

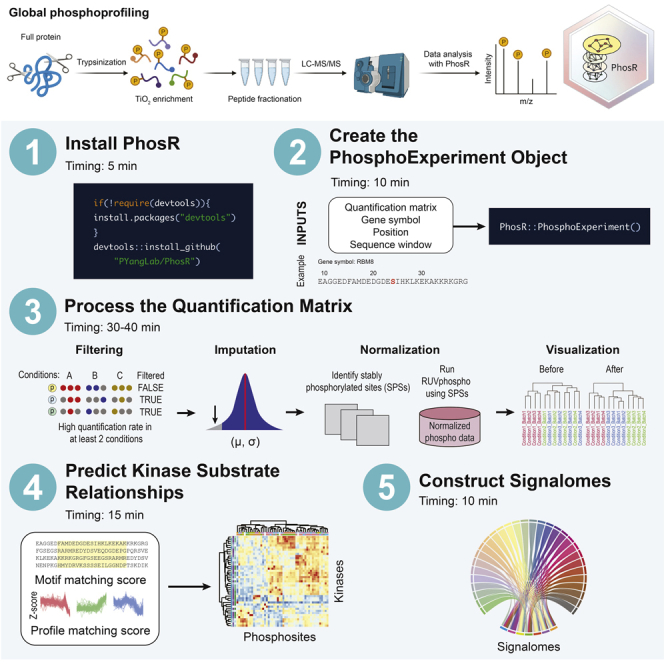
Summary
Analysis of phosphoproteomic data requires advanced computational methodologies. To this end, we developed PhosR, a set of tools and methodologies implemented in R to allow the comprehensive analysis of phosphoproteomic data. PhosR enables processing steps such as imputation, normalization, and functional analysis such as kinase activity inference and signalome construction. Together, PhosR facilitates interpretation and discovery from large-scale phosphoproteomic data sets.
For complete details on the use and execution of this protocol, please refer to Kim et al. (2021).
Subject areas: Bioinformatics, Proteomics, Systems biology
Graphical Abstract
Highlights
-
•
This protocol describes how to run and interpret the results of PhosR
-
•
PhosR performs filtering, imputation, and normalization of phosphoproteomic data
-
•
PhosR enables kinase-substrate predictions and signalome construction
-
•
The step-by-step protocol provides a comprehensive introduction to phospho-data analysis
Analysis of phosphoproteomic data requires advanced computational methodologies. To this end, we developed PhosR, a set of tools and methodologies implemented in R to allow the comprehensive analysis of phosphoproteomic data. PhosR enables processing steps such as imputation, normalization, and functional analysis such as kinase activity inference and signalome construction. Together, PhosR facilitates interpretation and discovery from large-scale phosphoproteomic data sets.
Before you begin
Raw mass spectrometry data pre-processing
PhosR is designed for the processing and functional analysis of data generated from mass spectrometry (MS)-based phosphoproteomic technologies. PhosR expects pre-processed data such as those generated from vendor provided software, such as Proteome Discoverer (Thermo Fisher Scientific) and ProteinPilot (AB Sciex), or those from third parties, such as MaxQuant (Cox and Mann, 2008).
The pre-processed data matrix is expected to have the phosphosites labeled with site position, “sequence window” that captures the amino acid sequence flanking the phosphorylation sites, and the official gene symbol in capital case.
CRITICAL: Position, residue, gene symbol, and sequence windows of phosphosites are compulsory information required to run various PhosR functions. Please ensure the final processed matrix from the proteomics software packages provide these labels.
Note: Note that consistent phosphosite labels across datasets are required if multiple datasets will be integrated in the study.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Rat L6 myoblasts | PRIDE | PRIDE: PXD019127 |
| ESC phosphoproteome | PRIDE | PRIDE: PXD010621 |
| Hepa 1-6 & FL83 phosphoproteome | PRIDE | PRIDE: PXD001792 |
| Adipocyte FGF2 phosphoproteome | PRIDE | PRIDE: PXD003631 |
| Adipocyte insulin, LY, and MK phosphoproteome | Humphrey et al., 2015 | N/A |
| Software and algorithms | ||
| PhosR | Kim et al., 2021 | https://github.com/PYangLab/PhosR |
| R | R Core Team, 2020 | https://www.r-project.org/ |
Materials and equipment
-
•
Pre-processed phosphoproteomic data (see Raw Mass Spectrometry Data Pre-processing in Before You Begin).
-
•R software and required packages. The required R packages deposited in CRAN will be automatically installed the first time you run PhosR. Bioconductor packages may need to be manually installed. While different version of the R software and associated packages may work correctly with PhosR, the authors use R version 4.0.3 and the following packages at the indicated versions at time of writing this protocol:
-
○PhosR (1.1.7)
-
○CRAN packages
-
•ruv (v0.9.7.1)
-
•e1071 (v1.7-6)
-
•dendextend (v1.14.0)
-
•RColorBrewer (v1.1-2)
-
•circlize (v0.4.12)
-
•dplyr (v1.0.5)
-
•igraph (v1.2.6)
-
•pheatmap (v1.0.12)
-
•tidyr (v1.1.3)
-
•rlang (v0.4.10)
-
•S4Vectors (v0.28.1)
-
•ggplot2 (v3.3.3)
-
•ggdendro (v0.1.22)
-
•ggpubr (v0.4.0)
-
•network (v1.16.1)
-
•reshape2 (v1.4.4)
-
•ggtext (v0.1.1)
-
•GGally (v2.1.1)
-
•
-
○Bioconductor packages
-
•limma (v3.46.0)
-
•pcaMethods (v1.82.0)
-
•preprocessCore (v1.52.1)
-
•SummarizedExperiment (v1.20.0)
-
•BiocGenerics (v0.36.0)
-
•
-
○Hardware
-
•Memory: 16 GB recommended
-
•Processors: 4 threads recommended
-
•
-
○
Pause point: If you want to pause at any time during this protocol, please save your work and R session using the following command within the R or R Studio console:
> save.image("Your_Experiment_Name_and_Date_Here.RData")
This session can be reloaded by using the following command:
> load("Your_Experiment_Name_and_Date_Here.RData")
Step-by-step method details
Step 1: Installing PhosR
Timing: 5 min
Full installation of PhosR includes downloading the PhosR package from GitHub or BioConductor. To use the latest developmental version of PhosR, install from GitHub. An example of how to perform all steps of this protocol using real data is available on the project GitHub at https://pyanglab.github.io/PhosR/articles/web/PhosR_STAR_protocols.html.
-
1.Install PhosR by running the following code:
- > if(!require(devtools)){
- > install.packages("devtools") # If not already installed
- > }
- > devtools::install_github("PYangLab/PhosR",
- build_opts = c("--no-resave-data", "--no-manual"),
- build_vignettes = TRUE,
- dependencies = TRUE)
- > library(PhosR)
-
2.To access the vignette directly from R console, run the following code after installation:
- > browseVignettes("PhosR")
Note: Note that all the necessary data needed to reproduce the code can be downloaded from https://github.com/PYangLab/PhosR_STAR_Protocols.
Step 2: Creating a PhosphoExperiment object from a MaxQuant output
To increase the usability of PhosR functions, we implement a “PhosphoExperiment” (ppe) object based on the “SummarizedExperiment” class. To create the PhosphoExperiment object, you will be required to provide a quantification matrix where columns refer to samples and rows refer to phosphosites. Additional annotation labels for phosphosites should be provided alongside the matrix, including the phosphosite residue and position, “sequence window” that captures the amino acids flanking the phosphorylation sites, and the official gene symbol of the host protein in capital letters.
Here, we will show the basic steps for generating a PhosphoExperiment object for an exemplar MaxQuant-processed phosphoproteomic data from two liver cell lines, FL83B and Hepa 1-6 cells (Humphrey et al., 2015). This dataset contains the phosphoproteomic quantifications of mouse hepatocyte cell lines that were treated with either PBS (mock) or insulin. Each condition includes six biological replicates.
-
3.Load the txt output file to an R environment
- > phospho_hepatocyte_raw <- read.delim("Data/PXD001792_raw_hepatocyte.txt", header = TRUE)
-
4.Clean and process the raw txt files to extract quantifications.
-
a.To delete reverse matches and potential contaminants.
- > del <- which(phospho_hepatocyte_raw[,"Reverse"]=="+" | phospho_hepatocyte_raw[,"Potential.contaminant"]=="+")
- > phospho_hepatocyte_clean <- phospho_hepatocyte_raw[-del,]
-
b.We subset the raw data to select columns with “Intensity” values.
- > PXD001792_raw_hepatocyte <- phospho_hepatocyte_clean[,grep("Intensity", colnames(phospho_hepatocyte_clean))]
-
a.
-
5.Create the PhosphoExperiment Object
-
a.Creating the base PhosphoExperiment object
- > ppe <- PhosphoExperiment(assays = list(Quantification = as.matrix(PXD001792_raw_hepatocyte)))
-
b.Add site annotations to the object
- > GeneSymbol <- toupper(sapply(strsplit(as.character(phospho_hepatocyte_clean[,"Gene.names"]), ";"), function(x){x[1]}))
- > Residue <- as.character(phospho_hepatocyte_clean [,"Amino.acid"])
- > Site <- as.numeric(phospho_hepatocyte_clean [,"Position"]) >
- Sequence <- sapply(strsplit(as.character(phospho_hepatocyte_clean[,"Sequence.window"]), ";"), function(x){x[1]})
- > ### add these annotations to respective ppe slots > ppe@GeneSymbol <- GeneSymbol
- > ppe@Residue <- Residue
- > ppe@Site <- Site
- > ppe@Sequence <- Sequence
-
c.Alternatively, we can create PhosphoExperiment object as following
- > ppe <- PhosphoExperiment(assays = list(Quantification = as.matrix(PXD001792_raw_hepatocyte)), Site = Site, GeneSymbol = GeneSymbol, Residue = Residue, Sequence = Sequence)
-
d.Lastly add colData information
- > sample_name <- strsplit(gsub("ˆIntensity.", "", colnames(ppe)), "_")
- > df <- S4Vectors::DataFrame(
- > cellline = sapply(sample_name, "[[", 1),
- > condition = sapply(sample_name, "[[", 2),
- > replicate = sapply(sample_name, "[[", 3)
- > )
- > rownames(df) <- colnames(ppe)
- > SummarizedExperiment::colData(ppe) <- df
-
e.Have a quick glance of the object
- > ppe
- > dim(ppe)
-
f.Additional arguments
-
i.UniprotID: Uniprot ID of the protein which has the phosphosite
-
ii.Localization: localization reliability of the phosphosite
-
i.
-
a.
The final output of “Step 2: Creating a PhosphoExperiment object from a MaxQuant output” is a PhosphoExperiment object containing the quantification matrix and site annotations of all phosphosites.
-
6.Requirements of the basic phosphosite features
-
a.Position of the phosphosite: An integer vector denoting the position of the phosphosite within the protein
-
b.Residue of the phosphosite: A character vector denoting the residue of the phosphosite as a single letter (“S”, “Y”, or “T”)
-
c.Gene symbol: A character vector containing gene symbols in upper case
-
d.Sequence window: A character vector denoting the sequence window, typically of 15–31 in length. The residues in the sequence window should be capitalized. Note that the predicted phosphosite should reside in the middle of the window. Any phosphosites that are found near the N or C terminus of the protein will require a placeholder (i.e., add “_” to either side of the flanking sequence) to position the phosphosite in concern at the middle of the sequence window (Most processing software will do this for you automatically).
-
a.
Step 3: Data pre-processing and differential phosphosite identification
The presence of missing values in quantitative phosphoproteomics reduces the completeness of data. Whilst imputation has been widely applied to handle missing values, it remains a major challenge when analyzing phosphoproteomic data and has significant impact on downstream analysis such as normalization. PhosR provides users with greater flexibility for imputation with imputation functions such as ‘scImpute’ and ‘tImpute’. Here, we will go through each function step by step to demonstrate their use in imputing phosphoproteomic data. We will call differentially phosphorylated sites between two cell lines FL83B and Hepa 1-6 to visualize the change in phosphorylation upon insulin simulation.
-
7.Log transformation
-
a.We perform log2 transformation of the data
- > logmat <- log2(SummarizedExperiment::assay(ppe, "Quantification"))
- > # mark any missing values as NA
- > logmat[is.infinite(logmat)] <- NA
- > SummarizedExperiment::assay(ppe, "Quantification") <- logmat
-
a.
-
8.Perform filtering
-
a.We first extract the grouping information for cell type and condition
- > grps <- paste0(SummarizedExperiment::colData(ppe)$cellline, "_", SummarizedExperiment::colData(ppe)$condition)
-
b.We filter for sites with at least 50% quantification rate (q ≥ 0.5) in one or more conditions
- > ppe <- selectGrps(ppe, grps, 0.5, n=1)
-
c.Check the filtering results
- > dim(ppe)
-
a.
Note: The ‘Quantification’ assay in PhosphoExperiment object is now filtered of phosphosites with high number of missing values. The user may wish to save the unfiltered matrix, `PXD001792_raw_hepatocyte, for future analysis.
-
9.Perform imputation of missing values
-
a.PhosR enables site- and condition-specific imputation. Here, for each phosphosite in each condition, we impute its missing values in that condition (if any) using site- and condition-specific imputation if the quantification rate within that condition is equal to or greater than a desired percentage (such as ≥ 50% in the example below).
- > set.seed(123)
- > ppe <- scImpute(ppe, 0.5, grps)
- > ppe
-
b.Lastly, we can impute the remaining sites using tail-based imputation
- > ppe <- tImpute(ppe, assay = "imputed")
-
a.
Note: At this stage, the imputed quantification matrix is stored as an assay called ‘imputed’ in the PhosphoExperiment object.
-
10.Centering data across their median
- > ppe <- medianScaling(ppe, scale = FALSE, assay = "imputed")
- > ppe
Note: The scaled quantification matrix can be found as the ‘scaled’ matrix in the PhosphoExperiment object.
-
11.Call differentially phosphorylated sites for the FL83B and Hepa 1-6 insulin stimulation dataset
-
a.We use limma package for calling for differentially phosphorylated sites between control and insulin-simulated conditions in the two cell types.
- > library(limma)
- > design <- model.matrix(~ grps - 1)
- > fit <- lmFit(ppe@assays@data$scaled, design)
- > contrast.matrix <- makeContrasts(grpsFL83B_Ins-grpsFL83B_Control, grpsHepa1.6_Ins-grpsHepa1.6_Control, levels=design)
- > fit2 <- contrasts.fit(fit, contrast.matrix)
- > fit2 <- eBayes(fit2)
-
b.Visualize differentially phosphorylated sites using a volcano plot (Figure 1).
- > par(mfrow=c(1,2))
- > FL83B.DE <- topTable(fit2, coef="grpsFL83B_Ins - grpsFL83B_Control", number = Inf)
- > plot(FL83B.DE[,"logFC"], -log10(FL83B.DE[,"adj.P.Val"]), main="FL83B", xlab="Log2 FC", ylab="-Log10(Adjust P)")
- > sel <- which(FL83B.DE[,"adj.P.Val"] < 0.05 & FL83B.DE[,"logFC"] > 0)
- > points(FL83B.DE[sel,"logFC"], -log10(FL83B.DE[sel,"adj.P.Val"]), pch=16, col="red")
- > sel <- which(FL83B.DE[,"adj.P.Val"] < 0.05 & FL83B.DE[,"logFC"] < 0)
- > points(FL83B.DE[sel,"logFC"], -log10(FL83B.DE[sel,"adj.P.Val"]), pch=16, col="blue")
- > Hepa1.6.DE <- topTable(fit2, coef="grpsHepa1.6_Ins - grpsHepa1.6_Control", number = Inf)
- > plot(Hepa1.6.DE[,"logFC"], -log10(Hepa1.6.DE[,"adj.P.Val"]), main="Hepa 1-6", xlab="Log2 FC", ylab="-Log10(Adjust P)")
- > sel <- which(Hepa1.6.DE[,"adj.P.Val"] < 0.05 & Hepa1.6.DE[,"logFC"] > 0)
- > points(Hepa1.6.a.DE[sel,"logFC"], -log10(Hepa1.6.DE[sel,"adj.P.Val"]), pch=16, col="red")
- > sel <- which(Hepa1.6.DE[,"adj.P.Val"] < 0.05 & Hepa1.6.DE[,"logFC"] < 0)
- > points(Hepa1.6.DE[sel,"logFC"], -log10(Hepa1.6.DE[sel,"adj.P.Val"]), pch=16, col="blue")
-
a.
-
12.After imputation, we calculate the ratio of each value against the mean phosphosite values of the control samples. The ratios were calculated independently for each of the two cell lines (Figure 2).
- > FL83B.ratio <- SummarizedExperiment::assay(ppe, "scaled")[, grep("FL83B_", colnames(ppe))] -
- rowMeans(SummarizedExperiment::assay(ppe, "scaled")[,grep("FL83B_Control", colnames(ppe))])
- > Hepa.ratio <- SummarizedExperiment::assay(ppe, "scaled")[, grep("Hepa1.6_", colnames(ppe))] -
- rowMeans(SummarizedExperiment::assay(ppe, "scaled")[,grep("Hepa1.6_Control", colnames(ppe))])
- > SummarizedExperiment::assay(ppe, "ratio")<- cbind(FL83B.ratio, Hepa.ratio)
- > par(mfrow=c(1,2))
- > boxplot(ppe@assays@data$scaled, ylab="Log2 LFQ", main="Normalized LFQ data", las=2, col=factor(rep(1:4, each=6)))
- > boxplot(ppe@assays@data$ratio, ylab="Log2 Fold Change", main="Ratio data", las=2, col=factor(rep(1:4, each=6)))
-
13.Lastly, we will save this processed ppe object for later use
- > PXD001792_ppe_hepatocyte <- ppe
- > save(PXD001792_ppe_hepatocyte, file = "Data/PXD001792_ppe_hepatocyte.RData")
Figure 1.

Volcano plots visualizing significantly up- and down-regulated phosphosites
Differential phosphosites can be called using normalized LFQ values and visualized using volcano plots. The above plots show up- (red) and down-regulated (blue) phosphosites in the two liver cell lines, respectively.
Figure 2.

Boxplot of quantifications before and after taking the ratio
Phosphoproteomic data from label-free quantification (LFQ) are typically converted to ratios against the control samples to generate fold changes (FCs). The boxplots of phosphoproteomic quantifications demonstrate the change in distribution of values before and after conversion for each sample.
Step 4: Identifying stably phosphorylated sites
Several commonly used data normalization approaches such as the 'removal of unwanted variation” (RUV) (Gagnon-Bartsch and Speed, 2012) require a set of internal standards whose expression are known to be unchanged in the samples measured. This is a challenge for phosphoproteomic data since phosphorylation is a highly dynamic biochemical process. Identifying a set of stably phosphorylated sites (SPSs) is a unique feature of PhosR which enables users to identify context-dependent sets of SPSs. We also included a set of 100 SPSs as a resource, identified from multiple high-quality datasets generated from different cell types and experimental conditions (Kim et al., 2021). As an example, we will use three datasets to demonstrate how SPSs can be identified from multiple phosphoproteomic datasets. Users may wish to replace the example datasets with their own collection of datasets.
-
14.Load and set up datasets
-
a.Load datasets
- > load("Data/PXD010621_ppe_ESC.RData", verbose = TRUE)
- > load("Data/PXD003631_ppe_adipocyte.RData",
- verbose = TRUE)
- > load("Data/phospho_ppe_adipocyte.RData",
- verbose = TRUE)
-
b.Simplify names of datasets
- > ppe1 <- PXD010621_ppe_ESC
- > ppe2 <- PXD003631_ppe_adipocyte
- > ppe3 <- phospho_ppe_adipocyte
-
c.Make a list of all PhosphoExperiment objects
- > ppe.list <- list(ppe1, ppe2, ppe3)
-
d.Make a list of grouping information of each dataset
- > cond.list <- list(
- grp1 = gsub("_.+", "", colnames(ppe1)),
- grp2 = gsub("_r[0-9]", "", colnames(ppe2)),
- grp3 = colnames(ppe3))
-
a.
Note: The regular expression used for defining ‘cond.list’ above is highly specific to the example dataset used in this tutorial. Users should adopt and modify these codes according to the specific experimental design and conditions in their own datasets.
-
15.Identify SPSs by calling getSPS()
- > generate a vector of the selected assays in each of the PhosphoExperiment objects
- > assays <- "Quantification"
- > inhouse_SPSs <- getSPS(phosData = ppe.list,
- assays = assays,
- conds = cond.list, num = 100)
Note: Please note that presence of sufficient overlapped phosphosites identified from the input datasets is critical to identify SPSs. You will receive an error if the number of overlapped sites is fewer than 200 in at least two datasets or fewer than 1,000 across all input datasets.
-
16.Required parameters:
-
a.phosData: a list of users' PhosphoExperiment objects from which generate SPSs. For creating PhosphoExperiment objects, please refer to “Step 2: Creating a PhosphoExperiment object from a MaxQuant output”. Please note that the datasets in the list require to be processed.
-
b.conds: a list of vectors contains the conditions or time-course labels of each sample in the phosphoExperiment objects.
-
c.assays: a vector indicating the assay to select, by default is “Quantification”.
-
d.num: the number of SPSs to identify, by default is 100.
-
a.
The output of “Step 4: Identifying stably phosphorylated sites” will be a set of stably phosphorylated sites identified across the input datasets that users provide. The SPSs will be used to perform batch correction.
Step 5: Normalization and batch correction of data sets
A common but critical step in phosphoproteomic data analysis is to correct for batch effect. Without batch effect correction, it is often not possible to analyze datasets in an integrative manner. To perform data integration and batch effect correction, we utilize a set of stably phosphorylated sites (SPSs) across a panel of phosphoproteomic datasets (defined from “Step 4: Identifying stably phosphorylated sites”) and implement normalization using RUV-III (Molania et al., 2019). To demonstrate batch effect correction, we will perform RUVphospho to normalize a SILAC data from L6 myotubes treated with two factors: 1) AICAR, an analog of adenosine monophosphate (AMP) that stimulates AMPK activity and/or 2) insulin.
-
17.Load PhosphoExperiment objects
- > load("Data/PXD019127_ratio_myoblast.RData",
- verbose = TRUE)
- > ppe <- PhosphoExperiment(assays = list(Quantification = as.matrix(PXD019127_ratio_myoblast)))
-
18.Add site annotations to PhosphoExperiment object
- > rowNames <- strsplit(rownames(ppe), "~")
- > ppe@GeneSymbol <- toupper(sapply(rowNames, "[[", 2))
- > ppe@Residue <- gsub("[0-9]","", sapply(rowNames, "[[", 3))
- > ppe@Site <- as.numeric(gsub("[A-Z]","", sapply(rowNames, "[[", 3)))
- > ppe@Sequence <- sapply(rowNames, "[[", 4)
-
19.Add colData information
- > sample_name <- strsplit(colnames(ppe), "_")
- >
- > df <- S4Vectors::DataFrame(
- > condition = sapply(sample_name, "[[", 1),
- > replicate = gsub("exp", "", sapply(sample_name, "[[", 2))
- > )
- > rownames(df) <- colnames(ppe)
- > SummarizedExperiment::colData(ppe) <- df
-
20.Diagnosing batch effect
-
a.Hierarchical clustering
- > plotQC(SummarizedExperiment::assay(ppe, "Quantification"), panel = "dendrogram",
- grps = SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe)) + ggplot2::ggtitle("before batch correction")
-
b.PCA plot
- > plotQC(SummarizedExperiment::assay(ppe, "Quantification"), panel = "pca",
- grps = SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe)) + ggplot2::ggtitle("before batch correction")
-
a.
-
21.Correcting batch effect
-
a.Construct a design matrix by condition
- > design <- model.matrix(~ SummarizedExperiment::colData(ppe)$condition - 1)
- > head(design) # observe first 6 rows of the design matrix
-
b.Define negative controls sites
- > # Given that the rownames of a matrix ppe is in a format 'GENESYMBOL;RESIDUE;SITE;'
- > sites <- paste(
- sapply(ppe@GeneSymbol, function(x)x),
- ";",
- sapply(ppe@Residue, function(x)x),
- sapply(ppe@Site, function(x)x),
- ";",
- sep = "")
- > data(SPSs)
- > ctl <- which(sites %in% SPSs)
-
c.Run RUVphospho
- > ppe <- RUVphospho(ppe, M = design, k = 3, ctl = ctl)
-
a.
-
22.Quality control to assess the removal of batch effect
-
a.Hierarchical clustering
- # plot before and after batch correction
- > p1 <- plotQC(SummarizedExperiment::assay(ppe,"Quantification"),
- grps= SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe),
- panel = "dendrogram")
- > p2 <- plotQC(SummarizedExperiment::assay(ppe,"normalized"),
- grps= SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe),
- panel="dendrogram")
- > ggpubr::ggarrange(p1, p2, nrow = 1)
-
b.PCA
- # plot before and after batch correction
- > p1 <- plotQC(SummarizedExperiment::assay(ppe,"Quantification"),
- grps= SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe),
- panel = "pca")
- > p2 <- plotQC(SummarizedExperiment::assay(ppe,"normalized"),
- grps= SummarizedExperiment::colData(ppe)$condition,
- labels = colnames(ppe),
- panel="pca")
- > ggpubr::ggarrange(p1, p2, nrow = 1)
-
a.
-
23.We can now save the final processed data for future use
- > PXD019127_ppe_myoblast = ppe
- > save(PXD019127_ppe_myoblast,
- file = "Data/PXD019127_ppe_myoblast.RData")
Step 6: Predicting kinase substrates
A key end-goal of phosphoproteomic data analysis is to identify kinases that are responsible for the phosphorylation of specific sites. The basic computational approach to annotate kinases to their substrates or phosphosites is to find consensus amino acid sequences around the phosphorylation site. We can go beyond this approach by considering cell type and/or treatment (perturbation) specificity of phosphorylation. PhosR implements a multi-step framework that contains two major components including (i) a kinaseSubstrateScore function which scores a given phosphosite using kinase recognition motif and phosphoproteomic dynamics, and (ii) a kinaseSubstratePred function which synthesize the scores generated from (i) for predicting kinase-substrate relationships using an adaptive sampling-based positive-unlabeled learning method (Yang et al., 2018).
In the original PhosR publication, we demonstrate the application of the scoring method to the myotube phosphoproteome and uncover potential kinase-substrate pairs and global relationships between kinases. We confirm well established substrates of AMPK in our publication: ACACA S79, AKAP1 S103, SMCR8 S488 (Hoffman et al., 2015) and MTFR1L S100 (Schaffer et al., 2015). Importantly, PhosR generates a list of potential candidates not included in the PhosphoSitePlus database for validation.
-
24.Prepare inputs
-
a.Load the PhosphoExperiment object
- > load("Data/PXD019127_ppe_myoblast.RData",
- verbose = TRUE)
- > ppe <- PXD019127_ppe_myoblast
- > mat <- SummarizedExperiment::assay(ppe, "normalized")CRITICAL: If you have not processed your data, please go through Steps 1–5 to perform the necessary processing prior to performing the downstream analysis in Steps 6 and 7.
-
b.Filter for up-regulated phosphosites
- > # filter for up-regulated phosphosites
- > mat.mean <- meanAbundance(mat,
- grps = SummarizedExperiment::colData(ppe)$condition)
- > aov <- matANOVA(mat=mat,
- grps = SummarizedExperiment::colData(ppe)$condition)
- > idx <- (aov < 0.05) & (rowSums(mat.mean > 0.5) > 0)
- > mat.reg <- mat[idx, ,drop = FALSE]
-
c.Standardize the matrix
- > mat.std <- standardize(mat.reg)
- > rownames(mat.std) <- sapply(strsplit(rownames(mat.std), "~"), function(x){gsub(" ", "", paste(toupper(x[2]), x[3], "", sep=";"))})CRITICAL: To calculate the profile matching score, we rely on the z-score transformed matrix to compare the profiles of phosphosites. Thus, the standardization step is critical.
-
a.
-
25.Kinase substrate scoring step integrates information from both kinase recognition motif (i.e., motif matching score) and experimental perturbation (i.e., profile matching score) for prioritizing kinases that may be regulating the phosphorylation level of each site quantified in the dataset.
-
a.The kinase-substrate prediction step requires the following specific inputs:
-
i.‘substrate.list’ denotes an object that contains all kinases and their substrate peptide sequences from the PhosphoSitePlus database. This is used to compute the position-specific motif matching score.
- > data('KinaseMotifs')
- > head(PhosphoSite.mouse)
-
ii.‘mat’ denotes the standardized matrix. PhosR uses a pre-defined number of substrates to compare the dynamic phosphorylation profiles of the substrate against that of their known kinases. Next, the profile matching scores of each phosphosite quantified in ‘mat’ are calculated by using Pearson’s correlation with respect to the averaged profiles of known substrates of each of all kinases.
- > head(mat.std)
-
iii.‘seqs’ denotes a vector containing all the phosphosites detected in the phosphoproteomic dataset at hand. Note that if any filtering was performed in the previous step, we must perform the same filtering here.
- > seqs <- ppe@Sequence[idx]
- > head(seqs)
-
i.
-
b.Run PhosR kinase-substrate prediction with the default parameters. PhosR generates the final combined scores of the motif matching score and the profile matching score by taking into account the number of sequences and substrates used for calculating the motif and profile of the kinase.
- > kssMat <- kinaseSubstrateScore(substrate.list = PhosphoSite.mouse,
- mat = mat.std,
- seqs = seqs,
- numMotif = 5,
- numSub = 1)
-
c.There are two arguments that are set to default in ‘kinaseSubstrateScore’
-
i.‘numMotif’ denotes the minimum number of sequences used for compiling the motif for each kinase. The default value is set to 5.
-
ii.‘numSub’ denotes the minimum number of phosphosites used for compiling the phosphorylation profile. The default value is set to 1.
-
i.
-
a.
Note: Please ensure that there are no duplicate phosphosites by removing or aggregating them before running the kinase-substrate prediction (for a workaround please refer to Problem 5 in Troubleshooting).
Note: Please be patient, as this step may take several minutes.
As the second and last step of kinase-substrate prediction, PhosR uses the ‘kinaseSubstratePred’ function to synthesize the scores generated from ‘kinaseSubstrateScore’ to predict the kinase-substrate relationships using an adaptive sampling-based positive-unlabeled learning method (Yang et al., 2018). This step prioritizes the kinases that are most likely to regulate a phosphosite.
-
26.Use the out of ‘kinaseSubstrateScore’ to run kinase substrate prediction
- > set.seed(1)
- > predMat <- kinaseSubstratePred(kssMat, top = 30)
Note: While the default values in ‘kinaseSubstratePred’ provide reliable performance of PhosR, the user may wish to optimize the parameters for each phosphoproteomic dataset. Listed below are the parameters to choose with default values and tips for each selection provided.
-
27.Selection of Additional Parameters
-
a.‘ensembleSize’ denotes the number of the support vector machine (SVM) classifiers to be used in an ensemble. The default value is set to 10. Decreasing the number may speed up the process but may compromise the accuracy.
-
b.‘top’ denotes the top number of kinase substrates to select. The top kinase substrates are selected based on the combined scores. They are used as positive examples for training SVMs for predicting substrates of that kinase. The default value is 50.
-
c.‘cs’ denotes the score threshold. The default value is 0.8. This argument is used to filter any of the top kinases that do not meet a certain threshold. This argument would override the ‘top’ argument in that any substrates among the top 50 that do not meet the score threshold are excluded.
-
d.‘inclusion’ denotes the minimal number of substrates required for a kinase to be selected. The default value is 20. Decreasing this number would increase the total number of kinases for which we generate kinase-substrate scores, and vice versa.
-
e.‘iter’ denotes the number of iterations for adaSampling. AdaSampling procedure is used to update the SVM training examples based on the model confidence on each phosphosite. The default value is 5.
-
f.‘verbose’ is set to TRUE by default, allowing the tracking of the analysis progress.
-
a.
Step 7: Constructing signaling networks (signalomes)
Constructing signaling networks referred to as “Signalomes” is a useful feature in PhosR that allows the users to obtain a global view of kinase regulation and to establish distinct modules of proteins that demonstrate similar kinase and dynamic regulation upon perturbation or across a time-course.
An important feature of PhosR signalomes is that the resultant signaling modules denote a set of proteins and not phosphosites. Proteins are frequently phosphorylated at multiple sites and these sites may not necessarily be targeted by the same kinases. Site- and protein-centric analyses of phosphoproteomic data lie at opposite ends of the spectrum, with the former treating phosphosites on the same protein independently and ignoring the host protein information, and the latter focusing on a specific protein, losing information from individual phosphosites. Using our global kinase-substrate scoring of phosphosites, we generate signalomes wherein dynamic changes in phosphorylation within and across proteins are conjointly analyzed, allowing us to detect proteins that are co-regulated across multiple phosphosites.
-
28.
Signalome construction
The signalome construction uses the outputs of ‘kinaseSubstrateScore’ and ‘kinaseSubstratePred’ functions for the generation of a visualization of the kinase regulation of discrete regulatory protein modules present in our phosphoproteomic data (Figure 3).-
a.Prepare inputs
-
i.‘KSR’ denotes the output of the kinase-substrate relationship scoring function.
-
ii.‘predMatrix’ denotes the output of the ‘kinaseSubstratePred’ function.
-
iii.‘exprsMat’ denotes the matrix with rows corresponding to phosphosites and columns corresponding to samples. Users may wish to perform various processing steps such as filtering, imputations, and normalization.
-
iv.‘KOI’ denotes a vector containing kinases of interest for which the expanded signalomes will be generated. The Signalomes function also outputs signalomes associated to any kinase of interest (referred to as extended signalome of a kinase). To facilitate assessment of proteins and phosphosites that are under similar regulation, the extended signalome of a kinase of interest combines cognate signalomes from other kinases that share a high degree of similarity in substrate regulation.
-
i.
-
b.Then to generate the signalomes, run:
- > kinaseOI = c("AKT1")
- > signalomesRes <- Signalomes(KSR = kssMat,
- predMatrix = predMat,
- exprsMat = mat.std,
- module_res = 6,
- KOI = kinaseOI)
-
c.There are a number of important arguments in the ‘Signalomes’ function.
-
i.‘module_res’ denotes the argument to control the number of final modules.
-
ii.‘threskinaseNetwork’ denotes the threshold used to select interconnected kinases for the expanded signalomes.
-
iii.‘signalomeCutoff’ refers to the cutoff used to filter kinase-substrate relationships based on scores from the ‘kinaseSubstratePred’ function. A cutoff of 0.5 is used as default. By increasing the threshold, you can reduce the number of phosphosites used to generate the signalomes by selecting high confidence phosphosites only.
-
iv.‘filter’ requires a Boolean argument. When set to TRUE, the function filters modules with fewer than 10 proteins.
-
i.
-
a.
-
29.Visualization of the Signalome as a Balloon Plot
-
a.We can visualize the signalomes as a balloon plot. Using the resulting visualization, we are better able to compare the differences in kinase regulation of the modules and the varying proportions of regulation. In the balloon plot, the size of the balloons denotes the percentage magnitude of kinase regulation in each module.
-
b.The code to generate the balloon plot is:
- > ### generate palette
- > my_color_palette <- grDevices::colorRampPalette(RColorBrewer::brewer.pal(8, "Accent"))
- > kinase_all_color <- my_color_palette(ncol(kssMat$combinedScoreMatrix))
- > names(kinase_all_color) <- colnames(kssMat$combinedScoreMatrix)
- > kinase_signalome_color <- kinase_all_color[colnames(predMat)]
- > plotSignalomeMap(signalomes = signalomesRes, color = kinase_signalome_color)
-
a.
-
30.Construction of the Kinase Network
-
a.Finally, we can also plot the signalome network that illustrates the connectivity between kinase signalome networks.
- > plotKinaseNetwork(KSR = kssMat,
- predMatrix = predMat,
- threshold = 0.95,
- color = kinase_all_color)
-
b.The edges refer to the Pearson’s correlation of kinase regulation between the two nodes linked by the edge. The wider the link, the stronger the correlation. You can control the links shown in the visualization by changing the ‘threshold’. Decreasing this value, which is set to a default of ‘0.9’, would reveal more links.
-
a.
Figure 3.

Visualizations of global kinase-substrate relationships and signalomes
(A) A heatmap of global kinase-substrate relationship scores. A clustered heatmap of the combined score from kinase-substrate scoring function for the top three phosphosites of all kinases evaluated in this study. A higher combined score denotes a better fit to a kinase motif and kinase-substrate phosphorylation profile of a phosphosite. Kinases are annotated to which kinase group they belong.
(B) Signalomes identified from the phosphoproteomic data. The branching nodes denote the kinases for which substrates have been predicted, and the stem nodes denote the protein modules that they regulate. Edges between nodes connect kinases to the protein modules they regulate.
Expected outcomes
Outputs and formatting
Outputs of PhosR can be derived at various points of the analysis by the user. The following descriptions of PhosR outputs provides details of the expected data in each output. All outputs are available in the relevant PhosR objects. We provide a sample code with which the users can access each expected outcome.
Filtered, imputed, and normalized data
At various stages of the processing, the users may wish to extract different outputs of the data using the following code:
> # original quantification matrix
> phosphoMat = SummarizedExperiment::assay(ppe, "Quantification")
> # imputed matrix
> phosphoImputedMat = SummarizedExperiment::assay(ppe, "imputed")
> # normalized matrix
> phosphoNormMat = SummarizedExperiment::assay(ppe, "normalized")
Set of stably phosphorylated sites
If the users have generated an SPS set using in-house datasets, we recommend that the users save the output for future use.
> inhouseSPS <- getSPS(ppe.list, assays = assays, conds = cond.list)
> save(inhouseSPS, file = "Data/inhouseSPS.RData")
Motif and phospho-profile matching matrices
PhosR predicts kinase substrate relationships on the basis of two scores: motif matching score and phosphorylation profile matching score. After running the kinase-substrate scoring, users can access the individual matching scores following this code:
> motifScore <- kssMat$motifScoreMatrix
> profileScore <- kssMat$profileScoreMatrix
> combinedScore <- kssMat$combinedScoreMatrix
> kinaseActivityScore <- kssMat$ksActivityMatrix
Predicted Kinase-Substrate Relationships
The above matrices provide the intermediate results to the final prediction scores. The final scores can be derived from running the following function and can be saved as a separate prediction result.
> set.seed(1)
> predMat <- kinaseSubstratePred(kssMat, top = 30)
> write.table(predMat, file = "Data/KS_prediction.txt", sep = "\t")
Signalome networks for kinase of interest
We can extract various outputs from running the Signalome construction function. First, users can access the predicted targets of all kinases.
> signalomeRes$kinaseSubstrates
Users can access the module groups.
> signalomeRes$proteinModules
> table(signalomeRes$proteinModules)
Lastly, if users are interested in the signaling networks regulated by a particular kinase of interest, PhosR outputs useful information related to this network, such as the quantification of phosphorylation sites of all the predicted phosphosites of the kinase of interest, the predicted kinase, and the predicted kinase substrate score. Note that by default, PhosR generates an extended signalomes to combine similar signaling networks together.
> kinaseOI = c("AKT1")
- > signalomesRes <- Signalomes(KSR = kssMat,
- predMatrix = predMat,
- exprsMat = mat.std,
- KOI = kinaseOI)
> str(signalomesRes$Signalomes)
Quantification and statistical analysis
For the differential analysis, differentially phosphorylated sites were identified using the two-sided moderated t test implemented in the limma R package. Analyses were done on log (base 2)-transformed data and p values were adjusted for multiple testing using BenjaminiHochberg FDR correction at a = 0:05.
Limitations
Kinase substrate annotations
The choice of kinase substrate annotation database is essential to the prediction of high confidence kinase-substrate relationships. Inaccurate kinase substrate annotations may lead to inconsistencies and false discoveries. PhosphoSitePlus (Hornbeck et al., 2012), one of the most popular annotation database, was used in this protocol but alternative annotation sources could be used together with or replacing PhosphoSitePlus in the analysis.
Limited number of known substrates for lesser studied kinases
Only a small percentage of the phosphoproteome have been functionally linked to a kinase. As PhosR relies on the presence of known kinase-substrate relationships in curated databases to derive kinase activities from phosphoproteomic data, naturally, kinases and substrates with more annotations will tend to show better predictions. We anticipate that as the number of experimentally validated kinase-substrate annotations increases, the prediction accuracy and our capacity to recapitulate the underlying signaling network will improve.
Species specificity of stably phosphorylated sites
The use of predefined (or a set of user-defined) stably phosphorylated sites to normalize datasets may pose a problem when the method is used to analyze phosphoproteomic data derived from different species. The users may consider using species-specific stably phosphorylated sites when such information is available. Results should be interpreted with caution if generalized across species.
Troubleshooting
Problem 1
You may receive the following error message when installing the PhosR package during “Step 1: Installing PhosR”.
Error: Failed to install 'PhosR' from GitHub :
(converted from warning) download of package 'reactome.db' failed
Potential solution
This error may arise because of package dependencies. In this example, the “reactome.db” package has failed to be installed. A potential workaround to this error is to set ‘build_vignettes’ and ‘dependencies’ as ‘FALSE’ to install the PhosR first and then install the required dependent packages such as ‘reactome.db’ independently when required.
- > devtools::install_github("PyangLab/PhosR",
- build_opts = c("--no-resave-data", "--no-manual"),
- build_vignettes = FALSE,
- dependencies = FALSE)
- > if (!requireNamespace("BiocManager", quietly = TRUE))
- install.packages("BiocManager")
- > BiocManager::install("reactome.db")
Problem 2
In “Step 2: Creating the PhosphoExperiment Object from a MaxQuant output”, you are receiving the following error message or output to the terminal when attempting to create a PhosphoExperiment object from a matrix.
> ppe = PhosphoExperiment(assays = list(Quantification = mat))
- Error in validObject(.Object) :
- invalid class "SummarizedExperiment" object:
- 'names(x)' must be NULL or a character vector with no attributes
Potential solution
This error can occur if your matrix contains rownames that are named. Running the following code should return NULL.
> names(rownames(mat))
However, if you find that the above code returns a string of characters (indicating named rownames), you can potentially overcome this error by removing the names.
> rName = rownames(mat)
> names(rName) = NULL
> rownames(mat) = rName
> ppe = PhosphoExperiment(assays = list(Quantification = mat))
Problem 3
Related to creating the PhosphoExperiment object, when dealing with outputs from software tools like Proteome Discoverer, ProteinPilot and MaxQuant, users should be aware that this software may generate outputs with subtle formatting differences. For example, software may generate one letter versus three letter residue codes (e.g., “Ser” versus “S”). They may also differ in terms of the length of the flanking sequence of the phosphosites or the way in which missing values are denoted. For users who may require help in converting the features to a format that is compatible with the PhosR pipeline, we provide some example code below.
Potential solutions
To convert a three-letter residue code into a single residue code:
> ## Note the length of 'residue' vector would equate to the number of phosphosites identified in your dataset.
> residue = c("Ser", "Thr", "Tyr")
>
> switchRes = function(x) { > x = toupper(x)
> x = switch(x, "SER" = "S", "THR" = "T", "TYR" = "Y")
> return(x)
> }
>
> newResidue = sapply(residue, switchRes)
> newResidue
Whilst PhosR is insensitive to the length of the flanking sequence, it is important that the phosphosite in concern is positioned in the middle of the sequence window. Preprocessing software will generally do this for you automatically. When the phosphosite is found close to either the N or C terminus of the protein, placeholder characters may be automatically added on either side of the sequence to position these phosphosites at the middle of the sequence window. Different software may use different placeholder symbols. Users may convert the sequence windows into a format that PhosR can handle.
> residues = c("A", "R", "N", "D", "C", "E", "Q", "G", "H",
> "I", "L", "K", "M", "F", "P", "S", "T", "W", "Y", "V")
> ppe@Sequence = sapply(ppe@Sequence, function(x) {
>
> window = strsplit(x, "")[[1]]
> seqWindow = paste0(unlist(lapply(window, function(y) {
> if (y %in% residues) {
> toupper(y)
> } else { "_" }
> })), collapse = "")
> return(seqWindow)
>
> })
PhosR will convert any missing values denoted as ‘0’ into ‘NA’. If any missing values are represented as a non-numeric value, PhosR will not recognize them. The users may wish to use the following example code to ensure all missing values are represented as ‘NA’.
> ### An example quantification matrix
> mat = matrix(rnorm(100), nrow = 10, ncol = 10)
> ### Artificially add a missing value denoted by "X"
> mat[4,10] = "X"
> ### Find any values that are not numeric
> sum(!is.numeric(mat))
> ### Replace missing values with NA
> mat[!is.numeric(mat)] = NA
Problem 4
Related to the above, when dealing with outputs from software tools like MaxQuant, users should be aware that a phosphosite may be assigned two or more positions with equal probability. Whilst this particular error may not introduce an obvious error message, these phosphosites may unwittingly be excluded during downstream analyses in PhosR.
Potential solution
As one of the first steps of analysis, the users may want to check that there are no double entries of amino acid position and ensure that only a single position is provided per phosphosite. For users that may be unfamiliar with R, we provide an example to illustrate how we may troubleshoot this. We note that it is beyond the scope of this protocol paper to determine which positions are more likely to denote the true position of the phosphosite. We arbitrarily select the first position in our example.
> # save phosphosites as rNames
> rNames = rownames(ppe)
> # split the names into individual letters
> rNames_split = strsplit(rNames, "")
> # identify which sites contain unusually long sites
> # note that these sites denote phosphosites with two annotated positions on the amino acid (separated by a comma)
> head(rNames[which(sapply(rNames_split, length) > 6)])
> [1] "424,145" "1193,122" "748,139" "373,119" "484,153" "1097,107"
> # we can now use the identified separator to select only the first position
> rNames = sapply(strsplit(rNames, ","), "[[", 1)
> # replace rownames with the new rownames
> rownames(ppe) = rNames
At this stage, the users may also like to ensure other key inputs, such as the sequence window and residue, are correctly provided. Often users erroneously provide predicted peptide sequence, as opposed to the sequence window. The peptide is the actual fragment identified from MS-proteomics, whilst the required “sequence window” referred the sequence wherein the phosphosite has been defined and at which the window is centered. Thus, for a sequence window, we would expect equal number of amino acids flanking the phosphosite.
Problem 5
In “Step 7: Constructing Signaling Networks (Signalomes)”, you are receiving the following error message or output to the terminal when running the ‘Signalomes’ function.
Error in `.rowNamesDF<-`(x, value = value) :
duplicate 'row.names' are not allowed
Potential solution
This error can occur if the rows of your matrix contain duplicate rownames as a result of multiplicity of phosphorylation sites. You can diagnose this by running the following code:
> sum(is.duplicated(rownames(ppe)))
Whilst we do not investigate the best way to deal with multiplicity of phosphosites in PhosR, as it beyond the scope of this report, we provide potential solutions to the error. PhosR implements a versatile function, ‘phosCollapse’, that can be used to generate a combined score for the multiply quantified values. The ‘stat’ argument denotes how we will summarize each phosphosite quantifications across conditions so that for each value in the multiplicity we derive a single quantification, and the ‘by’ argument denotes the method by which to select among the multiplicity.
> mat = SummarizedExperiment::assay(ppe, "Quantification")
- > mat = phosCollapse(mat, rownames(mat),
- stat = apply(abs(mat), 1, max),
- by='mid')
> # note that we no longer have duplicated rows
> sum(is.duplicated(rownames(mat)))
Users may follow an alternative solution to average across multiple sites for each individual sample.
> mat = SummarizedExperiment::assay(ppe, "Quantification")
- > mat = apply(mat, 2, function(x) {
- unlist(sapply(split(x, rownames(mat)), mean))
> })
> SummarizedExperiment::assay(ppe, "Quantification") = mat
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Pengyi Yang (pengyi.yang@sydney.edu.au).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The data and code generated during this study are available at https://pyanglab.github.io/PhosR/ and https://github.com/PYangLab/PhosR_STAR_Protocols.
Acknowledgments
The authors thank the colleagues at the Charles Perkins Centre and the School of Mathematics and Statistics, the University of Sydney, for their intellectual engagement. This work is supported by a National Health and Medical Research Council (NHMRC) Investigator Grant (1173469) to P.Y., an Australian Research Council (ARC) Postgraduate Research Scholarship to H.J.K., Children’s Medical Research Institute Postgraduate Scholarship to H.J.K. and D.X., and the Judith and David Coffey Life Lab Scholarship to T.K.
Author contributions
Writing, H.J.K., T.K., D.X., and P.Y.; development, H.J.K., T.K., D.X., and P.Y.; processing, H.J.K., T.K., D.X., and P.Y.; funding acquisition, P.Y.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Hani Jieun Kim, Email: hani.kim@sydney.edu.au.
Pengyi Yang, Email: pengyi.yang@sydney.edu.au.
References
- Beck S., Michalski A., Raether O., Lubeck M., Kaspar S., Goedecke N., Baessmann C., Hornburg D., Meier F., Paron I. The impact II, a very high-resolution quadrupole time-of-flight instrument (QTOF) for deep shotgun proteomics. Mol. Cell. Proteomics. 2015;14:2014–2029. doi: 10.1074/mcp.M114.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Gagnon-Bartsch J.A., Speed T.P. Using control genes to correct for unwanted variation in microarray data. Biostatistics. 2012;13:539–552. doi: 10.1093/biostatistics/kxr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman N.J., Parker B.L., Chaudhuri R., Fisher-Wellman K.H., Kleinert M., Humphrey S.J., Yang P., Holliday M., Trefely S., Fazakerley D.J. Global phosphoproteomic analysis of human skeletal muscle reveals a network of exercise-regulated kinases and AMPK substrates. Cell Metab. 2015;22:922–935. doi: 10.1016/j.cmet.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S.J., Azimifar S.B., Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015:1–7. doi: 10.1038/nbt.3327. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Kim T., Hoffman N.J., Xiao D., James D.E., Humphrey S.J., Yang P. PhosR enables processing and functional analysis of phosphoproteomic data. Cell Rep. 2021;34:108771. doi: 10.1016/j.celrep.2021.108771. [DOI] [PubMed] [Google Scholar]
- Molania R., Gagnon-Bartsch J.A., Dobrovic A., Speed T.P. A new normalization for Nanostring nCounter gene expression data. Nucleic Acids Res. 2019;47:6073–6083. doi: 10.1093/nar/gkz433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . R Foundation for Statistical Computing; 2020. R: A language and environment for statistical computing.https://R-project.org/ [Google Scholar]
- Schaffer B.E., Levin R.S., Hertz N.T., Maures T.J., Schoof M.L., Hollstein P.E., Benayoun B.A., Banko M.R., Shaw R.J., Shokat K.M. Identification of AMPK phosphorylation sites reveals a network of proteins involved in cell invasion and facilitates large-scale substrate prediction. Cell Metab. 2015;22:907–921. doi: 10.1016/j.cmet.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P., Ormerod J.T., Liu W., Ma C., Zomaya A.Y., Yang J.Y.H. AdaSampling for positive-unlabeled and label noise learning with bioinformatics applications. IEEE Trans. Cybern. 2018;49:1932–1943. doi: 10.1109/TCYB.2018.2816984. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data and code generated during this study are available at https://pyanglab.github.io/PhosR/ and https://github.com/PYangLab/PhosR_STAR_Protocols.