

Summary
Apolipoprotein E (apoE) is a major lipid carrier in the brain and closely associated with the pathogenesis of Alzheimer's disease (AD). Here, we describe a protocol for efficient knockout of APOE in human induced pluripotent stem cells (iPSCs) using the CRISPR-Cas9 system. We obtain homozygous APOE knockout (APOE-/-) iPSCs and further validate the deficiency of apoE in iPSC-derived cerebral organoids. APOE-/- cerebral organoids can serve as a useful tool to study apoE functions and apoE-related pathogenic mechanisms in AD.
For complete details on the use and execution of this protocol, please refer to Zhao et al. (2020).
Subject areas: CRISPR, Neuroscience, Stem Cells, Organoids
Graphical abstract
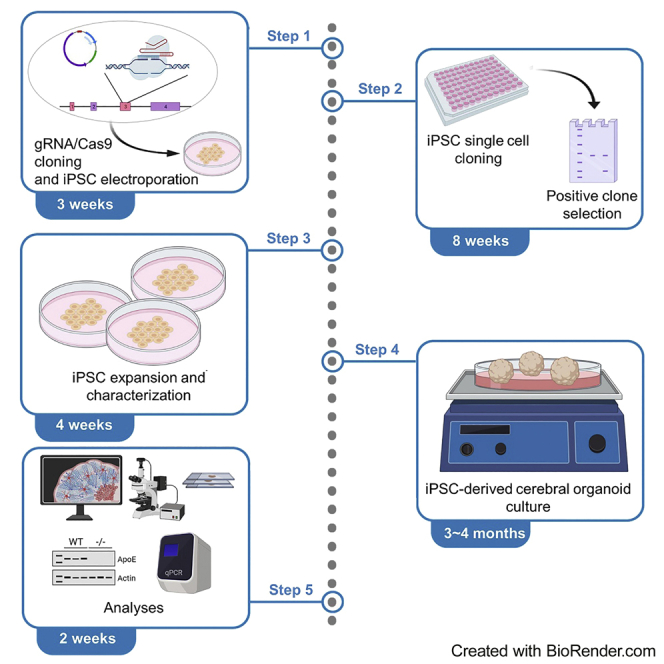
Highlights
-
•
Detailed protocol to generate APOE knockout human iPSC lines via CRISPR-Cas9 technology
-
•
Detailed protocol to generate cerebral organoids from parental and isogenic iPSC lines
-
•
Steps for quality control and assessment of apoE deletion in cerebral organoids
-
•
Provides a valuable tool for apoE function study using iPSC-derived cerebral organoids
Apolipoprotein E (apoE) is a major lipid carrier in the brain and closely associated with the pathogenesis of Alzheimer's disease (AD). Here, we describe a protocol for efficient knockout of APOE in human induced pluripotent stem cells (iPSCs) using the CRISPR-Cas9 system. We obtain homozygous APOE knockout (APOE-/-) iPSCs and further validate the deficiency of apoE in iPSC-derived cerebral organoids. APOE-/- cerebral organoids can serve as a useful tool to study apoE functions and apoE-related pathogenic mechanisms in AD.
Before you begin
Design of candidate guide RNAs (gRNAs) for APOE deletion
Timing: 1 Day
APOE (Apolipoprotein E, NM_001302688.1, NM_000041.4, NM_001302689.1, NM_001302690.1 and NM_001302691.1) (https://uswest.ensembl.org/Homo_sapiens/Gene/ Summary?db=core;g=ENSG00000130203;r=19:44905791-44909393;t=ENST00000252486) has 4 transcript isoforms and the longest transcript has 4 exons. To knockout the APOE gene, we target exon 3 of the longest transcript because it is conserved among all four primary transcripts. We analyze the exon 3 DNA sequence and design three gRNAs with high cutting efficiency and off-target scores using two online gRNA design tools (Broad Institute GPP & CRISPOR; Broad Institute GPP: https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design; CRISPOR: http://crispor.tefor.net/) (Figure 1). To knock out APOE, we use a deletion approach in which a pair of gRNAs will delete a portion of exon 3 to create a frame shift mutation. Three combinations of these gRNAs provide good potential to produce promising frame shift mutations (Table 1).
CRITICAL: Key factors to consider when selecting gRNAs (Joung et al., 2017; Ran et al., 2013).
-
1.
gRNAs located close to the N-terminus of the target gene are generally preferred for knockout to prevent translation of a truncated protein.
-
2.
gRNAs are selected based on high on-target efficiency and low off-target rate as determined from two different online gRNA design tools/algorithms.
-
3.
gRNA pairs should generate a frame shift deletion to create a nonsense mutation.
-
4.
gRNA pairs resulting in larger deletions will be more readily screened. In this study, a gRNA2+5 pairing generates only a 41 bp deletion, which might be relatively difficult to separate from the wildtype (WT) band (304 bp) by DNA electrophoresis.
-
5.
The gRNA binding region should also be validated by Sanger sequencing to make sure no SNPs in target iPSC are present.
Note: Here we discuss the usage of the PX459 plasmid (Addgene Plasmid ID 48139) (Ran et al., 2013) as the cloning vector for gRNAs. Once constructed, the gRNA plasmids are then used to introduce the deletion in APOE. This plasmid simultaneously expresses single guide RNA (sgRNA) and S. pyogenes Cas9 (SpCas9) and carries a puromycin expression cassette for selection. Plasmids PX330 (Addgene Plasmid ID 42230) or PX458 (Addgene Plasmid ID 48138) can be used as alternatives. Remember to add the appropriate sequences for cloning into these vectors when you design the gRNAs (Table 2).
Figure 1.

Selected gRNAs locations within APOE gene and their cutting efficiency and specificity (off-target scores)
Table 1.
Pairs of gRNAs and their predicted DNA deletion lengths to confirm frame shift
| gRNA combinations | Length of deletion (bp) | Frame shift | Truncated protein size |
|---|---|---|---|
| gRNA2+gRNA8 | 59 | Yes | 42 aa |
| gRNA5+gRNA8 | 100 | Yes | 44 aa |
| gRNA2+gRNA5 | 41 | Yes | 48 aa |
Table 2.
Guide RNA sequence for cloning in pX459 plasmid
| Guide RNA | gRNA sequence + adapter |
|---|---|
| gRNA2 F | CACCgAGCTGCGCCAGCAGACCGAG |
| gRNA2 R | aaacCTCGGTCTGCTGGCGCAGCTc |
| gRNA5 F | CACCgGGCCAAGGTGGAGCAAGCGG |
| gRNA5 R | aaacCCGCTTGCTCCACCTTGGCCc |
| gRNA8 F | CACCgACAGTGTCTGCACCCAGCGC |
| gRNA8 R | aaacGCGCTGGGTGCAGACACTGTc |
Order DNA oligos for each gRNA from suppliers such as IDT (Integrated DNA Technologies, https://www.idtdna.com/), and use them to construct the gRNA expression plasmids for the APOE knockout experiments. Each of these gRNAs is cloned into an expression vector and sequence verified. We will examine these gRNA pairs and select the one with the highest deletion efficiency for subsequent screening of knockout cell clones.
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Tuj1 | Sigma | Cat#: T2200; RRID: AB_262133 |
| Mouse monoclonal anti-Tuj1 (clone 2G10) | Abcam | Cat#: ab78078; RRID: AB_2256751 |
| Rabbit polyclonal anti-Sox2 | Abcam | Cat#: ab97959; RRID: AB_2341193 |
| Rat monoclonal anti-Ctip2 (clone 25B6) | Abcam | Cat#: ab18465; RRID: AB_2064130 |
| Rabbit polyclonal anti-Satb2 | Abcam | Cat#: ab34735; RRID: AB_2301417 |
| Mouse monoclonal anti-GFAP (clone GA5) | Millipore | Cat#: MAB360; RRID: AB_11212597 |
| Goat polyclonal anti-ApoE | Millipore | Cat#: AB947; RRID: AB_2258475 |
| Mouse monoclonal anti-actin (clone AC-74) | Sigma | Cat#: A2228; RRID: AB_476697 |
| Rabbit monoclonal anti-Nanog (clone D73G4) | Cell Signaling Technology | Cat#: 4903 S; RRID: AB_10559205 |
| Mouse monoclonal anti-TRA-1-60 (clone TRA-1-60) | Abcam | Cat#: ab16288; RRID: AB_778563 |
| Mouse monoclonal anti-Nestin (clone 2C1.3A11) | Abcam | Cat#: ab18102; RRID: AB_444246 |
| Mouse monoclonal anti-Sox17 (clone 3B10) | Abcam | Cat#: ab84990; RRID: AB_1861437 |
| Goat polyclonal anti-Brachyury | R&D Systems | Cat#: AF2085; RRID: AB_2200235 |
| Rabbit monoclonal anti-S100β | Abcam | Cat#: ab52642; RRID: AB_882426 |
| Rabbit polyclonal anti-Pax6 | BioLegend | Cat#: 901302; RRID: AB_2565003 |
| Rabbit polyclonal anti-Tbr2 | Abcam | Cat#: ab23345; RRID: AB_778267 |
| Mouse monoclonal anti-Nkx2.1 (clone 8G7G3/1) | Thermo Fisher Scientific | Cat#: MA5-13961; RRID: AB_10984070 |
| Donkey polyclonal anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Invitrogen | Cat#: A32790; RRID: AB_2762833 |
| Donkey polyclonal anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Invitrogen | Cat#: A32754; RRID: AB_2762827 |
| Donkey polyclonal anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Invitrogen | Cat#: A32766; RRID: AB_2762823 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Invitrogen | Cat#: A32744; RRID: AB_2762826 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#: A21208; RRID: AB_2535794 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | Cat#: A21209; RRID: AB_2535795 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#: A-11055; RRID: AB_2534102 |
| IRDye 800CW Donkey anti-Goat IgG antibody | LI-COR Biosciences | Cat#: 926-32214; RRID: AB_621846 |
| IRDye 680RD Donkey anti-Mouse IgG secondary antibody | LI-COR Biosciences | Cat#: 926-68072; RRID:AB_10953628 |
| Bacterial and virus strains | ||
| DH5α competent cells | Thermo Fisher Scientific | Cat#: 18265017 |
| Chemicals, peptides, and recombinant proteins | ||
| FastDigest BbsI | Thermo Fisher Scientific | Cat#: ER1011 |
| FastAP | Thermo Fisher Scientific | Cat#: EF0651 |
| Plasmid-Safe exonuclease | Lucigen | Cat#: E3101K |
| SOC medium | Thermo Fisher Scientific | Cat#: 15544034 |
| mTeSR1 | STEMCELL Technologies | Cat#: 85850 |
| Matrigel hESC-qualified | Corning | Cat#: 354277 |
| Y-27632 | STEMCELL Technologies | Cat#: 72304; CAS: 129830-38-2 |
| Dispase | STEMCELL Technologies | Cat#: 07923 |
| Accutase | STEMCELL Technologies | Cat#: 07920 |
| STEMdiff™ Cerebral Organoid Kit | STEMCELL Technologies | Cat#: 08570 |
| DMEM/F-12 | Thermo Fisher Scientific | Cat#: 11330-057 |
| Neurobasal | Thermo Fisher Scientific | Cat#: 21103-049 |
| B-27 Supplement | Thermo Fisher Scientific | Cat#: 17504044 |
| N2 supplement | Thermo Fisher Scientific | Cat#: 17502-048 |
| GlutaMAX | Thermo Fisher Scientific | Cat#: 35050061 |
| Nonessential amino acids (NEAA) | Thermo Fisher Scientific | Cat#: 11140076 |
| Ascorbic acid | STEMCELL Technologies | Cat#: 72132; CAS: 50-81-7 |
| DbcAMP | Sigma | Cat#: D0260; CAS: 16980-89-5 |
| Recombinant Human/Murine/Rat BDNF | PeproTech | Cat#: AF-450-02 |
| Animal-Free Recombinant Human GDNF | PeproTech | Cat#: AF-450-10 |
| Sodium pyruvate | Thermo Fisher Scientific | Cat#: 11360070 |
| Normal goat serum | Thermo Fisher Scientific | Cat#: 31872 |
| Bovine serum albumin (BSA) | Sigma | Cat#: A1470 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat#: 15140122 |
| Critical commercial assays | ||
| QIAquick® Gel Extraction Kit | QIAGEN | Cat#: 28704 |
| Phusion™ High-Fidelity DNA Polymerase | Thermo Fisher Scientific | Cat#: F530L |
| Quick Ligation™ Kit | NEB | Cat#: M2200S |
| Invitrogen Neon™ Transfection System | Thermo Fisher | Cat#: MPK5000 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725274 |
| Quick-DNA genomic extraction kit | Zymo Research | Cat#: D3025 |
| Gel Purification Kit | MACHEREY-NAGEL | Cat#: 740609.250 |
| RNase-Free DNase Set | QIAGEN | Cat#: 79254 |
| RNeasy Mini Kit | QIAGEN | Cat#: 74104 |
| ReverTra Ace® qPCR RT Master Mix | Toyobo | Cat#: FSQ-201 |
| Experimental models: cell lines | ||
| MC0192 human iPSC line | (Zhao et al., 2017) | N/A |
| Oligonucleotides | ||
| Primer pairs for qRT-PCR for APOE | (Meyer et al., 2019) | N/A |
| Primer pairs for qRT-PCR for ACTB | (Zhao et al., 2020) | N/A |
| Recombinant DNA | ||
| PX459 plasmid | (Ran et al., 2013) | Addgene_48139 |
| Software and algorithms | ||
| GPP | Broad Institute | https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| CRISPOR | Broad Institute | http://crispor.tefor.net/ |
| Other | ||
| 96-Well round-bottom ultra-low attachment plate | Corning | Cat#: 7007 |
| Organoid Embedding Sheet | STEMCELL Technologies | Cat#: 08579 |
Materials and equipment
Y27632 10 mM
Dissolve 5 mg of Y27632 in 1.56 mL of sterile distilled water to obtain a stock solution of 10 mM. Aliquot and store at −20°C for up to 1 year. Dilute 1000 times with mTeSR1 for iPSC passage.
mTeSR1
| Reagent | Final Concentration | Volume |
|---|---|---|
| mTeSR1 basal medium | n/a | 395 mL |
| mTeSR1 supplement (5×) | 1× | 100 mL |
| Penicillin-Streptomycin (100×) (optional) | 1× | 5 mL |
| Total | n/a | 500 mL |
Note: For regular human iPSC culture, Penicillin-Streptomycin is not necessary. Antibiotics are recommended for iPSC culture during the gene editing process. Complete mTeSR1 medium can be stored at 4°C for 2 weeks.
EB formation medium
| Reagent | Final Concentration | Volume |
|---|---|---|
| Cerebral organoid basal medium 1 | n/a | 40 mL |
| Cerebral organoid supplement A | n/a | 10 mL |
| Total | n/a | 50 mL |
Complete EB formation medium can be stored at 4°C for 1 week.
Neural induction medium
| Reagent | Final Concentration | Volume |
|---|---|---|
| Cerebral organoid basal medium 1 | n/a | 49.5 mL |
| Cerebral organoid supplement B | n/a | 0.5 mL |
| Total | n/a | 50 mL |
Complete Neural induction medium can be stored at 4°C for 1 week.
Expansion medium
| Reagent | Final Concentration | Volume |
|---|---|---|
| Cerebral organoid basal medium 2 | n/a | 24.25 mL |
| Cerebral organoid supplement C | n/a | 0.25 mL |
| Cerebral organoid supplement D | n/a | 0.5 mL |
| Total | n/a | 25 mL |
Complete Expansion medium can be stored at 4°C for 1 week.
Maturation medium for early stage cerebral organoid culture
| Reagent | Final Concentration | Volume |
|---|---|---|
| Cerebral organoid basal medium 2 | n/a | 98 mL |
| Cerebral organoid supplement E | n/a | 2 mL |
| Total | n/a | 100 mL |
Complete Maturation medium can be stored at 4°C for 2 weeks.
Neuron medium for late stage cerebral organoid culture
| Reagent | Final Concentration | Volume |
|---|---|---|
| DMEM/F-12 | n/a | 472 mL |
| Neurobasal | n/a | 472 mL |
| B27 supplement with vitamin A (50×) | 0.5× | 10 mL |
| N2 supplement (100×) | 0.5× | 5 mL |
| Glutamax (100×) | 1× | 10 mL |
| sodium pyruvate mixture (100×) | 1× | 10 mL |
| Ascorbic acid (50 mg/mL stock) | 200 ng/mL | 4 μL |
| NEAA (100×) | 1× | 10 mL |
| dbCAMP (5 mg/mL stock) | 500 ng/mL | 100 μL |
| Penicillin-Streptomycin | 1× | 10 mL |
| BDNF (50 μg/mL stock) | 10 ng/mL | 200 μL |
| GDNF (50 μg/mL stock) | 10 ng/mL | 200 μL |
| Total | ~ 1000 mL |
Neuron medium can be stored at 4°C for 2 weeks. Add BDNF and GDNF right before use.
Blocking buffer for staining
| Reagent | Final Concentration | Amount |
|---|---|---|
| Donkey serum | 4% | 4 mL |
| BSA | 2% | 2 g |
| Glycine | 1 M | 7.5 g |
| PBS | n/a | 96 mL |
| Total | n/a | ~ 100 mL |
Blocking buffer can be stored at 4°C for 1 week. Aliquot can be made and stored in −20°C for 1 year.
Antibody buffer for staining
| Reagent | Final Concentration | Volume |
|---|---|---|
| Blocking buffer | n/a | 10 mL |
| Triton-X100 | 0.25% | 25 μL |
| Total | n/a | ~ 10 mL |
Antibody buffer can be stored at 4°C for 1 week. Aliquot can be made and stored in −20°C for 1 year.
Step-by-step method details
Cloning gRNA oligos into Cas9 vectors
This step generates the plasmids that express candidate gRNAs and Cas9 to be tested for CRISPR/Cas9-mediated gene deletion.
-
1.
Digest 1 μg of PX459 plasmid with BbsI, prepare the reactions as in the following table and incubate for 30 min at 37°C,
| Component | Amount |
|---|---|
| Plasmid | 1 μg |
| FastDigest BbsI | 1 μL (10 U/μL) |
| FastAP | 1 μL (1 U/μL) |
| 10× FastDigest Buffer | 2 μL |
| ddH2O | Supplement to 20 μL |
| Total | 20 μL |
-
2.
Purify the digested plasmid using QIAquick® Gel Extraction Kit and elute with 30 μL elution buffer following the instructions of the supplier.
-
3.
Phosphorylate and anneal each pair of oligos, prepare the reactions as in the following table.
| Component | Amount |
|---|---|
| Oligo 1 (gRNA F, 100 μM) | 1 μL |
| Oligo 2 (gRNA R, 100 μM) | 1 μL |
| 10× T4 Ligation Buffer | 1 μL |
| T4 PNK | 0.5 μL |
| ddH2O | 6.5 μL |
| Total | 10 μL |
Anneal in a thermocycler using the following parameters:
37°C 30 min
95°C 5 min and then ramp down to 25°C at 5°C/min
-
4.
Set up ligation reaction, prepare the reactions as in the following table and incubate at room temperature (20°C–25°C) for 10 min.
| Component | Amount |
|---|---|
| BbsI digested plasmid from Step 2 | 50 ng |
| Phosphorylated and annealed oligo duplex from step 3 (1:200 dilution) | 1 μL |
| 2× Quick Ligation Buffer | 5 μL |
| Quick Ligase | 1 μL |
| ddH2O | Supplement to 10 μL |
| Total | 10 μL |
-
5.
(optional) Treat ligation reaction with Plasmid Safe exonuclease to prevent unwanted recombination products. Prepare the reactions as in the following table and incubate reaction at 37°C for 30 min.
| Component | Amount |
|---|---|
| Ligation reaction from Step 4 | 10 μL |
| 10× Plasmid Safe Buffer | 1.5 μL |
| 10 mM ATP | 1.5 μL |
| Exonuclease | 1 μL (10,000 U/mL) |
| ddH2O | 1 μL |
| Total | 15 μL |
-
6.Transformation:
-
a.Thaw 100 μL aliquots of DH5α competent cells from −80°C freezer on ice.
-
b.Add 10 μL of the products from Step 5 into 50 μL of ice-cold DH5α competent cells.
-
c.Incubate the mixture on ice for 30 min.
-
d.Heat shock the mixture at 42°C for 45 s and return immediately to ice for 2 min.
-
e.Add 600 μL SOC medium (without antibiotics) and incubate in 37°C shaker for 30-40 min.
-
f.Plate 200 μL of the mixture onto an LB plate containing 100 μg/mL ampicillin and spread around.
-
g.Incubate the plate for 16 to 24 h at 37°C.
-
a.
-
7.
Pick 2–4 colonies per construct. Grow colonies in LB/100 μg/mL ampicillin. Shake 16 to 24 h at 37°C.
-
8.
Miniprep each culture, extract genomic DNA using the Zymo genomic extraction kit following manufacturer’s instructions.
-
9.
Submit the extracted plasmids for Sanger sequencing to verify successful cloning of the candidate gRNAs using U6 forward primer (ACGATACAAGGCTG TTAGAGAG).
Pause point: Keep the glycerol stocks harboring the correctly cloned plasmids. At this point, the glycerol stocks at −80°C can be stored for long-term and you can proceed to the next steps later.
-
10.
Choose a sequence-verified colony and inoculate into a maxiprep culture. Store the plasmids at −80°C.
Electroporating plasmids into human iPSCs
This step tests the cutting efficiencies of the candidate gRNAs in human iPSCs. gRNAs that show no or low cutting efficiency will be excluded before single cell clone isolation. Here we use MC0192 human iPSC line generated in our lab, which has been reported previously (Zhao et al., 2017; Zhao et al., 2020). This iPSC line has been fully characterized and monitored for mycoplasma contamination regularly.
-
11.Culturing human iPSCs: Thaw and culture human iPSC line (ID: MC0192) in feeder-free conditions with mTesR1 medium in a humidified 37°C incubator with 5% CO2.
-
a.Coat 100 mm cell culture dish with Matrigel: Dilute Matrigel with 6 mL cold DMEM/F-12 according to the dilution factor provided in the manufacturer’s datasheet and dispense the solution onto the plate. The aliquot (dilution factor) volume of Matrigel is typically between 270 and 350 μL (refer to the Certificate of Analysis). Add one aliquot (dilution factor) of Matrigel to 18 mL of DMEM/F-12 to coat three 100 mm dishes. Move the plate around so that the bottom of the plate is completely covered. Leave it at room temperature (20°C–25°C) in the biosafety hood for at least 1 h. Always prepare Matrigel-coated plate freshly!
-
b.Thaw a vial of cells in 37°C water bath, transfer the cells to a 15 mL centrifuge tube, add 5 mL of mTeSR1 medium and centrifuge at 300 × g for 5 min at room temperature (20°C–25°C).
-
c.Aspirate the Matrigel coating medium and seed cells with 10 mL of mTeSR1 medium containing ROCK inhibitor (10 μM Y27632).
-
d.Change medium with regular mTeSR1 medium after 24 h and refeed daily.
-
a.
Note: Extensive passage might induce some alteration in the human iPSC line, it is recommended to use human iPSC lines with comparatively low passage number (< passage 30). Before electroporation, we recommend to passage the cells one time after thawing followed by culturing for at least 5 days. When the cells reach 70% confluency during this period, passage cells according to the following steps.
-
12.Passaging cells: Passage the cells before they reach 70% confluency.
-
a.Observe the iPSCs under the microscope. If any differentiated cells appear, scrape them off using a P2 pipette tip under the microscope (in the hood).
-
b.Aspirate the mTeSR1 medium and wash the cells once with 10 mL DPBS.
-
c.Add 6 mL Dispase to 100 mm cell culture dish and leave it in the incubator at 37°C for ~5 min. When the edge of the iPSC colonies starts to peel off the plate, aspirate the Dispase, and rinse the dish with 6 mL pre-warmed DMEM/F-12.
-
d.Aspirate DMEM/F-12, add 6 mL of mTeSR1 with 10 μM Y27632, and scrape the dish to detach the iPSC.
-
e.Transfer the cells to a 15 mL centrifuge tube and centrifuge at 300 × g for 5 min at room temperature (20°C–25°C).
-
f.Using 1 mL pipette, break the colonies into smaller pieces by pipetting up and down a few times.
-
g.Transfer the iPSC to a new plate at 1:3 to 1:6 ratio depending on your experimental plan.
-
a.
-
13.Electroporation: Use the Invitrogen Neon™ transfection system or similar systems according to the manufacturer’s instructions.
-
a.Aspirate the mTeSR1 medium and wash the cells once with DPBS.
-
b.Add 6 mL of Accutase to 100 mm cell culture dish and leave it in the incubator at 37°C for ~5 min. Using a pipettor, pipette the cell suspension up and down 3 - 5 times to dislodge remaining attached cells. Add 10 mL of DMEM/F-12 to the plate, and collect the cells into a 50 mL tube. Centrifuge at 300 × g for 5 min. Gently resuspend cell pellet with mTeSR1 with 10 μM Y27632. Count the number of cells needed for nucleofection (3 × 105 cells per electroporation) and spin down at 300 × g for 5 min at room temperature (20°C–25°C).
-
c.Remove the medium completely and resuspend cells in nucleofection solution.
-
d.Pipette the resuspended cells with 1.5 μg of each gRNA/Cas9 plasmid into electroporation cuvettes and electroporate according to the parameters in the following table. This electroporation parameter has been tested to ensure optimal transfection efficiency in several iPSC lines. However, we always recommend additional optimization for each particular iPSC line.
Pulse voltage (V) Pulse width (ms) Pulse number MC0192 1000 30 2 -
e.Gently plate the electroporated cells onto coated 100 mm plates in mTeSR1 supplemented with 10 μM Y27632. Typically 1 to 2 ×105 cells survive 24 h post nucleofection.
-
f.Refeed the cells daily with regular mTeSR1 medium beginning 24 h after nucleofection. Puromycin selection can be applied to remove non-transfected cells at a concentration of 0.5 μg/mL for 24 h.Note: The concentration of puromycin may vary depending on the cell line. The lowest concentration can be 0.25 μg/mL. The puromycin concentration can range from 0.25 to 1 μg/mL. In our experience, 0.5 μg/mL with 24 h treating time shows the best results. Treating cells with puromycin for longer time is not recommended since the cells are very sensitive to puromycin. Further optimization for each particular iPSC line is always recommended.CRITICAL: Human iPSCs can vary widely in their transfection efficiency, tolerance of single-cell dissociation, and maintenance conditions. For a given cell line of interest, relevant literature or the supplier should be consulted.
-
a.
-
14.
Evaluate gRNA pairs via PCR amplification: Each of the two gRNA pairs indicated in Table 1 are transfected into iPSCs. After puromycin selection, the pooled cells which contain a mixed population of Cas9/gRNA transfected cells are collected to assess the deletion efficiency of its gRNA pair. The following PCR primers are used to amplify the targeted region, which will be around 204 bp for deletion alleles, and 304 bp for wild type alleles.
APOE-F (AGGTACTAGATGCCTGGACGG)
APOE-R (GTATAGCCGCCCACCAGGAG)
Assemble the PCR reactions and run the cycling program as follows. Run 10 μL of PCR product on a 2% (wt/vol) agarose gel to check for size of the products using UV imaging. gRNA5+8 generates >40% deletion efficiency since a stronger edited PCR band is present compared to the wild-type band. These pooled cells are used for isolation of single cell clones (Figure 2).
Figure 2.

APOE deletion in pooled cells transfected with different gRNA combinations
| Component | Amount |
|---|---|
| Template genomic DNA | 1.0 μL |
| APOE PCR Forward and Reverse primers | 2.5 μL |
| Phusion® DNA polymerase | 0.3 μL |
| 5× Phusion® HF buffer | 4.0 μL |
| 10 mM dNTP | 0.4 μL |
| DMSO (optional) | 1.0 μL |
| ddH2O | 10.8 μL |
| Total Vol. (μL) | 20 μL |
| PCR Cycling Conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 98°C | 5 min | 1 |
| Denaturation | 98°C | 5 s | 30 |
| Annealing | 66.6°C | 10 s | |
| Extension | 72°C | 30 s | |
| Final extension | 72°C | 2 min | 1 |
| Hold | 4°C | ||
Single-cell clone isolation
-
15.
Once the desired deletion rate of pooled transfected cells is confirmed by PCR, dissociate the cells from the transfected cell pool using Accutase.
-
16.
Count the number of transfected iPSCs from each 6-well plate, and serially dilute them in culture medium supplemented with 10 μM Y27632 to a final concentration of 0.8 cells per 100 μL to reduce the likelihood of having multiple cells per well.
-
17.
Use 80 cells in 12 mL of culture medium for each fresh Matrigel-coated 96-well plate and use at least two 96-well plates for each transfected population.
-
18.
Observe each of 96-wells the next day and mark off the wells that may have been seeded with multiple cells. Allow the single cells to expand to a clonal appearance ~1 week after plating.
-
19.
Return the cells to the incubator and allow them to expand for 2–3 weeks. Refeed the single cell clones daily with regular mTeSR1 medium.
PCR analysis and DNA sequencing
-
20.
Single cells are placed in 96-well plates and cultured for about 14 days before expanding and culturing in 24-well plates.
-
21.
Each clone from the 24-well plates is isolated and genomic DNA is extracted using Zymo genomic extraction kit according to the manufacturer’s instructions. The genomic DNA is subsequently used for PCR amplification assay.
-
22.
The same PCR primers used in Step 14 are used to amplify the targeted region.
APOE-F (AGGTACTAGATGCCTGGACGG)
APOE-R (GTATAGCCGCCCACCAGGAG)
Assemble the PCR reactions and run the cycling program as in Step 14.
-
23.
Run 10 μL of PCR product on a 2% (wt/vol) agarose gel to check for size of the PCR products using UV imaging (Figure 3).
-
24.
Submit the purified PCR products (using Macherey-Nagel Gel Purification Kit or similar) for Sanger sequencing with one or both of the PCR primers to confirm the APOE deletion (Figure 4).
Note: Pick the clones that show normal pluripotent colony morphology with comparable growth rate to the parental cells. Confirm pluripotency by checking marker expression such as TRA-1-60, Nanog, and Sox2. If the differentiation rate is higher than 30%, it is recommended to do further selection following the regular iPSC handling procedure. Also confirm normal karyotypes of the selected clones before proceeding to the next steps. After pluripotency marker staining and three germ layer differentiation, we choose clone #11 to continue with the rest of the protocol (Figure 5).
Figure 3.

APOE deletion in cells after single cell isolation and amplification
Assay was run on genomic DNA samples from experimental clones and wildtype control. The gel images show that wildtype gives a 304 bp band. PCR products of clones with homozygous deletion (HM) have one band at around 204 bp, while heterozygous-deletion clones (HT) show both bands.
Figure 4.

Validation of APOE deletion in iPSCs via Sanger DNA sequencing
Clones #11, #34 and #40 show deletions in APOE gene compared with wildtype. Sanger DNA sequencing results of the 5’ and 3’ of deleted genomic region in APOE knockout clone #11 (A and B), #34 (C and D) and #40 (E and F).
Figure 5.

Characterization of iPSCs after gene editing
(A) Immunostaining of pluripotency markers (Nanog and TRA-1-60) in parental and APOE-/- iPSC lines.
(B) In vitro differentiation of parental and APOE-/- iPSC lines into cells of all three germ layers. Cells were immunostained for Sox17 (endoderm), Brachyury (mesoderm), Nestin (ectoderm), and DAPI (nucleus). Scale bar: 100 μm.
Karyotyping analysis
GTL-banding karyotype analysis is performed by a Cytogenetics Core at Mayo Clinic.
-
25.
Cells are treated with 0.0125 μg/mL colcemid (Life Technologies) for 16 to 24 h before being harvested using TrypLE (Gibco).
-
26.
Cell suspension is then treated with hypotonic solution (equal parts KCl and NaCitrate) followed by fixation using 2:1 methanol:glacial acetic acid before spotted onto a glass slide.
-
27.
Prepared slides are stained using Trypsin and Leishman’s staining solutions. The stained slides are scanned using GSL-120 and analyzed using CytoVision™ (Leica Biosystems).
Generation of cerebral organoid from APOE-/- isogenic human iPSC line
ApoE is produced primarily by astrocytes in the central nervous system as a carrier of cholesterol and other lipids to support membrane homeostasis, synaptic integrity and injury repair (Liu et al., 2013; Yamazaki et al., 2019; Yu et al., 2014). The cerebral organoid model system is highly reminiscent of human brain structure with diverse cell types (Lancaster and Knoblich, 2014; Lancaster et al., 2013; Renner et al., 2017), which can be a powerful human related model to investigate gene functions and crosstalk between different cell types. Here, we use STEMdiff™ Cerebral Organoid Kit from Stemcell Technologies for cerebral organoid differentiation. Cerebral organoids in this study are generated following the manufacturer’s guidelines with some in-house optimizations (Figure 6).
-
28.
Thaw Supplement(s) at room temperature (20°C–25°C). Add Supplement(s) to Basal Medium as indicated by the manufacturer (see Materials and Equipment).
Note: If not used immediately, aliquot Supplement(s) and store at −20°C. After thawing aliquots, use immediately. Do not re-freeze.
-
29.Embryonic body (EB) formationNote: This protocol is for the formation of EBs from a human iPSC culture in a 100 mm dish, adjust volumes for other cultureware accordingly.CRITICAL: Use a microscope to visually identify regions of differentiation in the iPSC culture. Remove regions of differentiation by scraping with a pipette tip.
-
a.Prewarm EB Formation medium aliquot to room temperature (20°C–25°C) and add ROCK inhibitor (final concentration: 10 μM).
-
b.Aspirate medium from human iPSC culture and wash the dish with 10 mL of prewarmed sterile DPBS.
-
c.Aspirate DPBS and add 6 mL of Accutase. Incubate at 37°C for 5 min.Note: Check the digest status under microscope. Incubation time may vary when using different cell lines. Other non-enzymatic cell dissociation reagents can also be used. Make sure all cells are dissociated into single cells. Dissociated cells can be filtered through 30 μm cell strainer when necessary.
-
d.Transfer cell suspension into a sterile 15 mL conical tube.
-
e.Rinse the dish with 6 mL of mTeSR1 and add this rinse to the tube containing cells. Centrifuge cells at 300 × g for 5 min at room temperature (20°C–25°C).
-
f.Aspirate the supernatant. Add 1–2 mL of EB medium with 10 μM Y27632 to resuspend cells.
-
g.Count cells and calculate the volume of cells required to obtain 150,000 cells/mL; add cells to extra EB medium with 10 μM Y27632.
-
h.Add 100 μL of cell suspension into 96-well round-bottom ultra-low attachment plate using multi-channel pipettor (15,000 cells/well). Centrifuge the plate at 500 × g for 5 min.
-
i.Incubate 96-well plate at 37°C for 24 h. Do not disturb plate during this period.Note: It is important to optimize the initial cell seeding densities for optimal EB formation. Cell density needs to be adjusted according to specific iPSC line. Increase the cell number if the EB size is too small (<100 μm). Preliminary experiments can be performed before culturing the experimental cohort.
-
j.Gently add 100 μl of EB Formation medium per well on day 2 and day 4 respectively. Add the medium directly, and do not change medium.
-
k.Observe EBs under microscope on Day 5. EBs should reach a diameter of > 300 μm (typically 400–600 μm) and exhibit round and smooth edges. At this stage, most cells are still positive for pluripotency markers (Figure 7).
-
a.
Figure 6.

Workflow for the generation of cerebral organoids (Created with BioRender.com)
Figure 7.

Characterization of EBs at early differentiation stages
Representative images of pluripotency (TRA-1-60 and Nanog) and neural stem cell (Nestin and Sox2) markers expression in EBs at different stages.
-
30.Neural induction
-
a.Prewarm neural induction medium to room temperature (20°C–25°C).
-
b.Add 250 μL of Induction Medium to each well of a 48-well ultra-low attachment plate.
-
c.Transfer 1 EB to each well of the 48-well plate using a wide-bore 200 μL pipette tip.Note: EBs can merge with each other during the induction period, which will influence the size evaluation of the cerebral organoid. Transfer of multiple EBs into one well is not recommended.
-
d.Incubate EBs in Neural induction medium for 48 h. Observe EBs under microscope, EBs should show smooth and optically translucent edges.CRITICAL: Induction time needs to be adjusted among different lines according to the morphology of EBs. Optically translucent edge is an important hallmark for successful neuroepithelia induction. Marker staining shows the emergence of Nestin positive cells (Figure 7).
-
a.
-
31.Cerebral organoid expansion
-
a.Prewarm Expansion medium aliquot to room temperature (20°C–25°C), thaw Matrigel on ice at 2°C–8°C for 1–2 h.Note: You will lose some Matrigel during embedding, thaw Matrigel slightly more than the needed amount (20 μL/EB; 96 EBs × 20 μL/EB ≈ 2 mL Matrigel). Keep Matrigel on ice to prevent premature polymerization. All plasticwares used for embedding should be kept at −20°C prior to use.
-
b.Place the Organoid Embedding Sheet into an empty sterile 100 mm dish on ice.
-
c.Use a pre-chilled wide-bore 200 μL pipette tip and set the pipet at 25 μL, draw up EBs from the 48-well plate and transfer to embedding surface.Note: Matrigel can get dry and polymerize prematurely, Embed no more than 12–16 EBs at a time as Matrigel can get dry and polymerized prematurely.
-
d.Carefully remove excessive medium from each EB. Add 20 μL of Matrigel dropwise onto each EB using pre-chilled 20 μL pipette tip.
-
e.Reposition the EB to the center of the droplet using pre-chilled pipette tip.
-
f.Place the plate in an incubator at 37°C for 15 min to polymerize Matrigel.
-
g.Use sterile forceps to position the embedding sheet directly above a 6-well ultra-low adherent plate. Gently wash Matrigel droplets off the sheet using a 1 mL pipettor (3 mL Expansion Medium/well). Repeat until all Matrigel droplets are washed off.
-
h.Incubate EBs in Expansion medium for 3 days. Embedded organoids will show budding of the EB surface (Nestin +/Sox2 +), which is a hallmark for neuroepithelia development (Figure 7).
-
a.
Troubleshooting (Problem 4)
-
32.Cerebral organoid maturation
-
a.Prewarm Maturation medium (Medium E).
-
b.Use a wide bore 1 mL pipette tip to carefully transfer 16–20 Matrigel droplets containing organoids to 100 mm dish.
-
c.Remove excess medium, add 15 mL Maturation medium to the dish.
-
d.Place plate of organoids on an orbital shaker in a 37°C incubator. Set the shaker speed at 40 rpm.
-
e.Change medium with Maturation medium every 3 days.
-
f.After culturing organoids in Medium E for 4 weeks, replace medium with Neuron medium, and change medium every 3 days until harvesting.
-
a.
Note: Heterogeneous morphologies will be apparent in cerebral organoid culture. During the differentiation process, some abnormal cerebral organoids will fall apart into small pieces in the orbital shaker, which can be eliminated during medium change. Collect cerebral organoids with normal size range (typically 2–4 mm in diameter) and exclude those with obviously smaller sizes (<1 mm) (Figure 8). The exclusion criteria should be adjusted for different iPSC lines, especially for those with disease-associated mutations.
Figure 8.

Representative image of cerebral organoids after 12 Weeks of differentiation
Those that are much smaller than others can be excluded from further analysis (an example is shown with red arrow).
Troubleshooting (Problem 5)
Cerebral organoid validation via specific neural marker immunostaining
During the long-term differentiation procedure, cerebral organoids can be collected at different time points to confirm the emergence of neural differentiation markers, which will help to evaluate the quality of the cerebral organoids. After a period of maturation, organoids display cortical-like regions such as the ventricular zone and the emergence of multiple neuronal layers, which is similar to in vivo brain development. Here we stain organoid sections with Sox2 (neural progenitor cell marker), Pax6 (dorsal region marker), Nkx2.1 (ventral region marker), Tbr2 (intermediate progenitors),Tuj1 (early neuronal marker), Ctip2 (deep cortical layer marker) and Satb2 (superficial cortical layer marker) at Week 4 and 12 to monitor the differentiation of cerebral organoids (Lancaster and Knoblich, 2014).
-
33.
Cut a 1 mL pipette tip using sterile scissors as needed according to the size of cerebral organoid. Transfer organoids to a new 6-well plate carefully.
-
34.
Remove excess medium from the well. Wash cerebral organoids 3× for 5 min with DPBS.
-
35.
Add 5 mL fresh 4% PFA per well of 6-well plate for 30 min at room temperature (20°C–25°C). Make sure all organoids are submerged in PFA solution.
-
36.
Discard PFA and wash cerebral organoids 3× for 5 min with DPBS.
Caution: PFA can be potentially harmful to eyes, skin, or respiratory system. Avoid repeated and prolonged exposure.
-
37.
Discard DPBS thoroughly. Add 30% Sucrose Solution at 5 mL per well of 6-well plate.
-
38.
Allow to equilibrate 24–48 h at 2°C–8°C. Adjust the equilibration time according to the size of organoid. Organoids should sink to the bottom of the well when equilibrated.
-
39.
Transfer organoids to Embedding mold and remove excess sucrose solution around the organoids.
-
40.
Add Tissue-Tek® optimum cutting temperature (O.C.T.) solutions to completely cover organoids, and use a 200 μL tip to adjust the location of the organoids. When embedding two or more organoids in one mold, separate organoids to avoid contact with each other.
-
41.
Freeze embedded organoids in O.C.T. on dry ice until completely frozen.
-
42.
Cut frozen organoid sections using Cryostat. Here we use 30 μm sections for marker staining. Thickness can be adjusted to suit imaging system.
-
43.
Dry slides completely on slide heater and store slides in −80°C freezer for long-term use.
-
44.
Move slides from −80°C freezer to room temperature (20°C–25°C). Use a PAP pen to outline the organoid sections. Proceed to the next step once the wax is completely dry.
-
45.
Wash slides with PBS in a Coplin staining jar 3× for 10 min to fully remove O.C.T. from slides.
-
46.
Incubate slides with 200 μL of 0.5% PBS-X in a humidified chamber at room temperature (20°C–25°C) for 15 min.
-
47.
Wash slides 3× for 5 min with DPBS in a Coplin staining jar.
-
48.
Incubate slides with 200 μL of Blocking buffer at room temperature (20°C–25°C) in a humidified chamber for 1 h.
-
49.
Prepare primary antibody solution in Antibody Dilution Buffer. Refer to Table 3 for recommended antibody concentrations.
-
50.
Aspirate Blocking Solution and add 200 μL of primary antibody mix to cover the sections completely. Incubate slides with primary antibody at 4°C in a humidified chamber for 16 to 24 h.
-
51.
Prepare secondary antibody solution in Antibody Dilution Buffer. Refer to Table 3 for recommended antibody concentrations.
-
52.
Wash slides 3× for 5 min with DPBS in a Coplin staining jar.
-
53.
Incubate slides with 200 μL of secondary antibody at room temperature (20°C–25°C) in a humidified chamber for 2 h.
-
54.
Wash slides 3× for 5 min with DPBS in a Coplin staining jar.
-
55.
Incubate slides with 200 μL of DAPI solution (1 μg/mL) at room temperature (20°C–25°C) in a humidified chamber for 30 s.
-
56.
Wash slides 3× for 5 min with DPBS in a Coplin staining jar.
-
57.
Remove excessive liquid from the slides using Kim wipes. Pipette Mounting medium to slide and mount slide with coverslip. Allow mounting medium to harden in 16 to 24 h and store at 4°C before imaging.
Table 3.
Antibodies for immunofluorescence staining
| Antibody | Dilution | Supplier |
|---|---|---|
| Primary antibodies | ||
| Tuj1 (immunostaining) | 1:1000 | Sigma, T2200 |
| Tuj1 (immunostaining) | 1:1000 | Abcam, ab78078 |
| Sox2 (immunostaining) | 1:300 | Abcam, ab97959 |
| Ctip2 (immunostaining) | 1:100 | Abcam, ab18465 |
| Satb2 (immunostaining) | 1:100 | Abcam, ab34735 |
| GFAP (immunostaining) | 1:300 | Millipore, MAB360 |
| Pax6 (immunostaining) | 1:300 | Biolegend, 901302 |
| Tbr2 (immunostaining) | 1:300 | Abcam, ab23345 |
| Nkx2.1 (immunostaining) | 1:300 | Thermo, MA5-13961 |
| S100β (immunostaining) | 1:300 | Abcam, ab52642 |
| Nanog (immunostaining) | 1:300 | Cell Signaling, 4903S |
| TRA-1-60 (immunostaining) | 1:300 | Abcam, ab16288 |
| Nestin (immunostaining) | 1:300 | Abcam, ab18102 |
| Sox17 (immunostaining) | 1:300 | Abcam, ab84990 |
| Brachyury (immunostaining) | 1:300 | R&D Systems, AF2085 |
| ApoE (western blotting) | 1:1000 | Millipore, AB947 |
| Actin (western blotting) | 1:5000 | Sigma, A2228 |
| Secondary antibodies | ||
| Alexa Fluor donkey anti-rabbit 488, 594 | 1:500 | Invitrogen: A32790, A32754 |
| Alexa Fluor donkey anti-mouse 488, 594 | 1:500 | Invitrogen: A32766, A32744 |
| Alexa Fluor donkey anti-rat 488, 594 | 1:500 | Invitrogen: A21208, A21209 |
| Alexa Fluor donkey anti-goat 488 | 1:500 | Invitrogen: A-11055 |
| IRDye® 800CW Donkey anti-Goat IgG Secondary Antibody | 1:5000 | LI-COR: 926-32214 |
| IRDye® 680RD Donkey anti-Mouse IgG Secondary Antibody | 1:5000 | LI-COR: 926-68072 |
Evaluation of APOE deletion efficiency in human iPSC-derived cerebral organoids: RT-qPCR
-
58.
Harvest cerebral organoids after 12 weeks of differentiation. Transfer organoids to 1.5 mL Eppendorf tube using 1 mL cut pipette tips. Remove excessive medium. Wash cerebral organoids with 1 mL DPBS for 3 times.
Note: Due to the heterogeneity of cerebral organoid, it is recommended to pool multiple cerebral organoids as one sample to reduce the variation. Here we pool 3 cerebral organoids as one sample.
-
59.
Aspirate DPBS. Add 0.5 mL of Trizol and mash with a pestle at room temperature (20°C–25°C).
Caution: Trizol will burn if spilt on skin/eyes. Conduct all experiments related with Trizol in the fume hood.
-
60.
Add another 0.5 mL of Trizol to obtain a final volume of 1 mL and gently pipette the mixture up and down until tissue is well lysed. Leave the tube at room temperature (20°C–25°C) for 2–3 min.
-
61.
Add 200 μL of Chloroform and shake each of the tubes 15 times. Leave the tubes undisturbed for 2–3 min. This causes the solution in the tube to separate into 2 distinct layers.
-
62.
Spin the tubes at 4°C for 10 min at maximum speed. Carefully remove the aqueous layer on the top into a 1.5 mL RNase-free Eppendorf tube containing 500 μL of 100% isopropanol.
Note: Do not disturb the layers! This method extracts large amount of RNA from the tissue and hence “less is better”. 300–400 μL of the aqueous layer should suffice.
-
63.
After transferring the aqueous layer into the Eppendorf tube, shake well. Spin at maximum speed for 10 min to obtain a pellet.
-
64.
Discard the supernatant and wash with 1 mL of 75% EtOH (using DEPC water to dilute the 100% Absolute EtOH to 75%). Flick tube to wash the pellet RNA.
-
65.
Spin the tubes at 4°C at maximum speed for 10 min to pellet RNA. Carefully pour off the EtOH. At this point it is important to change your gloves. Clean new gloves with EtOH and RNase zap. Leave tube upside down so 75% EtOH drains out, allowing the pellet to dry.
-
66.
Resuspend the pellet in 100 μL RNase free H2O.
-
67.
Treat RNA with DNase following the instructions of RNase-Free DNase Set and RNeasy Mini Kit from QIAGEN.
-
68.
Prepare cDNA with the iScript cDNA synthesis kit.
-
69.
Conduct Real-time (RT)-qPCR with Universal SYBR Green Supermix using an iCycler thermocycler.
Primers used to amplify target genes by RT-qPCR:
ACTB F (5’-CTGGCACCACACCTTCTACAATG-3’)
R (5’-AATGTCACGCACGATTTCCCGC-3’)
APOE F (5’-CGTTGCTGGTCACATTCCT-3’)
R (5’-CTCAGTTCCTGGGTGACCTG-3’).
-
70.
The 2exp (−ΔΔCt) method was used to determine the relative expression of each gene with ACTB gene coding β-actin as a reference.
Evaluation of APOE deletion efficiency in human iPSC-derived cerebral organoids: Western blotting
-
71.
Harvest cerebral organoids after 12 weeks of differentiation. Transfer organoids to 1.5 mL Eppendorf tube using 1 mL cut pipette tip. Remove excessive medium. Wash organoids with 1 mL DPBS 3 times.
Note: Due to the heterogeneity of cerebral organoids, it is recommended to pool multiple cerebral organoids as one sample to reduce the variation. Here we pooled 3 cerebral organoids as one sample.
Optional: Excessive Matrigel might influence sample quality for western blotting, which can be removed under microscope using forceps, especially for the samples collected at early stage of differentiation.
-
72.
Lyse organoids with RIPA Lysis and Extraction Buffer supplemented with Protease and Phosphatase Inhibitor Cocktails for Cell Lysis.
-
73.
Sonicate the lysate at 20 kHz (15 cycles, 1 s sonication/1 s rest for each cycle). Keep the sample on ice during the sonication. Then incubate for 30 min on ice.
-
74.
Samples were centrifuged in an ultracentrifuge at 45,000 × g, for 1 h at 4°C.
-
75.
Collect supernatants. Determine protein concentration in the supernatant using a Pierce BCA Protein Assay Kit.
-
76.
Mix samples with Western blotting loading buffer. Heat inactive samples at 95°C for 10 min.
-
77.
Load samples into a 4%–20% sodium dodecyl sulfate-polyacrylamide gel (Bio-Rad), and transferred to PVDF Immobilon FL membranes (Millipore).
-
78.
Block with 5% non-fat milk in PBS for 30 min, Blot membrane for 16 to 24 h with primary antibodies for human apoE and actin in 5% non-fat milk/PBS containing 0.01% Tween-20, and then probed with LI-COR IRDye secondary antibodies for 1 h at room temperature (20°C–25°C). Refer to Table 3 for recommended secondary antibody concentrations.
-
79.
Scan membrane using Odyssey scanner.
Expected outcomes
After the isolation of single iPSC clone and subsequent expansion, multiple clones are selected with each clone transferred to one well of 24-well plates. Some iPSC clones may not attach and survive after transfer to the new wells. Some clones may show aberrant growth kinetics or overt spontaneous differentiation. In those cases, abnormal clones are discarded, and only the remaining healthy clones are kept for further expansion and DNA extraction to evaluate the APOE deletion efficiency. PCR products of wild-type clones give a 304 bp band. PCR products of clones with homozygous deletion have one band at 204 bp, while heterozygous-deletion clones show both bands (Figure 3). Purified PCR products of 3 homozygous-deletion clones are submitted for Sanger sequencing to confirm the APOE deletion (Figure 4). Finally, we select APOE-deficient iPSC clones for further validation in the cerebral organoid models.
High quality and non-differentiated human iPSCs are used for cerebral organoid differentiation (Figures 5 and 6). After the single cell suspension is cultured for 24 h in EB formation medium, a single central EB with smooth surface should form and remain healthy in EB formation medium for additional 5 days. When cultured in Neural induction medium and Expansion medium, embedded organoids develop expanded neuroepithelia as evidenced by budding of the EB surface (Figure 7). Marker staining at different stages shows the diminishment of pluripotency markers (Nanog/TRA-1-60) and the emergence of neural lineage markers (Nestin/Sox2) (Figure 7) along with the differentiation. Organoids remain intact and continue to grow during further differentiation and maturation. The organoid size typically ceases to increase after 8 weeks of culturing (~ 3–4 mm in diameter, Figure 8). On Week 4, cerebral organoids show a predominantly dorsal forebrain region specification with Sox2/Pax6-positive neural progenitors in a ventricular/subventricular-like zone (VZ/SVZ) and beta-tubulin III (Tuj1)-positive neuroblasts in an outer layer (Figures 9A and 9B). Ctip2-positive neurons (deep cortical layer marker)and Satb2-positive neurons (superficial cortical layer marker) can be detected at Weeks 4 and 12 respectively (Figures 9C and 9D). Cerebral organoids also contain Tbr2-positive intermediate progenitors and Nkx2.1-positive cells (ventral region markers) at Week 4 (Figure 9E). These observations reveal the sequential emergence of different neuronal layers along with the differentiation, which is consistent with the previous publication (Lancaster et al., 2013). Since apoE is mainly produced by astrocytes in the brain, we assess the presence of astrocytes at different time points by immunostaining for S100β and glial fibrillary acidic protein (GFAP). At Week 4, only a small cluster of S100β/GFAP-positive astrocytes can be detected, which show an immature morphology with short processes. At Week 12 of differentiation, S100β/GFAP-positive astrocytes increased in number and migrated within the neuronal layers, displaying typical mature astrocyte morphology with long processes (Figure 10A). APOE-/- cerebral organoids show similar differentiation pattern and marker expression to parental cerebral organoids (Figure 10B). No significant size difference is observed between parental and APOE-/- organoids (Figure 10C). After 12 weeks of differentiation, cerebral organoids from both parental iPSC line and the APOE-/- isogenic line are collected to evaluate the expression of apoE, which is undetectable at both the mRNA and protein levels by RT-qPCR and Western blotting, respectively (Figures 10D and 10E).
Figure 9.

Characterization of cerebral organoids at different time points
(A and B) Representative images of the ventricular zone (VZ)-like structure by Tuj1, Sox2 and Pax6 staining at Week 4 of differentiation.
(C and D) Representative images of Ctip2-positive (deep cortical layer marker) and Satb2-positive (superficial cortical layer marker) neurons at Week 4 and 12, respectively. This figure is adapted from Figure 1 in Zhao et al. (2020) (Zhao et al., 2020).
(E) Representative images of Tbr2-positive (intermediate progenitor marker) and Nkx2.1-positive (ventral region marker) cells. Figure reprinted with permission from Zhao et al. 2020.
Figure 10.

Validation of APOE depletion in cerebral organoids
(A) Representative figures of astrocytes differentiation within cerebral organoids; the differentiation pattern of astrocytes in organoids were monitored by GFAP and S100β immunostaining (astrocytic markers) at Week 4 and 12, respectively. This figure is adapted from Figure 1 in Zhao et al. (2020) (Zhao et al., 2020).
(B) Representative images of markers staining within parental and APOE-/- cerebral organoids.
(C) Size evaluation of parental and APOE-/- cerebral organoids at Week 12. All data are expressed as mean ± SEM (n=6).
(D and E) Detection of apoE expression levels in cerebral organoids via both RT-qPCR (D) and Western blotting (E). All data are expressed as mean ± SEM (n=3). Student’s t test was performed to determine the significance. n.s., not significant, ∗p<0.05, ∗∗∗p<0.001. Figure reprinted with permission from Zhao et al. 2020.
Limitations
Cerebral organoid differentiation efficiency can be cell line dependent. Optimization may be needed for each specific line. Quality control tests should be carefully executed throughout the entire differentiation procedure. Due to the lack of vascular systems maintaining proper supplies of nutrition and gas exchange as well as immune cells to eliminate cell debris and toxic molecules, the core of cerebral organoids tend to show necrosis-like changes after long term differentiation, which may affect the final readouts. The lack of vascular system and microglia also limit the application of cerebral organoid models in related research fields. To address this caveat, emerging technology has further developed cerebral organoid models in which vascular cells (Cakir et al., 2019; Mansour et al., 2018; Robert et al., 2020; Zhang et al., 2021) or microglia are incorporated (Abreu et al., 2018; Lin et al., 2018; Ormel et al., 2018) .
Troubleshooting
Problem 1
Low efficiency of plasmid transfection efficiency
Potential solutions
The quality of human iPSCs should be carefully evaluated before electroporation. Human iPSCs should be maintained according to the standard culturing protocol.
Avoid any form of microbial contamination throughout the procedure.
Ensure all the reagents used are in the correct concentration.
Double check all gRNAs are designed correctly. gRNA pairs can be tested in other cell lines which are easier to handle (e.g., HEK293 T).
Problem 2
Differentiation of single cell clones after gene editing
Potential solutions
Ensure human iPSCs are undifferentiated and 100% positive for pluripotency markers.
Optimize electroporation parameters for specific iPSC lines.
Adjust the incubation time of iPSCs in ROCK inhibitor when necessary.
When the differentiation rate is higher than 30%, it is recommended to do further selection following the regular iPSC handling procedure
Problem 3
Failure in EB formation
Potential solutions
Avoid any form of microbial contamination throughout the procedure.
Ensure human iPSC clones amplified after gene editing are undifferentiated and 100% positive for pluripotency markers. Use human iPSCs with normal proliferation rate (doubling time<24 h).
ROCK inhibitor is needed for the first 5 days of differentiation. Make sure the correct concentration of Y-27632 is used.
Problem 4
Failure in EB budding
Potential solutions
Double check the quality of the human iPSCs used for differentiation.
Make sure there is a single embryoid body formed 24 h after plating. If not, it will likely impact downstream organoid development.
Increase starting cell concentration and adjust the culturing timeframe in different media for each specific line when necessary.
Problem 5
Failure in long-term differentiation of cerebral organoids
Potential solutions
Avoid any contamination in the culture system (e.g., bacteria, fungus, mycoplasma, etc.).
Ensure the medium is prepared correctly, and all the chemicals and cytokines supplemented are freshly made and added in the right concentration.
For cell lines that display low proliferation rate and poor organoid development, supplementation of other growth factors such as IGF and NT3 may be helpful.
Screen human iPSC lines with possible disease associated gene panels. Double-check whether the line carries any mutations that may influence cell viability and neural development.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Guojun Bu (Bu.Guojun@mayo.edu).
Materials availability
MC0192 and its APOE-/- isogenic iPSC line generated in this study will be made available upon request.
Data and code availability
This protocol does not generate or analyze any datasets or codes.
Acknowledgments
This work was supported by NIH grants R37AG027924, RF1AG046205, RF1AG057181, R01AG066395, P01NS074969, and P30AG062677 (to G.B.), a Cure Alzheimer’s Fund grant (to G.B.), and an Alzheimer’s Association Research Fellowship 2018-AARF-592302 (to J.Z.). This work was also partially supported by the Mayo Clinic Center for Regenerative Medicine.
Author contributions
J.Z. conceptualized the project and system, developed the methodology, performed the formal analysis, and wrote the protocol. G.B. conceptualized the project and system, acquired the resources and funding, and supervised the project. Y.M., C.-C.L., and T.K. helped to design the project and wrote the protocol. Y.A.M., R.T., G.L., X.Z., and S.X. helped to generated APOE-/- iPSC lines and wrote the protocol. W.L. helped J.Z. to execute the experiments using cerebral organoids. All authors reviewed the final draft of the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Guojun Bu, Email: bu.guojun@mayo.edu.
Jing Zhao, Email: zhao.jing@mayo.edu.
References
- Abreu C.M., Gama L., Krasemann S., Chesnut M., Odwin-Dacosta S., Hogberg H.T., Hartung T., Pamies D. Microglia increase inflammatory responses in iPSC-derived human brainspheres. Front. Microbiol. 2018;9:2766. doi: 10.3389/fmicb.2018.02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cakir B., Xiang Y., Tanaka Y., Kural M.H., Parent M., Kang Y.J., Chapeton K., Patterson B., Yuan Y., He C.S. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods. 2019;16:1169–1175. doi: 10.1038/s41592-019-0586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J., Konermann S., Gootenberg J.S., Abudayyeh O.O., Platt R.J., Brigham M.D., Sanjana N.E., Zhang F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.A., Knoblich J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster M.A., Renner M., Martin C.A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.T., Seo J., Gao F., Feldman H.M., Wen H.L., Penney J., Cam H.P., Gjoneska E., Raja W.K., Cheng J. APOE4 causes widespread molecular and cellular alterations associated with alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98:1141–1154 e1147. doi: 10.1016/j.neuron.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.C., Liu C.C., Kanekiyo T., Xu H., Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A.A., Goncalves J.T., Bloyd C.W., Li H., Fernandes S., Quang D., Johnston S., Parylak S.L., Jin X., Gage F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018;36:432–441. doi: 10.1038/nbt.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K., Feldman H.M., Lu T., Drake D., Lim E.T., Ling K.H., Bishop N.A., Pan Y., Seo J., Lin Y.T. REST and neural gene network dysregulation in iPSC models of alzheimer's disease. Cell Rep. 2019;26:1112–1127 e1119. doi: 10.1016/j.celrep.2019.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormel P.R., Vieira de Sa R., van Bodegraven E.J., Karst H., Harschnitz O., Sneeboer M.A.M., Johansen L.E., van Dijk R.E., Scheefhals N., Berdenis van Berlekom A. Microglia innately develop within cerebral organoids. Nat. Commun. 2018;9:4167. doi: 10.1038/s41467-018-06684-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M., Lancaster M.A., Bian S., Choi H., Ku T., Peer A., Chung K., Knoblich J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017;36:1316–1329. doi: 10.15252/embj.201694700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert J., Weilinger N.L., Cao L.P., Cataldi S., Button E.B., Stukas S., Martin E.M., Seibler P., Gilmour M., Caffrey T.M. An in vitro bioengineered model of the human arterial neurovascular unit to study neurodegenerative diseases. Mol. Neurodegener. 2020;15:70. doi: 10.1186/s13024-020-00418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki Y., Zhao N., Caulfield T.R., Liu C.C., Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 2019;15:501–518. doi: 10.1038/s41582-019-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J.T., Tan L., Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu. Rev. Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- Zhang S., Wan Z., Kamm R.D. Vascularized organoids on a chip: strategies for engineering organoids with functional vasculature. Lab Chip. 2021;21:473–488. doi: 10.1039/d0lc01186j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Davis M.D., Martens Y.A., Shinohara M., Graff-Radford N.R., Younkin S.G., Wszolek Z.K., Kanekiyo T., Bu G. APOE epsilon4/epsilon4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet. 2017;26:2690–2700. doi: 10.1093/hmg/ddx155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Fu Y., Yamazaki Y., Ren Y., Davis M.D., Liu C.C., Lu W., Wang X., Chen K., Cherukuri Y. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer's disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020;11:5540. doi: 10.1038/s41467-020-19264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This protocol does not generate or analyze any datasets or codes.