infiltrating T cells (n = 3 mice). (K and L) ACT tracking in mice with tumors in a dorsal skinfold chamber to enable multiphoton intravital imaging (L) and speed quantification of tumor-infiltrating T cells (M) (n = 8 mice). FOV, field of view. Experiments show mean values ± SEM and are representative of three independent experiments, except (G) and (H) that are representative of two independent experiments. P values for (C), (J), (L), and (M) are based on a two-sided unpaired t test. Analyses of differences between groups for (E) and (G) were performed using two-way analysis of variance (ANOVA) with correction for multiple testing by the Bonferroni method. Comparison of survival rates for (F) and (H) was performed with the log-rank (Mantel-Cox) test.
We next probed the hypothesis that forced expression of CCR8 in T cells would enable their migration toward CCL1, thereby enhancing tumor access. We first inserted this gene in a retroviral vector followed by a 2a-linker peptide fused to GFP and assessed that CCR8 was expressed efficiently upon retroviral transduction into primary T cells (Fig. 2B). There, CCR8 proved functional upon ligand binding, facilitating dose-dependent migration toward recombinant CCL1 (Fig. 2C). We then set up an in vivo experiment, on the Panc02-OVA model used previously, to assess the therapeutical efficacy of CCR8-transduced Teff cells (Fig. 2D). A single transfer of CCR8-transduced OT-I T cells resulted in significantly improved tumor growth control and prolonged survival of tumor-bearing mice compared to GFP-transduced OT-I T cells (Fig. 2, E and F). To test the impact of CCR8-expressing Teff cells on tumors that produce CCL1 themselves, we generated a Panc02-OVA variant overexpressing CCL1. Using the same treatment protocol as before, treatment of tumor-bearing mice with CCR8-transduced OT-I T cells almost completely abolished tumor growth and more markedly prolonged survival compared to control OT-I T cells (Fig. 2, G and H). To evaluate whether the observed phenotype did indeed rely on enhanced recruitment of T cells to the tumor site, we analyzed immune cell infiltrates by flow cytometry. Analysis of the tumor environment after transfer of an equal mix of CCR8-GFP– and mCherry-transduced T cells (Fig. 2I) revealed increased infiltration of CCR8 OT-I T cells compared to OT-I T cells in tumor tissues, which was further enhanced in tumors overexpressing CCL1 (Fig. 2J). Using the same experimental setup, we looked at the antigen-nonspecific infiltration of the ACT in multiple organs. We found CCR8-GFP T cells to preferentially infiltrate the liver, lung, and tumor relative to the spleen (fig. S1, B and C). We then wondered whether, in addition to amplifying the infiltration of tumors, CCR8 expression enhanced their intratumoral motility, which may promote tumor invasion and further increase their antitumor activity. For this purpose, we performed intravital microscopy that recapitulated the enhanced infiltrative capacity of CCR8-transduced T cells (Fig. 2, K and L). However, no significant changes in the motility of CCR8-transduced T cells compared to mCherry-transduced T cells were observed (Fig. 2M). Together, these data point toward enhanced tumor-directed migration, which is further enhanced by continued CCL1 production in the presence of a tumor target, as the mode of action of CCR8 in ACT.
Dominant-negative receptor–transduced T cells retain proliferative capacity despite TGF-β
As CCR8 is typically used by Treg cells for directed migration toward tumor tissues, effector cells recruited to the same site would be at increased risk of Treg suppression. We first quantified Treg cell infiltrates in Panc02 tumors (Fig. 3, A and B). The fraction of Treg cells in the immune cell infiltrate in this tumor model was higher than in lymphoid organs (Fig. 3B) and was essentially entirely composed of CD44hi effector Treg cells (Fig. 3C). Given that TGF-β is one of the main suppressive mechanisms used by Treg cells, and that tumor-infiltrating Treg cells produce this cytokine in the Panc02-OVA tumor model (Fig. 3D), we considered that shielding the transferred T cells from this suppressive cytokine would improve their tumor-ablating function. We also found large amounts of TGF-β in supernatants derived from in vitro cultures of Panc02 cells, further corroborating the immunosuppressive role of this cytokine in this model (Fig. 3E). To counteract the deleterious effects of this cytokine on gene-engineered cells, we used the well-described truncated version of the TGF-β receptor 2 [dominant-negative receptor (DNR); Fig. 3F]. This receptor lacks the intracellular signaling motifs required for TGF-β signal transduction and directly competes with wild-type TGF-β receptor 2 for binding to TGF-β. Binding of TGF-β to DNR-transduced T cells will scavenge TGF-β, preventing its activity on untransduced T cells (22, 23). Upon retroviral transduction, DNR was readily expressed in primary murine T cells (Fig. 3G). DNR efficiently shielded transduced T cells from TGF-β, as TGF-β did not negatively affect the proliferation of transduced T cells in contrast to control T cells (Fig. 3H). We next used DNR-transduced OT-I T cells to treat mice bearing Panc02-OVA tumors with the same treatment schedule as outlined previously (Fig. 3I). DNR-transduced OT-I T cells showed enhanced tumor control and prolonged survival of mice compared to GFP-transduced OT-I T cells (Fig. 3, J and K). In contrast to the chemokine receptor–engineered T cells, as expected, DNR-transduced cells did not show a preferential accumulation at the tumor tissue compared to control OT-I T cells (Fig. 3L). Together, these results indicate that DNR could be a suitable combination partner for CCR8 overexpression in antigen-specific T cells by shielding these cells from immune suppression.
Fig. 3. DNR-transduced T cells retain proliferative capacity despite TGF-β.
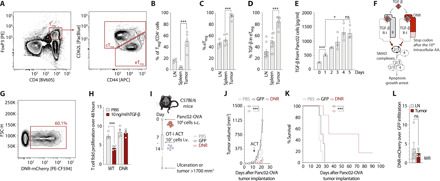
(A) CD45+CD3+CD4+FoxP3+ CD44high CD62L− eTreg and CD44low CD62L+ cTreg Panc02-OVA infiltrating cells (n = 5 mice). PE, Phycoerythrin. (B) Percentage of Treg/CD4+ in (A) (n = 5 mice). Treg cells: lymph nodes 15406 ± 5351, spleen 6992 ± 9702, and tumor 893 ± 244 (mean absolute cells ± SD). (C) Percentage eTreg cells in Treg cells. (D) Percentage TGF-β+ cells in eTreg (n = 10). (E) Murine TGF-β ELISA on 20.000 tumor cells supernatant (n = 3). (F) DNR downstream signaling. AA, amino acid. (G) Transduction efficiency of murine T cells with DNR. (H) T cell proliferation measured through flow cytometry (n = 3). (I) Experimental layout for tumor challenge with Panc02-OVA. (J) Tumor growth curve in mice treated with a single intravenous injection of PBS or 107 GFP-transduced or DNR-transduced OT-I T cells (n = 5). (K) Tumor survival curves of (J). (L) Flow cytometry ACT tracking in Panc02 tumor–bearing mice treated with an equal mix of DNR-mCherry T cells and GFP T cells (n = 3). Experiments show mean values ± SEM and are representative of three independent experiments, except (A) to (C), which was performed for a total of 5 mice, and (D) a total of 10 mice. P values for (B), (C), (D), (E), and (L) are based on two-sided unpaired t test. Analyses of differences between groups for (J) were performed using two-way ANOVA with correction for multiple testing by the Bonferroni method. Comparison of survival rates for (K) was performed with the log-rank (Mantel-Cox) test.
CCR8-DNR–transduced CAR T cells show in vivo efficacy in murine pancreatic solid tumors
Considering the enhanced tumor control we observed by generating conventional Teff cells expressing either ectopic CCR8 or DNR, we wanted to test whether this approach could also be used to improve the function of CAR T cells, and whether their combined expression produced synergistic effects. To this end, we developed polycistronic constructs allowing for coexpression of a CAR targeting the murine epithelial cell adhesion molecule (EpCAM), along with CCR8, DNR, or both in a single plasmid (Fig. 4A). We were able to efficiently introduce these receptor combinations into primary murine T cells (Fig. 4B). CCR8-equipped T cells migrated toward CCL1 gradients in vitro (Fig. 4C). T cells transduced with the DNR were shielded from TGF-β, allowing for TGF-β–unhindered proliferation (Fig. 4D). Last, the CAR conferred EpCAMantigen specificity as shown by interferon-γ (IFN-γ) release (Fig. 4E) and impedance-based killing assays (Fig. 4F) in coculture experiments. In vivo, tracking the infiltration of ACT in an antigen-specific setting revealed that CCR8-CAR T cells were enriched only in the tumor and not in any other of the organs analyzed when compared to CAR T cells (fig. S1D). Next, mice bearing Panc02-EpCAM tumors were treated with CAR, CCR8-CAR, DNR-CAR, or CCR8-DNR-CAR T cells (Fig. 4G). CAR and DNR-CAR T cells only modestly prolonged survival, and CCR8-CAR T cells led to one tumor rejection (10% of mice). CCR8-DNR-CAR T cells outperformed all other groups both in terms of antitumoral activity (with three of seven mice clearing the tumor) and prolongation of survival (Fig. 4, H and I). Together, these results indicate a synergy between CCR8 and DNR in the CAR setting, which warranted testing the applicability of this strategy to enable ACT in human solid tumor models.
Fig. 4. CCR8-DNR–transduced CAR T cells mediate superior in vivo efficacy in pancreatic solid tumors.
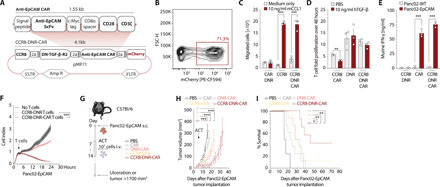
(A) Schematic representation of murine retroviral construct for T cell engineering. 5’ LTR, long terminal repeat; Amp R, Ampicilin resistance. (B) Transduction efficiency of primary murine T cells with CCR8-DNR-CAR. (C) Boyden chamber assay to assess in vitro migration of primary murine T cells to recombinant murine CCL1 measured by flow cytometry. Migrated cells were normalized for CCR8 expression. n = 3. (D) T cell fold expansion over 48 hours with or without TGF-β on the culture media, measured through flow cytometry (representative of n = 3 independent experiments). (E) ELISA for murine IFN-γ was performed on supernatants generated after 24-hour culture of T cells with Panc02-EpCAM tumor cells, in a ratio of 10 effector cells to 1 target cell (representative of n = 3 independent experiments). (F) Panc02-EpCAM tumor cell killing by different T cell conditions, in a 10-fold concentration, measured over time through xCELLigence. Representative of n = 3 independent experiments. (G) Experimental layout for the ACT experiments depicted in (H) and (I). (H) Growth curves of Panc02-OVA-EpCAM tumors in C57Bl/6 mice that were treated with a single intravenous injection of PBS (n = 5 mice) or 107 CAR-transduced, DNR-CAR–transduced, CCR8-CAR–transduced (n = 8 mice) or CCR8-DNR-CAR–transduced T cells (n = 7 mice). (I) Tumor survival curves of a tumor challenge experiment with Panc02-OVA-EpCAM tumors. Mice were treated with a single intravenous injection of PBS (n = 5 mice) or 107 CAR-transduced, DNR-CAR–transduced, CCR8-CAR–transduced (n = 7 mice), or CCR8-DNR-CAR–transduced T cells (n = 8 mice). Experiments show mean values ± SEM and are representative of three independent experiments. P values for (C) to (E) are based on two-sided unpaired t test. Analyses of differences between groups for (F) and (H) were performed using two-way ANOVA with correction for multiple testing by the Bonferroni method. Comparison of survival rates for (I) was performed with the log-rank (Mantel-Cox) test.
CCR8-DNR-CAR T cells are functional in human xenograft tumor models
To provide further rationale for the translational potential of our findings from murine models, we next probed our underlying hypothesis in publicly available databases: If CCR8 is a relevant chemokine receptor for the access of immunosupressive cells (Treg cells) to different cancer tissues, then this should be reflected within these samples. Mining The Cancer Genome Atlas(TCGA) database, we indeed found high expression of the genes CCR8, FOXP3 (expressed in Treg cells), and TGFB1 in pancreatic ductal adenocarcinoma (PDAC) (fig. S2A). Similar associations were found across the diverse disease entities studied such as breast invasive carcinoma, stomach adenocarcinoma, colon adenocarcinoma, rectum adenocarcinoma, skin cutaneous melanoma, lung adenocarcinoma, lung squamous cell carcinoma, and mesothelioma (fig. S2A). Along these lines, CCL1, the main ligand of CCR8, and also the other minor ligands (CCL4, CCL17, and CCL18) were found to be up-regulated in pancreatic cancer compared to healthy tissue (fig. S2B). CCR8 and TGFB1 expression were strongly associated with the Treg cell up-regulated marker, FOXP3, indicating that Treg cells indeed express CCR8 and TGFB1 across these studied tumor entities (fig. S2C).
We scrutinized PDAC patient data and found that high CCR8 expression correlated with more advanced tumor staging and thus a more rapid disease progression (fig. S3A). High CCR8 expression also correlated with a lower TIDE (Tumor immune dysfunction and exclusion) exclusion score and trended with a higher dysfunction score, which corroborates a previous analysis that CCR8 can induce recruitment of immune cells to the tumor microenvironment and is associated with lower effector functions on-site (fig. S3B) (15). This was accompanied by higher lymphocyte infiltration, Treg and TGFB1 scores, and a significantly lower proliferation score (fig. S3C). Together, these data strengthen the rationale that CCR8 is an attractive target molecule for immune cell recruitment to human cancer tissue and that Treg cells are an important source of TGF-β at the tumor site correlating with T cell proliferation arrest.
To translate these findings into a therapeutic application, we engineered primary human T cells with an anti-mesothelin (MSLN) CAR, previously described (24), together with human CCR8, DNR, or both (Fig. 5A). The constructs were successfully transduced into primary human T cells (Fig. 5B), where we observed comparable CAR transduction efficiency and surface expression independent of construct design (fig. S4A). The anti-MSLN CAR allowed effective recognition and elimination of MSLN antigen–positive cells, as assessed by impedance measurements of cell killing and by IFN-γ release (Fig. 5, C and D). As seen in the murine cells, activated human T cells produced CCL1 upon coculture with tumor cells, the amount of which correlated with the amount of IFN-γ released (Fig. 5E). DNR, in turn, improved proliferation of transduced T cells in the presence of TGF-β compared to control-transduced T cells (Fig. 5F). DNR functionality was further validated via phosphoflow, revealing no phosphorylation of SMAD2/3 downstream of TGF-β when DNR was expressed by T cells (fig. S4B). CCR8 enabled migration toward human CCL1 gradients compared to control-transduced T cells (Fig. 5G).
Fig. 5. CCR8-DNR-CAR T cells are effective in human tumor models.
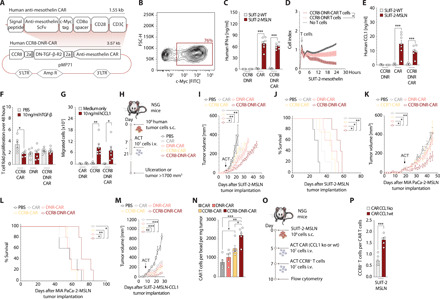
(A) Schematic representation of construct. (B) Transduction efficiency of human T cells with CCR8-DNR-CAR. (C) Human IFN-γ ELISA on 24-hour coculture supernatants. (D) SUIT-2-MSLN killing measured by xCELLigence. (E) Human CCL1 ELISA on 24-hour coculture supernatants. (C to E) 10:1 effector to target cells. (F) Forty-eight–hour T cell expansion, flow cytometry. (G) In vitro migration of T cells to CCL1, flow cytometry. (H) Experimental layout for (I) to (M). Tumor growth and survival curves of SUIT-2-MSLN (I and J), MIA PaCa-2-MSLN (K and L), and SUIT-2-MSLN-CCL1 (M) treated with a single intravenous injection of PBS or 107 CAR-, DNR-CAR–, CCR8-CAR–, or CCR8-DNR-CAR–transduced T cells (n = 5 mice per group). (N) Flow cytometry quantification of CAR T cells per bead per milligram on day 27 after tumor implantation (n = 5 mice). (O) Experimental layout for (P). (P) SUIT-2-MSLN tumor–bearing mice were administered with 107 CAR T cells that were either wild-type (wt) or CCL1 knockout (ko). This was followed by a treatment with 107 CCR8-transduced T cells (n = 7 mice). Experiments show mean values ± SEM of n = 7 healthy donors for (B) to (G), one experiment for (I) to (L) and (P), and two independent experiments for (M) and (N). P values for (C), (E), (F), (G), (N), and (P) are based on a two-sided unpaired t test. (D), (I), (K), and (M) were assessed through two-way ANOVA with correction for multiple testing by the Bonferroni method. (J and L) Survival rate comparison through the log-rank (Mantel-Cox) test.
We next sought to provide a more in-depth characterization of the phenotype and function of CCR8-DNR-CAR–modified T cells over the single- or double-modified T cells. We could observe that in vitro over a prolonged period with repeat antigen exposures, DNR-CAR and CCR8-DNR-CAR T cell conditions (for short, DNR conditions) had an increased and more durable proliferative capacity (fig. S4C). DNR conditions did not alter CD4 and CD8 ratios (fig. S4D) nor significantly change the effector or memory phenotype compared to CCR8-CAR or CAR conditions (fig. S4E). DNR conditions did not alter PD-1, TIM-3 (T-cell immunoglobulin and mucin-domain containing-3), or LAG-3 (Lymphocyte-activation gene 3) levels (fig. S4F). The impact of CCR8 and DNR expression on the secretion profile of these CAR T cells revealed that the DNR conditions show a modest increase in T helper 2 (TH2) cytokines [such as interleukin-4 (IL-4) and IL-10] and TH1 cytokines (such as IL-2 and granzyme B) (fig. S4G). DNR conditions also showed a more robust secretion of the chemokines probed (including CCL1, CCL3, and CCL4) (fig. S4G). As expected, TGF-β levels measured within this coculture gradually rose as T cells decreased in their proliferative capacity (fig. S4G).
To investigate whether T cell–derived CCL1 would indeed be necessary and sufficient to recruit CCR8+ T cells and mediate therapeutic benefits, we went on to assess the impact in CCL1-negative tumor cell models. We tested CCR8-DNR-CAR T cells in human pancreatic xenograft tumor models where SUIT-2-MSLN and MIA PaCa-2-MSLN tumors were implanted in nonobese diabetic (NOD)–scid (Severe Combined Immunodeficiency)–IL2rγnull (NSG) mice (Fig. 5H). CCR8-DNR-CAR T cells controlled tumor growth and prolonged survival in both human pancreatic tumor models, demonstrating the necessity of both CCR8 and DNR transduction into the CAR T cells for effective tumor control in solid tumors (Fig. 5, I to L). Together, these data indicate that T cell–derived CCL1 is sufficient to mediate CCR8-associated therapeutic benefit.
Given that CCL1 expression is elevated in patient cohorts of specific disease subtypes, including pancreatic cancer, we further engineered the abovementioned SUIT-2-MSLN pancreatic cancer xenograft model to overexpress CCL1. SUIT-2-MSLN-CCL1 tumors were implanted in NSG mice. We tested CCR8-DNR-CAR T cells in this model, where they outperformed single- or double-transduced T cells in controlling tumor growth (Fig. 5M).
Next, to test whether therapeutic advantage was indeed paralleled by increased T cell access to the tumor site, we analyzed infiltration into the implanted tumors following T cell transfer. At the probed time point, there was an enhanced accumulation of transferred T cells in the tumor, showing increased CCR8-DNR-CAR T cell numbers compared to all other groups (Fig. 5N). At the tumor site, we found a preferential accumulation of CD8+ T cells compared to the spleen where most of the cells were CD4+ T cells with a skew toward a central memory phenotype (fig. S4, H and I).
Last, to test whether increased T cell access was indeed due to CCL1 production from the transferred T cells, we used the SUIT-2-MSLN tumor model and a two-step ACT starting with CAR T cells (wild-type or CCL1 knockout) followed by CCR8-transduced T cells (Fig. 5O). CCL1 knockout using CRISPR-Cas9 editing on primary T cells was technically validated by measuring CCL1 levels 24 hours after T cell stimulation (fig. S4J). We show that tumor infiltration of CCR8-transduced T cells was mostly abolished when CAR T cells had lost the ability to produce CCL1 (Fig. 5P), highlighting that CCL1 produced by activated T cells is necessary to generate a robust positive feedback loop recruitment of CCR8+ T cells.
DISCUSSION
T cell therapies have shown remarkable results in hematological cancer (25), but this success has yet to be successfully translated to solid malignancies. Appropriate trafficking to the tumor site and resistance to the tumor microenvironment–induced suppression are major hurdles to the employment of T cell therapies in solid tumors (14). Improved trafficking of T cells through chemokine receptors has been shown to be a reliable approach in tumor models (26). At the same time, CAR T cell therapy has entered the clinical realm for selected hematological malignancies, with newer generations of CAR T cells in clinical trials. Nevertheless, only two clinical trials are using chemokine receptors, CXCR2 (NCT01740557) and CCR4 (NCT03602157), to improve trafficking of T cells to the tumor site, with results pending.
The plethora of chemokine receptors and their respective ligands, as well as their promiscuity, means that sophisticated screening of targets and careful patient selection are required. Chemokines expressed in tumors cells and in tumor stroma cells can be subject to posttranslational changes that modify their function (27). In addition, atypical chemokine receptors can scavenge secreted ligands rendering them unavailable to trigger migration of Teff cells (28). Furthermore, the chemokine receptor-ligand choice for engineering CAR T cells should not only aim at improving migration into the given tumor but also target critical axes for the tumor’s immune evasion. Here, we reason that the CCR8-CCL1 axis fulfills these criteria, as it has been demonstrated to be a pivotal receptor-ligand pair for recruiting Treg cells to the tumor and for potentiating their immunosuppressive capacity across different cancer entities, as well as being associated with poor disease prognosis (15, 16). Down-regulation or loss of an immunosuppression-enabling axis appears highly unlikely to occur in the tumor environment.
Our TCGA analysis confirms both the correlation of CCR8 and FOXP3 and the correlation of Treg cells with poor prognosis and further expands it to several solid tumors so far impervious to CAR T cell therapy. CCR8+ Treg cells have been proposed to be master drivers of immune regulation as specific binding of CCL1 to CCR8 increases the suppressive capacity of Treg cells. The CCL1-CCR8 axis is, however, incapable of inducing a switch from FOXP3− to FOXP3+ (16). Furthermore, the low endogenous CCR8 expression in Teff cells increases the likelihood of an improved migration upon overexpression.
Transduction of different chemokine receptors—namely, CXCR2 (29), CCR4 (26), CCR2 (30), and CX3CR1 (31)—has been used in the past to improve ACTs for solid tumor entities (26, 29). Out of these, CCR4 is the only other chemokine receptor expressed in Treg cells, thus making it an approach that is directly comparable to the strategy in the present study. CCR4 transduction improved CAR T cell efficacy in a model of Hodgkin lymphoma, which was dependent on the availability of CCL22 (26). CCR4 and CCR8 ACT highjack two distinct axes—CCL22 and CCL1, respectively—that can enable Treg cell trafficking to solid tumors. The CCL1-CCR8 therapeutic axis is more specific to tumors and can directly counterbalance Treg cell infiltration in solid tumors, making it a superior choice for chemokine receptor enhanced ACT in solid tumors when compared to the CCL22-CCR4 axis. Considering the tumor specificity and potential safety, our TCGA analysis on expression levels of CCL1 and CCL22 reveals that CCL22 is broadly expressed in different healthy tissues. This may lead to redirection of CCR4+ CAR T cells into off-target CCL22+ tissue, on the one hand diminishing the antitumor effect and on the other increasing the risk for off-tumor toxicity. On the contrary, CCL1 expression is lower and more restricted in healthy tissue, which can enable a more specific homing of CCR8-transduced CAR T cells to the tumor site.
Besides the need for improved tumor infiltration, there is also a fundamental need for CAR T cell activity and proliferation at the tumor site. TGF-β is a hallmark of the immunosuppressive tumor milieu in pancreatic and other solid tumors (32). It can control T cell homeostasis by inhibiting both T cell proliferation and activation (33). In addition, it can inhibit Teff cell activity through an anticytotoxic program of transcriptional repression, down-regulating the expression of perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ (34). The DNR is a well-characterized receptor in which binding by TGF-β will not trigger downstream signaling in the cell. Moreover, TGF-β scavenging will prevent its detrimental effects on other Teff cells. Such a receptor harbors multiple advantages when compared to anti–TGF-β antibody therapy as the effect with DNR is limited to the proximity of the DNR-expressing cell. It will also be specific for the tumor tissue expressing the CAR-targeted antigen. Thus, endogenous T cells with different TCR specificities will remain unaffected, reducing the risk of self-reactivity (23). In addition to TGF-β, other Treg cell–derived cytokines such as IL-10 can be found in abundance in the tumor microenvironment. IL-10 has been described to impair dendritic cell functionality and hamper Teff cell cytotoxicity. This cytokine may also play a role in promoting tumor rejection instead of inducing immunosuppression (35). Genome editing technologies such as CRISPR-Cas9 are an equally valid alternative approach for relieving T cell immunosuppression. However, the knowledge on safety and efficacy of these approaches is still limited, and more evidence will be needed for a comprehensive assessment. The advanced stage of testing, including use in clinical trials, as well as knowledge of the safety profile of current DNR-based therapies, makes it an attractive tool for the relief of T cell immunosuppression (23).
Similar to human breast tumors (15), we were able to show that FOXP3, CCR8 (as well as its ligands), and the TGFB1 genes are up-regulated in human pancreatic adenocarcinoma compared to healthy pancreatic tissue and to verify that there are strong correlations between the expression of these genes. This highlights the potential relevance of the CCL1-CCR8 axis and of TGF-β expression in this disease and provides the rationale for targeting them in pancreatic cancer and in other tumor entities with comparable expression profiles.
Despite the improvement that targeting this axis had on the efficacy of CAR T cell therapy, some limitations must be considered. Beyond improving migration to the tumor site and relieving immunosuppression on T cells, CAR T cell therapy still stands to benefit from continued improvement in the design (8), precise integration (36), and regulation of the CAR construct itself (37), as outlined by the recent studies. Furthermore, antigen-specificity is still a major hurdle and a pivotal requirement to ensure the feasibility of any tumor-targeted T cell therapy (38). Similar studies using the same murine Panc02 tumor cell line were able to show T cell memory formation (39); nonetheless, this analysis was not performed in the present study. Targeting the model antigen EpCAM in the murine tumor models of the present study did not reveal any signs of toxicity. However, C57Bl/6 mice are typically highly resistant to immune-mediated side effects, which may only occur with added modifications to the treatment schedule (40). Thus, toxicity associated with CCR8- and DNR-transduced CAR T cells could not be comprehensively assessed in this model. Furthermore, the herein presented human CCL1 tissue expression data, while suggestive of a safer T cell infiltration profile, cannot fully rule out potential side effects when treating patients. Nonetheless, the addition of chemokine receptors or DNR to T cells has not been associated with acute toxicity events, such as cytokine release syndrome and tumor lysis syndromes, which have been observed for CAR T cell therapy (41). Autoimmunity has been described as a long-term deleterious effect of T cell TGF-β deprivation for DNR-based T cell therapy (42), and transgenic mice expressing DNR have been reported to develop lymphoproliferative disorder (43). Currently, a clinical trial is assessing the safety and efficacy of DNR transduction in Epstein-Barr virus–specific T cells for lymphoma (NCT00368082) (23). A recent preclinical study investigating the potential of expressing DNR in CAR T cells similarly showed superior proliferation and antitumoral function in prostate cancer (18), with a clinical trial assessing its safety and feasibility currently underway (NCT03089203).
From the long-term in vitro coculture assays, we could better understand the effect the DNR has on the cytokine profile of T cells. We saw increased levels in a variety of cytokines associated with both the innate and adaptive immune response when the DNR was expressed. TH1- and TH2-associated cytokine levels were both elevated and more balanced, while chemokine levels were also increased. Overall, the expression of the DNR appears to confer Teff cells with greater functionality, and the expression of CCR8 does not seem to affect the phenotype of Teff cells.
The treatment experiments in human xenograft tumor models revealed that only in the presence of DNR could CCR8-CAR T cells mediate significant tumor control. This supports the notion that a feedback loop mechanism sustained by CCL1 from activated T cells stands to benefit from immunosuppression shielding. Beyond TGF-β, we postulate that relieving other immunosuppressive axes can further sustain this CCL1 feedback loop mechanism, thereby improving the antitumoral function of CCR8+ ACT. In the model using CCL1 overexpression, the treatment effect was more pronounced as continued recruitment of CCR8-DNR-CAR T cells no longer solely relied on T cell–derived CCL1 to effectively migrate into the tumor, given the tumor itself was overexpressing the chemokine ligand. A better understanding of other suppressive axes that could be additionally targeted could potentiate the effectiveness of this approach. Furthermore, anti–PD-1 checkpoint blockade therapy was recently shown to improve the proliferation and suppressive capacity of PD-1+ Treg cells (44). Added to the fact that CCL1 levels will rise in the tumor after checkpoint blockade therapy–induced T cell activation, it is reasonable to postulate that simultaneously targeting the CCR8-CCL1 axis might also prove beneficial.
Hence, given appropriate antigen targeting, we propose that CCR8- and DNR-transduced CAR T cells can exploit two critical biological axes to render CAR T cell therapy effective in solid tumors such as pancreatic cancer. The therapeutic potential of this approach could extend to other Treg-rich solid tumor entities where limited infiltration into the tumor and intratumoral T cell proliferation prevent therapeutic success. The novel combination described in the present study may harness the power of CAR T cell therapy for solid tumors with as dismal a prognosis as pancreatic cancer.
MATERIALS AND METHODS
Study design
This study was designed to evaluate the benefit of the addition of CCR8 and DNR to CAR T cell therapy in solid tumors using controlled laboratory experiments. We used 6- to 10-week-old female mice from OT-I, C57Bl/6, or NSG strains as donors or recipients of matching appropriate tumor cell lines as described for each experiment. Mice have been implanted with 106 tumor cells. Mice were randomized and the investigator was blinded when tumors reached a volume of at least 50 mm3. No time points or mice were excluded from the experiments presented in the study. For ethical reasons, endpoints of survival studies were defined as tumor ulceration, tumor sizes exceeding 15 mm in any dimension, weight loss above 20%, or clinical signs of distress. These endpoints were used as surrogate endpoints for survival and are used as such throughout the study. Peripheral blood mononuclear cells were collected from healthy individuals. The murine tumor cell lines used for this study were Panc02, Panc02-OVA, Panc02-OVA-EpCAM, Panc02-H2B-Cerulean, or Panc-02-OVA-CCL1 as described in the respective figures. The human cell lines used for this study were SUIT-2-WT, SUIT-2-MSLN, SUIT-2-MSLN-CCL1, or MIA PaCa-2-MSLN as described. Effects have been assessed through quantitative PCR, enzyme-linked immunosorbent assay (ELISA), tumor measurement, flow cytometry, or multiphoton intravital microscopy as described. No outliers have been excluded from any of the experiments. At least three biological replicates were performed for each experiment and n > 5 for each group unless otherwise indicated in the figure legends. One-way analysis of variance (ANOVA) with Tukey’s posttest, two-way ANOVA with Bonferroni posttest, or unpaired Student’s t tests were performed with an α value of <0.05 to detect differences between group means.
Mice
C57BL/6RJ and NSG (NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ) mice were purchased from Charles River Laboratories. OT-I mice were bred at the animal facilities at the Klinikum der Universität München or Massachusetts General Hospital (MGH). Animals were housed in specific pathogen–free facilities. All experimental studies were approved and performed in accordance with guidelines and regulations implemented by the Regierung von Oberbayern and the MGH Institutional Animal Care and Use Committee. All experiments were carried out randomized and performed blinded and with adequate controls. In accordance with the animal experiment application, tumor growth and health status of mice were checked every other day. For survival analyses, the above-defined criteria were taken as surrogates for survival and recorded in Kaplan-Meier plots.
Tumor growth studies and treatments
All tumor cell lines were subcutaneously injected in 100 μl of phosphate-buffered saline (PBS) into the flanks of mice. Animals were randomized into treatment groups according to tumor volumes. Tumor volumes were measured before and every other day after treatment was started and calculated as V = (length × width2)/2. For ACT studies, 107 T cells were injected intravenously in 100 μl of PBS once average group tumor volumes had reached at least 50 mm3.
Cell line generation, culture, and validation
Murine Panc02, Panc02-OVA, and Panc02-EpCAM (11) have been previously described. The human SUIT-2, SUIT-2-MSLN, and MIA PaCa-2-MSLN tumor cell lines have been previously described (11). The Panc02-OVA cell line has been modified with retroviruses to express the murine CCL1 chemokine (UniProt entry P10146) (Panc02-CCL1) or a blue fluorescent protein tagged to a Histone protein (Panc02-H2B-Cerulean). The SUIT-2-MSLN tumor cell line has been modified with retroviruses to express the human CCL1 chemokine (UniProt entry P22362) (SUIT-2-MSLN-CCL1). All tumor lines were grown as previously described (11) and used for experiments when in exponential growth phase. 293Vec-Galv, 293Vec-Eco, and 293Vec-RD114 were a gift of M. Caruso, Québec, Canada and have been previously described (45). For virus production, retroviral pMP71 (provided by C. Baum, Hannover) vectors carrying the sequence of the relevant receptor were stably introduced in packaging cell lines. Single-cell clones were generated and indirectly screened for the highest level of virus production by determining transduction efficiency of primary T cells. This method was used to generate the producer cell lines 293Vec-RD114 for GFP, mCherry, CCR8, -DNR, CAR-MSLN, CCR8-CAR-MSLN, DNR-CAR-MSLN, and CCR8-DNR-CAR-MSLN or 293Vec-Eco for GFP, mCherry, H2B-Cerulean, CCR8, DNR, CCR8-DNR, CAR-EpCAM, CCR8-CAR-EpCAM, DNR-CAR-EpCAM, CCR8-DNR-CAR-EpCAM. 293Vec-Galv, 293Vec-Eco, and 293Vec-RD114 were grown as previously described (45). Primary murine and human T cells were cultured according to previously described protocols (46). All cell lines used in experiments were regularly checked for mycoplasma species with the commercial testing kit MycoAlert (Lonza). Authentication of human cell lines by STR DNA profiling analysis was conducted in-house.
T cell generation, retroviral transduction, CRISPR, and culture
Murine T cells have been differentiated from splenocytes from donor mice. T cell isolation and transduction have been previously described (47) and then expanded or directly expanded with T cell medium supplemented with human IL-15 (PeproTech) every second day. Human T cells have been differentiated and transduced using previously described protocols (39) or directly taken into culture with human T cell medium in concentrations of 106 T cells per milliliter medium. CCL1 knockout human T cells were generated using a two-component guide RNA (gRNA) CRISPR method as previously described (48) after magnetic removal of DynaBeads, using the EH-115 pulse code on a 4D-Nucleofector (Lonza). gRNAs [crRNA (Trans-activating CRISPR RNA) CCL1 AA: TGTAACACAGGATTGCCCTCAGG; and crRNA CCL1 AF: CGGAGCAAGAGATTCCCCTGAGG) were selected from CHOPCHOPv3 (49), synthesized by IDT (Integrated DNA technologies), and used simultaneously for human CCL1 knockout. T cells were used in experiments 5 days after genetic modifications.
Migration assays
Cell migration was evaluated using Transwell plates (Corning) as previously described (39). A total of 5 × 105 T cells were placed on a 3-μm pore membrane in the upper chamber of a Transwell plate with the lower chamber containing different concentrations of recombinant murine or human CCL1 (BioLegend). After 3 hours of incubation at 37°C, the migrated cells in the lower chamber were analyzed by flow cytometry.
Cytotoxicity assays
For impedance-based real-time killing assays using an xCELLigence system (ACEA Biosciences, USA), previously described (11), 104 tumor cells were seeded per well in a 96-well plate. The cell number was monitored over the time frame of 10 hours for every 20 min. A total of 105 T cells transduced with the indicated receptors were added to the tumor cells.
Cytokine protein level quantification
Murine and human IFN-γ were quantified using ELISA sets from BD Biosciences (555138 and 555142, respectively). Murine and human CCL1 were quantified using DuoSet ELISA from R&D Systems (DY845 and DY272, respectively). Murine TGF-β was quantified using a DuoSet ELISA from R&D Systems (DY1679). Other cytokine quantifications are further described in Supplementary Methods.
Proliferation assays
Proliferation was measured using a flow cytometry–based assay that compared fold proliferation of T cells over a period of 48 hours normalized to the number of T cell per condition upon assay start. Recombinant human TGF-β (Cell Signaling Technology) or vehicle solution was added to concentrations of 20 ng/ml to test for proliferation arrest of T cells cultured with murine T cell medium supplemented with IL-15.
Preparation of single-cell suspensions, antibody staining, and flow cytometry
LNs (lymph nodes) and spleens were passed through 30-μm cell strainers, followed by erythrocyte lysis in the spleens. Tumors were digested with collagenase IV (1.5 mg/ml) and deoxyribonuclease I (50 U/ml) for 30 min at 37°C under agitation. Dead cells were stained using the fixable viability violet dye Zombie Red or Violet (BioLegend) for 15 min at room temperature, followed by blocking of Fc receptors with TruStain FcX (BioLegend) for 20 min at 4°C. Following this, cell surface proteins were stained for 20 min at 4°C with anti-CCR8 (SA214G2), anti-CD4 (GK1.5), anti-CD45.1 (A20), anti-CD45.2 (104) anti-CD8α (53-6.7), anti-CD90.1 (OX-7), anti-CD90.2 (30-H12), CD62L (MEL-14), and CD44 (IM7) (all from BioLegend) or anti–c-myc (SH1-26E7.1.6, Miltenyi Biotec) for detection of CAR constructs. Nuclear proteins were stained for 60 min at room temperature after permeabilization and fixation (Mouse Regulatory T cell Staining Kit, eBioscience) using Foxp3 (MF-14, BioLegend) and TGF-β (TW7-16B4, BioLegend). Antibodies used for flow cytometry of human samples are further detailed in Supplementary Methods. Cells were analyzed on a Canto or LSRFortessa flow cytometer (BD Biosciences), and data were analyzed with FlowJo software version 9.9.5 or version 10.3.
PCR and quantitative RT-PCR
All DNA constructs were generated by overlap extension PCR (50) and recombinant expression cloning into the retroviral pMP71 vector (51) using standard molecular cloning protocols (50). RNA was extracted from cells using the InviTrap Spin Universal RNA Extraction Kit (Stratec). cDNA was synthesized using the Superscript II kit (Life Technologies). PCR primers for real-time PCR were designed automatically from the National Center for Biotechnology Information GenBank sequences in the assay design center from the Roche Universal ProbeLibrary and have been previously published (39). Real-time PCR was performed using a Kapa Probe Universal MasterMix (VWR) in a LightCycler 480 instrument (Roche Diagnostics).
Immunofluorescence
Tissue samples obtained from tumors were embedded and frozen in OCT (Optimal cutting temperature compound). Sections of 5 μm were stained with a primary antibody for goat anti-CCL1 (R&D Systems) and an Alexa Fluor 488 (Life Technologies) secondary antibody and DAPI (4′,6-diamidino-2-phenylindole) (VECTASHIELD) according to previously described standard procedures (52).
Multiphoton intravital microscopy
A total of 5 × 106 CCR8-GFP–transduced T cells were coinjected with 5 × 106 mCherry-transduced T cells. Tumor cells that expressed the H2B-Cerulean fluorescent protein were implanted in the back of mice after removal of hair. Five days later, a dorsal skinfold chamber was implanted around engrafted tumors through an aseptic surgical procedure under general isoflurane inhalation anesthesia and buprenorphine analgesia, as previously described (53). Mice were monitored daily for tumor growth as well as for pain and local or systemic inflammatory signs. Imaging took take place every other day under isoflurane anesthesia, and Qtracker 655 nontargeted quantum dots (Invitrogen) were injected intravenously to visualize blood vessels. Multiphoton excitation was achieved with a MaiTai Ti:sapphire laser (Spectra-Physics) tuned to 950 nm to excite all fluorescent probes used. Stacks of 18 to 24 squares optical sections with 500-μm width and 4- to 5-μm z spacing were acquired on an Ultima In Vivo multiphoton microscope (Bruker) every 60 s. Emitted fluorescence was detected through 460/50, 525/50, 595/50, and 660/40 band-pass filters and nondescanned detectors to generate four-color images. Image processing and quantification of cell motility were performed with Imaris software (Bitplane).
Statistical analysis
Two-tailed Student’s t test was used for comparisons between two groups, while two-way ANOVA with Bonferroni posttest (multiple time points) or one-way ANOVA with Tukey’s posttest (single time points) was used for comparisons across multiple groups. A log-rank (Mantel-Cox) test was used to compare survival curves. All statistical tests were performed with GraphPad Prism 8 software, and P < 0.05 was considered statistically significant and represented as *P < 0.05, **P < 0.01, and ***P < 0.001. No statistical methods were used to predetermine sample size. Investigators were blinded to allocation during experiments and outcome assessment.
Acknowledgments
We acknowledge the reviewers of this manuscript for critical input that strengthened the data herein presented. The results published here are in part based on data generated by the TCGA Research Network: http://cancergenome.nih.gov/. We acknowledge Life Science Editors for editing assistance. We acknowledge the iFlow Core Facility of the LMU University Hospital for assistance with the generation of flow cytometry data. Funding: This study was supported by the International Doctoral Program i-Target: Immunotargeting of Cancer funded by the Elite Network of Bavaria (to S.K. and S.E.), by the Melanoma Research Alliance Grants 409510 (to S.K.), by the Marie-Sklodowska-Curie Program Training Network for the Immunotherapy of Cancer funded by the H2020 Program of the European Union (grant 641549 to S.E. and S.K.), by the Marie-Sklodowska-Curie Program Training Network for Optimizing Adoptive T Cell Therapy of Cancer funded by the H2020 Program of the European Union (grant 955575 to S.K.), by the Hector foundation (to S.K.), by the Else Kröner-Fresenius-Stiftung (to S.K.), by the German Cancer Aid (to S.K.), by the Ernst-Jung-Stiftung (to S.K.), by the LMU Munich’s Institutional Strategy LMUexcellent within the framework of the German Excellence Initiative (to S.E. and S.K.), by the Bundesministerium für Bildung und Forschung Project Oncoattract (to S.E. and S.K.), by the European Research Council Grant 756017, ARMOR-T (to S.K.), by the German Research Foundation (DFG to S.K.), by the NIH grant AI123349 (to T.R.M.), by the Deutsche Gesellschaft für Immun- und Targeted-therapie (to B.L.C.), by the Fritz-Bender Foundation (to S.K.), by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 866411) (to C.M.), by the Volkswagen Foundation (project OntoTime) (to M.T.), and the José-Carreras Foundation (to S.K.). Author contributions: B.L.C. and M.-R.B. designed and performed experiments, analyzed the data, and wrote the manuscript; K.D., A.O., T.L., H.O., M.V., M.D.P., J.N.P., S.S., D.H., F.M., M.S., K.M., J.S.-G., and Y.Z. performed experiments and analyzed the data; S.L., C.H.K, C.H., A.G., M.T., C.M., J.Z., D.P., T.F., and M.S. provided technical and theoretical assistance; T.R.M. and S.E. supervised the project and designed experiments; S.K. conceived the study, supervised the project, designed experiments, and wrote the manuscript. Competing interests: Parts of this work have been performed for the thesis of B.L.C., K.D., and Y.Z. B.L.C., S.E., and S.K. are inventors on a patent application filed by Klinikum der Universität München (no. WO 2020/182681 A1, filed 6 March 2020, published 17 September 2020). S.E. and S.K. have received research support from TCR2 Inc. and Arcus Biosciences for work unrelated to the present manuscript. S.K. has received honoraria from Novartis and TCR2 Inc. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabi5781/DC1
REFERENCES AND NOTES
- 1.Kalos M., Levine B. L., Porter D. L., Katz S., Grupp S. A., Bagg A., June C. H., T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian S. L., Hodi F. S., Brahmer J. R., Gettinger S. N., Smith D. C., McDermott D. F., Powderly J. D., Carvajal R. D., Sosman J. A., Atkins M. B., Leming P. D., Spigel D. R., Antonia S. J., Horn L., Drake C. G., Pardoll D. M., Chen L., Sharfman W. H., Anders R. A., Taube J. M., McMiller T. L., Xu H., Korman A. J., Jure-Kunkel M., Agrawal S., McDonald D., Kollia G. D., Gupta A., Wigginton J. M., Sznol M., Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roseberg S. A., Restifo N. P., Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benmebarek M.-R., Karches H. C., Cadilha L. B., Lesch S., Endres S., Kobold S., Killing mechanisms of chimeric antigen receptor (CAR) T cells. Int. J. Mol. Sci. 20, 1283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park J. H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K. J., Sauter C., Wang Y., Santomasso B., Mead E., Roshal M., Maslak P., Davila M., Brentjens R. J., Sadelain M., Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu S. S., Locke F. L., Bartlett N. L., Lekakis L. J., Miklos D. B., Jacobson C. A., Braunschweig I., Oluwole O. O., Siddiqi T., Lin Y., Timmerman J. M., Stiff P. J., Friedberg J. W., Flinn I. W., Goy A., Hill B. T., Smith M. R., Deol A., Farooq U., McSweeney P., Munoz J., Avivi I., Castro J. E., Westin J. R., Chavez J. C., Ghobadi A., Komanduri K. V., Levy R., Jacobsen E. D., Witzig T. E., Reagan P., Bot A., Rossi J., Navale L., Jiang Y., Aycock J., Elias M., Chang D., Wiezorek J., Go W. Y., Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah N. N., Fry T. J., Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 16, 372–385 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoiber S., Cadilha L. B., Benmebarek M.-R., Lesch S., Endres S., Kobold S., Limitations in the design of chimeric antigen receptors for cancer therapy. Cell 8, 472 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sullivan P. M., Arguedas-Jimenez L., Johnson A., Yokoyama J., Yuzefpolskiy Y., Jensen M., Kalia V., Sarkar S., A novel preclinical immunocompetent CAR T cell mouse model for solid tumors. J. Immunol. 202, 71.16 (2019). [Google Scholar]
- 10.Zitvogel L., Pitt J. M., Daillère R., Smyth M. J., Kroemer G., Mouse models in oncoimmunology. Nat. Rev. Cancer 16, 759–773 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Karches C. H., Benmebarek M.-R., Schmidbauer M. L., Kurzay M., Klaus R., Geiger M., Rataj F., Cadilha B. L., Lesch S., Heise C., Murr R., vom Berg J., Jastroch M., Lamp D., Ding J., Duewell P., Niederfellner G., Sustmann C., Endres S., Klein C., Kobold S., Bispecific antibodies enable synthetic agonistic receptor-transduced T cells for tumor immunotherapy. Clin. Cancer Res. 25, 5890–5900 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avanzi M. P., Yeku O., Li X., Wijewarnasuriya D. P., van Leeuwen D. G., Cheung K., Park H., Purdon T. J., Daniyan A. F., Spitzer M. H., Brentjens R. J., Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 23, 2130–2141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Viaud S., Ma J. S. Y., Hardy I. R., Hampton E. N., Benish B., Sherwood L., Nunez V., Ackerman C. J., Khialeeva E., Weglarz M., Lee S. C., Woods A. K., Young T. S., Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. U.S.A. 115, E10898–E10906 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim W. A., June C. H., The principles of engineering immune cells to treat cancer. Cell 168, 724–740 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plitas G., Konopacki C., Wu K., Bos P. D., Morrow M., Putintseva E. V., Chudakov D. M., Rudensky A. Y., Regulatory T cells exhibit distinct features in human breast cancer. Immunity 45, 1122–1134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barsheshet Y., Wildbaum G., Levy E., Vitenshtein A., Akinseye C., Griggs J., Lira S. A., Karin N., CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. U.S.A. 114, 6086–6091 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wieser R., Attisano L., Wrana J. L., Massagué J., Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol. Cell. Biol. 13, 7239–7247 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kloss C. C., Lee J., Zhang A., Chen F., Melenhorst J. J., Lacey S. F., Maus M. V., Fraietta J. A., Zhao Y., June C. H., Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 26, 1855–1866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller M. D., Hata S., De Waal Malefyt R., Krangel M. S., A novel polypeptide secreted by activated human T lymphocytes. J. Immunol. 143, 2907–2916 (1989). [PubMed] [Google Scholar]
- 20.Miller M. D., Krangel M. S., The human cytokine I-309 is a monocyte chemoattractant. Proc. Natl. Acad. Sci. U.S.A. 89, 2950–2954 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curiel T. J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J. R., Zhang L., Burow M., Zhu Y., Wei S., Kryczek I., Daniel B., Gordon A., Myers L., Lackner A., Disis M. L., Knutson K. L., Chen L., Zou W., Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Brand T., MacLellan W. R., Schneider M. D., A dominant-negative receptor for type beta transforming growth factors created by deletion of the kinase domain. J. Biol. Chem. 268, 11500–11503 (1993). [PubMed] [Google Scholar]
- 23.Bollard C. M., Tripic T., Cruz C. R., Dotti G., Gottschalk S., Torrano V., Dakhova O., Carrum G., Ramos C. A., Liu H., Wu M. F., Marcogliese A. N., Barese C., Zu Y., Lee D. Y., O’Connor O., Gee A. P., Brenner M. K., Heslop H. E., Rooney C. M., Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J. Clin. Oncol. 36, 1128–1139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adusumilli P. S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D. R., Sadelain M., Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brentjens R. J., Davila M. L., Riviere I., Park J., Wang X., Cowell L. G., Bartido S., Stefanski J., Taylor C., Olszewska M., Borquez-Ojeda O., Qu J., Wasielewska T., He Q., Bernal Y., Rijo I. V., Hedvat C., Kobos R., Curran K., Steinherz P., Jurcic J., Rosenblat T., Maslak P., Frattini M., Sadelain M., CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Stasi A., De Angelis B., Rooney C. M., Zhang L., Mahendravada A., Foster A. E., Heslop H. E., Brenner M. K., Dotti G., Savoldo B., T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113, 6392–6402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molon B., Ugel S., Del Pozzo F., Soldani C., Zilio S., Avella D., De Palma A., Mauri P., Monegal A., Rescigno M., Savino B., Colombo P., Jonjic N., Pecanic S., Lazzarato L., Fruttero R., Gasco A., Bronte V., Viola A., Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 208, 1949–1962 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D’Amico G., Frascaroli G., Bianchi G., Transidico P., Doni A., Vecchi A., Sozzani S., Allavena P., Mantovani A., Uncoupling of inflammatory chemokine receptors by IL-10: Generation of functional decoys. Nat. Immunol. 1, 387–391 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Kershaw M. H., Wang G., Westwood J. A., Pachynski R. K., Tiffany H. L., Marincola F. M., Wang E., Young H. A., Murphy P. M., Hwu P., Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 13, 1971–1980 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Craddock J. A., Lu A., Bear A., Pule M., Brenner M. K., Rooney C. M., Foster A. E., Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 33, 780–788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siddiqui I., Erreni M., van Brakel M., Debets R., Allavena P., Enhanced recruitment of genetically modified CX3CR1-positive human T cells into Fractalkine/CX3CL1 expressing tumors: Importance of the chemokine gradient. J. Immunother. Cancer 4, 21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Principe D. R., DeCant B., Mascariñas E., Wayne E. A., Diaz A. M., Diaz N., Hwang R., Pasche B., Dawson D. W., Fang D., Bentrem D. J., Munshi H. G., Jung B., Grippo P. J., TGFβ signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. 76, 2525–2539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gorelik L., Flavell R. A., Transforming growth factor-beta in T-cell biology. Nat. Rev. Immunol. 2, 46–53 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Thomas D. A., Massagué J., TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Rabinovich G. A., Gabrilovich D., Sotomayor E. M., Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25, 267–296 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S. J., Hamieh M., Cunanan K. M., Odak A., Gonen M., Sadelain M., Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feucht J., Sun J., Eyquem J., Ho Y. J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M., Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 25, 82–88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hinrichs C. S., Restifo N. P., Reassessing target antigens for adoptive T-cell therapy. Nat. Biotechnol. 31, 999–1008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rapp M., Grassmann S., Chaloupka M., Layritz P., Kruger S., Ormanns S., Rataj F., Janssen K.-P., Endres S., Anz D., Kobold S., C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Onco. Targets. Ther. 5, e1105428 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheadle E. J., Sheard V., Rothwell D. G., Bridgeman J. S., Ashton G., Hanson V., Mansoor A. W., Hawkins R. E., Gilham D. E., Differential role of Th1 and Th2 cytokines in autotoxicity driven by CD19-specific second-generation chimeric antigen receptor T cells in a mouse model. J. Immunol. 192, 3654–3665 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Brudno J. N., Kochenderfer J. N., Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 127, 3321–3330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorelik L., Flavell R. A., Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12, 171–181 (2000). [DOI] [PubMed] [Google Scholar]
- 43.Lucas P. J., Kim S. J., Melby S. J., Gress R. E., Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor β II receptor. J. Exp. Med. 191, 1187–1196 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kamada T., Togashi Y., Tay C., Ha D., Sasaki A., Nakamura Y., Sato E., Fukuoka S., Tada Y., Tanaka A., Morikawa H., Kawazoe A., Kinoshita T., Shitara K., Sakaguchi S., Nishikawa H., PD-1+ regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc. Natl. Acad. Sci. U.S.A. 116, 9999–10008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghani K., Wang X., de Campos-Lima P. O., Olszewska M., Kamen A., Riviere I., Caruso M., Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum. Gene Ther. 20, 966–974 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobold S., Grassmann S., Chaloupka M., Lampert C., Wenk S., Kraus F., Rapp M., Duwell P., Zeng Y., Schmollinger J. C., Schnurr M., Endres S., Rothenfußer S., Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J. Natl. Cancer Inst. 107, djv146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benmebarek M.-R., Cadilha B. L., Herrmann M., Lesch S., Schmitt S., Stoiber S., Darwich A., Augsberger C., Brauchle B., Rohrbacher L., Oner A., Seifert M., Schwerdtfeger M., Gottschlich A., Rataj F., Fenn N. C., Klein C., Subklewe M., Endres S., Hopfner K.-P., Kobold S., A modular and controllable T cell therapy platform for acute myeloid leukemia. Leukemia , 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh S. A., Seki A., Rutz S., Ribonucleoprotein transfection for CRISPR/Cas9-mediated gene knockout in primary T cells. Curr. Protoc. Immunol. 124, e69 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Labun K., Montague T. G., Krause M., Torres Cleuren Y. N., Tjeldnes H., Valen E., CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heckman K. L., Pease L. R., Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Sommermeyer D., Neudorfer J., Weinhold M., Leisegang M., Engels B., Noessner E., Heemskerk M. H., Charo J., Schendel D. J., Blankenstein T., Bernhard H., Uckert W., Designer T cells by T cell receptor replacement. Eur. J. Immunol. 36, 3052–3059 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Das S., Sarrou E., Podgrabinska S., Cassella M., Mungamuri S. K., Feirt N., Gordon R., Nagi C. S., Wang Y., Entenberg D., Condeelis J., Skobe M., Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J. Exp. Med. 210, 1509–1528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bauer C. A., Kim E. Y., Marangoni F., Carrizosa E., Claudio N. M., Mempel T. R., Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J. Clin. Invest. 124, 2425–2440 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., Liu J., Freeman G. J., Brown M. A., Wucherpfennig K. W., Liu X. S., Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorsson V., Gibbs D. L., Brown S. D., Wolf D., Bortone D. S., Yang T. H. O., Porta-Pardo E., Gao G. F., Plaisier C. L., Eddy J. A., Ziv E., Culhane A. C., Paull E. O., Sivakumar I. K. A., Gentles A. J., Malhotra R., Farshidfar F., Colaprico A., Parker J. S., Mose L. E., Vo N. S., Liu J., Liu Y., Rader J., Dhankani V., Reynolds S. M., Bowlby R., Califano A., Cherniack A. D., Anastassiou D., Bedognetti D., Mokrab Y., Newman A. M., Rao A., Chen K., Krasnitz A., Hu H., Malta T. M., Noushmehr H., Pedamallu C. S., Bullman S., Ojesina A. I., Lamb A., Zhou W., Shen H., Choueiri T. K., Weinstein J. N., Guinney J., Saltz J., Holt R. A., Rabkin C. S., Lazar A. J., Serody J. S., Demicco E. G., Disis M. L., Vincent B. G., Shmulevich I., The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabi5781/DC1