

Spread of tau pathology follows brain connectivity, is modulated by selective vulnerability, and is predicted by network analysis.
Abstract
Neuropathological staging studies have suggested that tau pathology spreads through the brain in Alzheimer’s disease (AD) and other tauopathies, but it is unclear how neuroanatomical connections, spatial proximity, and regional vulnerability contribute. In this study, we seed tau pathology in the brains of nontransgenic mice with AD tau and quantify pathology development over 9 months in 134 brain regions. Network modeling of pathology progression shows that diffusion through the connectome is the best predictor of tau pathology patterns. Further, deviations from pure neuroanatomical spread are used to estimate regional vulnerability to tau pathology and identify related gene expression patterns. Last, we show that pathology spread is altered in mice harboring a mutation in leucine-rich repeat kinase 2. While tau pathology spread is still constrained by anatomical connectivity in these mice, it spreads preferentially in a retrograde direction. This study provides a framework for understanding neuropathological progression in tauopathies.
INTRODUCTION
Neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), are estimated to affect over 60 million people worldwide (1, 2). Neurological symptoms and the presence of pathological protein inclusions are used to categorize the two diseases. However, there exists substantial overlap in both symptoms and pathologies, especially as these diseases progress (3, 4). Tau pathology, while diagnostic of AD and other primary tauopathies, appears prominently in PD, PD dementia (PDD), and dementia with Lewy bodies, where it correlates with α-synuclein pathological burden and cognitive decline (5, 6). These data suggest that multiple pathologies may act additively to influence disease progression and that underlying risk factors for one pathology may confer risk for additional pathologies.
Postmortem neuropathology studies have demonstrated that patients with more severe clinical AD and PDD have elevated levels of pathological tau in an increasing number of regions of the brain (5–8). The stages of observed tau pathology (beginning in the locus coeruleus, then transentorhinal and entorhinal cortex, and moving through the hippocampus and cortical regions) are suggestive of pathology “spreading” (7, 8). Pathological tau from human brains injected into nontransgenic (NTG) mice can be internalized into nearby neurons, initiating misfolding and hyperphosphorylation of endogenous mouse tau in a prion-like manner into intraneuronal inclusions resembling those from human disease tissue (9–11). Over time, tau pathology can be found in more regions of the mouse brain connected to the injection site (9–11), suggesting that tau is spreading. How tau pathology spreads has been a matter of debate because it has not been possible to disambiguate the contribution of neuroanatomical connectivity, spatial proximity of regions, and intrinsic neuronal vulnerability. Recent studies based on mathematical modeling of human brain suggest that anatomical connectivity serves as a strong predictor of brain atrophy or general pathology patterns in neurodegenerative diseases (12–17).
In the current study, we investigated processes underlying the development and spread of tau pathology. We developed a methodology for reproducibly quantifying tau pathology in 134 regions of the mouse brain following an intracranial injection of pathogenic tau. Tau pathology in this model begins slowly, first affecting the injection site and highly connected regions. As time progresses, more regions are affected. Once affected, regions show a nearly linear increase in tau pathology over time. While this atlas of pathology is informative, it is still not possible to discern mechanisms of pathology spread without considering the brain as a network of interconnected regions. We therefore used computational analysis of spatiotemporal tau pathology patterns. We found that tau pathology spread is best explained by diffusion along neuroanatomical connections in a bidirectional manner. Residual variance in the data that was not well explained by connectivity was used to generate estimates of differential vulnerability to tau in measured regions. Comparison of regional vulnerability to regional gene expression identified several previously unidentified candidate genes that may control susceptibility to tau pathology. To further assess the utility of quantitative pathology and network modeling, we analyzed a mouse expressing the LRRK2G2019S mutation. This mutation is a risk factor for PD, but many of these patients exhibit tau pathology. LRRK2G2019S mice exhibit a bias toward retrograde spread of tau pathology, providing insight into the network-level impact of cell biology events. This work provides a framework for understanding the spread of pathological tau throughout the brain and investigating the impact of genetic risk factors on pathology progression.
RESULTS
Quantitative immunohistochemistry to evaluate pathological tau spread
To investigate mechanisms underlying the spread of tau pathology, we used a recently developed seed-based model of tauopathy in which injection of AD brain–derived tau into NTG mice can induce the misfolding of endogenous tau into hyperphosphorylated tau inclusions in a time- and region-dependent manner (9). Previous studies have suggested that this spatiotemporal pattern of tau pathology induction is consistent with spread along neuroanatomical connections (9, 10), although these patterns were not quantitatively evaluated. Biochemical sequential detergent extraction of gray matter from AD patient brains was used to obtain an enriched fraction of paired helical filament (PHF) AD tau (Fig. 1A) (9). This extraction method yielded a final purity of 22.6 to 35.7% tau, with 0.01% or less α-synuclein and amyloid β (table S1). The purified tau fraction retains the pathogenic conformation present in human disease and induces the misfolding of tau in mice without the overexpression of tau (9). To investigate how tau pathology spreads through the brain, we injected NTG mice with PHF tau from individual extractions in the hippocampus and overlaying cortex (Fig. 1B). Mice were allowed to age 1, 3, 6, or 9 months post-injection (MPI) to capture the temporal dynamics of tau pathology spread (Fig. 1C). Brains were then sectioned, and representative sections (Fig. 1D) were selected and stained for phosphorylated tau pathology throughout the brain. We manually annotated 194 areas from 134 anatomical regions on the selected sections (Fig. 1E and fig. S1) so that pathology could be quantified as the percentage of each area occupied by pathology (Fig. 1, F and G). Brain region annotations were based on the Allen Brain Atlas (ABA) Common Coordinate Framework (CCF v3, 2017, brain-map.org), although smaller subregions with minimal pathology were grouped to minimize error and annotation time. To mitigate overinterpretation of individual sections, we similarly annotated and quantified a second set of nearby sections (16,684 annotations in total), and the average for each region was used for subsequent analyses.
Fig. 1. Quantitative immunohistochemistry to evaluate pathological tau spread.
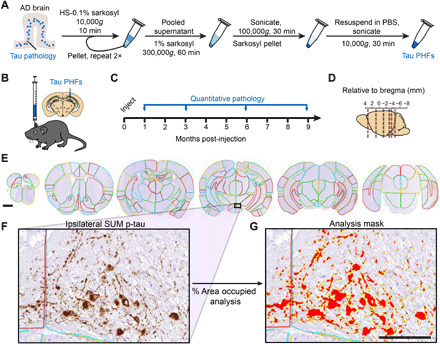
(A) AD brain with a high burden of tau pathology was chosen for extraction of pathological tau. Brains went through sequential extraction of tau PHFs as noted in the schematic. Final tau PHF preparations were used for all subsequent steps. PBS, phosphate-buffered saline. HS, high salt. (B) NTG mice were injected unilaterally with AD PHF tau in the hippocampus and overlaying cortex as shown at 3 to 4 months of age. (C) Mice were euthanized 1 (n = 4), 3 (n = 8), 6 (n = 6), or 9 (n = 6) months following injection. (D) Mouse brain was sectioned, and the sections representing the regions shown were stained for pathological tau. (E) Representative sections were selected, and 194 regions were annotated for each brain. A second set of nearby sections was similarly annotated to reduce selection bias. Scale bar, 1 mm. Annotation colors are arbitrary. (F) An enlarged image of the annotated supramammillary nucleus (SUM) is shown with the inclusions stained for pS202/T205 tau. (G) Annotations allow automated quantification of percentage of area occupied with pathology in specific regions of the brain. An analysis mask is overlaid on the image in (F) to demonstrate this quantification of pathology. Scale bar, 100 μm.
Quantitative pathology mapping reveals dynamic patterns of tau pathology spread over time
Because imaging of tau pathology in these mice can only be done after death, data are pseudo-longitudinal. That is, different time points are represented by separate groups of mice. Despite this fact, tau pathology patterns were highly reproducible across cohorts and time points (Fig. 2). One remarkable finding from this analysis was that once pathology seeds a region, it continues in an almost linear fashion over time in most regions (Fig. 2), suggesting that the process of pathology formation is similar across the brain. The lack of a plateau in most regions also suggests that this model is recapitulating the early disease process, before the saturation of pathology and profound neuron death. While the process of pathology formation was similar across regions, the delay between injection and pathology formation varied widely. Regions at the injection sites or with high connectivity to the injection sites developed pathology by 3 MPI (Fig. 2, A and B). Other regions showed a further delay, with only minimal pathology before 6 MPI (Fig. 2, C and D). Last, some regions did not develop pathology until 9 MPI (Fig. 2, E and F), and some regions did not develop substantial pathology during the course of the study (fig. S10).
Fig. 2. Brain regions show progressive accumulation of tau pathology.
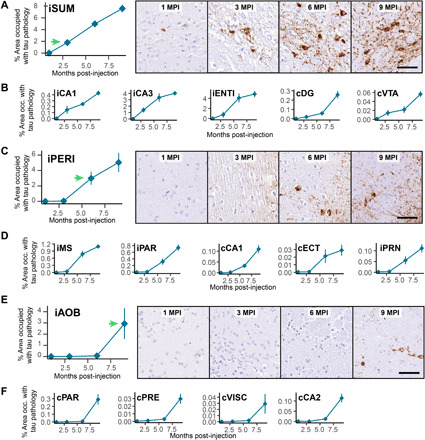
The percentage of area occupied with tau pathology is plotted as a function of time demonstrating three distinct onsets of pathological tau accumulation: 3 MPI (A and B), 6 MPI (C and D), and 9 MPI (E and F). Most of the other regions also fall into one of these patterns. Green arrows denote the time point with initial substantial pathology accumulation. (A) The ipsilateral supramammillary nucleus accumulates pathology by 3 MPI as demonstrated by the quantitative pathology plot and images at right. (B) Additional regions that show a similar onset of pathology at 3 MPI (iCA1, field CA1 of the hippocampus; iCA3, field CA3 of the hippocampus; iENTl, entorhinal area, lateral; cDG, dentate gyrus; cVTA, ventral tegmental area). (C) The ipsilateral perirhinal area (iPERI) accumulates pathology by 6 MPI as demonstrated by the quantitative pathology plot and images at right. (D) Additional regions that show a similar onset of pathology at 6 MPI (iMS, medial septal nucleus; iPAR, parasubiculum; cCA1, field CA1 of the hippocampus; cECT, ectorhinal area; iPRN, pontine reticular nucleus). (E) The ipsilateral accessory olfactory bulb (iAOB) accumulates pathology by 9 MPI as demonstrated by the quantitative pathology plot and images at right. (F) Additional regions that show a similar onset of pathology at 9 MPI (cPAR, parasubiculum; cPRE, presubiculum; cVISC, visceral area; cCA2, field CA2 of the hippocampus). Data are represented as means ± SEM. Scale bars, 50 μm.
While patterns of pathology in individual regions are informative, overall patterns throughout the brain are best observed as a heatmap overlaid on the mouse brain (Fig. 3) or as a region-by-time matrix (fig. S2). Several whole-brain patterns were apparent. At 1 MPI, there was minimal tau pathology outside of the injection sites. At 3 MPI, more pathology accumulated ipsilateral to the injection site including hippocampal and entorhinal regions. By 6 MPI, pathology spread to the contralateral hippocampus and associated cortical areas. By 9 MPI, pathology continued to spread, affecting more rostral regions, although certain rostral cortical and contralateral thalamic regions show minimal pathology. Representative images of all regions are available in the Supplementary Materials (fig. S10). Together, these patterns are highly suggestive of tau pathology spread throughout the brain, which may involve transmission along neuroanatomical connections or other regional factors.
Fig. 3. Quantitative pathology mapping reveals dynamic patterns of tau pathology spread over time.
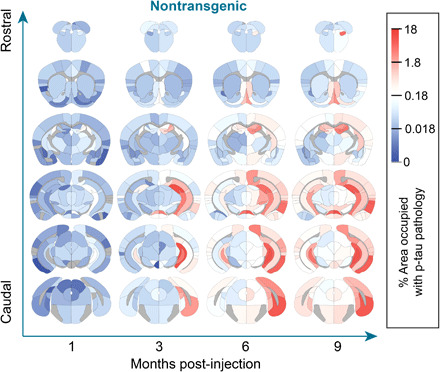
Regional pathology measures plotted on anatomical maps as a heatmap, with blue representing minimal pathology, white representing moderate pathology, and red representing substantial pathology. Note that tau pathology was not quantified in white matter regions, and these regions are gray. Representative images for all regions can be found in the Supplementary Materials. n = NTG-1 M: 4, NTG-3 M: 8, NTG-6 M: 6, and NTG-9 M: 6.
Tau pathology spread is predicted by diffusion through the neuroanatomical connectome
There is substantial evidence that tau can be released from neurons (18–20) in an activity-dependent manner (21, 22) and internalized by other neurons (23–26), likely leading to the spread of tau pathology throughout the brain. However, it is still unclear precisely how spread occurs. The main hypotheses are that tau pathology spreads between anatomically connected regions of the brain, between physically contiguous regions, or to selectively vulnerable populations of neurons. Of course, these mechanisms of spread are not mutually exclusive; thus, we sought to assess how each of these putative routes contributes to the observed pathology spread in mice.
We evaluated the ability of a network diffusion model with neuroanatomical connectivity (brain-map.org) (27) as a scaffold to predict the empirical measures of tau pathology over time. ABA regions at the injection sites (iDG, iCA1, iCA3, iRSPagl, and iVISam) were used as seed regions for initiation of the model. Direct connectivity of these regions was highest in hippocampal, septal, and entorhinal regions of the brain (fig. S3A), and direct connectivity to the injection site was highly correlated with tau pathology measures in those regions (fig. S3, B and C). Our model posits that tau spreads from the injection site along anatomical connections both in retrograde and anterograde directions, with the final amount of regional pathology determined by a weighted sum of these two independent processes. This bidirectional anatomical connectivity model weakly predicted tau pathology at 1 MPI, likely due to minimal spread at this time point, and strongly predicted tau pathology at 3, 6, and 9 MPI (Fig. 4A). In addition, this bidirectional anatomical connectivity model outperformed a model in which tau spread was proportional to the Euclidean distance between regions (Fig. 4B). In the Euclidean distance plots, the five outlying regions in the upper right were the injection sites, as noted (Fig. 4B); however, inclusion or exclusion of these sites during model optimization did not measurably affect the fit of the Euclidean distance model (fig. S4, A and B). Excluding the injection sites from the bidirectional model did not impair its predictivity over the Euclidean distance model (fig. S4C). Rewiring the network to disrupt the connectome but preserve basic network properties such as in-degree (fig. S5A) or out-degree connectivity (fig. S5B) also reduced model performance. To further validate the anatomical connectivity model, we ensured that the model’s performance was specific to the choice of the experimental injection site, compared with 500 randomly chosen sets of five regions with mean spatial proximity similar to that of the experimental injection sites (Fig. 4C). Models using the experimental seed regions were among the best performing models at all time points (Fig. 4C), confirming the specificity of our model to the experimental injection site. The performance of models using random sites could be partially explained by a generalized additive model relying on three variables: in-projection similarity between actual and alternate seeds, out-projection similarity between actual and alternate seeds, and Euclidean distance between actual injection sites and alternate seed sites (fig. S5C).
Fig. 4. Tau pathology spread is predicted by diffusion through neuroanatomical connectivity.
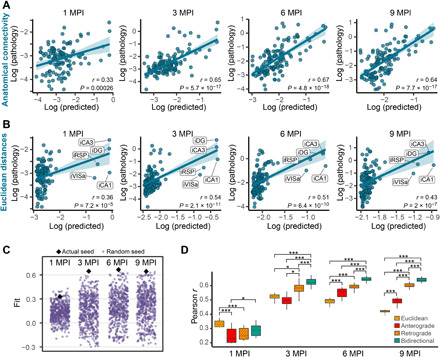
(A and B) Predictions of log tau pathology (x axis) from spread models based on (A) retrograde and anterograde anatomical connections or (B) Euclidean distance, plotted against log actual regional tau pathology values (y axis) at 1, 3, 6, and 9 MPI. The solid lines represent the lines of best fit, and the shaded ribbons represent the 95% prediction intervals. The r and P values for the Pearson correlation between model-fitted values and observed pathology are noted on the plots. In (B), the injection site regions are noted but did not affect model fit (fig. S4). (C) To evaluate the specificity of the seed sites to predict the pathology spread pattern, 500 alternate combinations of five seed sites (purple dots) were evaluated for their ability to predict pathology spread at 1, 3, 6, and 9 MPI. Using the actual five injection regions (black diamonds) produced among the best fits at all time points (1 MPI, P = 0.042; 3 MPI, P < 0.002; 6 MPI, P < 0.002; and 9 MPI, P < 0.002. (D) Distributions of model fits in 500 held-out samples using Euclidean, anterograde, retrograde, and bidirectional models. Fit differences were analyzed by pairwise two-tailed nonparametric tests for different models. All connectivity models outperform Euclidean distance after 3 MPI, and a bidirectional model outperforms either a retrograde or anterograde model alone (*P < 0.05 and ***P < 0.002). In box plots, box edges represent the 25th and 75th percentiles, the middle line shows the median, and whiskers extend from the box edges to the most extreme data point value that is at most 1.5× interquartile range (IQR). Data beyond the end of the whiskers are plotted individually as dots.
Last, we rigorously evaluated the out-of-sample performance of the bidirectional anatomical connectivity model compared to three other models in which spread was based on either Euclidean distance, anterograde spread alone, or retrograde spread alone. To compare the distributions of out-of-sample fits between each of the four models, we generated 500 train-test splits of the mice used in the study. Next, we obtained model parameters (diffusion rate constants and regression weights for anterograde and retrograde spread) in the training set and evaluated model performance in the test set at each time point. Our measure of performance was the spatial Pearson correlation coefficient between the observed pathology in the test set and the predicted pathology estimated by each model. This analysis revealed that the bidirectional model was superior to both the anterograde and retrograde models and that all three connectivity-based models were superior to the Euclidean distance model by 6 and 9 MPI (Fig. 4D). To understand whether the inclusion of Euclidean distance could improve predictivity of the bidirectional model, we evaluated a combined model that integrated bidirectional connectivity and Euclidian distance. We found that the bidirectional-alone model performed at least as well as the combined model at all time points (fig. S5D). To understand the contribution of anterograde and retrograde connections to the bidirectional model, we estimated the diffusion rate constant (fig. S5F) and standardized b (fig. S5G) from the model. We found a stronger contribution of retrograde connectivity to the spread model in both measures. In summary, these findings provide strong support for the notion that both anterograde and retrograde spread along anatomical connections independently contribute to the propagation of tau pathology, with retrograde spread being predominant.
Model residuals as an estimate of regional vulnerability
A third possible mediator of tau pathology spread is intrinsic neuronal vulnerability. While this concept is less easily parsed for tauopathies than for α-synucleinopathies where dopaminergic neurons appear particularly vulnerable, our network model of tau pathology spread can be used to infer regional vulnerability. We have previously validated this approach by showing that residuals from network model predictions of α-synuclein pathology correlated with α-synuclein gene expression, which was expected to contribute to regional vulnerability (28). Here, we estimated regional vulnerability to tau pathology spread by taking the residuals from the bidirectional anatomical connectivity model and averaging them over 3, 6, and 9 MPI (Fig. 5A). We excluded 1 MPI because of the low model predictivity and dissimilarity to residuals from other time points (fig. S6A). This estimate of regional vulnerability revealed that amygdalar, thalamic, and rostral cortical nuclei were resilient, while septal, mesencephalic, and caudal cortical regions were more vulnerable (Fig. 5A). We then tested whether tau gene (Mapt) expression from ABA in situ hybridization data was associated with vulnerability. Mapt is expressed quite broadly in the brain, including most gray matter and white matter regions, and showed no association with regional vulnerability estimates (fig. S6B), suggesting that tau expression is not a major limiting factor in the spread of tau pathology.
Fig. 5. Model residuals as a predictor of regional vulnerability.
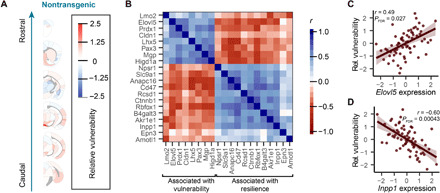
(A) Residuals between actual tau pathology levels and pathology levels predicted by the bidirectional model were averaged for each region over time and across hemispheres to give an average regional vulnerability to tau pathology. Here, those values are plotted as an anatomical heatmap. (B) Gene expression patterns that were statistically significantly associated with regional vulnerability (FDR-corrected P < 0.05 cutoff for inclusion) are plotted here with heatmap values indicating the similarity of these gene expression patterns to each other. The genes cluster into two groups, one associated with vulnerability and one associated with resilience, as noted by brackets at the bottom of the plot. (C) Normalized relative regional vulnerability is plotted as a function of normalized Elovl5 expression. (D) Normalized relative regional vulnerability is plotted as a function of normalized Inpp1 expression. For (C) and (D), the solid line represents the line of best fit, and the shaded ribbons represent the 95% prediction intervals. The r and P values for the Pearson correlation between vulnerability and gene expression are noted on the plots.
We sought to further investigate gene expression patterns associated with regional vulnerability by performing a genome-wide search for genes whose expression patterns measured by the ABA were spatially similar to our regional vulnerability measure. Many gene expression patterns correlated with regional vulnerability. After adjusting false discovery rate (FDR) to q < 0.05 (29), we found that 20 genes had expression patterns with a statistically significant spatial correlation with relative regional vulnerability to tau pathology (Fig. 5B). Twelve of these genes showed expression patterns that were negatively associated with regional vulnerability, and 8 of these genes showed expression patterns that were positively associated with vulnerability. Plotting the gene expression versus vulnerability of individual genes (Fig. 5, C and D, and fig. S6C) demonstrates that this association is not driven by outliers. In the future, gene expression patterns may be useful parameters of regional vulnerability in computational models of pathology spread.
In silico seeding from alternate sites
In addition to inferring mechanisms of spread through network modeling, we can also extend the value of our validated network models by generating predictions of tau spread patterns from alternate injection sites, assuming that the rates of spread and contributions of anterograde versus retrograde spread are the same for those injection sites. For example, one of the earliest sites with tau pathology outside of the brainstem in humans is the entorhinal cortex (7). However, this site is difficult to inject reproducibly in mice because of its lateral location. In silico modeling of tau pathology spread from this injection site shows a more lateralized spreading pattern that largely affects hippocampal and parahippocampal regions, with spread to contralateral regions occurring relatively late (Fig. 6A). We chose to also model a caudoputamen injection site (Fig. 6B) to compare tau pathology spread to previously published data modeling α-synuclein pathology spread. While tau pathology is predicted to spread to some conserved regions, including the substantia nigra and frontal cortical regions, there is also more engagement of thalamic and mesencephalic regions and less engagement of contralateral regions by tau than with α-synuclein pathology.
Fig. 6. In silico seeding from alternate sites.
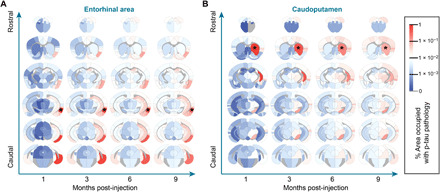
Using the diffusion rate constant estimated by model fitting to empirical tau pathology spread, we estimated the distinct spreading patterns that arise after injection into alternate sites in the entorhinal area (A) and caudoputamen (B). Estimated spread is plotted as a heatmap in anatomical space with blue indicating regions with minimal estimated pathology and red indicating regions with elevated pathology. *Sites of injections.
LRRK2G2019S mice have altered tau pathology patterns
The quantitative pathology-network modeling approach established in NTG mice allows the assessment of dynamics of tau pathology spread. To further investigate the utility of this approach, we performed similar analysis in mice expressing the G2019S mutation in LRRK2. This mutation is the most common cause of familial PD and a common risk factor for idiopathic PD (30). Although these patients show similar symptoms to idiopathic PD, neuropathologically, 21 to 54% of patients lack the hallmark α-synuclein Lewy bodies exhibited by patients with idiopathic PD (31–33). Notably, most of the LRRK2 mutation carriers exhibit tau pathology (33, 34). To assess alterations in tau pathology distribution and spread related to LRRK2, we performed quantitative analysis of pathology in LRRK2G2019S mice [B6.Cg-Tg(Lrrk2*G2019S)2Yue/J]. We first established that in the absence of pathological tau injection, LRRK2G2019S mice do not accumulate detectable tau pathology up to 12 months of age (fig. S7). Following pathological tau injection, LRRK2G2019S mice accumulate tau pathology in similar regions as NTG mice (Fig. 7A), suggesting that overall spreading is constrained by anatomical connectivity. However, there are clear differences in the regional distribution of tau pathology in LRRK2G2019S mice (Fig. 7B and figs. S8 and S9). In some regions, like the injected iDG and highly connected iSUM, tau pathology is almost identical in NTG and LRRK2G2019S mice (Fig. 7, C and D). In contrast, other regions, especially those that require extended periods of time to exhibit pathology, show elevated pathology in LRRK2G2019S mice (Fig. 7, C and D). These results suggest that LRRK2G2019S alters some aspect of tau pathology spread.
Fig. 7. LRRK2G2019S mice have altered tau pathology patterns.
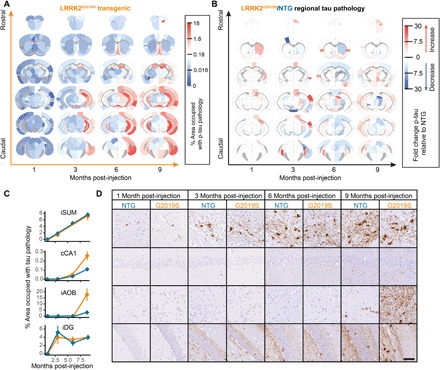
(A) Regional pathology measures for LRRK2G2019S mice plotted on anatomical scaffolds as a heatmap, with blue representing minimal pathology, white representing moderate pathology, and red representing substantial pathology. (B) The fold change between NTG and LRRK2G2019S mice is plotted on anatomical maps as a heatmap, with blue representing regions with higher pathology in NTG mice and red representing regions with higher pathology in LRRK2G2019S mice. (C) The percentage of area occupied with tau pathology is plotted as a function of time for four different regions (iSUM, cCA1, iAOB, and iDG) demonstrating four distinct patterns of pathology propagation in NTG and LRRK2G2019S mice. Some regions, like the iSUM and iDG, show similar tau pathology progression in NTG and LRRK2G2019S mice. In contrast, the cCA1 and iAOB, although similar at 1 and 3 MPI, show enhanced pathology in LRRK2G2019S mice, especially at 9 MPI. (D) Representative images of the regions plotted in (C) stained for p-tau and directly adjacent to the plots demonstrating the pathology patterns. Representative images for all regions can be found in the Supplementary Materials. n = NTG-1 M: 4, G2019S-1 M: 3, NTG-3 M: 8, G2019S-3 M: 7, NTG-6 M: 6, G2019S-6 M: 5, NTG-9 M: 6, and G2019S-9 M: 4. Scale bar, 50 μm.
The LRRK2G2019S genetic risk factor alters network dynamics of tau pathology spread
To gain a deeper understanding into what parameters of tau pathology spread may be altered, we assessed spread in LRRK2G2019S mice using network modeling. As suggested by the overall quantitative pathology pattern (Fig. 7A), tau spread can be well explained in LRRK2G2019S mice by anatomical connectivity (Fig. 8A). Similar to NTG mice, tau pathology spread in LRRK2G2019S mice is only moderately well fit at 1 MPI but shows improved fit at 3, 6, and 9 MPI, suggesting that anatomical connectivity is also a major factor driving tau pathology spread in LRRK2G2019S mice. We next sought to explain the regional differences in pathology in LRRK2G2019S mice. We first assessed the relationship of estimated regional vulnerability in NTG mice to the difference in pathology in LRRK2G2019S mice (Fig. 8B). There is a negative correlation between NTG vulnerability and the difference in pathology at 1 and 3 MPI, suggesting that at those time points, there is a shift in pathology in LRRK2G2019S mice from vulnerable regions to more resilient regions. At 6 and 9 MPI, the relationship between NTG vulnerability and pathology difference is diminished. Given the low levels of pathology in NTG and LRRK2G2019S mice at 1 and 3 MPI, the inverse relationship between the difference in pathology in LRRK2G2019S mice and NTG vulnerability may reflect not a shift in vulnerability of regions but early changes in the directionality of pathology movement in the brain.
Fig. 8. The LRRK2G2019S genetic risk factor alters network dynamics of tau pathology spread.
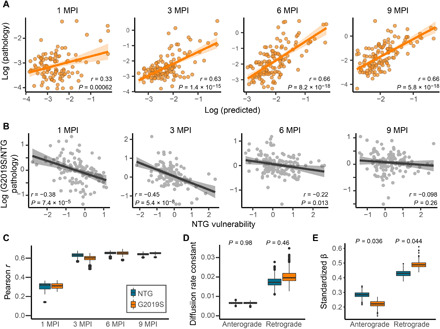
(A) Predictions of regional log tau pathology (x axis) in LRRK2G2019S mice from spread models based on retrograde and anterograde anatomical connections, plotted against log actual regional tau pathology values (y axis) at 1, 3, 6, and 9 MPI. (B) Plots of the NTG vulnerability measure versus log G2019S/NTG pathology, showing a negative correlation between the two measures especially at early time points (1 and 3 MPI) that levels off at later time points (6 and 9 MPI). Solid lines represent the lines of best fit, and the shaded ribbons represent the 95% prediction intervals. The r and P values for the Pearson correlation between G2019S/NTG pathology ratio and NTG vulnerability are noted on the plots. (C) Distributions of model fit (Pearson r) for fitting data to bootstrap samples of mice. NTG and G20 do not differ in model fit (nonparametric, two-tailed test). (D) Distributions of diffusion rate constants reveal greater intersample variability in retrograde constants compared to anterograde. LRRK2G2019S and NTG do not differ in diffusion rate constants. (E) Anterograde and retrograde betas differ between NTG and LRRK2G2019S mice, with LRRK2G2019S preferentially spreading in the retrograde direction. In box plots, box edges represent the 25th and 75th percentiles, the middle line shows the median, and whiskers extend from the box edges to the most extreme data point value that is at most 1.5× IQR. Data beyond the end of the whiskers are plotted individually as dots.
We next sought to determine whether the difference in regional pathology distribution in LRRK2G2019S mice is related to a difference in spread in anterograde or retrograde directions. To infer the mechanisms of network spread affected by the LRRK2G2019S mutation, we fit the bidirectional model on bootstrapped samples of NTG and LRRK2G2019S mice to obtain distributions of model parameters, namely, the diffusion rate constants and regression weights that measure the relative importance of anterograde and retrograde spread. We observed no statistically significant difference in the overall fit of NTG and LRRK2G2019S mice over time (Fig. 8C), suggesting that our network model adequately captures tau spread in LRRK2G2019S mice. In addition, the overall diffusion rate constant along anterograde connections was no different than that along retrograde connections (Fig. 8D), suggesting that the rates of anterograde and retrograde spread are similar. The contribution of anterograde and retrograde connections over time did show differences (Fig. 8E), such that anterograde spread was less important and retrograde spread more important for explaining tau spread patterns in LRRK2G2019S mice. These findings suggest that the LRRK2G2019S mutation may lead to increased shunting of misfolded tau into the already predominant retrograde pathways, which partially explains the differences in pathology patterns observed in these mice.
DISCUSSION
Neurodegenerative diseases progress symptomatically as pathologies appear in previously unaffected regions of the brain. Identification of the neuropathological proteins aggregated in these diseases and subsequent staging studies demonstrated that symptom progression is associated with the presence of aggregated proteins in more and more regions (7). The stages of disease suggest that either there is a fine gradient of regional vulnerability such that regions become sequentially affected or pathology can spread through the brain by transcellular means. The latter hypothesis is more parsimonious, and mounting evidence has supported this explanation in recent years. The current study demonstrates using an interdisciplinary approach bridging quantitative pathology and network analysis that tau pathology patterns can be predicted by linear diffusion through the anatomical connectome with modulations of that spread by regional vulnerability.
Our study is congruous with previous work using computational modeling to understand the distribution of tau pathology and related regional atrophy in tauopathies. These studies assessed the utility of computational models to predict semiquantitative regional tau pathology scores in transgenic mice (35), global atrophy magnetic resonance imaging in AD and frontotemporal dementia (12, 13), tau positron emission tomography signal in AD (14, 15), or general histopathological patterns of AD (16, 17). While each of these studies used different modeling parameters, each found that a model incorporating spread along anatomical connections was the most efficient and accurate predictor of tau pathology or related atrophy patterns. Our study has now extended these previous efforts in three important ways. First, the current study used tau seeding in an NTG mouse, giving our model a precise spatiotemporal starting point, as defined by the site and time of pathogenic tau injection. Second, we used quantitative measures of mouse tau pathology in 134 regions of the mouse brain, providing several log-fold depth of data throughout the brain. Third, mice were euthanized at four time points, following injection of pathogenic tau, providing pseudo-longitudinal data for model fitting. Our modeling of this data is consistent with previous studies, finding that tau pathology spreads in a pattern that is well described by anatomical connectivity. Further, we were able to delineate the contribution of anterograde and retrograde connections to this spread through a rigorous model comparison approach, assess the kinetics of spread, understand regional vulnerability to pathology, compare regional vulnerability to regional gene expression, and assess the impact of a genetic risk factor on the spread of tau pathology. Future studies could test in silico whether deleting connections would impact spread, which could be validated in vivo with lesioning experiments.
This study has several limitations. One is the resolution of tau pathology data and the reliance on mesoscale connectivity measures to model tau pathology spread. Tau pathology was assessed at a mesoscale to match the mesoscale connectivity atlas generated by the Allen Institute (27). However, this scale obscures more granular information, such as the cortical laminar distribution of pathology. The use of the connectivity atlas also imposes technical limitations of connectivity tracing, including a potential bias in cell types infected by tracer viruses, difficulty in distinguishing passing axons from terminals, and possible underestimation of connectivity due to the use of projection volume instead of fluorescence intensity (27). Higher-resolution connectivity maps are currently being optimized, and future studies would benefit from higher-resolution pathology maps to match the higher-resolution connectivity maps. A second limitation is the use of a transgenic mouse overexpressing LRRK2. When this study began, this model was desirable because LRRK2 is expressed on the endogenous mouse promoter, ensuring that its regional expression matched that of endogenous LRRK2. However, next-generation models with a knock-in of the LRRK2 mutation are now available (36–38), and future work should validate the effects of LRRK2 on tau spread in these models.
Despite recent efforts, the neuropathological substrate of LRRK2 PD has been elusive. While LRRK2 mutation carriers all have degeneration of substantia nigra neurons, many of them do not accumulate α-synuclein Lewy bodies in the brain (31, 32). This fact has left some question as to the neuropathological substrate of degeneration in these patients. In the search for an alternate pathology, it has now been recognized that most LRRK2 mutation carriers have tau pathology to varying degrees (31, 33, 34). Although it is not clear that tau pathology is responsible for dopaminergic neuron death in LRRK2 mutation carriers, it does raise the possibility that mutations in LRRK2 can predispose patients to developing tau pathology.
Hyperphosphorylated tau is a well-cataloged feature present in LRRK2 mutant mice (39, 40), flies (41), and induced pluripotent stem cell–derived neurons (42). It has also been demonstrated that LRRK2 mutations can lead to phosphorylation of tau (43–46). Although it is not clear that this hyperphosphorylated tau is pathological, it is possible that it represents a pool of tau that is more rapidly recruited upon the introduction of a pathogenic tau seed. The current study was conducted in mice up to 12 months of age, and the LRRK2G2019S mice in this study showed no evidence of phosphorylated tau accumulation without pathogenic tau injection. This observation suggests that the changes in tau pathology spread are likely due not to elevated phosphorylation but rather to alterations in cellular mechanisms such as protein release, internalization, transport, or degradation. This hypothesis is supported by another recent study showing enhanced neuronal transmission of virally expressed tau (47). Other studies found that overall tau pathology was not affected in transgenic mice that exhibit broad tau pathology controlled by the transgene promoter (47, 48).
One possible cellular mechanism regulating the change in tau pathology spread in LRRK2G2019S mice is elevated presynaptic vesicle release, a phenomenon that has been observed in LRRK2G2019S mice (38). Enhanced synaptic vesicle release is accompanied by enhanced membrane internalization, which could provide an increased opportunity for extracellular tau to be internalized into presynaptic boutons and thereby enhance retrograde transmission of pathology. Previous research has demonstrated that tau is readily released by neurons (18–20) in proportion to neuronal activity (21, 22). Future research incorporating functional connectivity strength and tau receptor distribution as regulators of tau spread may help clarify how tau pathology spread is regulated.
In conclusion, the current study demonstrates that tau pathology spreads from an initial injection site through the brain via neuroanatomical connectivity. This spread can be modulated by a genetic risk factor for PD. Future work should substantiate that this alteration is related to LRRK2 kinase activity and explore whether LRRK2 inhibitors would be a viable therapeutic treatment for tauopathies.
MATERIALS AND METHODS
Mice
All housing, breeding, and procedures were performed according to the National Institutes of Health (NIH) Guide for the Care and Use of Experimental Animals and approved by the University of Pennsylvania Institutional Animal Care and Use Committee. C57BL/6 J (NTG, the Jackson laboratory 000664, Research Resource Identifier (RRID): IMSR_JAX:000664) and B6.Cg-Tg(Lrrk2*G2019S)2Yue/J (G2019S, the Jackson laboratory 012467, RRID: IMSR_JAX:012467) mice have been previously described (49). The current G2019S bacterial artificial chromosome line was backcrossed to C57BL/6 J mice for >10 generations and bred to homozygosity at loci as determined by quantitative polymerase chain reaction and outbreeding. The expression level of LRRK2G2019S was thereby stabilized in this line of mice. All experiments shown use homozygous LRRK2G2019S mice. Both male (n = 24) and female (n = 19) mice were used and were 3 to 4 months old at the time of injection. No influence of sex was identified in the measures reported in this study.
Human tissue
All procedures were done in accordance with local institutional review board guidelines of the University of Pennsylvania. Written informed consent for autopsy and analysis of tissue sample data was obtained from either patients themselves or their next of kin. All cases used for extraction (table S1) of PHF tau were Braak stage VI and were selected on the basis of a high burden of tau pathology by immunohistochemical staining. Cases used for extraction were balanced by sex (female = 2; male = 2) and were frozen an average of 5 hours postmortem. Differences in sex were not assessed because these cases were only used for protein extraction.
Human brain sequential detergent fractionation
Frozen postmortem human frontal or temporal cortex brain tissue containing abundant tau-positive inclusions was selected for sequential extraction on the basis of immunohistochemistry examination of these samples as described (50) using previously established methods. These brains were sequentially extracted with increasing detergent strength as previously described (9). After thawing, meninges were removed, and gray matter was carefully separated from white matter. The gray matter was weighed and suspended in nine volumes (w/v) high-salt buffer [10 mM tris-HCl (pH 7.4), 800 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol, 1:1000 protease and 1:100 phosphatase inhibitors, and 1 mM phenylmethylsulfonyl fluoride] with 0.1% sarkosyl and 10% sucrose, followed by homogenization with a dounce homogenizer and centrifugation at 10,000g for 10 min at 4°C. The resulting pellet was re-extracted with the same buffer conditions, and the supernatants from all extractions were filtered and pooled.
Additional sarkosyl was added to the pooled supernatant to reach a final concentration of 1%, and the supernatant was nutated for 1 hour at room temperature. The samples were then centrifuged at 300,000g for 60 min at 4°C. The pellet, which contains pathological tau, was washed once with phosphate-buffered saline (PBS) and resuspended in 100 μl of PBS per gram of gray matter by passing through a 27-gauge/0.5-inch needle. The pellets were further suspended by brief sonication (QSonica Microson XL-2000; 20 pulses; setting 2; 0.5 s per pulse). The suspension was centrifuged at 100,000g for 30 min at 4°C. The pellet was suspended in one-fifth to one-half the precentrifugation volume, sonicated briefly (60 to 120 pulses; setting 2; 0.5 s per pulse), and centrifuged at 10,000g for 30 min at 4°C. The final supernatant was used for all studies and is referred to as AD PHF tau. All extractions were characterized by Western blotting for tau, sandwich enzyme-linked immunosorbent assay (ELISA) for tau, α-synuclein and Aβ 1–40, Aβ 1–42, and validated by immunocytochemistry in primary neurons from NTG mice. For the extractions used in this study, tau constituted 22.6 to 35.7% of the total protein, while α-synuclein and Aβ constituted 0.011% or less of total protein.
Sandwich ELISA
Characterization of tau, α-synuclein and Aβ 1–40, Aβ 1–42 from AD PHF preparations by sandwich ELISA has been previously described (9). Assays were performed on 384-well MaxiSorp clear plates (Thermo Fisher Scientific). Plates were coated with well-characterized capture antibodies (tau: Tau5; α-synuclein: Syn9027; Aβ 1–40/Aβ 1–42: Ban50) at 4°C overnight, washed, and blocked with Block Ace (AbD Serotec) overnight at 4°C. AD PHF preparations were diluted at 1:100 and added to plates alongside serial dilutions of recombinant α-synuclein, recombinant T40, or peptides for Aβ 1–40 and Aβ 1–42 monomeric standards. Plates were incubated overnight at 4°C and then washed and incubated with reporter antibodies (tau: BT2 + BT7; α-synuclein: MJF-R1; Aβ 1–40: BA27; Aβ 1–42: BC05) overnight at 4°C. Plates were washed and incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies for 1 hour at 37°C. Plates were developed with 1-Step Ultra TMB-ELISA Substrate Solution (Thermo Fisher Scientific) for 10 to 15 min. The reaction was quenched with 10% phosphoric acid and read at 450 nm on a plate reader (M5, SpectraMax).
AD PHF stereotaxic injection
All surgery experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. AD PHF tau from individual extractions was vortexed and diluted with Dulbecco’s Phosphate Buffered Saline to 0.4 mg/ml. Tau was sonicated (QSonica Microson XL-2000; 60 pulses; setting 1.5; 1 s per pulse). Mice were injected at 3 to 4 months old. Mice were deeply anesthetized with ketamine/xylazine/acepromazine and injected unilaterally by insertion of a single needle into the right forebrain (coordinates: −2.5 mm relative to bregma and +2.0 mm from midline) targeting the hippocampus (2.4 mm beneath the skull) with 1 μg of tau (2.5 μl). The needle was then retracted to 1.4 mm beneath the skull, targeting the overlaying cortex, and another 1 μg of tau (2.5 μl) was injected. The needle was left in place for 2 min following the injection. Injections were performed using a 25-μl syringe (Hamilton, NV) at a rate of 0.4 μl/min.
Immunohistochemistry
After 1 to 9 months, mice were perfused transcardially with PBS; brains were removed and underwent overnight fixation in 70% ethanol in 150 mM NaCl (pH 7.4). After perfusion and fixation, brains were processed into paraffin via sequential dehydration and perfusion with paraffin under vacuum (70% ethanol for 2 hours, 80% ethanol for 1 hour, 95% ethanol for 1 hour, 95% ethanol for 2 hours, three times 100% ethanol for 2 hours, xylene for 30 min, xylene for 1 hour, xylene for 1.5 hours, and three times paraffin for 1 hour at 60°C). Brains were then embedded in paraffin blocks, cut into 6-μm sections, and mounted on glass slides. Slides were then stained using standard immunohistochemistry as described below. Slides were deparaffinized with two sequential 5-min washes in xylenes, followed by 1-min washes in a descending series of ethanols: 100, 100, 95, 80, and 70%. Slides were then incubated in deionized water for 1 min before microwave antigen retrieval in the BioGenex EZ-Retriever System. Slides were incubated in antigen unmasking solution (Vector Laboratories, catalog no. H-3300) and microwaved for 15 min at 95°C. Slides were allowed to cool for 20 min at room temperature and washed in running tap water for 10 min. Slides were incubated in 7.5% hydrogen peroxide in water to quench endogenous peroxidase activity. Slides were washed for 10 min in running tap water, 5 min in 0.1 M tris (diluted from 0.5 M tris made from tris base and concentrated hydrochloric acid to pH 7.6), and then blocked in 0.1 M tris/2% fetal bovine serum (FBS) for 15 min or more. Slides were incubated in primary antibody in 0.1 M tris/2% FBS in a humidified chamber overnight at 4°C. For pathologically phosphorylated tau, pS202/T205 tau (AT8, Thermo Fisher Scientific, catalog no. MN1020) was used at 1:10,000.
Primary antibody was rinsed off with 0.1 M tris, and slides were incubated in 0.1 M tris for 5 min. Primary antibody was detected using a BioGenex Polymer detection kit (catalog no. QD440-XAK) per the manufacturer’s protocol as outlined below. Slides were incubated in 50% enhancer solution in 0.1 M tris/2% FBS for 20 min. Enhancer was rinsed off with 0.1 M tris, incubated in 0.1 M tris for 5 min, and incubated in 0.1 M tris/2% FBS for 5 min. Slides were then incubated in 50% poly-HRP in 0.1 M tris/2% FBS for 30 min. Poly-HRP was rinsed off with 0.1 M tris; slides were then incubated for 5 min with 0.1 M tris and then developed with ImmPACT diaminobenzidine (DAB) peroxidase substrate (Vector SK-4105) for 10 min. DAB was rinsed off with 0.1 M tris and incubated in distilled water for 5 min. Slides were then counterstained briefly with Harris hematoxylin (Thermo Fisher Scientific, catalog no. 6765001). Slides were washed in running tap water for 5 min, dehydrated in ascending ethanol for 1 min each (70, 80, 95, 100, and 100%), then washed twice in xylenes for 5 min, and coverslipped in Cytoseal Mounting Media (Thermo Fisher Scientific, catalog no. 23-244-256). Slides were scanned into digital format on a Lamina scanner (PerkinElmer) using a 20× objective (0.8 numerical aperture) into .mrxs files. Digitized slides were then used for quantitative pathology.
Quantitative pathology
All section selection, annotation, and quantification were done blinded to treatment. For quantification of tau pathology, coronal sections were selected to closely match the following coordinates, relative to bregma: 3.20, 0.98, −1.22, −2.92, −3.52, and − 4.48 mm. The digitized images were imported into HALO software to allow annotation and quantification of the percentage area occupied by tau pathology. Standardized annotations were drawn to allow independent quantification of 194 gray matter regions throughout the brain. Brain region annotations were largely based on the ABA (CCF v3, 2017; brain-map.org), although smaller subregions that are difficult to annotate by eye and have minimal pathology were grouped to minimize error and annotation time. For example, thalamic and midbrain nuclei, which accumulate minimal pathology, were grouped into larger regions (fig. S1). Each set of annotations was imported onto the desired section and modified by hand to match the designated brain regions. After annotation, the analysis scripts were applied to the brain to make sure that no nonpathology signal was detected. After annotation of all brains, analysis algorithms were applied to all stained sections, and data analysis measures for each region were recorded.
The total pathology analysis detects total signal above a minimum threshold. Specifically, the analysis included all DAB signal that was above a 0.099 optical density threshold, which was empirically determined to not include any background signal. This signal was then normalized to the total tissue area. A minimal tissue optical density of 0.02 was used to exclude any areas where tissue was split, and a tissue-edge thickness of 25.2 μm was applied to exclude any edge effect staining. Quantitative pathology measures for the 48 regions with substantial pathology were analyzed to determine whether there was any overall genotype effect or a time-dependent effect of genotype on pathology, using a cumulative logistic model. For each region, an interaction between genotype and months was initially investigated via a likelihood ratio test (LRT); this interaction was dropped for models with P > 0.15 based on the LRT. LRTs were again performed to determine whether there existed statistically significant genotype difference either across all time points or whether genotype differences changed over time. P values from these LRTs were adjusted using the Benjamini-Hochberg FDR to account for multiple testing of 48 regions. Post hoc tests were then performed via the emmeans package in R with, again, an FDR adjustment for multiple testing. Second-generation P values were generated, as described in (51), with a null interval of odds ratio within 0.9 to 1.1, were also calculated. In general, second-generation P values control the familywise error rate and FDR but can often identify additional differences when the number of comparisons is greater than 10. The null interval of 0.9 to 1.1 (i.e., within 10% difference), was chosen as a conservative range.
Computational models of pathological protein spread
Models of linear diffusion along white matter fibers have been used to predict the spread of misfolded α-synuclein in mice (28), as well as patterns of atrophy observed in various neurodegenerative diseases (12, 52). In the present work, we extended these models to the spread of tau between anatomically connected brain regions from an injection site in the right hippocampus.
We model pathological spread of tau as a diffusion process on a directed structural brain network G = {V, E} whose nodes V are Na = 426 cortical and subcortical gray matter regions and whose edges eij ∈ E represent an axonal projection initiating in Vi and terminating in Vj, where eij ≥ 0 for all E. Edge strength was quantified by the Allen Brain Institute using measures of fluorescence intensity from anterograde viral tract tracing (27). We define the weighted adjacency matrix of G as A = [Aij], such that row i contains the strengths of axonal projections from region i to all other Na − 1 regions. We define the current levels of simulated tau pathology of all Na nodes at time t as the vector x(t). We make empirical measurements of tau pathology at t = 1, 3, 6, and 9 MPI in an Nc = 134 region vector y(t), which is a spatially coarse-grained version of x(t). Note that y(t) = f(x(t)), where f is a linear transformation that sets each element (region) of y(t) equal to the arithmetic mean of the elements (regions) of x(t) that lie within the regions of y(t). This transformation is needed to avoid quantifying pathology in many of the smaller regions in the Na-dimensional space used by the Allen Brain Institute (27), which are difficult to identify reliably across mice in practice.
In the simplest case of our models of pathological network spread, we simulated the spread of tau pathology throughout the Na anatomically connected brain regions in A to compute the predicted pathology in Nc empirically assessed brain regions as a function of a set of seed regions s ∈ V using the form
where the retrograde, out-degree graph Laplacian
cr is a diffusion rate constant representing the global speed of retrograde spread, f is the linear transformation described above that converts the Na region space to the Nc region space, and t is in units of months. In this manuscript, s contains the ABA regions DG, CA1, CA3, VISam, and RSPagl, to account for experimental variability in targeting a hippocampal injection site. Note that cr tunes the time scale of the system, which is necessary because of the fact that the units of connection strength are arbitrary relative to the units of pathology. To fit this model, we swept through values of cr from 10−5 to 0.2 and chose the value of cr that maximized the average Pearson correlation coefficient between log10 y(t) and over t = [1 3 6 9]. Empirically, the value of this correlation plateaued for values of cr larger than 0.2, justifying this upper bound on cr. Note that L is the out-degree Laplacian, a version of the well-characterized graph Laplacian designed for directed graphs (53). Intuitively, this model posits that pathology spreads retrogradely from region i to other regions at a rate proportional to the number of synapses projecting onto i from those regions, while pathology at region i decays as a function of the sum of the strength of projections into region i.
Recent studies by this group (28) and others (54) have suggested that both anterograde and retrograde spread of pathology contribute to neurodegenerative disease progression. Thus, we expanded the retrograde model described above to a bidirectional model including anterograde spread, using the form
where the anterograde, out-degree graph Laplacian
ca is a diffusion rate constant representing the global speed of anterograde spread, bo is an intercept, ba is a weight for the importance of anterograde spread, br is a weight for the importance of retrograde spread, t is time, and ε is an error term. We used the “optim” function in R to solve for the combination of cr and ca that maximizes the average Pearson correlation coefficient between log10 y(t) and over t = [1 3 6 9], with a linear regression inside of the objective function to solve for bo, ba, and br aggregating across all time points for each cr and ca. The linear regression coefficients ba, and br, when standardized, capture the relative importance of anterograde and retrograde spread at time t, respectively, while controlling for the potentially ambiguous overlapping contributions of the two modes of transmission.
We also defined intrinsic regional vulnerability based on ε(t), the error term in the model above. Intuitively, if εi(t) is large, then this model underpredicted pathology at region i such that region i is more vulnerable to pathology than expected on the basis of bidirectional linear diffusion and vice versa for regions with small values of ε(t). We hypothesized that both static, intrinsic regional vulnerability and possible time-dependent vulnerability are captured by ε(t). Therefore, we averaged ε(t) over hemispheres and over t = [3 6 9] to capture static, intrinsic regional vulnerability as an vector vs and average out the effects of both time-dependent vulnerability and unexplainable measurement error. We excluded 1 MPI because pathology at this time point was poorly captured by the spread model, and ε(1) exhibited a distinct spatial pattern of vulnerability from the patterns observed in ε(t) at t = [3 6 9] (fig. S6).
Quantification of model specificity
In a previous study (28), we found that the substantial variance explained in misfolded α-synuclein spread by connectome-based linear diffusion models was specific to the use of ipsilateral caudoputamen as the seed site s, which defines the vector xo, over nearly every other region in the connectome. Here, we sought to replicate this finding in the study of tau spread. However, because of the use of multiple seed sites in the present study, additional considerations applied in ensuring a rigorous test of the model’s specificity to the experimentally motivated group of seed sites. The five chosen seed sites in s (DG, CA1, CA3, VISam, and RSPagl) were relatively close together (average distance of 1.96 mm), so we wanted to rule out the possibility that (i) choosing multiple random sites would trivially improve model performance and (ii) selecting multiple spatially clustered random seed sites would trivially improve model performance. Thus, we choose 500 random sets of five seed sites snull ∈ V, all of which had an average distance from one another within 1.96 ± 0.196 mm, and fit the bidirectional model detailed above using each set of distance-constrained random points snull to define the initial vector xo. We computed a one-tailed, nonparametric P value for the specificity of s by computing the percentage of times snull yielded a better fit to the observed pathology data than s.
Model comparison
The data presented in the body of the paper use values for the diffusion rate constants, cr and ca, and time-dependent weights on retrograde and anterograde spread, bo, ba, and br, obtained using data from all mice at every time point (yfull(t)). To ensure that this approach did not result in overfitting and to rigorously test whether including both anterograde and retrograde connections improved model performance, we randomly sampled without replacement the available mice at each time point to generate ytrain(t) and ytest(t) for each time point. These parameters were determined by applying the model fitting procedure described above on ytrain(t), and the model was evaluated on the basis of its fit with ytest(t). This process was repeated 500 times for models based on Euclidean distance, anterograde connections alone, retrograde connections alone, or both anterograde and retrograde connections, generating a distribution of out-of-sample fits for each time point for each model, allowing us to statistically compare the out-of-sample performance between each of the candidate models. Using a similar approach, we generated distributions of model parameters for the bidirectional model using bootstrapped samples of NTG and LRRK2 G2019S mice.
Network null models
To ensure that our results were specific to the retrograde spread of misfolded synuclein along neuronal processes, we repeated our analyses using several network null models. To demonstrate a general specificity of the model for the topology of the anatomic connectome represented by A, we carried out a procedure that rewires the edges of G while exactly preserving either the out-degree or the in-degree sequence, i.e.,
This rewiring approach tests whether the model fit is due to a relatively basic structural property of the graph, i.e., degree, as opposed to unique, higher-order topological features of the synaptic connectome. Last, we tested the null model that spread of misfolded protein occurs simply because of diffusion through tissue based on closeness in Euclidean space. To test this model, we reconstructed A = [Aij] such that the edges represented the inverse Euclidean distance between region i and region j. We used the initial procedure for retrograde model fitting to test this model, because Euclidean distance is symmetric and thus the bidirectional model cannot be applied.
Assessment of regional gene expression patterns
To assess the cellular and molecular characteristics of intrinsically vulnerable or resilient regions, we compared the spatial alignment between our vector of regional vulnerability measurements, vs, with microarray gene expression levels obtained from the Allen Mouse Brain Atlas (brain-map.org). After applying a previously validated quality control approach to hone in on genes with the most reliable expression measurements (55), we obtained G, an matrix of gene expression values across each brain region. Because of the non-normality of both vs and gene expression in G, we applied a rank inverse normalization transform (56) to each column of G to control type I error and maximize statistical power. Next, we computed 4277 spatial Pearson correlations between vs and each column of G. We performed multiple comparisons correction by controlling the FDR at q < 0.05.
Quantification and statistical analysis
The number of animals analyzed in each experiment, the statistical analysis performed, and the P values for all results <0.05 are reported in the figure legends. In vivo pathological spread data were analyzed, and all computations were performed in R (www.R-project.org/) (57) as described.
Acknowledgments
We would like to thank members of the laboratory for feedback in developing this manuscript and the Van Andel Research Institute Bioinformatics and Biostatistics Core, especially Z. Madaj, for assistance with statistical analysis of tau pathology. Funding: This study was supported by the Michael J. Fox Foundation for Parkinson’s Research grant 16879 (to M.X.H.); NIH grants T32-AG000255 (to V.M.Y.L.), P30-AG10124 (to J.Q.T.), U19-AG062418 (to J.Q.T.), P50-NS053488 (to J.Q.T.), R01-NS099348 (to D.S.B.), F30 MH118871-01 (to E.J.C.); and NSF grants PHY-1554488 (to D.S.B.) and BCS-1631550 (to D.S.B.). D.S.B. also acknowledges support from the John D. and Catherine T. MacArthur Foundation, the ISI Foundation, the Alfred P. Sloan Foundation, and the Paul G. Allen Foundation. Author contributions: Conceptualization: M.X.H. and V.M.Y.L. Methodology: M.X.H., E.J.C., and V.M.Y.L. Software: E.J.C. and D.S.B. Formal analysis: M.X.H. and E.J.C. Investigation: M.X.H., E.J.C., H.L.L., L.C., B.Z., H.J.B., R.J.G., and M.F.O. Resources: L.C. Writing—original draft, M.X.H. and E.J.C. Writing—review and editing, all. Visualization: M.X.H. and E.J.C. Supervision: M.X.H., D.S.B., J.Q.T., and V.M.Y.L. Funding acquisition: M.X.H., D.S.B., J.Q.T., and V.M.Y.L. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Primary data and code used to generate the spread modeling are available on GitHub (https://github.com/ejcorn/tau-spread).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg6677/DC1
REFERENCES AND NOTES
- 1.GBD 2016 Parkinson’s Disease Collaborators , Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.GBD 2016 Dementia Collaborators , Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 88–106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robinson J. L., Lee E. B., Xie S. X., Rennert L., Suh E. R., Bredenberg C., Caswell C., van Deerlin V. M., Yan N., Yousef A., Hurtig H. I., Siderowf A., Grossman M., McMillan C. T., Miller B., Duda J. E., Irwin D. J., Wolk D., Elman L., McCluskey L., Chen-Plotkin A., Weintraub D., Arnold S. E., Brettschneider J., Lee V. M. Y., Trojanowski J. Q., Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141, 2181–2193 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coughlin D. G., Hurtig H. I., Irwin D. J., Pathological influences on clinical heterogeneity in Lewy body diseases. Mov. Disord. 35, 5–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coughlin D., Xie S. X., Liang M., Williams A., Peterson C., Weintraub D., McMillan C. T., Wolk D. A., Akhtar R. S., Hurtig H. I., Coslett H. B., Hamilton R. H., Siderowf A. D., Duda J. E., Rascovsky K., Lee E. B., Lee V. M.-Y., Grossman M., Trojanowski J. Q., Irwin D. J., Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann. Neurol. 85, 259–271 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irwin D. J., Grossman M., Weintraub D., Hurtig H. I., Duda J. E., Xie S. X., Lee E. B., van Deerlin V. M., Lopez O. L., Kofler J. K., Nelson P. T., Jicha G. A., Woltjer R., Quinn J. F., Kaye J., Leverenz J. B., Tsuang D., Longfellow K., Yearout D., Kukull W., Keene C. D., Montine T. J., Zabetian C. P., Trojanowski J. Q., Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol. 16, 55–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H., Braak E., Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 8.Braak H., Alafuzoff I., Arzberger T., Kretzschmar H., Del Tredici K., Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo J. L., Narasimhan S., Changolkar L., He Z., Stieber A., Zhang B., Gathagan R. J., Iba M., McBride J. D., Trojanowski J. Q., Lee V. M. Y., Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 213, 2635–2654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Narasimhan S., Guo J. L., Changolkar L., Stieber A., McBride J. D., Silva L. V., He Z., Zhang B., Gathagan R. J., Trojanowski J. Q., Lee V. M. Y., Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J. Neurosci. 37, 11406–11423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McAllister B. B., Lacoursiere S. G., Sutherland R. J., Mohajerani M. H., Intracerebral seeding of amyloid-β and tau pathology in mice: Factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci. Biobehav. Rev. 112, 1–27 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Raj A., Kuceyeski A., Weiner M., A network diffusion model of disease progression in dementia. Neuron 73, 1204–1215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown J. A., Deng J., Neuhaus J., Sible I. J., Sias A. C., Lee S. E., Kornak J., Marx G. A., Karydas A. M., Spina S., Grinberg L. T., Coppola G., Geschwind D. H., Kramer J. H., Gorno-Tempini M. L., Miller B. L., Rosen H. J., Seeley W. W., Patient-tailored, connectivity-based forecasts of spreading brain atrophy. Neuron 104, 856–868.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ossenkoppele R., Iaccarino L., Schonhaut D. R., Brown J. A., la Joie R., O’Neil J. P., Janabi M., Baker S. L., Kramer J. H., Gorno-Tempini M. L., Miller B. L., Rosen H. J., Seeley W. W., Jagust W. J., Rabinovici G. D., Tau covariance patterns in Alzheimer’s disease patients match intrinsic connectivity networks in the healthy brain. NeuroImage Clin. 23, 101848 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel J. W., Iturria-Medina Y., Strandberg O. T., Smith R., Levitis E., Evans A. C., Hansson O.; Alzheimer’s Disease Neuroimaging Initiative; Swedish BioFinder Study , Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat. Commun. 11, 2612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fornari S., Schäfer A., Jucker M., Goriely A., Kuhl E., Prion-like spreading of Alzheimer’s disease within the brain’s connectome. J. R. Soc. Interface 16, 20190356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weickenmeier J. J., Jucker M., Goriely A., Kuhl E., A physics-based model explains the prion-like features of neurodegeneration in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. J. Mech. Phys. Solids 124, 264–281 (2019). [Google Scholar]
- 18.Saman S., Kim W. H., Raya M., Visnick Y., Miro S., Saman S., Jackson B., McKee A. C., Alvarez V. E., Lee N. C. Y., Hall G. F., Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y., Balaji V., Kaniyappan S., Krüger L., Irsen S., Tepper K., Chandupatla R. R., Maetzler W., Schneider A., Mandelkow E., Mandelkow E. M., The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12, 5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katsinelos T., Zeitler M., Dimou E., Karakatsani A., Müller H. M., Nachman E., Steringer J. P., Ruiz de Almodovar C., Nickel W., Jahn T. R., Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep. 23, 2039–2055 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Wu J. W., Hussaini S. A., Bastille I. M., Rodriguez G. A., Mrejeru A., Rilett K., Sanders D. W., Cook C., Fu H., Boonen R. A. C. M., Herman M., Nahmani E., Emrani S., Figueroa Y. H., Diamond M. I., Clelland C. L., Wray S., Duff K. E., Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holth J. K., Fritschi S. K., Wang C., Pedersen N. P., Cirrito J. R., Mahan T. E., Finn M. B., Manis M., Geerling J. C., Fuller P. M., Lucey B. P., Holtzman D. M., The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363, 880–884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holmes B. B., DeVos S. L., Kfoury N., Li M., Jacks R., Yanamandra K., Ouidja M. O., Brodsky F. M., Marasa J., Bagchi D. P., Kotzbauer P. T., Miller T. M., Papy-Garcia D., Diamond M. I., Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. U.S.A. 110, E3138–E3147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Usenovic M., Niroomand S., Drolet R. E., Yao L., Gaspar R. C., Hatcher N. G., Schachter J., Renger J. J., Parmentier-Batteur S., Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J. Neurosci. 35, 14234–14250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauch J. N., Chen J. J., Sorum A. W., Miller G. M., Sharf T., See S. K., Hsieh-Wilson L. C., Kampmann M., Kosik K. S., Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci. Rep. 8, 6382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rauch J. N., Luna G., Guzman E., Audouard M., Challis C., Sibih Y. E., Leshuk C., Hernandez I., Wegmann S., Hyman B. T., Gradinaru V., Kampmann M., Kosik K. S., LRP1 is a master regulator of tau uptake and spread. Nature 580, 381–385 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oh S. W., Harris J. A., Ng L., Winslow B., Cain N., Mihalas S., Wang Q., Lau C., Kuan L., Henry A. M., Mortrud M. T., Ouellette B., Nguyen T. N., Sorensen S. A., Slaughterbeck C. R., Wakeman W., Li Y., Feng D., Ho A., Nicholas E., Hirokawa K. E., Bohn P., Joines K. M., Peng H., Hawrylycz M. J., Phillips J. W., Hohmann J. G., Wohnoutka P., Gerfen C. R., Koch C., Bernard A., Dang C., Jones A. R., Zeng H., A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henderson M. X., Cornblath E. J., Darwich A., Zhang B., Brown H., Gathagan R. J., Sandler R. M., Bassett D. S., Trojanowski J. Q., Lee V. M. Y., Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat. Neurosci. 22, 1248–1257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B. Methodol. 57, 289–300 (1995). [Google Scholar]
- 30.Healy D. G., Falchi M., O’Sullivan S. S., Bonifati V., Durr A., Bressman S., Brice A., Aasly J., Zabetian C. P., Goldwurm S., Ferreira J. J., Tolosa E., Kay D. M., Klein C., Williams D. R., Marras C., Lang A. E., Wszolek Z. K., Berciano J., Schapira A. H., Lynch T., Bhatia K. P., Gasser T., Lees A. J., Wood N. W.; International LRRK2 Consortium , Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol. 7, 583–590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poulopoulos M., Levy O. A., Alcalay R. N., The neuropathology of genetic Parkinson’s disease. Mov. Disord. 27, 831–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalia L. V., Lang A. E., Hazrati L. N., Fujioka S., Wszolek Z. K., Dickson D. W., Ross O. A., van Deerlin V. M., Trojanowski J. Q., Hurtig H. I., Alcalay R. N., Marder K. S., Clark L. N., Gaig C., Tolosa E., Ruiz-Martínez J., Marti-Masso J. F., Ferrer I., López de Munain A., Goldman S. M., Schüle B., Langston J. W., Aasly J. O., Giordana M. T., Bonifati V., Puschmann A., Canesi M., Pezzoli G., Maues de Paula A., Hasegawa K., Duyckaerts C., Brice A., Stoessl A. J., Marras C., Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 72, 100–105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henderson M. X., Sengupta M., Trojanowski J. Q., Lee V. M. Y., Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 7, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blauwendraat C., Pletnikova O., Geiger J. T., Murphy N. A., Abramzon Y., Rudow G., Mamais A., Sabir M. S., Crain B., Ahmed S., Rosenthal L. S., Bakker C. C., Faghri F., Chia R., Ding J., Dawson T. M., Pantelyat A., Albert M. S., Nalls M. A., Resnick S. M., Ferrucci L., Cookson M. R., Hillis A. E., Troncoso J. C., Scholz S. W., Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol. Aging 76, 214.e1–214.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mezias C., LoCastro E., Xia C., Raj A., Connectivity, not region-intrinsic properties, predicts regional vulnerability to progressive tau pathology in mouse models of disease. Acta Neuropathol. Commun. 5, 61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schapansky J., Khasnavis S., DeAndrade M. P., Nardozzi J. D., Falkson S. R., Boyd J. D., Sanderson J. B., Bartels T., Melrose H. L., LaVoie M. J., Familial knockin mutation of LRRK2 causes lysosomal dysfunction and accumulation of endogenous insoluble α-synuclein in neurons. Neurobiol. Dis. 111, 26–35 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matikainen-Ankney B. A., Kezunovic N., Mesias R. E., Tian Y., Williams F. M., Huntley G. W., Benson D. L., Altered development of synapse structure and function in striatum caused by Parkinson’s disease-linked LRRK2-G2019S mutation. J. Neurosci. 36, 7128–7141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beccano-Kelly D. A., Kuhlmann N., Tatarnikov I., Volta M., Munsie L. N., Chou P., Cao L.-P., Han H., Tapia L., Farrer M. J., Milnerwood A. J., Synaptic function is modulated by LRRK2 and glutamate release is increased in cortical neurons of G2019S LRRK2 knock-in mice. Front. Cell. Neurosci. 8, 301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y., Liu W., Oo T. F., Wang L., Tang Y., Jackson-Lewis V., Zhou C., Geghman K., Bogdanov M., Przedborski S., Beal M. F., Burke R. E., Li C., Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 12, 826–828 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melrose H. L., Dächsel J. C., Behrouz B., Lincoln S. J., Yue M., Hinkle K. M., Kent C. B., Korvatska E., Taylor J. P., Witten L., Liang Y. Q., Beevers J. E., Boules M., Dugger B. N., Serna V. A., Gaukhman A., Yu X., Castanedes-Casey M., Braithwaite A. T., Ogholikhan S., Yu N., Bass D., Tyndall G., Schellenberg G. D., Dickson D. W., Janus C., Farrer M. J., Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin C. H., Tsai P. I., Wu R. M., Chien C. T., LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J. Neurosci. 30, 13138–13149 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohta E., Nihira T., Uchino A., Imaizumi Y., Okada Y., Akamatsu W., Takahashi K., Hayakawa H., Nagai M., Ohyama M., Ryo M., Ogino M., Murayama S., Takashima A., Nishiyama K., Mizuno Y., Mochizuki H., Obata F., Okano H., I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased tau phosphorylation through the AKT/GSK-3β signaling pathway. Hum. Mol. Genet. 24, 4879–4900 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Kawakami F., Yabata T., Ohta E., Maekawa T., Shimada N., Suzuki M., Maruyama H., Ichikawa T., Obata F., LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLOS ONE 7, e30834 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailey R. M., Covy J. P., Melrose H. L., Rousseau L., Watkinson R., Knight J., Miles S., Farrer M. J., Dickson D. W., Giasson B. I., Lewis J., LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 126, 809–827 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shanley M. R., Hawley D., Leung S., Zaidi N. F., Dave R., Schlosser K. A., Bandopadhyay R., Gerber S. A., Liu M., LRRK2 facilitates tau phosphorylation through strong interaction with tau and cdk5. Biochemistry 54, 5198–5208 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Hamm M., Bailey R., Shaw G., Yen S. H., Lewis J., Giasson B. I., Physiologically relevant factors influence tau phosphorylation by leucine-rich repeat kinase 2. J. Neurosci. Res. 93, 1567–1580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen A. P. T., Daniel G., Valdés P., Islam M. S., Schneider B. L., Moore D. J., G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum. Mol. Genet. 27, 120–134 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Mikhail F., Calingasan N., Parolari L., Subramanian A., Yang L., Flint Beal M., Lack of exacerbation of neurodegeneration in a double transgenic mouse model of mutant LRRK2 and tau. Hum. Mol. Genet. 24, 3545–3556 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Li X., Patel J. C., Wang J., Avshalumov M. V., Nicholson C., Buxbaum J. D., Elder G. A., Rice M. E., Yue Z., Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irwin D. J., White M. T., Toledo J. B., Xie S. X., Robinson J. L., van Deerlin V., Lee V. M. Y., Leverenz J. B., Montine T. J., Duda J. E., Hurtig H. I., Trojanowski J. Q., Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 72, 587–598 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Blume J. D., Greevy R. A., Welty V. F., Smith J. R., Dupont W. D., An introduction to second-generation P values. Am. Stat. 73, 157–167 (2019). [Google Scholar]
- 52.Pandya S., Mezias C., Raj A., Predictive model of spread of progressive supranuclear palsy using directional network diffusion. Front. Neurol. 8, 692 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.W. H. R. Chan, M. Wildemeersch, T. Q. S. Quek, Characterization and control of diffusion processes in multi-agent networks. arXiv:https://arxiv.org/abs/1508.06738 [cs.SI] (27 August 2015).
- 54.Mezias C., Rey N., Brundin P., Raj A., Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 134, 104623 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fulcher B. D., Murray J. D., Zerbi V., Wang X. J., Multimodal gradients across mouse cortex. Proc. Natl. Acad. Sci. U.S.A. 116, 4689–4695 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Auer P. L., Reiner A. P., Leal S. M., The effect of phenotypic outliers and non-normality on rare-variant association testing. Eur. J. Hum. Genet. 24, 1188–1194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team, R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2020); www.R-project.org/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg6677/DC1