

Abstract
A decline in mitochondrial quality and activity has been associated with normal aging and correlated with the development of a wide range of age-related diseases. Here, we review the evidence that a decline in the levels of mitochondrial-derived peptides contributes to aging and age-related diseases. In particular, we discuss how mitochondrial-derived peptides, humanin and MOTS-c, contribute to specific aspects of the aging process, including cellular senescence, chronic inflammation, and cognitive decline. Genetic variations in the coding region of humanin and MOTS-c that are associated with age-related diseases are also reviewed, with particular emphasis placed on how mitochondrial variants might, in turn, regulate MDP expression and age-related phenotypes. Taken together, these observations suggest that mitochondrial-derived peptides influence or regulate a number of key aspects of aging and that strategies directed at increasing mitochondrial-derived peptide levels might have broad beneficial effects.
Keywords: Mitochondrial-derived peptides, Mitochondria, Humanin, MOTS-c, Aging, Age-related diseases
Introduction
Mitochondria are multifaceted organelles. They are the primary energy-generating system and, additionally, they participate in intermediary metabolism, calcium signaling, apoptosis, and retrograde signaling [1–4]. Given these well-established functions, it might be expected that mitochondrial dysfunction would give rise to a predictable set of defects in all tissues. Indeed, mitochondrial dysfunction has been associated with normal aging (Fig. 1) and correlated with the development of a wide range of age-related diseases, such as cardiovascular disease, diabetes, and neurodegeneration [5–8]. Mitochondrial membrane potential is the central bioenergetic parameter that controls respiratory rate, ATP synthesis, and the generation of reactive oxygen species [9]. Decreases of mitochondrial membrane potential have been found in a variety of aging cell types [9]. During ATP synthesis, the tricarboxylic acid (TCA) cycle and oxidative phosphorylation generate metabolic intermediates, which play an important role in regulating the transcriptional and epigenetic states of cells [1]. For example, TCA metabolism in the mitochondria matrix produces alpha-ketoglutarate, which is important in maintaining the pluripotency of embryonic stem cells [10]. The NAD+/NADH ratio is important for lysosome function and epigenetic modification in the nuclear genome [1, 11]. Levels of these metabolites decline in tissues as they age [12]. Mitochondria are crucial for storing calcium to maintain calcium homeostasis [2]. Calcium transfer to mitochondria is finely regulated, but excess calcium disturbs oxidative phosphorylation and leads to cell death that is observed during aging [13]. In addition to calcium, mitochondria release ATP, ROS, Ac-CoA, and NAD+, which serve as retrograde signals that have a variety of cellular effects [4, 14].
Fig. 1.

Mitochondrial alteration during aging. MPTP, mitochondrial permeability transition pore; MCU, mitochondrial calcium uniporter; MDPs, mitochondrial-derived peptides
Mitochondrial-derived peptides (MDPs) are newly discovered entities in the retrograde signaling from the mitochondria [15]. These molecules are essential components of mitochondria that activate signaling pathways and modulate nuclear gene expression [15, 16]. During aging, the levels of these peptides decline, leading to loss of physiological function [17–19]. The levels of MDPs are also associated with age-related diseases [20–22]. Modifying mitochondrial biology, therefore, has been proposed as a therapeutic target for preventing age-related disease. For example, metformin—a drug that acts on the mitochondria—is a leading therapeutic candidate for aging that is currently being studied in large-scale clinical trials [23]. However, despite data showing mitochondria can be modified to attenuate aging, the precise molecules that mediate these mitochondrial effects remain unclear. In this review, we discuss how MDPs contribute to the process of aging and age-related diseases. We will review the discovery of MDPs, their function, and their relation to aging and age-related diseases, with specific emphasis on the two most studied peptides called humanin and MOTS-c.
Mitochondrial-derived peptides
Traditionally, it has been known that mitochondrial DNA encodes 13 mRNAs, 22 tRNAs, and 2 rRNAs. Emerging evidence suggests that small open reading frames in the mitochondrial DNA encode small peptides that exert biological function in various tissues and conditions [24]. The peptides encoded from the mitochondrial small open reading frames are called mitochondrial-derived peptides (MDPs) [25]. Since the first MDP named humanin was discovered in 2001, seven more peptides have been identified that play important roles in both physiology and pathophysiology [26]. The levels of some MDPs declined with age in mice and humans, and the administration of some MDPs exert beneficial effects in in vitro and in vivo models of age-related disease including Alzheimer’s disease, cardiovascular disease, and type 2 diabetes [17–19].
Humanin and age-related diseases
Humanin, which is encoded in the 16S rRNA of the mitochondrial DNA, is the first discovered MDP. Humanin has historically been shown to have strong neuroprotective effects against Alzheimer’s disease (AD). In its initial discovery, a pool of cDNA was isolated from the relatively intact brain area called the occipital lobe from the postmortem brain of an AD patient to screen for a cDNA that protects against AD-specific neurotoxicity. A cDNA carrying humanin was found to have provided protection against AD-specific neurotoxicity, specifically through its interactions with the AD-associated protein, amyloid beta (Aβ) [27, 28].
Since the first discovery, studies have shown that humanin may play an important protective role against cognitive decline and associated neurological disorders such as Alzheimer’s disease (AD). The humanin pathway involved in cognitive function is complex. Briefly, the functional pathway involves inhibiting the release of cytochrome c by interacting with the pro-apoptotic proteins Bax, Bid, and Bim [29]. Additionally, humanin localizes to the lysosomal membrane surface and thus increases the activation of chaperone-mediated autophagy [30]. Humanin has also been shown to have an overlapping function with insulin-like growth factor 1 (IGF-1) and its associated binding partners, which may impact its participation in associated cell survival pathways [31]. More specific to AD, humanin also mediates signaling cascades associated with AD pathology, which will be discussed later in more detail.
Upon further analysis, many additional studies have demonstrated the neuroprotective role of humanin in Aβ-induced toxicity [32–34]. More specifically, humanin may suppress Aβ-induced toxicity and fibril formation by activating pro-survival signaling cascades associated with kinases including extracellular signal-regulated kinases 1 and 2 (ERK1/2), AKT1 kinase, and tyrosine kinases associated with STAT3 and caspase-3 activation [35, 36]. Studies using in vivo mouse models have further confirmed the observed protective association between humanin and AD. Previous models studied this association by first injecting mice with Aβ25–35 to induce AD behavior, then introducing the potent humanin derivative S14G-HN to demonstrate its effects [37]. Currently, through the use of transgenics, more prominent models have been created. One such model, the double transgenic mouse APPswe/PS1de9, is commonly used for AD research, as it shows the Aβ-induced neuropathology and many other cognitive characteristics associated with the disease. Treating APPswe/PS1de9 mice with S14G-HN has demonstrated the direct neuroprotective effects of humanin, through its reduction of cerebral plaque deposition, neuroinflammation, and levels of insoluble Aβ protein [38]. Other studies have used S14G-HN treatment in the triple transgenic mouse model containing the mutations APPswe/taup310L/PS1de9 and have generated similar results [39].
While humanin has been directly associated with AD pathology, the peptide has also recently been shown to prevent overall cognitive decline associated with aging. More specifically, in vivo rat models have shown that humanin release from astrocytes may regulate synaptic plasticity associated with cognitive function [40]. Additionally, a recent study has demonstrated that the humanin derivative S14G-HN inhibits diazepam (DZP)–induced cognitive impairment in mice. This suggests a modulatory relationship between humanin and cholinergic neurotransmitters or GABA-associated neurons that are impacted by DZP exposure and linked to cognitive function [41]. Taken together, humanin plays an important neuroprotective role that may improve the treatment of age-related cognitive decline and neurological disorders such as AD.
Evidence shows that humanin may also play an important protective role in cardiovascular disease (CVD), which represents the leading cause of death worldwide in both developed and developing countries [42]. Age represents the largest risk factor for CVD, resulting in diseases such as cardiac fibrosis, atrial fibrillation, and heart failure [43]. Circulating levels of humanin have been shown to decrease in both humans and mice in aging populations [44]. Interestingly, patients with coronary endothelial dysfunction have been shown to have significantly lower humanin levels compared with those with normal coronary endothelial function. Humanin administration showed protective roles in CVD-related phenotypes. An analog of humanin (HNG) has been shown to have protective effects against myocardial fibrosis in aged C57BL/6N mice [45]. Humanin also attenuated renal microvascular remodeling, inflammation, and apoptosis in the early stage of kidney disease in hypercholesterolemic ApoE(−/−) mice [46]. Treatment with HNG resulted in the reversal of cardiomyocyte apoptosis in aged mice, as well as a reduction in collagen deposits and fibroblast proliferation [45]. Similarly, in rats with myocardial ischemic/reperfusion (I/R) injury, high doses of HNG resulted in increased humanin production in damaged myocardium, as well as significantly decreased cardiac arrhythmia, cardiac mitochondrial dysfunction, myocardial infarct size, and left ventricular dysfunction [47]. These effects are largely due to humanin’s ability to attenuate mitochondrial dysfunction by protecting against oxidative stress [47–49]. Congruently, another study showed that HNG directly protects cardiac mitochondrial function against H2O2 by decreasing complex I activity, offering a mechanism for how HNG maintains proper cardiac mitochondrial function in myocardial I/R injury [48, 50]. Collectively, these results indicate that humanin plays a cardioprotective role that may be diminished as aging occurs and endogenous humanin levels are decreased.
Evidence for humanin as an anti-aging peptide
Humanin levels have been associated with many age-related diseases, and treatment with humanin has been able to protect against many age-related diseases, such as AD, cancer, fibrosis, heart disease, and age-related macular degeneration models [33, 45, 51–54]. Humanin also affects and is affected by known anti-aging pathways such as IGF-I [55]. Over an organism’s lifespan, humanin levels decrease in many different organisms including mice, monkeys, and humans [19]. Interestingly, the naked mole rat, a model of negligible senescence [56], has only a small trend towards a decrease in humanin levels over its 30-year lifespan, further supporting the idea that humanin is related to biological aging [19].
The relationship between aging and humanin has been further elucidated in two recent papers [57]. In mice, humanin administration to female mice beginning in midlife (18 months of age) was able to increase healthspan as measured by a number of different parameters [57]. In these experiments, humanin decreased functional cognitive decline as measured by Barnes maze, Y-maze, and rotarod, and decreased overall inflammation as measured by IL-6, IBA-1, and IL-10. This improvement in cognition was not likely due to an increase in neurogenesis as there was no significant difference between the groups. Furthermore, humanin treatment had a positive effect on metabolic aging and reduced midlife adiposity by decreasing visceral fat and increasing lean body mass [57]. The levels of IGF-I were also reduced in these mice. Perhaps due to an inadequate dose or late start of the injections, there was no significant change in overall survival, contrary to what we saw in healthspan.
To further support that humanin is a bona fide anti-aging peptide, we generated two transgenic models, mice and the worm model C. elegans. Humanin was able to induce a small, but significant, increase in lifespan in transgenic worms that was dependent on daf-16/FOXO, suggesting that the insulin/IGF signaling pathway is critical for the increase in lifespan by humanin. The humanin transgenic mice phenocopied several of the worms’ phenotypes and aging studies are currently ongoing in our lab. The phenotype of these mice is similar to mice with reduced IGF-I, including a reduction in body size and reduced fecundity [58].
Turning to humans, we looked at humanin levels in Alzheimer’s disease patients and found a decrease in CSF levels. We next examined the circulating humanin levels in children of centenarians, who have a greater chance of becoming a centenarian themselves [19]. Remarkably, the offspring of centenarians have a much higher level of humanin compared with age-matched controls, further supporting the idea that humanin plays a role in aging in humans. Although humanin has been associated with a number of age-related diseases and its use has been able to prevent many age-related diseases, until recently, the link between humanin and aging was only a hypothesis. With these new series of papers, we have shown that humanin is sufficient to increase lifespan in worms and is likely to increase lifespan in mammals as well.
MOTS-c
Another mitochondrial-derived peptide that has been characterized as an aging modulator is MOTS-c. MOTS-c is encoded by a small open reading frame within the mitochondrial 12S rRNA gene [17]. MOTS-c was initially characterized as an “exercise mimetic,” as it improves blood glucose regulation in age-dependent insulin-resistant mice models [59]. The effects of MOTS-c on insulin sensitivity is mostly observed in muscle tissue. In addition to the AMPK-specific effects of MOTS-c, this peptide has been reported to translocate to the nucleus and interact with NRF2 during cellular distress [16]. The MOTS-c/NRF2 interaction promotes the upregulation of genes involved in mitochondrial protection, and MOTS-c overexpression potentiates NRF2 signaling. However, during replicative senescence, MOTS-c levels decrease, suggesting that one-way aging progresses is through reduced MOTS-c expression [60]. Indeed, MOTS-c levels decline with age in mice skeletal muscle and circulation [17].
MOTS-c levels also decrease with age in humans. In middle-aged (45–55 years) and old-aged (70–81) individuals, circulating MOTS-c levels were 11% and 21% lower compared with those in younger individuals (18–30 years), respectively [61]. Furthermore, MOTS-c levels were reported to be strongly correlated to the diabetic Matsuda and Homeostatic Model Assessment (HOMA) indexes. That is, MOTS-c levels were higher in lean individuals with less-efficient glucose regulation, perhaps as a compensatory mechanism that is also weight-dependent [21]. In addition, levels of MOTS-c were lower in human subjects with coronary endothelial dysfunction, and elevating MOTS-c levels in mice models of endothelial dysfunction attenuated pathology [20]. However, unlike in reports focused on mice, MOTS-c was highest in skeletal muscle in aged humans [61]. This data specifically showed that, in young individuals, plasma MOTS-c positively correlates with muscle MOTS-c, but in older individuals, such a correlation was not observed. That MOTS-c is increased in human skeletal muscle and decreased in human plasma suggest human tissue–specific regulation occurring during the aging process.
The decline of MOTS-c levels during aging and the correlation between MOTS-c levels and pathological conditions suggest that higher MOTS-c levels may protect against age-related diseases. Indeed, MOTS-c administration to mice provides beneficial effects in multiple pathological conditions such as insulin resistance [17], ovariectomy-induced metabolic dysfunction [62], and bone loss [63].
Exercise is one of the interventions that prevents age-related adverse effects in mice and humans. A recent study demonstrated that acute high-intensity aerobic exercise upregulated MOTS-c expression in the skeletal muscle, which increased plasma MOTS-c levels as well [64]. Though the detailed mechanism has not been investigated, skeletal muscle contraction may upregulate MOTS-c expression similar to humanin expression in isolated mouse skeletal muscle [61, 65]. Therefore, it suggests that skeletal muscle is an important organ for MOTS-c secretion and that exercise increases MOTS-c expression as well as humanin.
It is now widely accepted that regular aerobic exercise is one of the therapeutic and preventive methods for metabolic disorders [66–68], and its beneficial effects are mediated by aerobic exercise-induced signaling, such as AMPK and SIRT1 [69]. The first report on MOTS-c by Lee et al. has demonstrated that the metabolic-protective effect of MOTS-c was achieved in an AMPK-dependent manner, suggesting that aerobic exercise and MOTS-c share the same signaling pathway in the skeletal muscle [17]. Moreover, a recent study demonstrated that 2 weeks of MOTS-c injection increases running capacity in both young and old mice [64]. Though the molecular targets of MOTS-c have not yet been identified, these observations suggest that MOTS-c has potential aerobic exercise training mimetic effects.
Mitochondrial single nucleotide polymorphisms and MDPs
Mitochondrial single nucleotide polymorphisms (SNPs) have also been associated with various age-related diseases. Previous studies suggest that mitochondrial SNPs could influence various mitochondrial functions related to mitochondrial oxidative phosphorylation, mitochondrial reactive oxygen species, and mitochondrial pH [70]. Recent studies focusing on the mitochondrial SNPs at the region of humanin and MOTS-c provide new insights regarding how mitochondrial SNPs impact the mitochondrial-derived peptides, which in turn contribute to age-related diseases. A MT-2607 A>G polymorphism is located in the coding region of humanin and is associated with lower circulating levels of humanin and higher cognitive decline in the African-American population [57]. These results are well aligned with previous studies that show that humanin administration improves cognitive function. Another mitochondrial SNP in the MOTS-c coding region, the MT-1382 A>C polymorphism, which causes a K14Q amino acid replacement, was found in 5–10% of the East-Asian populations [71]. We found that the C allele carriers of this SNP showed a significantly higher visceral fat area, which is a risk factor for T2D, than the A allele carriers in a small Japanese cohort. Additionally, a meta-analysis of three independent Japanese cohorts (n = 27,527) demonstrated that male subjects with the C allele of MT-1382 A>C polymorphism exhibited a higher prevalence of T2D than those with the A allele of the same polymorphism [72]. Interestingly, men with the C allele of the MT-1382 A>C polymorphism exhibited a 65% greater rate of T2D only in the sedentary group, demonstrating a kinesio-genomic interaction [72]. Moreover, SNP carriers exhibited dramatically elevated circulating MOTS-c levels in Japanese populations, implying that K14Q MOTS-c acts as a bioinactive form of MOTS-c, which is similar to bioinactive leptin associated with obesity and bioinactive insulin leading to MODY diabetes [73, 74]. To support these human observations, MOTS-c administration in high-fat-fed mice resulted in reduced weight and improved glucose tolerance in male mice, but not in K14Q MOTS-c–treated mice [17, 72]. K14Q MOTS-c also did not elicit the insulin-sensitization that is observed with wild-type MOTS-c in vitro [72]. This combination of human, mice, and cell line data suggests that the MT-1382 A>C polymorphism is one of the genetic risk factors for T2D in the East-Asian population as the SNP carriers produce K14Q MOTS-c, a bioinactive form of MOTS-c.
Conclusions
Recent work aimed at establishing hallmarks of aging may contribute to building a framework for future studies on the molecular mechanisms of aging, as well as aid in designing interventions to improve human healthspan [75]. Emerging studies related to mitochondrial-derived peptides indicate that MDPs may be closely related to mitochondrial dysfunction, deregulated nutrient-sensing, cellular senescence, and loss of proteostasis (Fig. 2). MDPs have beneficial effects on these processes, and yet they progressively decrease during aging, which can negatively impact said processes. In vivo studies with gain- or loss-of-function will be necessary for moving beyond correlative analyses and for providing causal evidence in favor of these proposed MDPs and their role in the aging process and age-related diseases. Technical advances in CRISPR and other gene editing targeting mitochondria will help develop the animal models and will eventually resolve many of the pending issues. Hopefully, combined approaches will allow for a detailed understanding of the mechanisms underlying the MDPs in aging and will facilitate future interventions for improving human healthspan and longevity.
Fig. 2.
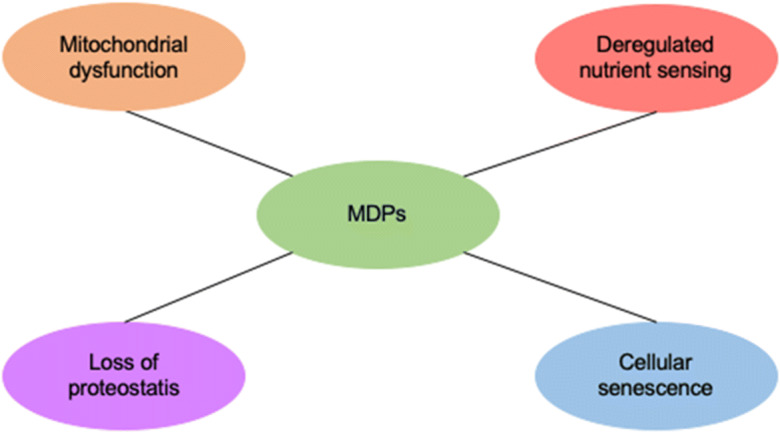
MDPs are associated with some hallmarks of aging
Acknowledgments
This work was supported by an Ellison/AFAR Postdoctoral Fellowship in Aging Research Program to SK.
Author contributions
SK, BM, HK, AS, MF, and KY all participated in the writing of this manuscript.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102–111. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giorgi C, Agnoletto C, Bononi A, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion. 2012;12:77–85. doi: 10.1016/j.mito.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hockenbery DM. Mitochondria and cell death. Humana Press; 2016.
- 4.Butow RA, Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell. 2004;14:1–15. doi: 10.1016/s1097-2765(04)00179-0. [DOI] [PubMed] [Google Scholar]
- 5.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson’s disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 7.Schrauwen-Hinderling VB, Kooi ME, Schrauwen P. Mitochondrial function and diabetes: consequences for skeletal and cardiac muscle metabolism. Antioxid Redox Signal. 2016;24:39–51. doi: 10.1089/ars.2015.6291. [DOI] [PubMed] [Google Scholar]
- 8.Siasos G, Tsigkou V, Kosmopoulos M, et al. Mitochondria and cardiovascular diseases-from pathophysiology to treatment. Ann Transl Med. 2018;6:256. doi: 10.21037/atm.2018.06.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholls DG. Mitochondrial membrane potential and aging. Aging Cell. 2004;3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 10.Carey BW, Finley LWS, Cross JR, et al. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baixauli F, Acin-Perez R, Villarroya-Beltri C, et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22:485–498. doi: 10.1016/j.cmet.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma R, Ramanathan A. The aging metabolome-biomarkers to hub metabolites. Proteomics. 2020;20:e1800407. doi: 10.1002/pmic.201800407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Müller M, Ahumada-Castro U, Sanhueza M, et al. Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front Neurosci. 2018;12:470. doi: 10.3389/fnins.2018.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohovych I, Khalimonchuk O. Sending out an SOS: mitochondria as a signaling hub. Front Cell Dev Biol. 2016;4:109. doi: 10.3389/fcell.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee C, Yen K, Cohen P. Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab. 2013;24:222–228. doi: 10.1016/j.tem.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KH, Son JM, Benayoun BA, Lee C. The mitochondrial-encoded peptide MOTS-c translocates to the nucleus to regulate nuclear gene expression in response to metabolic stress. Cell Metab. 2018;28:516–524.e7. doi: 10.1016/j.cmet.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C, Zeng J, Drew BG, et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21:443–454. doi: 10.1016/j.cmet.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cobb LJ, Lee C, Xiao J, et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging (Albany NY) 2016;8:796–809. doi: 10.18632/aging.100943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yen K, Mehta HH, Kim SJ, et al. The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY). 2020. 10.18632/aging.103534. [DOI] [PMC free article] [PubMed]
- 20.Qin Q, Delrio S, Wan J, et al. Downregulation of circulating MOTS-c levels in patients with coronary endothelial dysfunction. Int J Cardiol. 2018;254:23–27. doi: 10.1016/j.ijcard.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 21.Du C, Zhang C, Wu W, et al. Circulating MOTS-c levels are decreased in obese male children and adolescents and associated with insulin resistance. Pediatr Diabetes. 2018. 10.1111/pedi.12685. [DOI] [PubMed]
- 22.Gong Z, Tas E, Muzumdar R. Humanin and age-related diseases: a new link? Front Endocrinol (Lausanne) 2014;5:210. doi: 10.3389/fendo.2014.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23:1060–1065. doi: 10.1016/j.cmet.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saghatelian A, Couso J-P. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat Chem Biol. 2015;11:909–916. doi: 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yen K, Lee C, Mehta H, Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J Mol Endocrinol. 2013;50:R11–R19. doi: 10.1530/JME-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SJ, Xiao J, Wan J, et al. Mitochondrially derived peptides as novel regulators of metabolism. J Physiol Lond. 2017;595:6613–6621. doi: 10.1113/JP274472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto Y, Niikura T, Tajima H, et al. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc Natl Acad Sci U S A. 2001;98:6336–6341. doi: 10.1073/pnas.101133498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hashimoto Y, Ito Y, Niikura T, et al. Mechanisms of neuroprotection by a novel rescue factor humanin from Swedish mutant amyloid precursor protein. Biochem Biophys Res Commun. 2001;283:460–468. doi: 10.1006/bbrc.2001.4765. [DOI] [PubMed] [Google Scholar]
- 29.Ma Z-W, Liu D-X. Humanin decreases mitochondrial membrane permeability by inhibiting the membrane association and oligomerization of Bax and Bid proteins. Acta Pharmacol Sin. 2018;39:1012–1021. doi: 10.1038/aps.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong Z, Tasset I, Diaz A, et al. Humanin is an endogenous activator of chaperone-mediated autophagy. J Cell Biol. 2018;217:635–647. doi: 10.1083/jcb.201606095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao J, Kim SJ, Cohen P, Yen K. Humanin: functional interfaces with IGF-I. Growth Hormon IGF Res. 2016;29:21–27. doi: 10.1016/j.ghir.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chai G-S, Duan D-X, Ma R-H, et al. Humanin attenuates Alzheimer-like cognitive deficits and pathological changes induced by amyloid β-peptide in rats. Neurosci Bull. 2014;30:923–935. doi: 10.1007/s12264-014-1479-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan L, Liu X-J, Han W-N, et al. [Gly14]-humanin protects against amyloid β peptide-induced impairment of spatial learning and memory in rats. Neurosci Bull. 2016;32:374–382. doi: 10.1007/s12264-016-0041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maftei M, Tian X, Manea M, et al. Interaction structure of the complex between neuroprotective factor humanin and Alzheimer's β-amyloid peptide revealed by affinity mass spectrometry and molecular modeling. J Pept Sci. 2012;18:373–382. doi: 10.1002/psc.2404. [DOI] [PubMed] [Google Scholar]
- 35.Kim SJ, Guerrero N, Wassef G, et al. The mitochondrial-derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age- dependent signaling differences in the hippocampus. Oncotarget. 2016;7(30):46899–46912. doi: 10.18632/oncotarget.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hashimoto Y, Suzuki H, Aiso S, et al. Involvement of tyrosine kinases and STAT3 in Humanin-mediated neuroprotection. Life Sci. 2005;77:3092–3104. doi: 10.1016/j.lfs.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 37.Tajima H, Kawasumi M, Chiba T, et al. A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. J Neurosci Res. 2005;79:714–723. doi: 10.1002/jnr.20391. [DOI] [PubMed] [Google Scholar]
- 38.Zhang W, Zhang W, Li Z, et al. S14G-humanin improves cognitive deficits and reduces amyloid pathology in the middle-aged APPswe/PS1dE9 mice. Pharmacol Biochem Behav. 2012;100:361–369. doi: 10.1016/j.pbb.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Niikura T, Sidahmed E, Hirata-Fukae C, et al. A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PLoS One. 2011;6:e16259. doi: 10.1371/journal.pone.0016259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zárate SC, Traetta ME, Codagnone MG, et al. Humanin, a mitochondrial-derived peptide released by astrocytes, prevents synapse loss in hippocampal neurons. Front Aging Neurosci. 2019;11:123. doi: 10.3389/fnagi.2019.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murakami M, Nagahama M, Maruyama T, Niikura T. Humanin ameliorates diazepam-induced memory deficit in mice. Neuropeptides. 2017;62:65–70. doi: 10.1016/j.npep.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Hui-Fang L, Cai L, Wang X-M, Golden AR. Ethnic disparities in prevalence and clustering of cardiovascular disease risk factors in rural Southwest China. BMC Cardiovasc Disord. 2019;19:1–8. doi: 10.1186/s12872-019-1185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steenman M, Lande G. Cardiac aging and heart disease in humans. Biophys Rev. 2017;9(2):131–137. doi: 10.1007/s12551-017-0255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muzumdar RH, Huffman DM, Atzmon G, et al. Humanin: a novel central regulator of peripheral insulin action. PLoS One. 2009;4:e6334. doi: 10.1371/journal.pone.0006334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin Q, Mehta H, Yen K, et al. Chronic treatment with the mitochondrial peptide humanin prevents age-related myocardial fibrosis in mice. Am J Physiol Heart Circ Physiol. 2018;315:H1127–H1136. doi: 10.1152/ajpheart.00685.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang X, Urbieta-Caceres VH, Eirin A, et al. Humanin prevents intra-renal microvascular remodeling and inflammation in hypercholesterolemic ApoE deficient mice. Life Sci. 2012;91:199–206. doi: 10.1016/j.lfs.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thummasorn S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. High-dose humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc Ther. 2017;35:e12289. doi: 10.1111/1755-5922.12289. [DOI] [PubMed] [Google Scholar]
- 48.Yang Y, Gao H, Zhou H, et al. The role of mitochondria-derived peptides in cardiovascular disease: recent updates. Biomed Pharmacother. 2019. 10.1016/j.biopha.2019.109075. [DOI] [PubMed]
- 49.Zapała B, Kaczyński Ł, Kieć-Wilk B, et al. Humanins, the neuroprotective and cytoprotective peptides with antiapoptotic and anti-inflammatory properties. Pharmacol Rep. 2010;62:767–777. doi: 10.1016/S1734-1140(10)70337-6. [DOI] [PubMed] [Google Scholar]
- 50.Thummasorn S, Shinlapawittayatorn K, Khamseekaew J, et al. Humanin directly protects cardiac mitochondria against dysfunction initiated by oxidative stress by decreasing complex I activity. Mitochondrion. 2018;38:31–40. doi: 10.1016/j.mito.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 51.Lue Y, Swerdloff R, Wan J, et al. The potent humanin analogue (HNG) protects germ cells and leucocytes while enhancing chemotherapy-induced suppression of cancer metastases in male mice. Endocrinology. 2015;156:4511–4521. doi: 10.1210/en.2015-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu W-W, Wang S-R, Liu Z-H, et al. Gly[14]-humanin inhibits ox-LDL uptake and stimulates cholesterol efflux in macrophage-derived foam cells. Biochem Biophys Res Commun. 2017;482:93–99. doi: 10.1016/j.bbrc.2016.10.138. [DOI] [PubMed] [Google Scholar]
- 53.Nashine S, Cohen P, Chwa M, et al. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017;8:e2951. doi: 10.1038/cddis.2017.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sreekumar PG, Ishikawa K, Spee C, et al. The mitochondrial-derived peptide humanin protects RPE cells from oxidative stress, senescence, and Mitochondrial Dysfunction. Invest Ophthalmol Vis Sci. 2016;57:1238–1253. doi: 10.1167/iovs.15-17053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee C, Wan J, Miyazaki B, et al. IGF-I regulates the age-dependent signaling peptide humanin. Aging Cell. 2014;13:958–961. doi: 10.1111/acel.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruby JG, Smith M, Buffenstein R. Naked mole-rat mortality rates defy Gompertzian laws by not increasing with age. Elife. 2018;7:844. doi: 10.7554/eLife.31157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yen K, Wan J, Mehta HH, et al. Humanin prevents age-related cognitive decline in mice and is associated with improved cognitive age in humans. Sci Rep. 2018. 10.1038/s41598-018-32616-7. [DOI] [PMC free article] [PubMed]
- 58.Lorenzini A, Salmon AB, Lerner C, et al. Mice producing reduced levels of insulin-like growth factor type 1 display an increase in maximum, but not mean, life span. J Gerontol A Biol Sci Med Sci. 2014;69:410–419. doi: 10.1093/gerona/glt108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee C, Kim KH, Cohen P. MOTS-c: a novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic Biol Med. 2016;100:182–187. doi: 10.1016/j.freeradbiomed.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim SJ, Mehta HH, Wan J, et al. Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging (Albany NY) 2018;10:1239–1256. doi: 10.18632/aging.101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D'Souza RF, Woodhead JST, Hedges CP, et al. Increased expression of the mitochondrial derived peptide, MOTS-c, in skeletal muscle of healthy aging men is associated with myofiber composition. Aging (Albany NY) 2020;12:5244–5258. doi: 10.18632/aging.102944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu H, Wei M, Zhai Y, et al. MOTS-c peptide regulates adipose homeostasis to prevent ovariectomy-induced metabolic dysfunction. J Mol Med. 2019;50:19–13. doi: 10.1007/s00109-018-01738-w. [DOI] [PubMed] [Google Scholar]
- 63.Ming W, Lu G, Xin S, et al. Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochem Biophys Res Commun. 2016;476:412–419. doi: 10.1016/j.bbrc.2016.05.135. [DOI] [PubMed] [Google Scholar]
- 64.Reynolds J, Lai RW, Woodhead JST, et al. MOTS-c is an exercise-induced mitochondrial-encoded regulator of age-dependent physical decline and muscle homeostasis. bioRxiv 2019; 25:2019.12.22.886432. doi: 10.1101/2019.12.22.886432. [DOI] [PMC free article] [PubMed]
- 65.Woodhead JST, D’Souza RF, Hedges CP, et al. High-intensity interval exercise increases humanin, a mitochondrial encoded peptide, in the plasma and muscle of men. J Appl Physiol. 2020;128:1346–1354. doi: 10.1152/japplphysiol.00032.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kokkinos P, Myers J, Kokkinos JP, et al. Exercise capacity and mortality in black and white men. Circulation. 2008;117:614–622. doi: 10.1161/CIRCULATIONAHA.107.734764. [DOI] [PubMed] [Google Scholar]
- 67.Blair SN, Kohl HW, Paffenbarger RS, et al. Physical-fitness and all-cause mortality - a prospective-study of healthy-men and women. JAMA. 1989;262:2395–2401. doi: 10.1001/jama.1989.03430170057028. [DOI] [PubMed] [Google Scholar]
- 68.Warburton DER, Nicol CW, Bredin SSD. Health benefits of physical activity: the evidence. CMAJ. 2006;174:801–809. doi: 10.1503/cmaj.051351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fan W, Evans RM. Exercise mimetics: impact on health and performance. Cell Metab. 2017;25:242–247. doi: 10.1016/j.cmet.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kazuno A-A, Munakata K, Nagai T, et al. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2006;2:e128. doi: 10.1371/journal.pgen.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fuku N, Pareja-Galeano H, Zempo H, et al. The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell. 2015;14:921–923. doi: 10.1111/acel.12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zempo H, Kim SJ, Fuku N, et al. A pro-diabetogenic mtDNA polymorphism in the mitochondrial-derived peptide, MOTS-c. bioRxiv. 2019;53:695585. doi: 10.1101/695585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edghill EL, Flanagan SE, Patch A-M, et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes. 2008;57:1034–1042. doi: 10.2337/db07-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wabitsch M, Funcke J-B, Schnurbein v J, et al. Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J Clin Endocrinol Metab. 2015;100:3227–3230. doi: 10.1210/jc.2015-2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]