

Abstract
Hepatocellular carcinoma (HCC) belongs to the most frequent cancer with a high death rate worldwide. Thousands of long non-coding RNAs (lncRNAs) have been confirmed to influence the development of human cancers, including HCC. Nevertheless, the biological role of PRR34 antisense RNA 1 (PRR34-AS1) in HCC remains obscure. Here, we observed via quantitative real-time reverse transcriptase polymerase chain reaction (quantitative real-time RT-PCR) that PRR34-AS1 was highly expressed in HCC cells. Functional assays revealed that PRR34-AS1 promoted HCC cell proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) process in vitro and facilitated tumor growth in vivo. In addition, western blot analysis and TOP Flash/FOP Flash reporter assays verified that PRR34-AS1 stimulated Wnt/β-catenin pathway in HCC cells. Furthermore, RNA immunoprecipitation (RIP), RNA pull-down, and luciferase reporter assays uncovered that PRR34-AS1 sequestered microRNA-296-5p (miR-296-5p) to positively modulate E2F transcription factor 2 (E2F2) and SRY-box transcription factor 12 (SOX12) in HCC cells. Importantly, chromatin immunoprecipitation (ChIP) and luciferase reporter assays uncovered that E2F2 transcriptionally activated PRR34-AS1 in turn. Further, rescue experiments reflected that PRR34-AS1 affected HCC progression through targeting miR-296-5p/E2F2/SOX12/Wnt/β-catenin axis. Our findings found that PRR34-AS1 elicited oncogenic functions in HCC, which indicated that PRR34-AS1 might be a novel therapeutic target for HCC.
Keywords: PRR34-AS1, E2F2, SOX12, Wnt/β-catenin, hepatocellular carcinoma
Graphical abstract
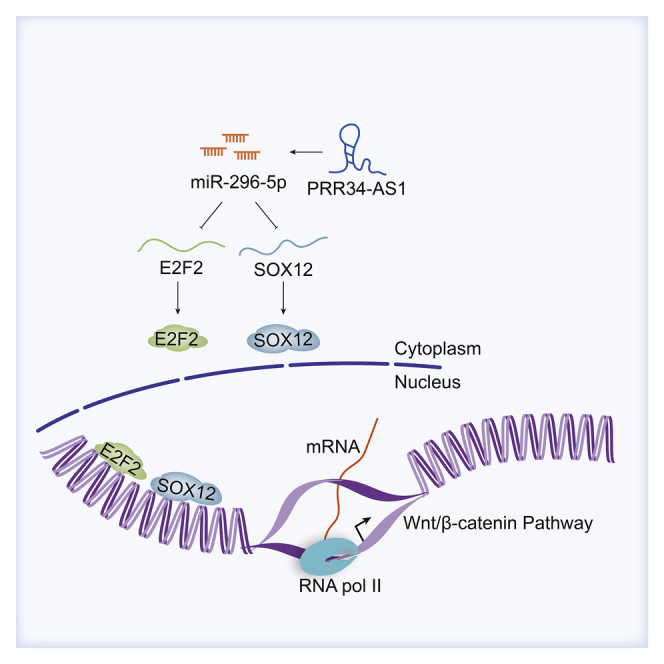
Huang and colleagues unmasked that long non-coding RNA PRR34-AS1 sequesters miR-296-5p to elevate E2F2 and SOX12, therefore activating Wnt/β-catenin pathway and facilitating hepatocellular carcinoma development. The findings supported PRR34-AS1 as a possible novel target for hepatocellular carcinoma treatment.
Introduction
Hepatocellular carcinoma (HCC) pertains to a type of most frequently diagnosed malignancies among men and women. Although main therapeutic methods including liver resection, hepatic resection, and radiofrequency ablation are broadly applied, HCC patients still suffer from poor survival rates and high recurrence.1 Besides, for the absences of a systemic surveillance program and advanced screening tool, HCC cases are usually diagnosed at advanced stages, which brings a great challenge for treatment.2 Therefore, to improve the treatment effect for prolonging the survival time of HCC patients, it is urgent to explore the potential molecules involved in HCC development.
In recent years, with the development of RNA sequencing technology and biological science, increasing attention has been paid to lncRNAs.3 Long non-coding RNAs (lncRNAs), as a group of ncRNAs with the length exceeding 200 nucleotides, play a key role in gene regulation and disease development.4 Accumulating reports have indicated that aberrant lncRNAs impact tumor formation and progression of human cancers by regulating cell proliferation, apoptosis, drug resistance, and metastasis, including those of HCC.5, 6, 7 For instance, a study from Xiao et al.8 has demonstrated that ribosomal protein L13a pseudogene 20 (HANR) upregulation promotes HCC cell proliferation and tumor growth. Another study revealed that SBF2-AS1 overexpression promotes epithelial-mesenchymal transition (EMT) process in HCC cells and causes short overall survival of HCC patients.9 Recently, a novel lncRNA, PRR34-AS1 has been discovered to affect Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway,10 which is closely linked to cancer development.11 However, its role has been rarely illustrated in cancer. Therefore, in this study, we aimed at investigating the role and regulatory mechanism of PRR34-AS1 in HCC.
As we all know, many important signaling pathways such as Notch signaling pathway, Wnt/β-catenin and Hedgehog pathway are closely related to the development of cancers.12, 13, 14 At present, mounting studies have demonstrated that the activation of Wnt/β-catenin pathway accelerates tumor growth and dissemination in HCC.15,16 In addition, Zhu et al.17 have revealed that CRNDE silencing inhibits the Wnt/β-catenin pathway to block the EMT process in HCC cells. Based on these data, this study analyzed the association between PRR34-AS1 and Wnt/β-catenin pathway in HCC.
Moreover, studies have indicated that lncRNAs can exert as a competing endogenous RNA (ceRNA) via competitively binding to microRNAs (miRNAs) to regulate mRNAs.18,19 For instance, MCM3AP-AS1 targets miR-194-5p to upregulate FOXA1 and thereby facilitate HCC cell growth.20 Similarly, CDKN2BAS modulates miR-153-5p/ARHGAP18 axis to contribute to HCC metastasis.21 In this study, miR-296-5p was specified as the potential miRNA interacting with PRR34-AS1 through LncBase prediction and mechanistic investigation. Nowadays, miR-296-5p has been documented to depress EMT process in HCC cells through inhibiting NRG1.22 Thereafter, SRY-box transcription factor 12 (SOX12) and E2F transcription factor 2 (E2F2) were recognized as miR-296-5p targets in HCC cells through bioinformatics analyses and experimental confirmation. Interestingly, Wan et al.23 have indicated that SOX12 has activating influence on Wnt/β-catenin pathway. However, the precise relationship among PRR34-AS1, miR-296-5p, SOX12, and Wnt/β-catenin in HCC has not been well defined yet. As for E2F2, it is a key member of E2F family that involves in cancer progression.24 In recent years, accumulating studies have showed that transcription factors can affect the transcription of lncRNAs in diverse cancers.25 For instance, STAT3 promotes HOXD-AS1 transcription in HCC.26 Nevertheless, whether there is certain association between E2F2 and PRR34-AS1 in HCC cells remains to be explored.
In brief, the current study principally explored the expression and biological functions of PRR34-AS1 in HCC. Meanwhile, the relationships among PRR34-AS1, miR-296-5p, E2F2, SOX12, and Wnt/β-catenin signaling pathway in HCC cells were also analyzed, respectively.
Results
PRR34-AS1 is overexpressed in HCC cells
lncRNA PRR34-AS1 located at chr22: 46449726–46454402 is the antisense of PRR34 gene in genome, while its association with HCC development is still unknown. To probe the role of lncRNA PRR34-AS1 in HCC, we first used University of California Santa Cruz database (UCSC Data: http://genome.ucsc.edu/) to observe the expression of PRR34-AS1 in multiple human normal tissues. As disclosed in Figure 1A, the PRR34-AS1 expression was low in normal liver tissues. The similar result was exhibited in NONCODE (NONCODE data: http://www.noncode.org/) database that the level of PRR34-AS1 in liver is relatively lower than that in other organs (Figure 1B). Subsequently, we searched for the expression pattern of PRR34-AS1 in liver HCC (LIHC) tissues based on Gene Expression Profiling Interactive Analysis v2.0 database (GEPIA2 data: http://gepia.cancer-pku.cn/). The result reflected that PRR34-AS1 expression was elevated in 369 LIHC tissues compared to 160 normal liver tissues (Figure 1C). Besides, we validated that PRR34-AS1 expressed higher in 50 HCC samples than in paired non-cancerous tissues (Figure S1A). Moreover, with quantitative real-time reverse transcriptase polymerase chain reaction (quantitative real-time RT-PCR) analysis, elevated expression of PRR34-AS1 was also observed in HCC cells (Hep 3B, SK-HEP-1, Huh7, MHCC97-H, and HCCLM3) compared to human normal hepatocyte cells (THLE-3; Figure 1D). Based on this, MHCC97-H/HCCLM3 cells with higher PRR34-AS1 expression and Hep 3B cells with lowest PRR34-AS1 expression were chosen for follow-up assays. To determine the underlying mechanism of PRR34-AS1 in HCC cells, we adopted subcellular fractionation and fluorescence in situ hybridization (FISH) assays to observe its distribution in HCCLM3 cells. Results suggested that PRR34-AS1 was chiefly scattered in the cytoplasm of HCCLM3 cells (Figures 1E and 1F), which reflected that PRR34-AS1 might function in gene regulation at the post-transcriptional level. In order to estimate the biological function of PRR34-AS1 in HCC cells, MHCC97-H and HCCLM3 cells were transfected with sh-PRR34-AS1#1/#2/#3 to silence PRR34-AS1 for loss-of-function experiments. Meanwhile, Hep 3B cells were transfected with pcDNA3.1/PRR34-AS1 to overexpress PRR34-AS1 for gain-of-function assays. As expected, the results of quantitative real-time RT-PCR uncovered that PRR34-AS1 expression was stably depleted in MHCC97-H and HCCLM3 cells with the transfection of sh-PRR34-AS1#1/#2/#3 and was elevated in Hep 3B cells after the transfection of pcDNA3.1/PRR34-AS1 (Figure 1G). Notably, sh-PRR34-AS1#1 and sh-PRR34-AS1#2 were selected for later experiments on account of their obvious inhibitory efficiency.
Figure 1.

PRR34-AS1 expression is elevated in HCC cells
(A) UCSC database presented PRR34-AS1 expression profile in multiple human normal tissues, including liver tissues. (B) NONCODE database indicated the profile of PRR34-AS1 expression in different normal tissue samples. (C) GEPIA2 database exhibited PRR34-AS1 expression pattern in 369 cases of LIHC tissues and 160 cases of normal tissues. (D) Quantitative real-time RT-PCR analyzed PRR34-AS1 expression in HCC cell lines (Hep 3B, SK-HEP-1, Huh7, MHCC97-H, and HCCLM3) and human normal hepatocyte cell line (THLE-3). (E and F) Subcellular fractionation and FISH experiments determined PRR34-AS1 location in HCC cells. (G) Knockdown efficiencies of sh-PRR34-AS1#1&2&3 in MHCC97-H and HCCLM3 cells, as well as overexpression efficiency of pcDNA3.1/PRR34-AS1 in Hep 3B cells, were evaluated via quantitative real-time RT-PCR. ∗p < 0.05, ∗∗p < 0.01.
PRR34-AS1 facilitates HCC cell proliferation in vitro and tumor growth in vivo
To identify how PRR34-AS1 affected the biological behaviors of HCC cells, we then conducted functional experiments. First, the results of Cell Counting Kit-8 (CCK-8) assays demonstrated that the proliferation ability of MHCC97-H and HCCLM3 cells was repressed after PRR34-AS1 depletion, and that of Hep 3B cells was enhanced under PRR34-AS1 upregulation (Figure 2A). Similarly, data from colony formation assays presented that the colonies were less formed in PRR34-AS1-silenced MHCC97-H and HCCLM3 cells, while the number of colonies was increased in Hep 3B cells with PRR34-AS1 overexpression (Figure 2B). Subsequently, TUNEL assay and flow cytometry analysis were applied to detect the influence of PRR34-AS1 on HCC cell apoptosis. It manifested that compared with short hair negative control (sh-NC) group, the ratio of TUNEL-positive cells was overtly increased in HCCLM3 and MHCC97-H cells after silencing PRR34-AS1 (Figure 2C; Figure S1B). Synchronously, flow cytometry analysis also indicated that the apoptosis rate was prominently increased by PRR34-AS1 silencing in both cells (Figure 2D; Figure S1C). Above data demonstrated that PRR34-AS1 promoted HCC cell proliferation while it restrained cell apoptosis.
Figure 2.

PRR34-AS1 facilitates HCC cell proliferation in vitro and tumor growth in vivo
(A) CCK-8 assays tested the changes in cell proliferation ability induced by PRR34-AS1 silencing in MHCC97-H and HCCLM3 cells and induced by PRR34-AS1 upregulation in Hep 3B cells. (B) Colony formation assays measured the number of colonies formed under PRR34-AS1 silencing in MHCC97-H, HCCLM3 cells, or under PRR34-AS1 upregulation in Hep 3B cells. (C and D) TUNEL assays and flow cytometry analysis assessed the apoptosis rate when PRR34-AS1 was downregulated in HCCLM3 cells (E) Representative pictures of xenografts derived from HCCLM3 cells transfected with sh-NC or sh-PRR34-AS1#1. (F) The grow curves of tumors from two different groups. (G and H) The final tumor volume and weight were measured. ∗p < 0.05, ∗∗p < 0.01.
Meanwhile, to determine the impacts of PRR34-AS1 on HCC tumorigenesis in vivo, HCCLM3 cells transfected with sh-NC or sh-PRR34-AS1#1 and Hep 3B cells transfected with pcDNA3.1 control or pcDNA3.1/PRR34-AS1 were respectively implanted into nude mice by subcutaneous injection. The results showed that tumor size was decreased due to PRR34-AS1 silencing (Figure 2E). Contrarily, when PRR34-AS1 was overexpressed, the tumor size was increased compared with that in the control group (Figure S1D). In addition, the tumor growth curve depicted that in comparison with corresponding controls, tumors treated with sh-PRR34-AS1#1 grew slower while those treated with pcDNA3.1/PRR34-AS1 grew faster (Figure 2F; Figure S1E), which indicated that PRR34-AS1 facilitated tumor growth in vivo. Resultantly, the final volume and weight of tumors were decreased under PRR34-AS1 depletion but augmented upon PRR34-AS1 upregulation (Figures 2G and 2H; Figures S1F and S1G). In summary, we concluded that PRR34-AS1 contributes to tumorigenesis in HCC.
PRR34-AS1 strengthens cell migration, invasion, and EMT process in HCC via activating the Wnt/β-catenin pathway
After identifying the impacts of PRR34-AS1 on HCC tumorigenesis, we continued to explore its influence on the metastasis of HCC. In this case, wound healing experiments and Transwell assays were first conducted in vitro. Interestingly, the results of wound healing assays showed that PRR34-AS1 silencing hampered the migration of HCCLM3 and MHCC97-H cells, while its overexpression expedited the migration of Hep 3B cells (Figure 3A; Figure S2A). Moreover, the results from Transwell assays displayed that the numbers of migrated and invaded HCCLM3 and MHCC97-H cells were effectively declined by PRR34-AS1 depletion, while the opposite phenomena were seen in Hep 3B cells after PRR34-AS1 upregulation (Figures 3B and 3C; Figures S2B and S2C). Furthermore, we assessed the changes in EMT, a hallmark of metastasis, in HCC cells under above conditions. From the results of western blot, we observed that PRR34-AS1 interference increased the protein level of E-cadherin while decreased that of N-cadherin and Vimentin in HCCLM3 and MHCC97-H cells. Conversely, PRR34-AS1 overexpression declined the protein level of E-cadherin, whereas induced that of N-cadherin and Vimentin in Hep 3B cells (Figure 3D; Figures S2D and S2E). These data supported that PRR34-AS1 exerted a promoting effect on HCC cell migration, invasion, and EMT process. To further certify the impact of PRR34-AS1 on tumor metastasis in HCC, we induced in vivo metastasis models. Results exhibited that reduced metastatic nodules were formed in lung tissues from mice injected with PRR34-AS1-depleted HCCLM3 cells (Figure 3E), while increased such nodules were observed in lungs from mice with PRR34-AS1-overexpressed Hep 3B cells (Figure S2F). All these data suggested that PRR34-AS1 facilitated metastasis in HCC.
Figure 3.

PRR34-AS1 contributes to cell migration, invasion, and EMT process in HCC cells and activates the Wnt/β-catenin pathway
(A) Wound healing assays detected the migration ability of indicated HCC cells when PRR34-AS1 was knocked down or upregulated. (B and C) Transwell experiments analyzed the migration and invasion properties in PRR34-AS1 silenced MHCC97-H and HCCLM3 cells, as well as in PRR34-AS1 upregulated Hep 3B cells. (D) Western blot assay examined the protein levels of E-cadherin, N-cadherin, and Vimentin in HCC cells under PRR34-AS1 depletion or overexpression. (E) H&E staining evaluated the metastatic ability of HCCLM3 cells after PRR34-AS1 depletion. (F) TOP Flash/FOP Flash reporter assays assessed the activity of Wnt/β-catenin in HCC cells with PRR34-AS1 depletion or overexpression. (G–I) Western blot evaluated the levels of indicated proteins in HCC cells after PRR34-AS1 depletion or overexpression. ∗p < 0.05.
Considering the key roles of Wnt/β-catenin signaling in cancer progression, we then investigated the relationship between PRR34-AS1 and this pathway in HCC cells. Through conducting TOP Flash/FOP Flash reporter experiments, we discovered that the TCF reporter plasmid/Mutant TCF binding sites (TOP/FOP) ratio was remarkably lowered by PRR34-AS1 silencing while increased by PRR34-AS1 upregulation (Figure 3F), indicating that PRR34-AS1 could positively regulate the activity of Wnt/β-catenin pathway. To further identify the impacts of PRR34-AS1 on this pathway, we applied western blot analysis to detect its influence on β-catenin. The results showed that loss of PRR34-AS1 promoted the level of p-β-catenin in HCCLM3 and MHCC97-H cells, while the upregulation of PRR34-AS1 reduced p-β-catenin level in Hep 3B cells (Figure 3G; Figures S3A and S3B). In depth, the level of nuclear β-catenin was reduced and that of cytoplasmic β-catenin elevated in HCCLM3 and MHCC97-H cells under the absence of PRR34-AS1, whereas overexpressing PRR34-AS1 in Hep 3B cells led to opposite results (Figure 3H; Figures S3C and S3D), suggesting that PRR34-AS1 accelerated the nuclear translocation of β-catenin in HCC cells. Moreover, western blot further analyzed the regulatory effect of PRR34-AS1 on several downstream target genes of Wnt/β-catenin pathway. As anticipated, PRR34-AS1 silencing lessened the protein levels of c-Myc, Cyclin D1, and MMP-7 in HCCLM3 and MHCC97-H cells, while PRR34-AS1 upregulation obviously elevated the levels of them in Hep 3B cells (Figure 3I; Figures S3E and S3F). To sum up, these findings demonstrated that PRR34-AS1 promotes HCC cell metastasis via activating the Wnt/β-catenin pathway.
miR-296-5p is targeted by PRR34-AS1 in HCC cells
Previous data showed that PRR34-AS1 was mainly a cytoplasmic lncRNA that might elicit post-transcriptional regulations in HCC cells. Hence, we assumed PRR34-AS1 might serve as a ceRNA to regulate the expression of its downstream genes. To confirm our assumption, we utilized LncBase database for predicting miRNAs interacting with PRR34-AS1.27 Results exposed that there were five miRNAs (miR-296-5p, miR-3614-5p, miR-3617-5p, miR-498, and miR-670-3p), which possibly combined with PRR34-AS1 (Figure 4A). Afterward, PRR34-AS1 was biotinylated to perform RNA pull-down assay in HCC cells to further select the target miRNA from above five candidates. The results unmasked that the enrichment of miR-296-5p was relatively stronger in Bio-PRR34-AS1 groups than in control group, while that of other miRNAs had no significant difference compared with the control group (Figure 4B). Likewise, we also observed abundant enrichment of PRR34-AS1 in Bio-miR-296-5p-MT groups rather than Bio-miR-296-5p-Mut groups in three kinds of HCC cells (Figure S4A). Therefore, miR-296-5p was selected for subsequent assays. The Encyclopedia of RNA Interactomes websites (ENCORI data: http://starbase.sysu.edu.cn/) predicted the underlying binding sites between PRR34-AS1 and miR-296-5p, and we also constructed the mutant PRR34-AS1 based on these sites (Figure 4C). To confirm the role of miR-296-5p in HCC, quantitative real-time RT-PCR was used to assess its expression in HCC tissues and cells. The data disclosed that the expression level of miR-296-5p was lower in HCC tissues relative to adjacent non-tumor ones (Figure S4B), and it was found in HCC cells with low expression compared to THLE-3 cells (Figure 4D). Besides, we also found that miR-296-5p had a negative correlation with PRR34-AS1 in their expression in 50 HCC tissues (Figure S4C). Then RNA immunoprecipitation (RIP) assay along with luciferase reporter experiments was conducted to determine the interaction between PRR34-AS1 and miR-296-5p. As indicated in Figure 4E, both PRR34-AS1 and miR-296-5p were abundantly concentrated in anti-AGO2 groups relative to that in anti-immunoglobulin G (IgG) groups in HCC cells, indicating PRR34-AS1 sponged miR-296-5p in RNA-induced silencing complex (RISC). Additionally, luciferase reporter experiments showed that the luciferase intensity of PRR34-AS1-wild-type (WT) was reduced by miR-296-5p overexpression, while that of PRR34-AS1-mutant (Mut) was not apparently changed (Figure 4F). Collectively, these data meant that PRR34-AS1 is a sponge of miR-296-5p in HCC cells.
Figure 4.

PRR34-AS1 sponges miR-296-5p in HCC cells
(A) LncBase database was used to predict underlying miRNAs that could combine with PRR34-AS1. (B) RNA pull-down assays assessed the abundance of candidate miRNAs in biotinylated PRR34-AS1 group in Hep 3B cells. (C) ENCORI database predicted the underlying binding sites between PRR34-AS1 and miR-296-5p. (D) Quantitative real-time RT-PCR analyzed miR-296-5p expression pattern in HCC cells (Hep 3B, SK-HEP-1, Huh7, MHCC97-H, and HCCLM3) and human normal hepatocyte cells (THLE-3). (E) RIP assays assessed the abundance of PRR34-AS1 and miR-296-5p in AGO2 complex. (F) Luciferase reporter assays detected the luciferase activity of PRR34-AS1-WT or PRR34-AS1-Mut under miR-296-5p upregulation in HCC cells. ∗p < 0.05, ∗∗p < 0.01.
PRR34-AS1 accelerates cell proliferation, migration, invasion, and EMT process in HCC via targeting miR-296-5p
To further explore whether miR-296-5p mediated the regulation of PRR34-AS1 on biological behaviors of HCC cells, we designed a series of rescue assays in HCCLM3 cells. First, quantitative real-time RT-PCR analysis testified that PRR34-AS1 deletion caused no obvious change to the expression of miR-296-5p, while the expression of miR-296-5p was greatly decreased after the co-transfection of miR-296-5p inhibitors or antagomir-296-5p in HCCLM3 cells (Figure 5A). Subsequently, the outcomes of CCK-8 and colony formation assays revealed that inhibiting miR-296-5p could restore the inhibitory effect of PRR34-AS1 silencing on cell proliferation (Figures 5B and 5C). Besides, the results of TUNEL assay and flow cytometry analysis reflected that miR-296-5p silencing could partly reverse the facilitating effect of PRR34-AS1 knockdown on cell apoptosis (Figures 5D and 5E). Moreover, data from wound healing and Transwell assays suggested that PRR34-AS1 depletion weakened cell migration and invasion, whereas its impact was then counteracted under the inhibition of miR-296-5p (Figure 5F–5H). Meanwhile, western blot analysis exhibited that the influences of PRR34-AS1 silencing on the protein level of E-cadherin, N-cadherin, and Vimentin were offset in a partial way after the miR-296-5p knockdown (Figure 5I; Figure S4D). The data demonstrated that miR-296-5p silencing rescued the blocked EMT process owing to PRR34-AS1 depletion in HCC cells. All in all, these findings suggested that PRR34-AS1 accelerated the malignant behaviors of HCC cells via a miR-296-5p-mediated manner.
Figure 5.

PRR34-AS1 aggravates HCC cell proliferation, migration, and EMT through targeting miR-296-5p
(A) Quantitative real-time RT-PCR detected the expression of miR-296-5p in HCCLM3 cells under indicated transfections. (B and C) CCK-8 and colony formation assays examined the proliferation ability of indicated HCCLM3 cells. (D and E) TUNEL and flow cytometry experiments analyzed the apoptosis rate of indicated HCCLM3 cells. (F–H) Wound healing and Transwell assays detected the migratory and invasive capacities of indicated HCCLM3 cells. (I) Western blot examined the protein levels of E-cadherin, N-cadherin, and Vimentin in HCCLM3 cells under different conditions. ∗p < 0.05.
E2F2 and SOX12 are targeted by miR-296-5p in HCC cells
To further probe the target genes of miR-296-5p, we adopted mirDIP (microRNA Data Integration Portal), microT,28 PITA (predicted miRNA targets), and miRmap (miRNAmap) databases to screen mRNAs that potentially combined with miR-296-5p (Figure 6A). Depending on the predicted results, quantitative real-time RT-PCR analyzed the expression of candidate genes (ZNF76, HMGA1, FAM53B, FGFR3, E2F2, HIPK1, SOX12, CDK16, BMF, and SLC16A3) in HCCLM3 cells transfected with sh-PRR34-AS1#1. The data indicated that the knockdown of PRR34-AS1 markedly declined the expression of E2F2 and SOX12, whereas it did not affect that of other mRNAs (Figure 6B). Additionally, data from RNA pull-down assays further showed the high enrichments of E2F2 and SOX12 in Bio-miR-296-5p-WT groups rather than Bio-miR-296-5p-Mut groups, compared with Bio-NC groups (Figure S4E). Moreover, we used ENCORI software and found that both E2F2 and SOX12 could bind to the seed region of miR-296-5p (Figure 6C). In light of above findings, E2F2 and SOX12 were chosen for performing later experiments. To figure out the functions of E2F2 and SOX12 in HCC, we explored their expression in HCC tissues and cells. Results showed that both E2F2 and SOX12 exhibited high expression trends in HCC tissues in comparison with matched non-cancerous controls (Figure S5A). Importantly, both of them were negatively correlated with miR-296-5p but positively related to PRR34-AS1 in their expressions in HCC samples (Figures S5B and S5C). Furthermore, quantitative real-time RT-PCR presented that E2F2 and SOX12 were both highly expressed in HCC cells compared to THLE-3 cells (Figure 6D). To verify the influence of PRR34-AS1 on E2F2 or SOX12, we carried out western blot analyses. The results suggested that interfering PRR34-AS1 evidently reduced the protein levels of E2F2 and SOX12 in HCCLM3 and MHCC97-H cells, whereas overexpressing PRR34-AS1 increased the levels of E2F2 and SOX12 in Hep 3B cells (Figure 6E; Figure S5D). So were the changes in the mRNA levels of them in HCC cells (Figure S5E). The above data indicated that PRR34-AS1 positively regulated E2F2 and SOX12 in HCC cells. Furthermore, to verify the correlation among PRR34-AS1, miR-296-5p, and E2F2/SOX12, we performed RIP assays. As expected, above four kinds of RNAs were all enriched in anti-AGO2 groups compared to anti-IgG groups, revealing that they co-existed in RISC (Figure 6F). Furthermore, upregulation of miR-296-5p effectively reduced the luciferase activities of E2F2-3′-UTR-WT and SOX12-3′-UTR-WT in HEK293T cells, while co-transfection of pcDNA3.1/PRR34-AS1 counteracted such effects (Figure 6G). Moreover, luciferase reporter experiments further validated the binding ability of miR-296-5p to both of the two sites predicted in SOX12 3′ UTR (Figure S5F). In conclusion, E2F2 and SOX12 are the downstream targets of PRR34-AS1/miR-296-5p axis in HCC cells.
Figure 6.

E2F2 and SOX12 are identified as the targets of miR-296-5p in HCC cells
(A) The mirDIP, microT, PITA, and miRmap databases were used to screen underlying mRNAs that potentially combined with miR-296-5p. (B) Quantitative real-time RT-PCR analyzed the expression of candidate targets (ZNF76, HMGA1, FAM53B, FGFR3, E2F2, HIPK1, SOX12, CDK16, BMF, and SLC16A3) in PRR34-AS1 silenced HCCLM3 cells. (C) ENCORI predicted the binding sites between E2F2/SOX12 and miR-296-5p. (D) Quantitative real-time RT-PCR detected E2F2 and SOX12 mRNA levels in HCC cells (Hep 3B, SK-HEP-1, Huh7, MHCC97-H, and HCCLM3) and THLE-3 cells. (E) Western blot assay analyzed the protein levels of E2F2 and SOX12 in PRR34-AS1 silenced or overexpressed HCC cells. (F) RIP assays assessed the enrichments of PRR34-AS1, miR-296-5p, and E2F2/SOX12 in anti-AGO2 complex. (G) Luciferase reporter experiments assessed the luciferase activity of E2F2-3′ UTR-WT/Mut and SOX12-WT/Mut in HEK293T cells with different transfections. ∗p < 0.05, ∗∗p < 0.01.
Upregulation of PRR34-AS1 is induced by E2F2 in HCC cells
Subsequently, we planned to probe the upstream mechanism of PRR34-AS1 in HCC. Considering that E2F2 and SOX12 were transcription factors, we wondered whether they could modulate PRR34-AS1 in return. Hence, we stably silenced the expression of E2F2/SOX12 in HCCLM3 and MHCC97-H cells while we overexpressed E2F2/SOX12 expression in Hep 3B cells (Figure 7A; Figures S6A–S6C). Interestingly, we found that PRR34-AS1 expression was significantly modulated when E2F2 was respectively knocked down or overexpressed, whereas it was not affected when SOX12 was knocked down or overexpressed (Figures 7B and 7C; S6D). Moreover, chromatin immunoprecipitation (ChIP) assay data manifested that compared to control IgG groups, PRR34-AS1 promoter could be enriched by anti-E2F2 but not by anti-SOX12 (Figure 7D; Figure S6E), implying that E2F2 might be the regulator of PRR34-AS1 in HCC cells. Thereafter, by utilizing the databases of UCSC and JASPAR (JASPAR data: http://jaspar.genereg.net/),29 we predicted that E2F2 could bind to PRR34-AS1 promoter based on its binding motif (Figure 7E). Furthermore, the results of luciferase reporter assays suggested that the luciferase activity of PRR34-AS1-Pro-WT was lessened after silencing E2F2 in HCCLM3 and MHCC97-H cells and was notably increased when overexpressing E2F2 in Hep 3B cells (Figure 7F), indicating the positive regulation of E2F2 on PRR34-AS1 transcription in HCC cells. To sum up, PRR34-AS1 upregulation is transcriptionally induced by E2F2 in HCC cells.
Figure 7.

E2F2 activates PRR34-AS1 transcription in HCC cells
(A) Western blot analyzed the knockdown or overexpression efficiencies of E2F2 in HCC cells. (B and C) The levels of E2F2 and PRR34-AS1 in indicated HCC cells were examined via quantitative real-time RT-PCR. (D) ChIP experiments measured the connection between PRR34-AS1 promoter and E2F2 in HCC cells. (E) The binding motif of E2F2 were predicted by JASPAR. (F) Luciferase reporter experiments assessed the luciferase activities of PRR34-AS1-Pro-WT and PRR34-AS1-Pro-Mut in HCC cells after E2F2 expression was increased or silenced. ∗∗p < 0.01.
Overexpression of E2F2 or SOX12 reverses the effect of PRR34-AS1 silencing on HCC cell biological behaviors
To further determine whether PRR34-AS1 relied on E2F2/SOX12 to function in HCC, we designed rescue assays. First, we utilized quantitative real-time RT-PCR and western blot analyses to assess the expression of E2F2/SOX12. Results proved that the mRNA and protein levels of E2F2 and SOX12 were initially sharply decreased by the transfection of sh-PRR34-AS1#1 and then recovered by respective co-transfection of pcDNA3.1/E2F2 or pcDNA3.1/SOX12 (Figures 8A–8B; Figure S6F). Subsequently, the results of CCK-8 and colony formation assays demonstrated that the cell proliferation inhibited by PRR34-AS1 silencing was then restored under E2F2 or SOX12 overexpression (Figures 8C and 8D). In addition, TUNEL assays and flow cytometry analyses displayed that cell apoptosis was sharply elevated due to PRR34-AS1 silencing, whereas such an elevating effect was then offset by co-transfection of pcDNA3.1/E2F2 or pcDNA3.1/SOX12 (Figures 8E and 8F). Furthermore, we discovered that the reduced cell migration and invasion mediated by sh-PRR34-AS1#1 transfection was reversed by co-transfection of pcDNA3.1/E2F2 or pcDNA3.1/SOX12 (Figures 8G–8I). At last, western blot analysis showed E2F2 or SOX12 upregulation absolutely promoted the inhibited EMT process caused by PRR34-AS1 knockdown in HCC cells (Figure 8J; Figure S6G). Taken together, PRR34-AS1 promoted HCC progression by upregulating E2F2 and SOX12 expression.
Figure 8.

PRR34-AS1 promotes the progression of HCC via upregulating E2F2 and SOX12
(A and B) Quantitative real-time RT-PCR and western blot assay examined the expression of E2F2 and SOX12 in HCCLM3 cells under diverse transfections. (C and D) CCK-8 and colony formation experiments assessed the proliferation of indicated HCCLM3 cells. (E and F) TUNEL assays and flow cytometry analysis detected HCCLM3 cell apoptosis rate in different groups. (G–I) Wound healing and Transwell assays assessed the migration and invasion capacities of indicated HCCLM3 cells. (J) Western blot assay analyzed the protein levels of E-cadherin, N-cadherin, and Vimentin in HCCLM3 cells with diverse transfections. ∗p < 0.05.
PRR34-AS1 affects the Wnt/β-catenin pathway via regulating SOX12
Since SOX12 is a recently recognized activator of Wnt/β-catenin signaling, we wanted to check whether PRR34-AS1 affected this pathway through targeting SOX12. In this case, rescue assays were conducted in HCCLM3 cells. As unveiled by TOP Flash/FOP Flash reporter assays, TOP/FOP ratio was remarkably inhibited by PRR34-AS1 silencing, whereas such inhibition was rescued by co-transfection of pcDNA3.1/SOX12 or the treatment of CHIR99021 (activator for Wnt/β-catenin; Figure 9A). In the meantime, western blot analysis showed that PRR34-AS1 deficiency promoted the protein levels of p-β-catenin in HCCLM3 cells, while SOX12 overexpression or CHIR99021 treatment counteracted the above effect (Figure 9B; Figure S7A). Besides, PRR34-AS1 silencing reduced the protein level of nuclear β-catenin and increased that of cytoplasmic β-catenin, while such phenomena were reversed by SOX12 overexpression or CHIR99021 treatment (Figure 9C; Figure S7B). Furthermore, it was also uncovered that the protein levels of c-Myc, Cyclin D1, and MMP-7 were suppressed in HCCLM3 cells transfected with sh-PRR34-AS1#1 but were then recovered due to SOX12 overexpression or CHIR99021 treatment (Figure 9D; Figure S7C). Altogether, PRR34-AS1 activated Wnt/β-catenin signaling by upregulating SOX12 in HCC cells.
Figure 9.

PRR34-AS1 activates the Wnt/β-catenin pathway by upregulating SOX12
(A) TOP Flash/FOP Flash reporter assays detected the activity of Wnt/β-catenin pathway in HCCLM3 cells with diverse transfections or treatments. (B–D) Western blot evaluated the protein levels of total β-catenin, p-β-catenin, nuclear β-catenin, cytoplasmic β-catenin, c-Myc, Cyclin D1, and MMP-7 in indicated HCCLM3 cells. ∗p < 0.05.
The mechanism of PRR34-AS1 in HCC
lncRNA PRR34-AS1 promoted HCC development via modulating Wnt/β-catenin pathway by absorbing miR-296-5p to upregulate E2F2 and SOX12.
Discussion
Increasing evidence has proven that lncRNAs function as potential and valuable therapeutic targets due to the vital importance of them in the occurrence and progression of human cancers.6 HCC, as one of malignant tumors, threatens human health in modern society. At present, research has shown that a handful of lncRNAs are abnormally expressed in HCC,30 leading to promoting or tumor-suppressive effects on HCC development. For instance, GHET1, as an oncogene, promotes cell proliferation and depresses cell apoptosis in HCC.31 CASC2 overexpression can restrain the tumorigenesis of HCC.32 In addition, our study found that PRR34-AS1 expression was elevated in HCC cells. Further, in vitro and in vivo findings here proved that PRR34-AS1 promoted tumorigenesis and metastasis in HCC, which were accordant with the discoveries of a recent report.33 These findings supported that PRR34-AS1 might be an underlying therapeutic target for HCC treatment.
As is well known, lncRNA-miRNA and mRNA-miRNA interactions are usually related to diverse cellular biological processes in HCC.34 For instance, SNHG8 accelerates HCC tumorigenesis and metastasis by inhibiting miR-149.35 In this work, we unveiled miR-296-5p as the miRNA sponged by PRR34-AS1 in HCC cells. Previously, miR-296-5p has been mentioned in the study proposed by Li et al.36 They uncover that NEAT1 promotes the proliferation and migration of HCC cells via inhibiting miR-296-5p. This study also unveiled that PRR34-AS1 facilitated HCC cell progression through absorbing miR-296-5p. Moreover, E2F2 and SOX12 were targeted by miR-296-5p. E2F2 has been recognized as an important factor in HCC development.37,38 Also, Huang et al.39 have indicated that SOX12 promotes EMT process in HCC cells. Song et al.40 further disclosed that SOX12 can be a novel cancer stem cells marker for HCC. In accordance with these bases, rescue assays validated that PRR34-AS1 facilitated HCC progression through upregulating E2F2 and SOX12.
Formerly, E2F2 and SOX12, as key members of transfection factors, have been reported to be involved in the course of HCC.41,42 Interestingly, here we discovered that E2F2 could modulate PRR34-AS1 transcription and induce PRR34-AS1 upregulation in HCC cells in return. At the same time, the present study also certified that PRR34-AS1 stimulated Wnt/β-catenin pathway by upregulating SOX12. This observation was consistent with several reports. For instance, Gao et al.43 have indicated that SOX12 regulates β-catenin expression and TCF/LEF transcriptional activity to promote development of multiple myeloma. Also, Wan et al.23 and Duquet et al.44 verified that SOX12 positively regulates β-catenin level to activate Wnt/β-catenin pathway in acute myeloid leukemia and colon cancer.
In summary, the present study indicated that PRR34-AS1 was upregulated in HCC and it promoted HCC progression via miR-296-5p/E2F2/SOX12 axis. In depth, PRR34-AS1 acted as a sponge of miR-296-5p to upregulate E2F2 and SOX12 in HCC cells. Further, PRR34-AS1 activated the Wnt/β-catenin pathway by enhancing SOX12. Intriguingly, E2F2 induced the transcription of PRR34-AS1. All in all, these findings disclosed that PRR34-AS1 might be a promising therapeutic target for HCC patients. Finally, it should be noted that the current study has some limitations in the lack of detailed investigations on Wnt/β-catenin pathway and metastasis, which needs comprehensive studies in the future.
Materials and methods
Clinical sample collection
The 50 pairs of clinical samples were collected from HCC patients in this research under the approval of the Ethics Committee of People’s Hospital of Baise. Following being sharply frozen with liquid nitrogen, all the tissues were then preserved at −80°C until application. Each patient involved in this study was treated with no other therapies and signed the written informed consent before operation.
Cell culture
Human HCC cell lines (Hep 3B and SK-HEP-1), human normal hepatocyte cell line (THLE-3), and human embryonic kidney cell line (HEK293T) were acquired from American Type Culture Collection (ATCC; Manassas, VA, USA). Human HCC cell line (Huh7) was derived from Chinese Academy of Sciences (Shanghai, China). Human HCC cell line (HCCLM3) was obtained from Procell Life Science & Technology (Wuhan, China). Human HCC cell line (MHCC97-H) was purchased from Cellcook (Guangzhou, China). THLE-3 cells were cultured in bronchial epithelial growth medium (BEGM) medium (LONZA, Basel, Switzerland) with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA). Other cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM; GIBCO, Rockville, MD, USA) adding 10% FBS plus 1% penicillin/streptomycin (Thermo Fisher Scientific). All media were utilized for cell culture at 37°C with 5% CO2.
Cell transfection
MHCC97-H and HCCLM3 cells were transfected with specific sh-RNAs to PRR34-AS1 (sh-PRR34-AS1#1/#2/#3), E2F2 (sh-E2F2#1/2), or SOX12 (sh-SOX12#1/2) or corresponding non-specific control (sh-NC). All these shRNAs were synthesized by Genepharma (Shanghai, China). Besides, miR-296-5p mimics, NC mimics, miR-296-5p inhibitors, and NC inhibitors were synthesized by RiboBio (Guangzhou, Guangdong, China). The pcDNA3.1 vectors (Invitrogen, Carlsbad, CA, USA) covering PRR34-AS1, E2F2, or SOX12 cDNA sequences were structured by Genepharma to overexpress indicated genes, with the empty vector as the negative control. Cells were collected after 48 h of transfection of appropriate plasmids by use of Lipofectamine 3000 (Invitrogen). The experiment was conducted three times. The sequences of transfection plasmids were presented in Table S1.
RNA extraction and quantitative real-time PCR
TRIzol reagent (Invitrogen) was used for the extraction of RNA. Reverse transcription of total RNA to complementary DNA (cDNA) was accomplished by the application of cDNA Reverse Transcription Kit (Invitrogen). The quantitative real-time RT-PCR was then performed via the SYBR Select Master Mix (Roche, Mannheim, Germany). GAPDH or U6 was used for standardization. The 2–ΔΔCT method was employed for detecting expression levels. The experiment was conducted three times. The primer sequences were shown in Table S2.
Subcellular fractionation assay
Cells were separated into nuclear or cytoplasm fractions via the Nuclear/Cytosol Fractionation Kit (Biovision, San Francisco, CA, USA). Then, RNAs in indicated fractions were subjected to quantitative real-time RT-PCR analysis, with U6 and GAPDH as several internal references for nuclear and cytoplasm. The experiment was conducted three times.
FISH assay
Synthesized PRR34-AS1-FISH probe was procured from Ribobio for culturing with hybridization buffer in presence of the air-dried cell samples. Samples were visualized using fluorescence microscope (Olympus, Tokyo, Japan) after double-staining cell nuclei with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, MO, USA). The experiment was conducted three times.
CCK-8 assay
Cell proliferation was measured by a CCK-8 kit (Dojindo, Kyushu Island, Japan). Transfected MHCC97-H, HCCLM3 and Hep 3B cells were grown in 96-well plates (Thermo Fisher Scientific) for 24, 48 or 72 h. Subsequently, CCK-8 reaction mixture (10 μL) was added into each well for 4 h of incubation. Afterward, the microplate reader (Thermo Fisher Scientific) was used to measure the absorbance at a wavelength of 450 nm. The experiment was conducted three times.
Colony formation assay
Transfected MHCC97-H, HCCLM3, and Hep 3B cells were added into 6-well cell plates (Thermo Fisher Scientific) and grown for around 2 weeks for colony formation assays. The plates were subsequently fixed by methanol and stained by crystal violet. After that, the stained colonies (over 50 cells) were counted manually. The experiment was conducted three times.
TUNEL assay
The transfected MHCC97-H, HCCLM3, and Hep 3B cells were prepared and washed in PBS and then processed with 4% PFA. The apoptotic cell samples were stained using TUNEL reagents (Merck KGaA, Darmstadt, Germany), following the user guide. After DAPI staining, samples were analyzed with optical microscopy (Olympus). The experiment was conducted three times.
Flow cytometry analysis
MHCC97-H, HCCLM3, and Hep 3B cells were inoculated in a 6-well plate (Thermo Fisher Scientific) at 37°C, 5% CO2 and saturated humidity for 48 h. After that, cells were collected and washed with PBS. Based on the instructions, the transfected cells stained by Annexin V/PI (Invitrogen) in the dark and the apoptosis rate of each group was detected by fortessa flow cytometer (BD Biosciences, Franklin Lake, New Jersey, USA). The experiment was conducted three times.
Xenograft tumor and in vivo metastasis models
In vivo models were established using the male BALB/C nude mice (Slac Laboratories, Shanghai, China), and acquired the ethical authorization from the Animal Research Ethics Committee of People’s Hospital of Baise. HCCLM3 transfected with sh-PRR34-AS1#1 or sh-NC, and Hep 3B cells transfected with pcDNA3.1/PRR34-AS1 or empty vector, were respectively suspended in PBS. For xenograft tumor assay, the suspensions were then subcutaneously injected into nude mice. Meanwhile, tumor volume monitored every fourth day. After 4 weeks, mice were killed by cervical decapitation, and tumors were excised and weighed. For in vivo metastasis assay, above suspensions were injected into mice from tail vein, and the lung tissues were excised from each mouse 40 days later for H&E staining.
Wound healing assay
Transfected MHCC97-H, HCCLM3, and Hep 3B cells were seeded into 6-well plates (Thermo Fisher Scientific). After the cells had grown to 80% of the plate, the tip of a sterile pipette was used to scrape the cell layer to make a scratch. The wound gaps were monitored at 0 and 24 h. Cell migration ability was assessed through measuring the recovery rate of wound areas. Experiments were conducted three times.
Transwell assay
Briefly, MHCC97-H, HCCLM3, and Hep 3B cells (1 × 104 cells per well) were transfected and then added to the upper chamber with serum-free culture medium. Meanwhile, the bottom chamber was added with culture medium with 10% FBS (Thermo Fisher Scientific). The upper chamber covered with Matrigel (BD Biosciences) was applied for invasion assay. After 24 h hatch at 37°C, the migrated or invaded cells were fixed with methanol and stained by crystal violet. Afterward, a photomicroscope (Olympus) was used to observe and count the cells in five random fields. The experiment was conducted three times.
ChIP assay
Based on the producer’s instructions, the EZ-Magna ChIP kit (Millipore, Billerica, MA, USA) was used for ChIP assay. Simply put, 2 μg anti-IgG antibody (CST, Boston, MA, USA), anti-SOX12 (Abcam), or anti-E2EF antibody (Shanghai Anyan Biological, Shanghai, China) was mixed with cell lysates containing ultrasonically fragmented chromatins overnight. Then, the enrichment of the bound DNA was analyzed by quantitative real-time RT-PCR. Experiments were conducted three times.
TOP/FOP flash assay
TOP/FOP Flash (Genechem) was co-transfected into MHCC97-H, HCCLM3, or Hep 3B cells along with indicated plasmids as needed. The luciferase activity of TOP Flash or FOP Flash was monitored via Dual Luciferase Report Assay System (Promega, Madison, WI, USA). The TOP/FOP ratio was then calculated to assess the activity of Wnt/β-catenin pathway. Experiments were conducted three times.
Luciferase reporter assay
PRR34-AS1-WT, PRR34-AS1-Pro-WT, E2F2-3′-UTR-WT, and SOX12-3′-UTR-WT and their respective mutants (PRR34-AS1-Mut, PRR34-AS1-Pro-Mut, E2F2-3′-UTR-Mut, and SOX12-3′-UTR-Mut) were synthesized by Genepharma (Shanghai, China). Then, these sequences were appropriately constructed to pGL3 or pmirGLO reporter vectors (Promega), and the recombinant plasmids were then co-transfected with indicated transfection plasmids (including miR-296-5p mimics or NC mimics, sh-E2F2#1&2 or sh-NC, pcDNA3.1/E2F2 or pcDNA3.1, sh-SOX12#1&2 or sh-NC, pcDNA3.1/SOX12, or pcDNA3.1) into HCC cells or HEK293T cells by Lipofectamine 3000 (Invitrogen). After 48 h transfection, the luciferase activity was monitored and calculated via Dual Luciferase Report Assay System (Promega). Assays were conducted three times.
RIP assay
Imprint RIP kit (Millipore, Billerica, MA, USA) was adopted for RIP assay. RIP lysis buffer including protease inhibitor (Thermo Fisher Scientific) was used to lyse MHCC97-H, HCCLM3, and Hep 3B cells. Afterward, magnetic beads (Thermo Fisher Scientific) conjugated with anti-AGO2 antibody (Abcam) or control anti-IgG antibody (Abcam) were added. Proteinase K (Absin, Shanghai, China) was selected to digest the proteins in the immunoprecipitated complex. Finally, quantitative real-time RT-PCR was carried out to detect RNA in the complex. Experiments were conducted three times.
RNA pull-down assay
Biotin-labeled miRNAs (Bio-miR-296-5p-WT, Bio-miR-miR-296-5p-Mut) and the negative control (Bio-NC) were transcribed with the Biotin RNA Labeling Mix (Roche, Mannheim, Germany). After 48 h, the transfected cells were processed with 4% paraformaldehyde and then terminated by glycine. After that, cells were scraped off and then lysed after washing with PBS. Afterward, streptavidin magnetic beads were added into the cell lysates. After 48 h, the beads were washed and incubated with the elution buffer at 85°C for 10 min, and then proteinase K was supplemented and cultured at 65°C for 2 h. In the end, the RNA was extracted via Trizol (Invitrogen), followed by analysis through quantitative real-time RT-PCR. Experiments were conducted three times.
Western blot analysis
Radioimmunoprecipitation assay (RIPA) lysis buffers (CST, Danvers, MA, USA) containing proteinase and phosphatase inhibitors were used to lyse the transfected MHCC97-H, HCCLM3, and Hep 3B cells. After being centrifuged for 30 min, a Pierce Bicinchoninic acid (BCA) Protein detection kit (Bio-Rad Laboratories, Hercules, CA, USA) was used for assessing the concentration of the proteins. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) used to separate proteins in each sample, and the proteins were then transferred to the polyvinylidene difluoride membranes (PVDF; Bio-Rad Laboratories, Hercules, CA, USA). The membranes were disposed with 5% non-fat milk and then cultured with primary antibodies E-cadherin (#3195, 1:1,000; CST), N-cadherin (#13116, 1:1,000; CST), Vimentin (#5741, 1:1,000; CST), GAPDH (ab9485, 1/10,000; Abcam), β-catenin (#8480, 1:1,000; CST), c-Myc (#18583, 1:1,000; CST), Cyclin D1 (#55506, 1:1,000; CST), MMP-7 (#71031, 1:1,000; CST), β-actin (#4970, 1:1,000; CST), E2F2 (ab138515, 1/1,000; Abcam), and SOX12 (cat. no. 23939-1-AP, 1:500; Proteintech) at 4°C overnight. Later, membranes were cultured with secondary antibodies (ab205718, 1/10,000; Abcam) at the room temperature for 1 h. The bands were visualized via enhanced chemiluminescence (ECL; Millipore, USA). GAPDH or β-actin was employed as the internal control. Experiments were conducted three times.
Statistical analyses
All experiments were repeated three times or more. Data were presented as mean ± standard deviation (SD) and analyzed by Student’s t test or analysis of variance (ANOVA) using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA, USA). The relationships among molecules were determined via Pearson’s correlation analysis. p <0.05 was defined as the threshold of statistical significance.
Acknowledgments
We are very grateful to all individuals and groups involved in this study. This study was supported by Guangxi Clinic Medicine Research Center of Hepatobiliary Disease under grant number AD17129025 and Natural Science Youth Science Fund Project of Guangxi Province under grant number 2017GXNSFBA198027.
Author contributions
Conceptualization, Y.M. and C.L.; methodology, Y.M. and C.L.; software, S.H. and F.Q.; validation, Y.Y. and J.H.; formal analysis, S.H. and F.Q.; investigation, H.Z. and J.H.; resources, Z.D. and Z.Q.; data curation, Y.Q. and T.Y.; writing – original draft preparation, M.Q.; writing – review & editing, Z.H.; visualization, G.H.; supervision, H.P.; project administration, M.Q.; funding acquisition, M.Q.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.04.016.
Contributor Information
Zansong Huang, Email: 1019846481@qq.com.
Tingzhuang Yi, Email: 410511515@qq.com.
Supplemental information
References
- 1.Zhu Z.X., Huang J.W., Liao M.H., Zeng Y. Treatment strategy for hepatocellular carcinoma in China: radiofrequency ablation versus liver resection. Jpn. J. Clin. Oncol. 2016;46:1075–1080. doi: 10.1093/jjco/hyw134. [DOI] [PubMed] [Google Scholar]
- 2.Francica G., Borzio M. Status of, and strategies for improving, adherence to HCC screening and surveillance. J. Hepatocell. Carcinoma. 2019;6:131–141. doi: 10.2147/JHC.S159269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qian X., Zhao J., Yeung P.Y., Zhang Q.C., Kwok C.K. Revealing lncRNA Structures and Interactions by Sequencing-Based Approaches. Trends Biochem. Sci. 2019;44:33–52. doi: 10.1016/j.tibs.2018.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Bhan A., Soleimani M., Mandal S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017;77:3965–3981. doi: 10.1158/0008-5472.CAN-16-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renganathan A., Felley-Bosco E. Long Noncoding RNAs in Cancer and Therapeutic Potential. Adv. Exp. Med. Biol. 2017;1008:199–222. doi: 10.1007/978-981-10-5203-3_7. [DOI] [PubMed] [Google Scholar]
- 7.Yang G., Lu X., Yuan L. LncRNA: a link between RNA and cancer. Biochim. Biophys. Acta. 2014;1839:1097–1109. doi: 10.1016/j.bbagrm.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 8.Xiao J., Lv Y., Jin F., Liu Y., Ma Y., Xiong Y., Liu L., Zhang S., Sun Y., Tipoe G.L. LncRNA HANR Promotes Tumorigenesis and Increase of Chemoresistance in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2017;43:1926–1938. doi: 10.1159/000484116. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y.T., Li B.P., Zhang B., Ma P., Wu Q.L., Ming L., Xie L.M. LncRNA SBF2-AS1 promotes hepatocellular carcinoma metastasis by regulating EMT and predicts unfavorable prognosis. Eur. Rev. Med. Pharmacol. Sci. 2018;22:6333–6341. doi: 10.26355/eurrev_201810_16044. [DOI] [PubMed] [Google Scholar]
- 10.Fang H., Zhang F.X., Li H.F., Yang M., Liao R., Wang R.R., Wang Q.Y., Zheng P.C., Zhang J.P. PRR34-AS1 overexpression promotes protection of propofol pretreatment against ischemia/reperfusion injury in a mouse model after total knee arthroplasty via blockade of the JAK1-dependent JAK-STAT signaling pathway. J. Cell. Physiol. 2020;235:2545–2556. doi: 10.1002/jcp.29158. [DOI] [PubMed] [Google Scholar]
- 11.Groner B., von Manstein V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017;451:1–14. doi: 10.1016/j.mce.2017.05.033. [DOI] [PubMed] [Google Scholar]
- 12.Li L., Tang P., Li S., Qin X., Yang H., Wu C., Liu Y. Notch signaling pathway networks in cancer metastasis: a new target for cancer therapy. Med. Oncol. 2017;34:180. doi: 10.1007/s12032-017-1039-6. [DOI] [PubMed] [Google Scholar]
- 13.Serafino A., Sferrazza G., Colini Baldeschi A., Nicotera G., Andreola F., Pittaluga E., Pierimarchi P. Developing drugs that target the Wnt pathway: recent approaches in cancer and neurodegenerative diseases. Expert Opin. Drug Discov. 2017;12:169–186. doi: 10.1080/17460441.2017.1271321. [DOI] [PubMed] [Google Scholar]
- 14.Zeng X., Ju D. Hedgehog Signaling Pathway and Autophagy in Cancer. Int. J. Mol. Sci. 2018;19:2279. doi: 10.3390/ijms19082279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perugorria M.J., Olaizola P., Labiano I., Esparza-Baquer A., Marzioni M., Marin J.J.G., Bujanda L., Banales J.M. Wnt-β-catenin signalling in liver development, health and disease. Nat. Rev. Gastroenterol. Hepatol. 2019;16:121–136. doi: 10.1038/s41575-018-0075-9. [DOI] [PubMed] [Google Scholar]
- 16.Khalaf A.M., Fuentes D., Morshid A.I., Burke M.R., Kaseb A.O., Hassan M., Hazle J.D., Elsayes K.M. Role of Wnt/β-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J. Hepatocell. Carcinoma. 2018;5:61–73. doi: 10.2147/JHC.S156701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu L., Yang N., Du G., Li C., Liu G., Liu S., Xu Y., Di Y., Pan W., Li X. LncRNA CRNDE promotes the epithelial-mesenchymal transition of hepatocellular carcinoma cells via enhancing the Wnt/β-catenin signaling pathway. J. Cell. Biochem. 2018;120:1156–1164. doi: 10.1002/jcb.26762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y., Yang L., Chen T., Liu X., Guo Y., Zhu Q., Tong X., Yang W., Xu Q., Huang D., Tu K. A novel lncRNA MCM3AP-AS1 promotes the growth of hepatocellular carcinoma by targeting miR-194-5p/FOXA1 axis. Mol. Cancer. 2019;18:28. doi: 10.1186/s12943-019-0957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J., Huang X., Wang W., Xie H., Li J., Hu Z., Zheng Z., Li H., Teng L. LncRNA CDKN2BAS predicts poor prognosis in patients with hepatocellular carcinoma and promotes metastasis via the miR-153-5p/ARHGAP18 signaling axis. Aging (Albany NY) 2018;10:3371–3381. doi: 10.18632/aging.101645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi D.M., Li L.X., Bian X.Y., Shi X.J., Lu L.L., Zhou H.X., Pan T.J., Zhou J., Fan J., Wu W.Z. miR-296-5p suppresses EMT of hepatocellular carcinoma via attenuating NRG1/ERBB2/ERBB3 signaling. J. Exp. Clin. Cancer Res. 2018;37:294. doi: 10.1186/s13046-018-0957-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wan H., Cai J., Chen F., Zhu J., Zhong J., Zhong H. SOX12: a novel potential target for acute myeloid leukaemia. Br. J. Haematol. 2017;176:421–430. doi: 10.1111/bjh.14425. [DOI] [PubMed] [Google Scholar]
- 24.Attwooll C., Lazzerini Denchi E., Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23:4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan H., Wang Q., Shen Q., Li Z., Tian J., Jiang Q., Gao L. Identification of potential transcription factors, long noncoding RNAs, and microRNAs associated with hepatocellular carcinoma. J. Cancer Res. Ther. 2018;14:S622–S627. doi: 10.4103/0973-1482.204846. [DOI] [PubMed] [Google Scholar]
- 26.Wang H., Huo X., Yang X.R., He J., Cheng L., Wang N., Deng X., Jin H., Wang N., Wang C. STAT3-mediated upregulation of lncRNA HOXD-AS1 as a ceRNA facilitates liver cancer metastasis by regulating SOX4. Mol. Cancer. 2017;16:136. doi: 10.1186/s12943-017-0680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paraskevopoulou M.D., Vlachos I.S., Karagkouni D., Georgakilas G., Kanellos I., Vergoulis T., Zagganas K., Tsanakas P., Floros E., Dalamagas T., Hatzigeorgiou A.G. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–D238. doi: 10.1093/nar/gkv1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paraskevopoulou M.D., Georgakilas G., Kostoulas N., Vlachos I.S., Vergoulis T., Reczko M., Filippidis C., Dalamagas T., Hatzigeorgiou A.G. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169-73. doi: 10.1093/nar/gkt393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48(D1):D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y., Chen L., Gu J., Zhang H., Yuan J., Lian Q., Lv G., Wang S., Wu Y., Yang Y.T. Recurrently deregulated lncRNAs in hepatocellular carcinoma. Nat. Commun. 2017;8:14421. doi: 10.1038/ncomms14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin L., He Y., Tang S., Huang S. LncRNA GHET1 predicts poor prognosis in hepatocellular carcinoma and promotes cell proliferation by silencing KLF2. J. Cell. Physiol. 2018;233:4726–4734. doi: 10.1002/jcp.26257. [DOI] [PubMed] [Google Scholar]
- 32.Fan J.C., Zeng F., Le Y.G., Xin L. LncRNA CASC2 inhibited the viability and induced the apoptosis of hepatocellular carcinoma cells through regulating miR-24-3p. J. Cell. Biochem. 2018;119:6391–6397. doi: 10.1002/jcb.26479. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z., Li Z., Xu B., Yao H., Qi S., Tai J. Long Noncoding RNA PRR34-AS1 Aggravates the Progression of Hepatocellular Carcinoma by Adsorbing microRNA-498 and Thereby Upregulating FOXO3. Cancer Manag. Res. 2020;12:10749–10762. doi: 10.2147/CMAR.S263619. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Lin C., Yuan G., Hu Z., Zeng Y., Qiu X., Yu H., He S. Bioinformatics analysis of the interactions among lncRNA, miRNA and mRNA expression, genetic mutations and epigenetic modifications in hepatocellular carcinoma. Mol. Med. Rep. 2019;19:1356–1364. doi: 10.3892/mmr.2018.9728. [DOI] [PubMed] [Google Scholar]
- 35.Dong J., Teng F., Guo W., Yang J., Ding G., Fu Z. lncRNA SNHG8 Promotes the Tumorigenesis and Metastasis by Sponging miR-149-5p and Predicts Tumor Recurrence in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2018;51:2262–2274. doi: 10.1159/000495871. [DOI] [PubMed] [Google Scholar]
- 36.Li Y., Ding X., Xiu S., Du G., Liu Y. LncRNA NEAT1 Promotes Proliferation, Migration And Invasion Via Regulating miR-296-5p/CNN2 Axis In Hepatocellular Carcinoma Cells. OncoTargets Ther. 2019;12:9887–9897. doi: 10.2147/OTT.S228917. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Zeng Z., Cao Z., Tang Y. Increased E2F2 predicts poor prognosis in patients with HCC based on TCGA data. BMC Cancer. 2020;20:1037. doi: 10.1186/s12885-020-07529-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan L., Huang C., Meng X.M., Song Y., Wu X.Q., Miu C.G., Zhan X.S., Li J. Promising roles of mammalian E2Fs in hepatocellular carcinoma. Cell. Signal. 2014;26:1075–1081. doi: 10.1016/j.cellsig.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 39.Huang W., Chen Z., Shang X., Tian D., Wang D., Wu K., Fan D., Xia L. Sox12, a direct target of FoxQ1, promotes hepatocellular carcinoma metastasis through up-regulating Twist1 and FGFBP1. Hepatology. 2015;61:1920–1933. doi: 10.1002/hep.27756. [DOI] [PubMed] [Google Scholar]
- 40.Zou S., Wang C., Liu J., Wang Q., Zhang D., Zhu S., Xu S., Kang M., He S. Sox12 Is a Cancer Stem-Like Cell Marker in Hepatocellular Carcinoma. Mol. Cells. 2017;40:847–854. doi: 10.14348/molcells.2017.0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang Z.Q., Li M.C., Zhang Y.Q., Liu X.G. MiR-490-5p inhibits the metastasis of hepatocellular carcinoma by down-regulating E2F2 and ECT2. J. Cell. Biochem. 2018;119:8317–8324. doi: 10.1002/jcb.26876. [DOI] [PubMed] [Google Scholar]
- 42.Jiang T., Guan L.Y., Ye Y.S., Liu H.Y., Li R. MiR-874 inhibits metastasis and epithelial-mesenchymal transition in hepatocellular carcinoma by targeting SOX12. Am. J. Cancer Res. 2017;7:1310–1321. [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Y., Li L., Hou L., Niu B., Ru X., Zhang D. SOX12 promotes the growth of multiple myeloma cells by enhancing Wnt/β-catenin signaling. Exp. Cell Res. 2020;388:111814. doi: 10.1016/j.yexcr.2020.111814. [DOI] [PubMed] [Google Scholar]
- 44.Duquet A., Melotti A., Mishra S., Malerba M., Seth C., Conod A., Ruiz i Altaba A. A novel genome-wide in vivo screen for metastatic suppressors in human colon cancer identifies the positive WNT-TCF pathway modulators TMED3 and SOX12. EMBO Mol. Med. 2014;6:882–901. doi: 10.15252/emmm.201303799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.