

Abstract
Although remarkable advances have been reported in high-throughput sequencing, the ability to aptly analyze a substantial amount of rapidly generated biological (DNA/RNA/protein) sequencing data remains a critical hurdle. To tackle this issue, the application of natural language processing (NLP) to biological sequence analysis has received increased attention. In this method, biological sequences are regarded as sentences while the single nucleic acids/amino acids or k-mers in these sequences represent the words. Embedding is an essential step in NLP, which performs the conversion of these words into vectors. Specifically, representation learning is an approach used for this transformation process, which can be applied to biological sequences. Vectorized biological sequences can then be applied for function and structure estimation, or as input for other probabilistic models. Considering the importance and growing trend for the application of representation learning to biological research, in the present study, we have reviewed the existing knowledge in representation learning for biological sequence analysis.
Keywords: Natural language processing, Representation learning, Sequence analysis, Word2vec, BERT
1. Introduction
Considerable advances in high-throughput sequencing have resulted in rapid data accumulation [1]. Although these modern technologies produce a considerable amount of data, they do not provide interpretation or biological information. Thus, the analysis of biological sequences, such as DNA/RNA/protein sequences, to realize biological discoveries has become more critical and challenging. To tackle this issue, the application of natural language processing (NLP) to sequence analysis has attracted considerable attention in terms of treating biological sequences as sentences and k-mers in these sequences as words [2], [3].
NLP aims to allow computers to understand the content of natural language, including the context, to accurately extract information, and to provide valuable insights [4]. Natural language is composed of characters, such as the alphabet, and the meaning is deduced and constructed using grammar and semantics. In the same manner, biological sequences can be regarded as sentences with different letters, and biophysical and biochemical rules can be used to define properties, such as the function and structure [5]. Biological sequences are consistent with natural language where characters are used to define their meaning, and the meaning depends on the neighboring sequence. For example, whether the word “bank” in a sentence refers to a financial institution or raised portion of seabed depends on the context. Similarly, whether a part of an RNA sequence forms a secondary structure depends on its neighboring sequences. Thus, considering the similarities between natural language and biological sequences, the application of NLP has the ability to provide a comprehensive understanding of the function and structure encoded in the biological sequence.
Representation learning is an essential step in NLP and indicates automatic systems to explore the representation of raw data, such as words or characters [6]. In general, the representation is provided as a real-valued vector known as distributed representation. Successful representation learning may convert words into vectors while preserving their semantic similarity. For example, the names of foods, like “sushi” and “pizza,” should be converted into similar vectors and the names of organisms, such as “frog,” should be assigned entirely different vectors (Fig. 1). In biological sequences, N-methyl-d-aspartate receptor and -amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, which are both ionotropic glutamate receptors, may be converted into similar vectors, whereas green fluorescent protein may be converted into a completely different vector. Thus, representation learning indicates the transformation from words to vectors while preserving the similarities and differences between words.
Fig. 1.
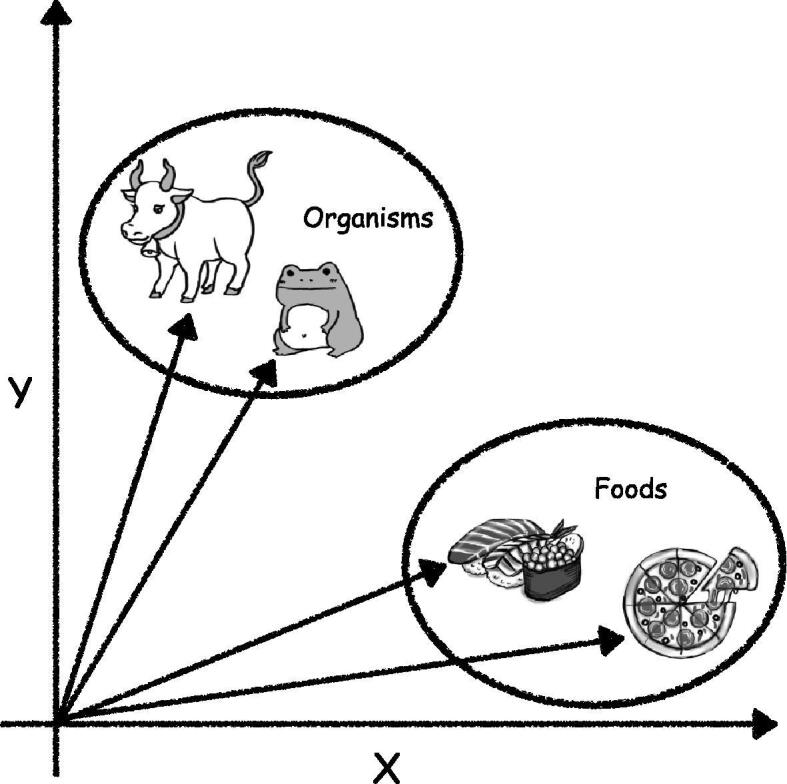
Ideal representation learning should perform the conversion of the names of foods, such as “sushi” and “pizza,” into similar vectors and assign different vectors to the names of organisms, such as “cow” and “frog.”.
Biological sequences vectorized by representation learning can be directly used for biological tasks, such as function and structure prediction [7], [8]. If the vector similarity between proteins is high, it can be inferred that they possess similar functions and structures. Note that vector similarity/distance can be calculated using linear algebra operations, such as dot product, Euclidian distance, and cosine similarity. Particularly, the successful encoding of words via representation learning has been recognized as an essential research area because the performance of NLP and deep learning depends on the quality of the representation [6]. Thus, a good representation of a biological sequence is critical for clustering, function, structure, and disorder prediction [2].
Considering the significance and growing trend in the application of representation learning in biology (Fig. 2), in the present study, we have described a review of representation learning for biological sequence analysis. It should be noted that this review covers concepts on the application of representation learning to biological sequence analysis, while its use in biological literature and medical records is beyond the scope of this review. This review is organized as follows: Section 2 introduces the basic representation techniques for NLP. Section 3 provides a comprehensive survey of representation learning approaches for sequence analysis. Section 4 presents a summary and an outlook of representation learning applications in biological sequence analysis.
Fig. 2.

Change in the number of hits for the search term “representation learning” (with double quotation) in PubMed ( https://pubmed.ncbi.nlm.nih.gov/).
2. Representation learning techniques
Currently, the acquisition of distributed representations of biological sequences is mainly achieved using neural networks developed in NLP. In representation learning for NLP, it is assumed that the words that appear in the same context have similar meanings according to the distribution hypothesis [9]. Representation learning methods based on the distribution hypothesis are used with an aim to vectorize words or phrases by training the neural networks with architectures specialized for understanding the relationships among words from a corpus (a set of documents. Various representation learning methods presented in this review are based on neural-network-based language models specialized for biological sequences; thus, it is essential to understand the underlying architecture of the neural networks developed for NLP. In this section, we have briefly summarized the development of basic representation learning techniques.
word2vec was the first successful method used to obtain distributed representations using a neural network [10], [11]. There are two types of neural networks used in word2vec and they are as follows: a skip-gram model, that predicts the words around the input word, and a continuous bag-of-words model, that predicts the target word from the surrounding words. Until the advent of word2vec, researchers used neural networks to describe the syntactic structure [12], [13]. The skip-gram model proposed by Mikolov attracted attention owing to its ability to capture not only grammatical correctness but also semantic features, as described in the introduction. word2vec with the skip-gram model acquires a distributed representation for each word by training the three-layer neural network, as shown in Fig. 3. Considering a sentence with T words and the t-th word , the model predicts the words present in the vicinity of in that sentence. Pre-defined vicinity is a hyper-parameter that is denoted as a constant, c. It shows the number of words that should be included in the prediction around .The parameters to be estimated in the skip-gram model include the weight matrix X to predict the d-dimensional hidden layer from the one-hot encoded input layer and weight matrix Y to predict the output from h. They are predicted using the formula described below:
| (1) |
Fig. 3.

Skip-gram model used in word2vec. This neural network model includes the following three fully connected layers: the input, hidden, and output layers. In this case, it attempts to learn the features from the sentence, “I am majoring in biology,” and to predict the words surrounding , “majoring. ”.
The model performs the same operation for all sentences and repeats multiple epochs to complete training. In this case, the weight matrix X is a matrix, where V represents the number of words in the vocabulary. If is the v-th word in the vocabulary, we can obtain the distributed representation of the word as the v-th vector of the predicted X (i.e., ). The word2vec representation has additive compositionality and has garnered fame for allowing intuitive operations, such as , as shown previously [11]. Hence, the use of word2vec succeeded in obtaining highly interpretable distributed representations for the first time and helped to direct subsequent development in representation learning.
The fact that word2vec captures semantic features is a remarkable breakthrough in representation learning, which has prompted the proposal of various extended models based on word2vec. GloVe uses word co-occurrence matrices, which have been used in classical latent semantic analysis, such as singular value decomposition [14]. It shows higher semantic accuracy than word2vec. FastText is an embedding method based on the skip-gram model [15]. It considers sub-word information that allows for the prediction of words that do not appear in the training data. Additionally, several methods have been developed to obtain a distributed representation for each sentence (not word) based on the word2vec concept. doc2vec utilizes the paragraph vectors, which captures the context for each paragraph and provides the features for each sentence [16].
Although word2vec has enabled considerable progress in representation learning, it cannot be used to express the semantic polysemy of words as it yields a single d-dimensional vector for a single lexicon, as mentioned above. For example, “right” that appears in “right to vote” and “turn right” differ in meaning; however, they are embedded at the same point using word2vec. The approach to solving this problem is known as word sense disambiguation in NLP [17], and it prescribes architecture for considering the context and meaning of a sentence. In biological sequences, the context of a word in a sentence is equivalent to the role of a particular nucleic/amino acid in the whole sequence. Hence, the polysemy in biological sequences is critical, similar to that observed in natural languages. Here, we have introduced the following two methods that can allow the consideration of such contexts: one method that can be performed to achieve this by rendering the neural network recursive using a recurrent neural network (RNN) or long short-term memory (LSTM) [18] and another method that uses the attention mechanism.
RNN and LSTM are developments of the classical autoregressive language models that have been primarily utilized for sequential tasks, such as document generation and machine translation [19], [20]. In the language model with a forward LSTM, as shown in Fig. 4, the occurrence probability of the t-th word in a sentence, , depends on the set of words that appear before (denoted as ). The model trains the parameters to maximize the joint probability for all words, . To calculate , LSTM uses the hidden layer of (the output for which is denoted by ), which depends on and . As the hidden layer is computed recursively depending on the word order, LSTM-based models allow context-aware learning. Currently, most LSTM-based language models are based on bidirectional-LSTM (bi-LSTM), which can be used to consider the context not only in the forward but also in the reverse direction. In a backward LSTM, the hidden layer of and its output depend on and . By considering word dependency in the backward direction, bi-LSTM can incorporate relationships among words that cannot be captured by using the forward LSTM alone. In bi-LSTM, all hidden layers are trained to maximize the joint probability of generating the entire sentence as follows:
| (2) |
Fig. 4.

Graphical representation of a forward LSTM. Input shows the embedding of the t-th word , and the output is transformed to a probability using a softmax function. For example, if is “majoring”, the model is trained to increase the possibility that “majoring” is output from , which is calculated from words up to , “am”.
Embeddings from language models (ELMo) represent the distributed representations provided by the model stacked with multiple bi-LSTM [21]. This model is referred to as bidirectional language model (bi-LM), and contains a stack of L bi-LSTM modules. ELMo is obtained by estimating the weighted-sum of outputs from layers, which are hidden layers for both forward and backward LSTM modules and an input embedding layer. ELMo avoids polysemy as it refers to the hidden layers of LSTM which considers the context for the input sentence, in addition to the input embedding layer which depends only on the lexicon. In fact, ELMo successfully embeds the same lexicon to different points in a high-dimensional space, depending on the context.
Another approach for addressing the polysemy issue is to use the attention mechanism. Briefly, attention quantifies the degree of dependency between words [22], [23]. Neural networks with attention mechanisms comprise an attention weight that is obtained by calculating the association of hidden layers (e.g., using the inner product) for arbitrary combinations of words in sentences. If the two words used to compute the attention weight originate from different sentences, this attention is referred to as the source-target-attention. On the other hand, if they originate from an identical sentence, it is designated as self-attention. Models that are based on the use of attention weights in the forward propagation are extremely expressive, allowing for a natural introduction of an attention mechanism to representation learning. Transformer, which implements the attention mechanism and positional encoding [24] in Key-Value Memory neural network [25], [26] without conventional context-aware architectures, such as RNN or LSTM, has demonstrated achievement of a state-of-the-art accuracy in several tasks, including machine translation [27].
Bidirectional encoder representations from transformers (BERT) is the model with multiple stacks of transformers (see Fig. 5) [28]. In the pre-training of BERT, the input is a set of tokens connecting two sentences. A part of the input words is randomly masked. When the masked word is the t-th word, , the model predicts what is considered as the context before and after . This language model is called Masked Language Model (MLM). Compared to the traditional autoregressive language models, MLM can “jointly”, rather than “independently”, consider the context before and after. That is, the occurrence probability of , cannot be factorized into ; this modification contributes to the improved accuracy. Therefore, in contrast to the Eq. (2), MLM maximizes the following joint likelihood:
| (3) |
where shows the set of masked tokens. Additionally, the model performs a binary classification of whether the two input sentences are semantically consecutive. Similar to the approaches used in other methods, we can use the outputs of pre-trained transformer layers as the distributed representations of input sentences.
Fig. 5.

The graphical representation of Bidirectional encoder representations from transformers (BERT) architecture. Preparation of special tokens ([CLS], [MASK] and [SEP]) enables the model to extract features based on the self-attention of the whole sentence. BERT is trained with the following two tasks: masked language model (MLM) and next sentence prediction (NSP). In pre-training for MLM, the model predicts the masked tokens original meaning (e.g., predicting “have” and “dollars” from ) considering the context before and after the masked tokens.
Neural networks with attention mechanisms, such as transformer and BERT, capture distal word associations better than conventional recursive models represented by RNN and LSTM [22], [29]. This is because, in recursive models, a hidden layer of a certain word depends on the hidden layers of the neighboring words only, and the contribution of distal words becomes small or converges to zero. In contrast, the use of attention is robust against such weight loss since the model always refers to its association with all words. This feature of BERT is attractive from the biological perspective since distal interactions are important for structural predictions and other purposes. Meanwhile, the calculation of the all-against-all attention always involves substantial computational complexity, for a sentence with T words. Thus, it is important to reduce this complexity, for which various approximation methods have been proposed [30], [31]. Another advantage of using BERT is task-independent versatility. For instance, when we use ELMo, it is necessary to prepare a task-specific model to transfer the obtained distributed representations to other tasks. In such cases of transfer learning, the new model may forget the features learned in pre-training, thus necessitating the conduction of careful retraining of the model in a sophisticated manner represented by ULMfit [32]. In contrast, with BERT, we can utilize the same architecture used in pre-training (as shown in Fig. 5) without modification. Fine-tuning, which uses pre-trained hidden layers for initialization and optimizes the parameters for each task, has achieved state-of-the-art accuracy in several NLP tasks [28].
The main advantage of obtaining features through unsupervised learning is that it can retain versatility for the transfer learning to various tasks. However, to build a specialized model for a specific task, representation learning in a supervised manner is also useful. StarSpace is a supervised learning method [33], which uses labeled documents as the training dataset, and embeds words and labels in the same space so that a label is close to words associated with it. Embedding with StarSpace allows for text classification, that is, the prediction of labels used in the course of learning with higher accuracy than the other unsupervised methods, and it provides highly interpretable vectors. As shown by this example, supervised representation learning is also a practical option if the correct labels are known.
Since the development of word2vec in 2013, the field of representation learning in NLP has been expanding at an astonishing pace. Considering the models based on transformer or BERT, several modern improved methods have continued to provide increased accuracy [34], [35]. Furthermore, similar to the considerable impact of the attention mechanism, the emergence of new concepts may also help reconstruct the current paradigm of language modeling. These substantial developments in machine learning will be useful for bioinformatics and sequence analyses. As numerous examples are introduced in later sections, we believe that application of the latest representation learning techniques to biological sequences will lead to a discovery or elucidation of novel information in this domain.
3. Survey of representation learning applications in sequence analysis
We conducted an exhaustive survey, as shown in Table 1 and supplementary data, for articles that met the following criteria: (i) peer-reviewed and published in PubMed, except for BERT, which was recently published with a limited number of peer-reviewed articles; (ii) explicitly used a language model, such as word2vec or BERT; (iii) provided the source code or the model for repeatability or verification.
Table 1.
Comprehensive survey of representation learning application in biological sequences
| Method name | Model | Training data | Task | Avail. and repr. | Ref. |
|---|---|---|---|---|---|
| ProtVec | word2vec | 547 K proteins | family classification, disorder prediction | + | [36] |
| HLA-vec | word2vec | HLA-I binding/non-binding peptides | HLA-I binding prediction | ++ | [37] |
| m-NGSG | word2vec | 0.1 K–3 K proteins | protein classification | ++ | [38] |
| ene2vec | word2vec | 89 K positive and 495 K negative mRNAs | N6-methyladenosine site prediction | ++ | [39] |
| – | word2vec | 3 K–101 K of 300 bp genomic regulatory regions | regulatory region prediction | ++ | [40] |
| ProtVecX | word2vec | 371–44 K proteins | venom toxin prediction, enzyme prediction | +++ | [41] |
| MHCSeqNet | word2vec | 228 K peptide-MHC pairs | MHC binding prediction | +++ | [42] |
| – | word2vec | 1 M 16S rRNAs | sample class (e.g., body part) prediction | +++ | [43] |
| fastDNA | word2vec | 356–3 K bacterial genomes | species identification | ++ | [44] |
| NucleoNN | word2vec | 86/72 SNPs in the control/exposure samples | investigating allele-interactions | ++ | [45] |
| – | word2vec | 3 K–22 K CPI pairs | CPI prediction | +++ | [46] |
| FastTrans | word2vec | 1 K membrane transporter and 1 K membrane non-transporter proteins | substrate prediction of transport proteins | ++ | [47] |
| INSP | word2vec | 78 nuclear proteins | nuclear localization prediction | ++ | [48] |
| – | word2vec | 9 M proteins | function prediction | ++ | [49] |
| Its2vec | word2vec | 126 K ITSs | species identification | ++ | [50] |
| 4mCNLP-Deep | word2vec | C. elegans genome (WBcel235/ce11) | N4-methylcytosine sites prediction | ++ | [51] |
| – | doc2vec | 525 K proteins | localization, T50, absorption, enantioselectivity prediction | +++ | [52] |
| EP2vec | doc2vec | 650 K enhancers and 93 K promotors | enhancer-promoter interaction prediction | ++ | [53] |
| IDP-Seq2Seq | Seq2Seq | 3 K proteins | disorder prediction | ++ | [54] |
| – | Glove | 244 K–504 K chromatin accessible regions | chromatin accessibility prediction | ++ | [55] |
| CircSLNN | Glove | 37 dataset of RBP-binding sites on circular RNAs | RBP-binding sites prediction of circRNAs | + | [56] |
| – | FastText | 3 K promoters and 3 K non-promoters | promoter stregnth classification | ++ | [57] |
| iEnhancer-5Step | FastText | 1 K human enhancers and 1 K human non-enhancers | enhancer prediction | ++ | [58] |
| TNFPred | FastText | 18 tumor and 133 non-tumor necrosis factors | tumor necrosis factors classification | ++ | [59] |
| eDNN-EG | FastText | 518 essential and 1 K non-essential genes | essential gene prediction | + | [60] |
| ProbeRating | FastText | 440 K proteins and 274 K nucleic acids | nucleic acid-binding proteins binding preference prediction | ++ | [61] |
| CSCS | bi-LSTM | 4 K–58 K viral proteins | viral escape mutation prediction | +++ | [62] |
| UniRep | mLSTM | 24 M proteins | structure and function prediction | +++ | [63] |
| UDSMProt | AWD-LSTM language model | 499 K proteins | enzyme class prediction, gene ontology prediction, remote homology, fold detection | +++ | [64] |
| USMPep | AWD-LSTM language model | 23 K–120 K MHC binding peptides | MHC binding affinity prediction | ++ | [65] |
| BindSpace | StarSpace | 505 K TF-associated and 505 K non-associated DNA | TF-binding prediction | ++ | [66] |
| MutSpace | StarSpace | cancer mutation sites | cancer type prediction | ++ | [67] |
| SeqVec | ELMo | 33 M proteins | 3-state secondary structure prediction, disorder prediction, localization prediction, membrane prediction | ++ | [68] |
| NuSpeak | ULMfit | 92 K RNAs | designing RNA toehold switches | ++ | [69] |
| DNA- transformer | transformer | E. coli genome (MG1655) | transcription start sites, translation initiation sites, 4mC methylation sites prediction | ++ | [70] |
| TAPE | BERT | 31 M proteins | 3-state secondary structure prediction, contact prediction, remote homology detection, fluorescence prediction, stability prediction | +++ | [71] |
| ESM-1b | BERT | 27 M–250 M proteins | remote homology detection, 8-state secondary structure prediction, contact map prediction, quantitative prediction of mutational effects | ++ | [72] |
| ProtBert | BERT | 216 M–2B proteins | 3-/8-state secondary structure prediction, subcellular localization prediction, membrane-boundness prediction | ++ | [73] |
| DNABERT | BERT | H. sapiens genome (GRCh38.p13) | promoter prediction, TF-binding site prediction, splicing site prediction, functional variant analysis | +++ | [74] |
| BERT4Bitter | BERT and bi-LSTM | 256 bitter and 256 non-bitter peptides | prediction of bitter peptides | ++ | [75] |
| BERT- Enhancer | BERT and CNN | 1 K human enhancers and 1 K human non-enhancers | enhancer prediction | ++ | [76] |
| BERT-RBP | BERT | 10 K RBP-bound and 10 K RBP-unbound RNA sequences | RNA-RBP interaction prediction | ++ | [77] |
Avail. and repr. indicate availability and reproductivity, respectively. (+++) The source code for the generation of the model, pre-trained model, and for conducting detailed documentation, including data links and installation instructions, are available. (++) Either the source code for the generation of the model or the pre-trained model is available, and detailed documentation, including data links and installation instructions, are available. (+) Either the source code for the generation of the model or the pre-trained model is available, but the documentation is limited. Model indicates a general model (described in Section 2) utilized in the method. K, kilo; M, mega; B, billion; HLA, human leukocyte antigen; MHC, major histocompatibility complex; CPI, compound–protein interaction; ITS, internal transcribed spacer; RBP, RNA binding protein; TF, transcription factor.
3.1. Applications for structure/function prediction
ProtVec is the first model to use the embedding method for biological sequences [36]. This method regarded 3-mers of amino acids as words and used data on 546,790 protein sequences obtained from the Swiss-Prot database as the training dataset. Subsequently, word2vec using the skip-gram model was applied to the dataset, and 100-dimensional protein vectors were calculated. Originally, ProtVec was evaluated based on protein family classification and disordered protein prediction accuracies and it achieved high performance in both. Currently, ProtVec has also been utilized for predicting kinase activity [78] and gene function [79]. As ProtVec is a straightforward model, various extensions have been proposed. One of the extensions is seq2vec, which embeds not the k-mers of amino acids but embeds the whole protein sequences [80]. Seq2vec utilizes doc2vec [16], an NLP method that embeds documents instead of words, which showed a higher performance than ProtVec in terms of protein family classification performance. Another extension is dna2vec [81], which embeds variable-length k-mers rather than fixed-length DNA k-mers using word2vec. ProtVecX is a similar method that uses word2vec to embed variable-length amino acid k-mers [41].
SeqVec is the first model that uses ELMo to achieve amino acid representation based on the whole protein sequence [68]. ELMo was applied to the UniRef50 dataset, which contains 33 M proteins with 9.6G residues, regarding single amino acids as words. The extracted features were then used as input into the per-residue prediction and per-protein prediction. With and without the evolutionary information, the model could accurately predict the secondary structure, disorder, localization, and membrane binding. The performance did not exceed that of the state-of-the-art methods [82], [83]. However, it was better than ProtVec [36] which is a context-independent model. In certain tasks, such as protein function prediction, it outperformed one-hot encoding of k-mer-based embeddings and provided competitive results obtained using ELMo [84].
UDSMProt is another language model representation extractor using a variant of LSTM [64]. The structure used is called AWD-LSTM [85], which is a three-layered bi-LSTM that introduces different types of dropout methods to achieve accurate word-level language modeling. UDSMProt was initially applied to the Swiss-Prot database and then fine-tuned for specific tasks, such as enzyme commission classification, gene ontology prediction, and remote homology detection. UDSMProt showed that upon pre-training with external data, the model performed in a manner that was comparable to the existing methods that were tailored to the task using a position-specific scoring matrix (PSSM) and outperformed them in two out of the three tasks conducted. Additionally, it demonstrated that utilization of pre-training information could compensate for the lack of data, compared to the case where PSSM information was provided. These results and extensions, such as USMPep, which revealed the ability to successfully predict MHC class I binding [65], imply that language models can be used to efficiently contextualize and achieve word-based representation.
ESM-1b is a BERT-based model trained on a massive biological corpus, particularly amino acid sequences [72]. The study presented a series of BERT models with varying parameter sizes. After conducting pre-training using up to 250 million protein sequences, where each amino acid residue in a sequence was treated as a word, models could accurately predict the structural characteristics of proteins, including remote homology, secondary structure, and residue–residue contact. Representations put forth by the pre-trained 34-layer model were merged with multiple sequence alignments (MSAs), which were considered as the original input of the existing secondary structure or contact prediction methods, and data on their prediction accuracy was improved. This result indicated that embedded representations based on the pre-trained BERT incorporated more information than the MSAs. Furthermore, the 34-layer model was fine-tuned to predict the quantitative effect of mutations and was found to outperform the state-of-the-art methods. Apart from the model trained on individual sequences, Rao et al. proposed a model trained on the sets of amino acid sequences in the form of MSAs [86]. As an attractive alternative, other protein BERT models, such as TAPE transformer and ProtBert, have also been developed [71], [73]. Meticulous inspection of the TAPE transformer revealed that data on attention maps extracted from the pre-trained model reflected the context of input amino acid sequences [87]. For instance, one attention module, which specializes in deciphering residue–residue interactions, exhibited a significant correlation with experimental labels although no structural information was provided. This phenomenon was later investigated by reconstructing protein contact maps using data obtained from the attention maps of pre-trained ESM-1b [7]. The collection of studies illustrates that BERT-based models are highly interpretable and widely applicable to protein-related bioinformatics problems.
DNABERT, in contrast, is the only model currently available that can be used to pre-train BERT-based models using a whole human reference genome [74]. During preprocessing, the genome, whose gaps and unannotated regions were excluded, was split into 5 to 510 consequent nucleotide sequences without overlapping and subsequently converted to 3- to 6-mer representations. In a simple sense, each subsequence of length 3 to 6 was regarded as a word. BERT models were pre-trained using k-mers with a masked language modeling objective and applied to downstream tasks. Upon performing task-specific fine-tuning, DNABERT demonstrated state-of-the-art or comparative performance in predicting promoter regions, binding sites of transcription factors (TFs), and splice sites. Attention analysis revealed that fine-tuned models captured the characteristics of each set of target sequences. For example, DNABERT fine-tuned using splicing datasets exhibited high attention weights in intronic regions in addition to the target splice sites, indicating the ability of the model to learn the contextual significance of splicing enhancers or silencers in predicting splice sites. The study further applied DNABERT to predict promoters in the mouse genome and reported higher performance than those of existing deep learning methods. Additionally, we recently adapted DNABERT for predicting RNA–protein interactions and demonstrated that the fine-tuned model could translate transcript region type and RNA secondary structure through attention analysis [77]. Overall, two-step training of the BERT architecture demonstrated its broad application to translate various genomic features in a cross-organism manner.
3.2. Applications for molecular interactions
Tsubaki et al. proposed a model by combining a graph neural network for compounds and a convolutional neural network (CNN) for proteins to predict compound–protein interactions (CPIs) [46]. Representations of compounds and proteins were obtained in an end-to-end manner. The word embeddings in the protein were learned from the training dataset using word2vec (3-mer of amino acids as words). To obtain protein vector representation, the average value of a set of hidden vectors was used with d-dimensional embedding after a hierarchical convolutional filter. Extensive evaluations were conducted for three CPI datasets (human, C. elegans [88] and DUD-E dataset [89]). The results showed that using the raw amino acid sequence as the input, the proposed approach significantly outperformed the existing methods utilizing traditional chemical and biological features. They also established that the model could highlight 3D structural interaction sites between the compounds and proteins through an attention mechanism similar to that observed with words in sentences.
ProbeRating is a neural network-based recommender system utilizing word embeddings in NLP to infer binding profiles for unexplored nucleic acid-binding proteins (NBPs) [61]. ProbeRating achieves this goal using a two-stage framework. In the first stage, representation learning is performed using a package called FastBioseq, implementing FastText. Thus, data on the input feature vectors are extracted from the NBP sequences and nucleic acid probes. The authors previously selected 3-mers amino acids for proteins and 5-mers for nucleic acids as words. Three datasets (Uniprot400k [90], RRM3k [91], and Homeo8k [92]) were used to pre-train the FastBioseq protein embedding models, whereas RNA embedding models were trained directly from the RRM162 dataset [91]. In contrast, 8-mer frequency features were used for the DNA sequences in the Homeo215 dataset [93]. In the second stage, prediction of the NBP binding preference was redefined as a recommender system formulation, where NBPs are considered as users and RNAs or DNAs are considered as products to be recommended. When no preference was available for a given user, the authors adapted and extended a strategy that converted the binding intensity prediction problem into a similarity prediction problem, solved it, and then converted it back. Extensive evaluation experiments were conducted for the following two tasks: RBP–RNA interaction and TF–DNA interaction. The results showed that ProbeRating outperformed three baseline methods (Nearest-Neighbor, Co-Evo [94] and AffinityRegression [93]). Further analysis suggested that this advantage was beneficial using both the neural network approach and data on input features extracted via word embeddings.
3.3. Applications in synthetic biology
Valeri et al. proposed a model that could predict synthetic riboregulators called toehold switches [95]. The model comprised a language model for toehold switch classification and a CNN-based model for toehold switch performance regression. In the language model, a sequence of toehold switches was embedded using ULMfit regarding a nucleotide as a word. They trained the model using toehold switches experimentally characterized by Angenent-Mari et al. [96]. The results showed that the model exhibited good and robust performance even for sparse training data and that the features obtained by the model revealed unknown properties of the toehold switches. They also showed that the trained model is easily fine-tuned by transfer learning using small external data [97], [98], and the fine-tuned model exhibited superior performance compared to an existing model. Finally, they showed that the fine-tuned model could help in the efficient design of toehold switches for various applications, such as SARS-CoV2 detection.
UniRep is a representation that comprehensively summarizes the semantics of arbitrary proteins and can be useful for various types of prediction tasks [99]. A protein sequence is embedded into UniRep using multiplicative LSTM (mLSTM), trained with 24 M UniRef50 sequences [100], where an amino acid is regarded as a word. UniRep is used to recapitulate biophysical properties, phylogenetics, and secondary structures of proteins. The authors also showed that UniRep outperformed other representations for predicting the structural and functional properties of de novo proteins, single point mutants, and natural proteins. These results suggest that UniRep is useful for the rational design of proteins. As a proof-of-concept, UniRep re-trained using deep mutational scanning data of GFP [101] was shown to effectively extrapolate GFP brightness outside the training domain. Therefore, UniRep was suggested to markedly reduce the cost for the rational design of GFP. Collectively, UniRep embodies various known protein characteristics and may be a versatile representation for protein bioinformatics.
3.4. Applications for other tasks
StarSpace is a supervised embedding method, which is different from the unsupervised embedding methods that we have introduced in Section 2 [33]. Although StarSpace was originally developed for general NLP tasks, such as text classification, there are currently two bioinformatics applications available. The first application is BindSpace, which is used to predict the binding sites of TFs [66]. BindSpace uses HT-SELEX experiments as the training dataset and applies StarSpace to the dataset by considering 8-mers and TFs as words and labels, respectively. In performance evaluation using the ENCODE ChIP-seq dataset, BindSpace achieved high classification performance even between paralogous TFs, which contain highly similar binding motifs. The second application is MutSpace, which is used to estimate the cancer types of patients from somatic mutation patterns [67]. This method regarded mutation patterns and cancer types as words and labels, respectively. MutSpace shows state-of-the-art performance in a breast cancer subclass classification problem. The high performance of these two applications means that StarSpace is likely to perform well in countering other bioinformatics problems.
A constrained semantic change search (CSCS) is a method for discovering word changes that significantly alter the semantics from an original sentence based on embedding techniques [102]. The key feature of this method is that it does not detect word changes that would abolish the grammar of the sentence but those that preserve the grammatical structure. For example, in an NLP task, CSCS can change “winegrowers revel in good season” to “winegrowers revel in flu season.” We define x and as the original and mutated sentences, respectively. The embedded representations of x and are defined as z and , respectively. Here, the semantic change is modeled as the distance between these embedded representations, that is, . Additionally, the preservation of the grammatical structure is evaluated by , which is also modeled using embedding techniques. Finally, maximizing , where is a scaling factor. One biological application of CSCS is the modeling of viral evolution [62]. This application considered viral proteins, preservation of the infectivity, and escape from antibody recognition as sentences, preservation of grammar, and semantic change, respectively, and detected escape mutations from immune systems as a result of the CSCS analysis. The analyses of HIV-1 and influenza viruses showed that mutations detected by the CSCS were in good agreement with the experimental mutation results.
Woloszynek et al. applied word2vec to a metagenomic dataset by regarding 4–15-mers in sequencing reads as words [43]. They trained word2vec with a skip-gram model using 2,262,986 full-length 16S rRNA amplicon sequences from GreenGenes [103], a microbial 16S rRNA sequence database obtained using metagenomic analysis. They verified the robustness of the model in a taxonomic identification task using an independent dataset of 16,699 full-length 16S rRNA sequences from the KEGG REST server [104] as a validation dataset. The embedding features exhibited superior performance to the k-mer frequency features. Additionally, the embedding has also performed using the American Gut project dataset [105], which comprises 11,341 partial 16S rRNA sequences from three body sites (the gut, skin, and oral cavity), and showed comparable performance to conventional methods, such as sequence alignment in the body site classification task. These results suggest the availability of embedding with pre-trained models instead of sequence alignment for metagenomic sequence profiling.
4. Summary and outlook
In this study, we introduced basic algorithms and reviewed the recent literature concerning representation learning applications in sequence analysis. Heinzinger, et al. highlighted three difficulties in biological sequence modeling with NLP [68] as follows: (i) proteins range from approximately 30 to 33,000 residues, which is markedly longer than the average English sentence, which consists of 15 to 30 words [106]; (ii) proteins use only 20 amino acids in most cases; if we consider one amino acid as a word, the word repertoire is 1/100,000 of English language, and if we consider 3-mer as a word, the word repertoire is 1/10 to 1/100 of English language; (iii) UniProt [90] is 10 times larger than the size of Wikipedia in terms of data repository size, and extracting information from a very large biological database may require the use of a commensurate model. Embedding of biological sequences using NLP overcomes these difficulties and outperforms existing methods in several tasks, such as function, structure, localization, and disorder prediction (Table 1). In addition to these general biological tasks, representation learning has also been used to solve specific problems, such as RNA aptamer optimization [107], viral mutation prediction [62], and venom toxin prediction [41]. In these studies, representation learning of biological sequences could capture biophysical and biochemical properties of the biological systems.
The development of novel representation learning methods has been actively studied in machine learning research. For example, hyperbolic embedding methods have been pursued in recent years [108], [109]. These methods allow embedding of the data not in Euclidean space, which is utilized in all the studies introduced in this review, but in the hyperbolic space. The hyperbolic space exhibits constant negative curvature; thus, it shows characteristic geometric features not observed in Euclidean space, such as the sum of the interior angles of a triangle being less than 180. Changes in the embedding space can considerably alter the efficiency of representation learning, while theoretical and experimental analyses have shown that hyperbolic embedding methods are suitable for data with hierarchical latent structure. Furthermore, research on embedding into more complex spaces, such as mixed-curvature spaces, has also attracted attention [110]. Although these non-Euclidean embedding methods have recently been used for various biological analyses, such as phylogenetic [111], and single-cell RNA-seq analyses [112], no applications exist for the biological sequence analysis emphasized in this review. Thus, the development of such an application is warranted.
Data on new approaches are published daily in this field, and the scientific community is engaging relentless efforts to compare their accuracy and to validate their potential uses [71], [113], [114]. It is, therefore, important to ensure the models are available in an easy-to-use format with documentation. Considering that powerful computer resources are required for the establishment of large-scale language models, such as transformer-based models, researchers without access to these resources will be unable to reproduce them even with the source code. Additionally, considering the rapid growth of biological databases, the source code for creating models should be made available for future updates. Only a limited number of studies have published data on both the source code and the pre-trained model with the relevant documentation. Finally, participants in this community must publish their papers in a reproducible and verifiable format.
In this study, we comprehensively surveyed and reviewed the application of representation learning to biological sequence analysis. Although NLP-based biological sequence analysis is in its early stages and warrants further development, in the light of novel challenges in biology, such as single-cell analysis, genome design, and personalized medicine, representation learning may contribute to the progression of bioinformatics studies thus revealing the grammar of life.
CRediT authorship contribution statement
Hitoshi Iuchi: Conceptualization, Writing - original draft. Taro Matsutani: Writing - original draft. Keisuke Yamada: Writing - original draft. Natsuki Iwano: Writing - original draft. Shunsuke Sumi: Writing - original draft. Shion Hosoda: Writing - original draft. Shitao Zhao: Writing - original draft. Tsukasa Fukunaga: Writing - original draft. Michiaki Hamada: Supervision, Writing - review & editing.
Conflict of Interest
The authors have no conflicts of interest directly relevant to the content of this article.
Acknowledgements
The illustrations in Fig. 1 were kindly provided by Kae Namie. This work was supported by the Ministry of Education, Culture, Sports, Science, and Technology (KAKENHI) [Grant Nos.: JP17K20032, JP16H05879, JP16H06279 JP19H01152 and JP20H00624 to MH, JP19K20395 to TF, JP19J20117 to SH, JP20J20016 to TM and JP21K15078 to HI] and JST CREST [Grant Nos.: JPMJCR1881 and JPMJCR21F1 to MH].
Footnotes
Supplementary data associated with this article can be found, in the online version, athttps://doi.org/10.1016/j.csbj.2021.05.039.
Contributor Information
Hitoshi Iuchi, Email: hitosh.iuchi@gmail.com.
Michiaki Hamada, Email: mhamada@waseda.jp.
Supplementary data
The following are the Supplementary data to this article:
References
- 1.Cunningham F., Achuthan P., Akanni W., Allen J., Amode M.R., Armean I.M. Ensembl 2019. Ensembl Nucleic Acids Res. 2019;47(D1):2019. doi: 10.1093/nar/gky1113. pp. D745–D751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Angermueller C., Pärnamaa T., Parts L., Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zou J., Huss M., Abid A., Mohammadi P., Torkamani A., Telenti A. A primer on deep learning in genomics. Nat Genet. 2019;51(1):12. doi: 10.1038/s41588-018-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowdhury G.G. Natural language processing. Annu Rev Inf Sci Technol. 2005;37(1):51–89. doi: 10.1002/aris.1440370103. [DOI] [Google Scholar]
- 5.Yu L., Tanwar D.K., Penha E.D.S., Wolf Y.I., Koonin E.V., Basu M.K. Grammar of protein domain architectures. Proc Natl Acad Sci USA. 2019;116(9):3636–3645. doi: 10.1073/pnas.1814684116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bengio Y., Courville A., Vincent P. Representation learning: a review and new perspectives. IEEE Trans Pattern Anal Mach Intell. 2013;35(8):1798–1828. doi: 10.1109/TPAMI.2013.50. arXiv:1206.5538. [DOI] [PubMed] [Google Scholar]
- 7.Rao R, Meier J, Sercu T, Ovchinnikov S, Rives A. Transformer protein language models are unsupervised structure learners, International Conference on Learning Representations (2021).
- 8.Littmann M., Heinzinger M., Dallago C., Olenyi T., Rost B. Embeddings from deep learning transfer GO annotations beyond homology. Sci Rep. 2021;11(1):1160. doi: 10.1038/s41598-020-80786-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris Z.S. Distributional Structure. WORD. 1954;10(2–3):146–162. doi: 10.1080/00437956.1954.11659520. [DOI] [Google Scholar]
- 10.Mikolov, T, Chen, K, Corrado, G, Dean, J. Efficient Estimation of Word Representations in Vector Space, 1st Int. Conf. Learn. Represent. ICLR 2013 - Work. Track Proc. (2013). arXiv:1301.3781.
- 11.Mikolov T., Sutskever I., Chen K., Corrado G., Dean J. Adv. Neural Inf. Process; Syst: 2013. Distributed representations of words and phrases and their compositionality. arXiv:1310.4546. [Google Scholar]
- 12.Bengio Y., Ducharme R., Vincent P., Jauvin C. A neural probabilistic language model. J Mach Learn Res. 2003;3:1137–1155. [Google Scholar]
- 13.Collobert R., Weston J. Proceedings of the 25th International Conference on Machine Learning. Association for Computing Machinery; New York, NY, USA: 2008. A unified architecture for natural language processing: Deep neural networks with multitask learning; pp. 160–167. [DOI] [Google Scholar]
- 14.Pennington, J, Socher, R, Manning, R. Glove: Global Vectors for Word Representation, in: Proc. 2014 Conf. Empir. Methods Nat. Lang. Process., Association for Computational Linguistics, Stroudsburg, PA, USA, 2014, pp. 1532–1543. doi:10.3115/v1/D14-1162. url http://aclweb.org/anthology/D14-1162.
- 15.Bojanowski P., Grave E., Joulin A., Mikolov T. Enriching word vectors with subword information. Trans Assoc Comput Linguist. 2017;5:135–146. arXiv:1607.04606. [Google Scholar]
- 16.Le Q, Mikolov T. Distributed representations of sentences and documents, in: 31st Int. Conf. Mach. Learn. ICML 2014, 2014. arXiv:1405.4053.
- 17.Weaver W. Translation. Mach Transl Lang. 1955;14(15–23):10. [Google Scholar]
- 18.Hochreiter S., Schmidhuber J. Long Short-Term Memory. Neural Comput. 1997;9(8):1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 19.Mikolov T., Karafiát M., Burget L., Černocký J., Khudanpur S. Eleventh annual conference of the international speech communication association. 2010. Recurrent neural network based language model. [Google Scholar]
- 20.Sutskever I., Vinyals O., Le Q.V. Adv. Neural Inf. Process; Syst: 2014. Sequence to Sequence Learning with Neural Networks. arXiv:1409.3215. [Google Scholar]
- 21.Peters M, Neumann M, Iyyer M, Gardner M, Clark C, Lee K, Zettlemoyer L. Deep Contextualized Word Representations, in: Proc. 2018 Conf. North Am. Chapter Assoc. Comput. Linguist. Hum. Lang. Technol. Vol. 1 (Long Pap., Association for Computational Linguistics, Stroudsburg, PA, USA, 2018, pp. 2227–2237. arXiv:1802.05365, doi:10.18653/v1/N18-1202. http://aclweb.org/anthology/N18-1202.
- 22.Bahdanau D, Cho K, Bengio Y. Neural Machine Translation by Jointly Learning to Align and Translate, 3rd Int. Conf. Learn. Represent. ICLR 2015 – Conf. Track Proc. (2014). arXiv:1409.0473.
- 23.Kim Y., Denton C., Hoang L., Rush A.M., Attention Structured. Networks. 2017 arXiv:1702.00887. [Google Scholar]
- 24.Gehring J, Auli M, Grangier D, Yarats D, Dauphin YN. Convolutional Sequence to Sequence Learning, 34th Int. Conf. Mach. Learn. ICML 2017 (2017). arXiv:1705.03122.
- 25.Sukhbaatar S, Szlam A, Weston J, Fergus R. End-to-end memory networks, in: Adv. Neural Inf. Process. Syst., 2015. arXiv:1503.08895.
- 26.Miller AH, Fisch A, Dodge J, Karimi AH, Bordes A, Weston J. Key-value memory networks for directly reading documents, in: EMNLP 2016 - Conf. Empir. Methods Nat. Lang. Process. Proc., 2016. arXiv:1606.03126, doi:10.18653/v1/d16-1147.
- 27.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser L., Polosukhin I. Attention Is All You Need. Adv Neural Inf Process Syst. 2017 arXiv:1706.03762. [Google Scholar]
- 28.Devlin J, Chang MW, Lee K, Toutanova K. BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding, NAACL HLT 2019 - 2019 Conf. North Am. Chapter Assoc. Comput. Linguist. Hum. Lang. Technol. - Proc. Conf. (2018). arXiv:1810.04805.
- 29.Luong MT, Pham H. C.D. Manning, Effective Approaches to Attention-based Neural Machine Translation, arXiv:1508.04025 [cs] (Sep. 2015). arXiv:1508.04025.
- 30.Choromanski K, Likhosherstov V, Dohan D, Song X, Gane A, Sarlos T, et al. Rethinking Attention with Performers, arXiv:2009.14794 [cs, stat] (Mar. 2021). arXiv:2009.14794.
- 31.Kitaev N, Kaiser Ł, Levskaya A. Reformer: The Efficient Transformer, arXiv:2001.04451 [cs, stat] (Feb. 2020). arXiv:2001.04451.
- 32.Howard J, Ruder S. Universal language model fine-tuning for text classification, arXiv (2018).
- 33.Wu L, Fisch A, Chopra S, Adams K, Bordes A, Weston J. StarSpace: Embed All The Things! (2017). arXiv:1709.03856.
- 34.Yang Z, Dai Z, Yang Y, Carbonell J, Salakhutdinov R, Le QV. Xlnet: Generalized autoregressive pretraining for language understanding, arXiv preprint arXiv:1906.08237 (2019). arXiv:1906.08237.
- 35.Raffel C, Shazeer N, Roberts A, Lee K, Narang S, Matena M, et al. Exploring the limits of transfer learning with a unified text-to-text transformer, arXiv preprint arXiv:1910.10683 (2019). arXiv:1910.10683.
- 36.Asgari E., Mofrad M.R.K. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS One. 2015;10(11) doi: 10.1371/journal.pone.0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vang Y.S., Xie X. HLA class I binding prediction via convolutional neural networks. Bioinformatics. 2017;33(17):2658–2665. doi: 10.1093/bioinformatics/btx264. arXiv:1701.00593. [DOI] [PubMed] [Google Scholar]
- 38.Islam S.M., Heil B.J., Kearney C.M., Baker E.J. Protein classification using modified n-grams and skip-grams. Bioinformatics. 2018 doi: 10.1093/bioinformatics/btx823. [DOI] [PubMed] [Google Scholar]
- 39.Zou Q., Xing P., Wei L., Liu B. Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from mRNA. RNA. 2019;25(2):205–218. doi: 10.1261/rna.069112.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mejía-Guerra M.K., Buckler E.S. A k-mer grammar analysis to uncover maize regulatory architecture. BMC Plant Biol. 2019;19(1):103. doi: 10.1186/s12870-019-1693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Asgari E., McHardy A.C., Mofrad M.R.K. Probabilistic variable-length segmentation of protein sequences for discriminative motif discovery (DiMotif) and sequence embedding (ProtVecX) Sci Rep. 2019;9(1):3577. doi: 10.1038/s41598-019-38746-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phloyphisut P., Pornputtapong N., Sriswasdi S., Chuangsuwanich E. MHCSeqNet: a deep neural network model for universal MHC binding prediction. BMC Bioinf. 2019;20(1):270. doi: 10.1186/s12859-019-2892-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woloszynek S., Zhao Z., Chen J., Rosen G.L. 16S rRNA sequence embeddings: Meaningful numeric feature representations of nucleotide sequences that are convenient for downstream analyses., PLoS Comput. Biol. 2019;15(2) doi: 10.1371/journal.pcbi.1006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menegaux R., Vert J.-P. Continuous Embeddings of DNA Sequencing Reads and Application to Metagenomics. J Comput Biol. 2019;26(6):509–518. doi: 10.1089/cmb.2018.0174. [DOI] [PubMed] [Google Scholar]
- 45.Shim H. Feature Learning of Virus Genome Evolution With the Nucleotide Skip-Gram Neural Network. Evol Bioinform. 2019;15 doi: 10.1177/1176934318821072. 117693431882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsubaki M., Tomii K., Sese J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics. 2019 doi: 10.1093/bioinformatics/bty535. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen T.-T.-D., Le N.-Q.-K., Ho Q.-T., Phan D.-V., Ou Y.-Y. Using word embedding technique to efficiently represent protein sequences for identifying substrate specificities of transporters. Anal Biochem. 2019;577:73–81. doi: 10.1016/j.ab.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 48.Guo Y., Yang Y., Huang Y., Shen H.B. Discovering nuclear targeting signal sequence through protein language learning and multivariate analysis. Anal Biochem. 2020 doi: 10.1016/j.ab.2019.113565. [DOI] [PubMed] [Google Scholar]
- 49.Buchan D.W.A., Jones D.T. Learning a functional grammar of protein domains using natural language word embedding techniques. Proteins. 2020;88(4):616–624. doi: 10.1002/prot.25842. [DOI] [PubMed] [Google Scholar]
- 50.Wang C., Zhang Y., Han S. Its2vec: Fungal Species Identification Using Sequence Embedding and Random Forest Classification. Biomed Res Int. 2020:2468789. doi: 10.1155/2020/2468789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wahab A., Tayara H., Xuan Z., Chong K.T. DNA sequences performs as natural language processing by exploiting deep learning algorithm for the identification of N4-methylcytosine. Sci Rep. 2021;11(1):212. doi: 10.1038/s41598-020-80430-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang K.K., Wu Z., Bedbrook C.N., Arnold F.H. Learned protein embeddings for machine learning. Bioinformatics. 2018 doi: 10.1093/bioinformatics/bty178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng W., Wu M., Jiang R. Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics. 2018;19(Suppl 2):84. doi: 10.1186/s12864-018-4459-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang Y.-J., Pang Y.-H., Liu B. IDP-Seq2Seq: Identification of Intrinsically Disordered Regions based on Sequence to Sequence Learning. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa667. [DOI] [PubMed] [Google Scholar]
- 55.Min X., Zeng W., Chen N., Chen T., Jiang R. Chromatin accessibility prediction via convolutional long short-term memory networks with k-mer embedding. Bioinformatics. 2017;33(14):i92–i101. doi: 10.1093/bioinformatics/btx234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ju Y., Yuan L., Yang Y., Zhao H. CircSLNN: Identifying RBP-Binding Sites on circRNAs via Sequence Labeling Neural Networks. Front Genet. 2019;10:1184. doi: 10.3389/fgene.2019.01184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Le N.Q.K., Yapp E.K.Y., Nagasundaram N., Yeh H.-Y. Classifying Promoters by Interpreting the Hidden Information of DNA Sequences via Deep Learning and Combination of Continuous FastText N-Grams. Front Bioeng Biotechnol. 2019;7 doi: 10.3389/fbioe.2019.00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Le N.Q.K., Yapp E.K.Y., Ho Q.-T., Nagasundaram N., Ou Y.-Y., Yeh H.-Y. iEnhancer-5Step: Identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal Biochem. 2019;571:53–61. doi: 10.1016/j.ab.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 59.Nguyen T.-T.-D., Le N.-Q.-K., Ho Q.-T., Phan D.-V., Ou Y.-Y. TNFPred: identifying tumor necrosis factors using hybrid features based on word embeddings., BMC Med. Genomics. 2020;13(Suppl 10):155. doi: 10.1186/s12920-020-00779-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le N.Q.K., Do D.T., Hung T.N.K., Lam L.H.T., Huynh T.-T., Nguyen N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int J Mol Sci. 2020;21(23) doi: 10.3390/ijms21239070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang S., Liu X., Ng R.T. ProbeRating: a recommender system to infer binding profiles for nucleic acid-binding proteins. Bioinformatics. 2020;36(18):4797–4804. doi: 10.1093/bioinformatics/btaa580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hie B., Zhong E.D., Berger B., Bryson B. Learning the language of viral evolution and escape. Science. 2021;371(6526):284–288. doi: 10.1126/science.abd7331. [DOI] [PubMed] [Google Scholar]
- 63.Alley E.C., Khimulya G., Biswas S., AlQuraishi M., Church G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods. 2019;16(12):1315–1322. doi: 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strodthoff N., Wagner P., Wenzel M., Samek W. UDSMProt: universal deep sequence models for protein classification. Bioinformatics. 2020;36(8):2401–2409. doi: 10.1093/bioinformatics/btaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vielhaben J., Wenzel M., Samek W., Strodthoff N. USMPep: universal sequence models for major histocompatibility complex binding affinity prediction. BMC Bioinform. 2020;21(1):279. doi: 10.1186/s12859-020-03631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan H., Kshirsagar M., Zamparo L., Lu Y., Leslie C.S. BindSpace decodes transcription factor binding signals by large-scale sequence embedding. Nat Methods. 2019 doi: 10.1038/s41592-019-0511-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y., Xiao Y., Yang M., Ma J. Cancer mutational signatures representation by large-scale context embedding. Bioinformatics. 2020;36(Supplement_1):i309–i316. doi: 10.1093/bioinformatics/btaa433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinzinger M., Elnaggar A., Wang Y., Dallago C., Nechaev D., Matthes F., Rost B. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinform. 2019;20(1):723. doi: 10.1186/s12859-019-3220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Valeri J.A., Collins K.M., Ramesh P., Alcantar M.A., Lepe B.A., Lu T.K., Camacho D.M. Sequence-to-function deep learning frameworks for engineered riboregulators. Nat Commun. 2020;11(1):5058. doi: 10.1038/s41467-020-18676-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clauwaert J., Waegeman W. Novel transformer networks for improved sequence labeling in genomics. IEEE/ACM Trans Comput Biol Bioinform. 2020 doi: 10.1109/TCBB.2020.3035021. 1-1. [DOI] [PubMed] [Google Scholar]
- 71.Rao R., Bhattacharya N., Thomas N., Duan Y., Chen P., Canny J., Abbeel P., Song Y. Evaluating protein transfer learning with tape. Adv Neural Inf Process Syst. 2019;32 [PMC free article] [PubMed] [Google Scholar]
- 72.Rives A., Meier J., Sercu T., Goyal S., Lin Z., Guo D. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. bioRxiv. 2020 doi: 10.1101/622803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, et al. ProtTrans: Towards cracking the language of life’s code through self-supervised deep learning and high performance computing, bioRxiv (2020). doi:10.1101/2020.07.12.199554.
- 74.Ji Y., Zhou Z., Liu H., Davuluri R.V. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charoenkwan P., Nantasenamat C., Hasan M.M., Manavalan B., Shoombuatong W. BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics. 2021 doi: 10.1093/bioinformatics/btab133. [DOI] [PubMed] [Google Scholar]
- 76.Le N.Q.K., Ho Q.-T., Nguyen T.-T.-D., Ou Y.-Y. A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Briefings Bioinform. 2021 doi: 10.1093/bib/bbab005. [DOI] [PubMed] [Google Scholar]
- 77.Yamada K, Hamada M. Prediction of rna-protein interactions using a nucleotide language model, bioRxiv (2021). doi:10.1101/2021.04.27.441365. [DOI] [PMC free article] [PubMed]
- 78.Deznabi I., Arabaci B., Koyutürk M., Tastan O. DeepKinZero: zero-shot learning for predicting kinase-phosphosite associations involving understudied kinases. Bioinformatics. 2020;36(12):3652–3661. doi: 10.1093/bioinformatics/btaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai Y., Wang J., Deng L. SDN2GO: An integrated deep learning model for protein function prediction. Front Bioeng Biotechnol. 2020;8:391. doi: 10.3389/fbioe.2020.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimothi D, Soni A, Biyani P, Hogan JM. Distributed representations for biological sequence analysis, arXiv (2016) 1608.05949.
- 81.Ng P. dna2vec: Consistent vector representations of variable-length k-mers, arXiv (2017) 1701.06279.
- 82.Klausen M.S., Jespersen M.C., Nielsen H., Jensen K.K., Jurtz V.I., Sønderby C.K. Netsurfp-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins: Struct Funct Bioinf. 2019;87(6):520–527. doi: 10.1002/prot.25674. [DOI] [PubMed] [Google Scholar]
- 83.Almagro Armenteros J.J., Sønderby C.K., Sønderby S.K., Nielsen H., Winther O. Deeploc: prediction of protein subcellular localization using deep learning. Bioinformatics. 2017;33(21):3387–3395. doi: 10.1093/bioinformatics/btx431. [DOI] [PubMed] [Google Scholar]
- 84.Villegas-Morcillo A., Makrodimitris S., van Ham R.C.H.J., Gomez A.M., Sanchez V., Reinders M.J.T. Unsupervised protein embeddings outperform hand-crafted sequence and structure features at predicting molecular function. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Merity S, Keskar NS, Socher R. Regularizing and optimizing lstm language models, arXiv (2017).
- 86.Rao RM, Liu J, Verkuil R, Meier J, Canny J, Abbeel P, et al. MSA transformer, bioRxiv (2021). doi:10.1101/2021.02.12.430858.
- 87.Vig J, Madani A, Varshney LR, Xiong C, Socher R, Rajani N. BERTology meets biology: Interpreting attention in protein language models, International Conference on Learning Representations (2021).
- 88.Liu H., Sun J., Guan J., Zheng J., Zhou S. Improving compound–protein interaction prediction by building up highly credible negative samples. Bioinformatics. 2015;31(12):i221–i229. doi: 10.1093/bioinformatics/btv256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mysinger M.M., Carchia M., Irwin J.J., Shoichet B.K. Directory of useful decoys, enhanced (dud-e): better ligands and decoys for better benchmarking. J Med Chem. 2012;55(14):6582–6594. doi: 10.1021/jm300687e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Uniprot: The universal protein knowledgebase in 2021, Nucleic Acids Research 49 (D1) (2021) D480–D489. doi:10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed]
- 91.Ray D., Kazan H., Cook K.B., Weirauch M.T., Najafabadi H.S., Li X. A compendium of rna-binding motifs for decoding gene regulation. Nature. 2013;499(7457):172–177. doi: 10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158(6):1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pelossof R., Singh I., Yang J.L., Weirauch M.T., Hughes T.R., Leslie C.S. Affinity regression predicts the recognition code of nucleic acid–binding proteins. Nat Biotechnol. 2015;33(12):1242–1249. doi: 10.1038/nbt.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang S., Wang J., Ng R.T. Inferring rna sequence preferences for poorly studied rna-binding proteins based on co-evolution. BMC Bioinform. 2018;19(1):1–12. doi: 10.1186/s12859-018-2091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Valeri J.A., Collins K.M., Ramesh P., Alcantar M.A., Lepe B.A., Lu T.K., Camacho D.M. Sequence-to-function deep learning frameworks for engineered riboregulators. Nat Commun. 2020;11(1):1–14. doi: 10.1038/s41467-020-18676-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Angenent-Mari N.M., Garruss A.S., Soenksen L.R., Church G., Collins J.J. A deep learning approach to programmable rna switches. Nat Commun. 2020;11(1):1–12. doi: 10.1038/s41467-020-18677-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Green A.A., Silver P.A., Collins J.J., Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159(4):925–939. doi: 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pardee K., Green A.A., Takahashi M.K., Braff D., Lambert G., Lee J.W. Rapid, low-cost detection of zika virus using programmable biomolecular components. Cell. 2016;165(5):1255–1266. doi: 10.1016/j.cell.2016.04.059. [DOI] [PubMed] [Google Scholar]
- 99.Alley E.C., Khimulya G., Biswas S., AlQuraishi M., Church G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat Methods. 2019;16(12):1315–1322. doi: 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, Consortium U. Uniref clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015:31(6):926–932. doi:10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed]
- 101.Sarkisyan K.S., Bolotin D.A., Meer M.V., Usmanova D.R., Mishin A.S., Sharonov G.V. Local fitness landscape of the green fluorescent protein. Nature. 2016;533(7603):397–401. doi: 10.1038/nature17995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hie B., Zhong E., Bryson B., Berger B. Learning mutational semantics. Adv Neural Inf Process Syst. 2020;33 [Google Scholar]
- 103.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tenenbaum D. Keggrest: Client-side rest access to kegg. R package version. 2016;1(1) [Google Scholar]
- 105.McDonald D., Hyde E., Debelius J.W., Morton J.T., Gonzalez A., Ackermann G. American Gut: an Open Platform for Citizen Science Microbiome Research. mSystems. 2018;3(3) doi: 10.1128/mSystems.00031-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schils E. Characteristics of Sentence Length in Running Text. Lit Linguist Comput. 1993;8(1):20–26. doi: 10.1093/llc/8.1.20. [DOI] [Google Scholar]
- 107.Iwano N,, Adachi T, Aoki K, Nakamura Y, Hamada M. RaptGen: A variational autoencoder with profile hidden Markov model for generative aptamer discovery, bioRxiv (2021) 2021.02.17.431338. doi:10.1101/2021.02.17.431338.
- 108.Nickel M, Kiela D. Poincaré embeddings for learning hierarchical representations, Advances in Neural Information Processing Systems (2017).
- 109.Ganea OE, Bécigneul G, Hofmann T. Hyperbolic neural networks, Advances in Neural Information Processing Systems (2018).
- 110.Gu A, Sala F, Gunel B. C. Ré, Learning mixed-curvature representations in product spaces, in: International Conference on Learning Representations, 2018. https://openreview.net/forum?id=HJxeWnCcF7.
- 111.Matsumoto H, Mimori T, Fukunaga T. Novel metric for hyperbolic phylogenetic tree embeddings, bioRxiv (2020). doi:10.1101/2020.10.09.334243. [DOI] [PMC free article] [PubMed]
- 112.Klimovskaia A., Lopez-Paz D., Bottou L., Nickel M. Poincaré maps for analyzing complex hierarchies in single-cell data. Nat Commun. 2020;11(1):2966. doi: 10.1038/s41467-020-16822-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Duong D., Uppunda A., Gai L., Ju C., Zhang J., Chen M., Eskin E., Li J.J., Chang K.-W. Evaluating representations for gene ontology terms. bioRxiv. 2020 doi: 10.1101/765644. [DOI] [Google Scholar]
- 114.Unsal S., Ata H., Albayrak M., Turhan K., Acar A.C., Doan T. Evaluation of methods for protein representation learning: a quantitative. Analysis. 2020 doi: 10.1101/2020.10.28.359828. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.