

SUMMARY
The e4 allele of apolipoprotein E (APOE4) is a genetic risk factor for many diseases including late onset Alzheimer’s Disease (AD). We investigated the cellular consequences of APOE4 in human iPSC-derived astrocytes. We observed an endocytic defect in APOE4 astrocytes compared to their isogenic APOE3 counterparts. Given the evolutionarily conserved nature of endocytosis, we built a yeast model to identify genetic modifiers of the endocytic defect associated with APOE4. In yeast, only expression of APOE4 exhibits dose-dependent defects in both endocytosis and growth. We discovered that increasing expression of the early endocytic adaptor protein, Yap1802p, homolog of the human AD risk factor PICALM, rescued the APOE4-induced endocytic defect. In iPSC-derived human astrocytes increasing expression of PICALM similarly reverses endocytic disruptions. Our work identifies a functional interaction between two AD genetic risk factors—APOE4 and PICALM—centered on the conserved biological process of endocytosis.
INTRODUCTION
Defective intracellular trafficking is a hallmark of neurodegenerative diseases including Alzheimer’s disease (AD) (Neefjes and van der Kant, 2014; Nixon, 2005). Multiple genes identified in genome-wide association studies for late-onset Alzheimer’s disease implicate endocytosis as a key risk-related process (Lambert et al., 2009; 2013; Naj et al., 2011). Endocytic defects are evident in preclinical stages of sporadic AD, particularly in APOE4 carriers (Cataldo et al., 2010). Impaired endocytosis in AD is most commonly associated with dysregulated production or poor clearance of amyloid-beta (Koo and Squazzo, 1994; Lah and Levey, 2000; Thinakaran and Koo, 2008; Vassar et al., 1999). However endocytic defects appear prior to significant amyloid deposition (Cataldo et al., 2010), suggesting that endocytic processes represent a critical node and possible target for therapeutic intervention.
The ε4 allele of the APOE gene (APOE4) encoding apolipoprotein E is a major genetic risk factor for late onset Alzheimer’s disease (AD) (Lambert et al., 2009; Strittmatter et al., 1993). The human APOE gene exists in three common alleles that differ from each other at just two amino acid positions (112 and 158). These alleles are present in the population at the following frequencies: 8.4% (APOE2, Cys112/Cys158), 77.9% (APOE3, Cys112/Arg168), and 13.7% (APOE4, Arg112/Arg158). Individuals carrying one APOE4 allele are 3–4 times more likely to develop AD than those without (Corder et al., 1993; Liu et al., 2013).
Previous studies on the role of APOE4 in AD risk implicate both amyloid-β-dependent and -independent pathways (Cataldo et al., 2010; Liu et al., 2013; Mahley and Rall, 2000; Yu et al., 2014). APOE4 has been reported to interact with the microtubule-associated protein tau (Shi et al., 2017), a key player in many neurodegenerative diseases like AD and Parkinson’s disease (Simón-Sánchez et al., 2009). APOE4 also increases risk for Dementia with Lewy Bodies (Tsuang et al., 2013). In addition, APOE4 impairs recovery from traumatic brain injury (Friedman et al., 1999; Houlden, 2006), and has been implicated in blood-brain-barrier dysfunction (Blanchard et al., 2020; Zlokovic, 2013).
Although the strong genetic association between this allele and disease risk has been recognized for over 20 years, the question of how APOE4 increases disease risk remains unresolved (Yamazaki et al., 2019). APOE4’s profound effect on AD risk and the early appearance of disrupted endocytosis in the pathogenesis of AD motivated us to (1) characterize the impact of APOE4 expression on disrupted endocytosis, and (2) identify genetic means to reverse these disruptions.
RESULTS
APOE4 disrupts endocytosis in iPSC-derived astrocytes
Induced pluripotent stem cells (iPSCs)-derived cells have been used to recapitulate many molecular aspects of various human diseases (Devine et al., 2011; Park et al., 2008; The HD iPSC Consortium, 2012) including AD-relevant consequences of APOE4 (Lin et al., 2018). We used two independent pairs of cells which are isogenic at all loci except for APOE where they are either APOE3 or APOE4. The pairs were generated using one APOE3 homozygous parental and one APOE4 homozygous parental line (Figure S1A). All iPSC lines used in this study exhibited a normal karyotype (Figure S1B). Our previous work has shown that APOE expression in APOE4 astrocytes is lower than in isogenic APOE3 astrocytes (Lin et al., 2018). We have also previously reported abnormal EEA1 staining in iPSC-derived APOE4 neurons (Lin et al., 2018), suggesting APOE4 could impact endocytic processes.
We chose to focus our study on iPSC-derived astrocytes (Chen et al., 2014) (Figure 1A), as astrocytes are the predominant source of APOE in the human brain (Liu et al., 2013). We confirmed that our astrocytes were positive for the astrocytic markers S100β and GFAP (Figure 1B) and do not express markers of other potentially contaminating cell types (Figure S1C). We systematically interrogated multiple stages of the endocytic pathway to identify whether APOE4 astrocytes displayed endocytic perturbations when compared to their isogenic APOE3 counterparts (Figure 1C). First, we performed immunofluorescence staining of Rab proteins and Rab effector proteins that specifically mark early, late, and recycling endosomes. We visualized the number of early endosomes directly using immunofluorescent probes against the early endosome-associated proteins, Rab5 and EEA1. We observed that the number of both Rab5 positive and EEA1 positive puncta were decreased in APOE4 compared to APOE3 astrocytes (Figure 1D–F). We used a similar approach to examine late endosomes (Rab7-positive) and recycling endosomes (Rab11-positive). In contrast to our finding in early endocytic effectors, we noted no difference in Rab11 (recycling endosome) levels between cells of the APOE3 and APOE4 genotype (Figure S2A,B). APOE4 astrocytes also exhibited a trend towards lower late endosomal and lysosomal number when compared to APOE3 astrocytes in both isogenic pairs (Figures S2C–F). These findings were not a result of differential expression of these endosomal markers between APOE3 and APOE4 astrocytes (Figure S2G). However, EEA1 levels detected on western blot were considerably lower in APOE4 astrocytes than in their APOE3 counterparts (Figure S2H), suggesting that changes in levels of endocytic proteins are result of altered recruitment and turnover of these proteins. These results reveal that early endocytosis is the most impacted when comparing APOE4 and APOE3 astrocytes.
Figure 1. APOE4-expressing iPSC-derived astrocytes exhibit a defect in early endocytosis.

(A) Differentiation of iPSCs to astrocytes. (B) Astrocytes stained for S100b and GFAP. Scale bar 100 μm. (C) A schematic diagram of endocytosis steps probed in human APOE astrocytes. (D) Levels of Rab5 (left) and EEA1 (right) staining an isogenic pair of APOE astrocytes. Mean ± SD shown of 80–100 cells per sample for n=3 samples. Representative images of (E) Rab5 staining and (F) EEA1 staining in APOE astrocytes. (G) Quantification of microscopy images of the number of EGF puncta per cell (left) or the internalization of transferrin per cell (right) in one isogenic pair of APOE astrocytes (APOE3 parental line). Mean ± SD shown of 30–50 cells per sample for n=12 samples. Representative images displaying (H) EGF endocytosis and (I) transferrin-647 endocytosis in human APOE astrocytes. Scale bar is 50 μm. See also Figures S1 and S2.
To determine whether the differences observed in the accumulation of early endocytic vesicles resulted in functional defects in early endocytosis, we used fluorescently-tagged ligands (epidermal growth (EGF) factor and transferrin (Tf)) to examine the early stages of clathrin-mediated internalization (Harding et al., 1983). By exposing cells to a 5-minute pulse of fluorescent EGF, we directly visualized the EGF taken up by the early endocytic machinery (Figure 1C,G,H). APOE4 astrocytes trend towards internalizing less EGF (Figure 1G,H). Using fluorescently-tagged Tf, we again observed that APOE4 astrocytes internalized less Tf than APOE3 astrocytes. Together these data suggest that APOE4 astrocytes display lower rates of early endocytosis than APOE3 astrocytes (Figure 1G,I). Previously generated transcriptomic data on these cells confirmed that effects were not due to decreased expression of the EGF or Tf receptors in the APOE4 astrocytes when compared to their APOE3 counterparts (Figure S2G) (Lin et al., 2018). These results were confirmed in astrocytes derived from an independent iPSC line (Figure S1A) (Lin et al., 2018), where we again observed that APOE4 astrocytes exhibit a similar genotype-dependent decrease in early clathrin-mediated endocytosis (Figure S2I) as well as a trend towards decrease in late endosome number (Figure S2C). Taken together, our results show that APOE4 disrupts early endocytosis in human astrocytes.
In order to assess whether the perturbations to early endocytosis were due to a toxic gain-of-function or loss-of-function, we utilized an APOE knockout line generated from the parental APOE3 line used in our study. This knockout line resulted from an early stop codon and produced no detectable APOE mRNA or protein (Blanchard et al., 2020; Lin et al., 2018). In stark contrast to our APOE4 lines, APOE knockout-derived astrocytes did not exhibit any early endocytic perturbations (Figure S2J–M). Therefore, we conclude that the endocytic dysfunction results from a toxic gain-of-function of the APOE4 isoform.
A yeast APOE model displays endocytic abnormalities in processes disrupted in human astrocytes
To explore the consequences of APOE4 on endocytosis in greater detail, we constructed a model of APOE biology in baker’s yeast, taking advantage of the evolutionary conservation of mediators and modulators of endomembrane trafficking within eukaryotes (Boettner et al., 2012; Goode et al., 2015; Kachroo et al., 2015). Our yeast model contained stably integrated constructs at known genomic loci under the control of an estradiol-inducible promoter (Aranda-Díaz et al., 2016; McIsaac et al., 2012) expressing human APOE isoforms (Figure 2A, S3A,B). We engineered the construct to include a secretory pathway signal sequence to ensure the APOE4 protein would experience an environment in yeast that is similar to what it encounters in human cells. APOE isoforms tagged with GFP within our yeast model display ER localization with some puncta in the case of APOE4 yeast (Figure S3D). Using semi-native gel electrophoresis analysis, we confirmed that, in contrast another yeast model expressing the toxic Aβ42 peptide using the same promoter and signal sequence, APOE4 does not accumulate in protein aggregates (Figure S3E). This finding suggests that the cellular phenotypes we observed are not a response to aberrant protein aggregation and therefore reflect biology relevant to APOE4.
Figure 2. A yeast model faithfully recapitulates APOE4-associated endocytic defects.
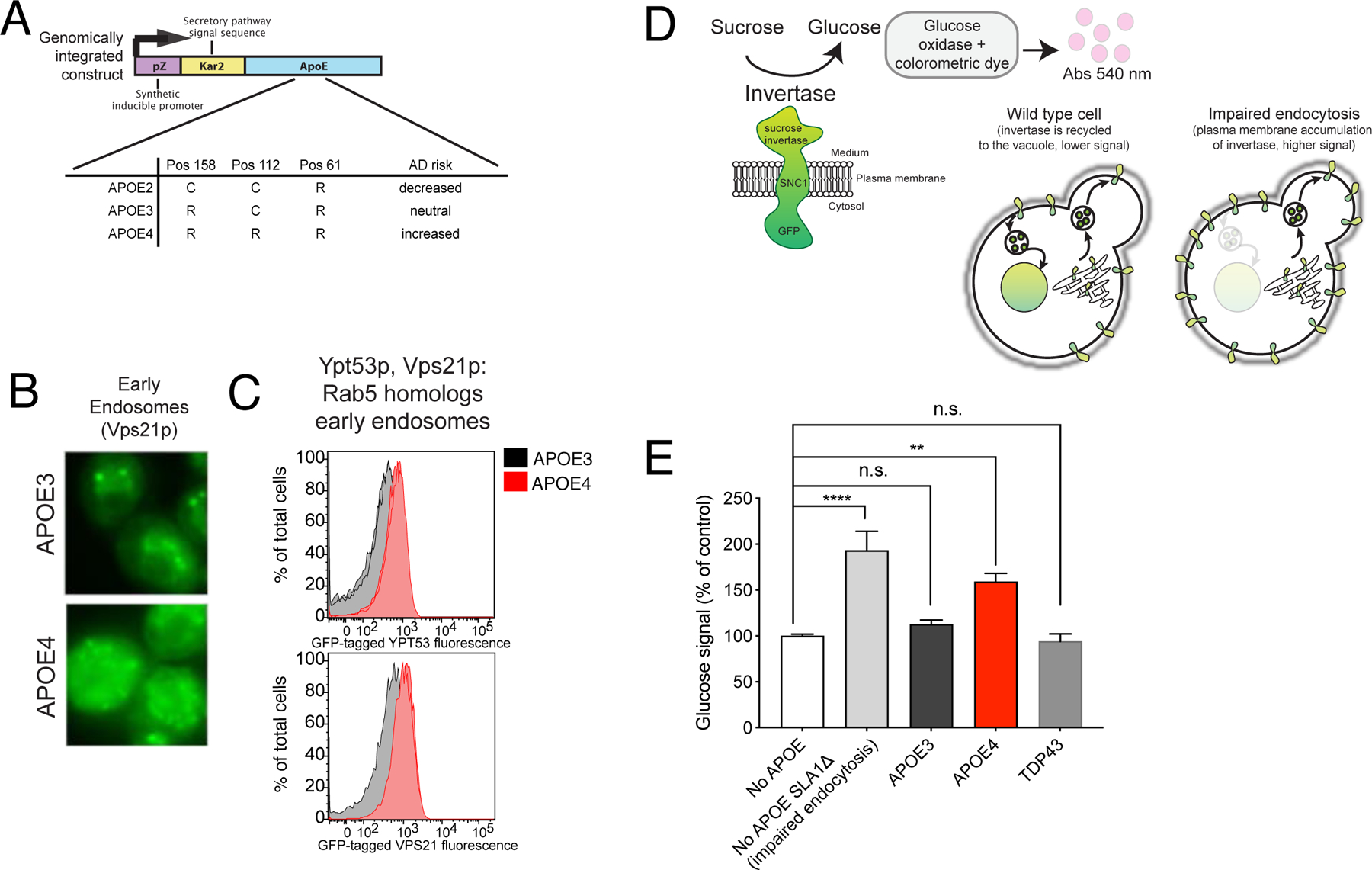
(A) The estradiol-inducible promoter system used in the APOE yeast model. (B) Fluorescence microscopy of the mNeonGreen-tagged Vps21p. (C) Mean fluorescent signal from mNeonGreen-tagged Vps21p and Ypt53p quantified by flow cytometry, 10,000 cells per sample. (D) A schematic of the GSS invertase assay adapted. (E) Glucose signal from invertase assays of the indicated APOE-expressing strains and a TDP43-expressing strain. One-way ANOVA followed by Tukey’s multiple comparisons test (**** p-value<0.0001). Mean ± SD for n=3 independent colonies. See also Figure S3.
Given our observation in iPSC-derived astrocytes, we asked whether our yeast model showed endocytic defects. Early endosomes were indeed abnormal in our APOE4 yeast model, which displayed disrupted levels and localization of endogenously tagged Rab5 homologs, YPT5 and VPS21 (Figure 2B,C), compared to APOE3 yeast. Using the trafficking of endogenously tagged Ste3p and Mup1p, which serve as sentinels for early endocytic disruption, we again observed defects associated only with expression of APOE4 (Figure S3E–I). We also observed disruptions to late endosomal levels and localization in APOE4 yeast (Figure S3J). Interestingly, while we observed a decrease in Rab5 and EEA1 in human iPSC-derived APOE4 astrocytes (Figure 1D–F), the levels of the yeast homologs were increased in APOE4 yeast (Figure 2C,D). We have previously shown that human iPSC-derived APOE4 neurons present increased EEA1 staining (Lin et al., 2018), and others have demonstrated a range of endocytic defects associated with APOE4 (Nuriel et al., 2017) suggesting that cell type can greatly influence APOE4 defects and our yeast model may reflect aspects of multiple human cell types.
In order to specifically interrogate early endocytosis, we used a quantitative invertase assay (Burston et al., 2009) to assess endocytic efficiency in APOE3- and APOE4-expressing yeast. This assay utilizes a transmembrane chimeric protein reporter (GSS) made of an extracellular sucrose invertase enzyme, the yeast protein Snc1p, and a cytosolic GFP. Accumulation of GSS on the cell surface due to impaired endocytosis leads to increased invertase activity, while an exocytosis or recycling defect results in a reduction in GSS at the cell surface and thus lower invertase activity. Endocytic efficiency is assessed via an enzymatic assay with a colorimetric readout (Burston et al., 2009; Dalton et al., 2015; Darsow et al., 2000) (Figure 2D). Deletion of a key early endocytosis factor, SLA1, resulted in increased cell surface GSS and therefore higher invertase signal (Figure 2E). APOE4-expressing yeast display a clear increase in the invertase signal compared to APOE3 or wild type yeast (Figure 2E). Yeast expressing another neurodegenerative disease-associated protein, TDP-43 (Johnson et al., 2008), displayed no significant reduction in invertase activity, suggesting the defect in endocytosis is a specific to APOE4.
Our yeast APOE4 model thus reflects disruption in endocytic processes, particularly in endocytic uptake, similar to those observed in our human iPSC-derived astrocytes and may therefore be used as a tool to identify genetic and chemical factors that could modify this effect of APOE4. However, validation and further analysis of specific mechanisms and pathways must be done in a human cell type specific manner. We utilized our yeast model to identify potential genetic modifiers of the early endocytosis disruption in APOE4, which we could then investigate in our human iPSC astrocyte system.
YAP1802, the yeast homolog of PICALM, reverses APOE4-induced endocytic disruptions
In our yeast model, a growth defect accompanied APOE4-induced perturbations to endocytosis (Figure 3A,B, Figure S4A,B). Despite being expressed at similar levels, APOE3 did not display a growth defect (Figure 3A,B Figure S3A,B, Figure S4A,B).
Figure 3. APOE4-associated endocytic defects in yeast can be rescued by increased expression of YAP1802.
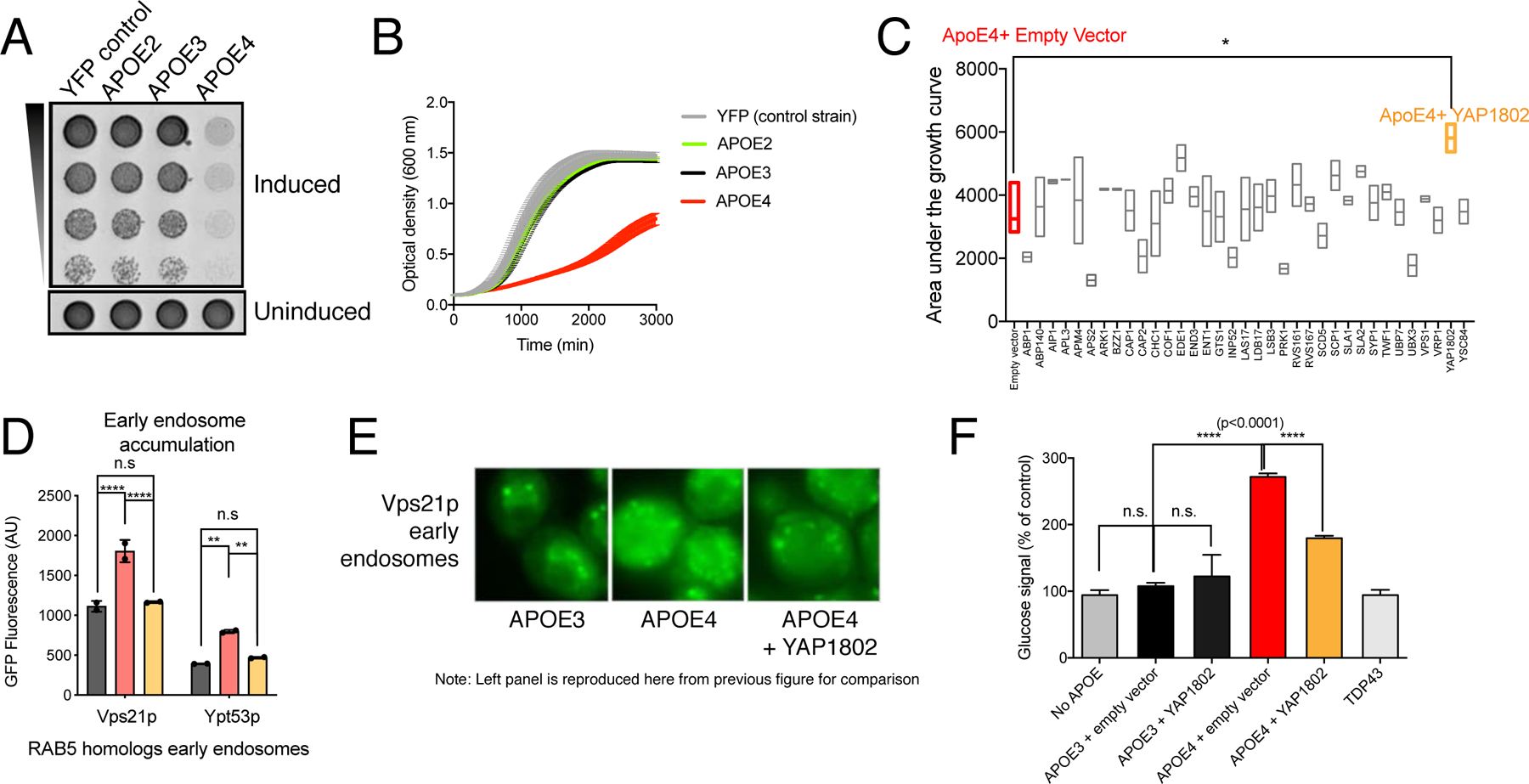
(A) Growth of APOE- yeast strains on (A) solid inducing (75 nM estradiol) or non-inducing media (no estradiol) and (B) liquid inducing (7.5 nM estradiol) or uninduced (no estradiol) media. Error bars are SD of n=4 trials. (C) Area under the growth curves for overexpressed endocytosis-related proteins on an APOE4 background. Ordinary one-way ANOVA followed by Dunnett’s test for multiple comparisons, * p<0.05, n=4 transformants per modifier, min to max with line at median. (D) Mean fluorescent signal of mNeonGreen-tagged Rab5 homologs, quantified by flow cytometry. 10,000 cells per sample, 2 samples per condition shown. Two-way ANOVA followed by Tukey’s multiple comparisons test (**** p<0.0001, **, p<0.01). (E) Fluorescence microscopy of mNeonGreen-tagged Vps21p. (F) Glucose signal from invertase assays of the indicated APOE-expressing strains with and without co-expression of YAP1802 and a TDP43-expressing strain. Value for “no APOE” is reproduced from Figure 2. One-way ANOVA followed by Tukey’s multiple comparisons test (**** p-value<0.0001). Mean ± SD for n=3 independent colonies. See also Figure S4 and Table S1.
We performed a screen for suppressors of the APOE4-induced growth defect and then examined their effects on our endocytic phenotypes. First, we profiled a set of thirty-four proteins involved in all stages of endocytic trafficking (Table S1) (Lu et al., 2016). When overexpressed, only Yap1802p, significantly impacted the APOE4 growth phenotype (Figure 3C, highlighted in yellow). Yap1802p is an early endocytic adaptor protein that participates in the budding off of early endocytic vesicles. We also observed that two other early endocytic proteins, Ede1p and Aps2p, displayed non-significant trends towards modification of the APOE4 yeast growth phenotype. Together, these suggest that disruptions to the endocytic pathway observed in our yeast and iPSC models arise at least in part from defects in early endocytosis, and that, in yeast, this is sufficient to modulate growth.
We asked whether increasing expression of YAP1802 altered levels of APOE4 by immunoblotting or fluorescence of a GFP-tagged APOE construct (Figure S4D,E), We did not observe a change in APOE4 levels, thus increasing YAP1802 expression is able to uncouple the presence of APOE4 from its detrimental effects.
We examined whether overexpression of Yap1802p could modify the dysregulation observed in Rab proteins. Increasing expression of Yap1802p restored the accumulated levels of Rab5 homologs observed in the APOE4-expressing strains to lower, APOE3-like levels (Figure 3D). Increasing Yap1802p expression restored the localization of Vps21p, a Rab5 homolog, to a less diffuse, APOE3-like pattern (Figure 3E).
Finally, we asked whether the growth defect rescue by Yap1802p was accompanied by a rescue of the APOE4-induced defect in early endocytosis. Indeed, overexpression of Yap1802p resulted in significant reduction of the GSS invertase signal, indicating that Yap1802p expression can functionally rescue APOE4-induced trafficking defects (Figure 3F).
PICALM overexpression rescues an APOE4-induced disruption of endocytosis in iPSC-derived astrocytes
Our APOE4 yeast model suggested YAP1802 is a robust genetic modifier of the APOE4-induced endocytic defect. The human homolog of YAP1802 is PICALM, which is itself implicated in AD. We therefore examined whether PICALM could modify APOE4 phenotypes in human astrocytes.
Having confirmed that endogenous expression of PICALM was not significantly different between APOE3 and APOE4 astrocytes (Figure S4F), we treated APOE3 and APOE4 astrocytes with a lentivirus driving the expression of PICALM under an astrocyte-specific GFAP promoter (Figure 4A). Since modifying endocytosis may enhance or reduce rates of internalization, recycling or degradation of proteins (Antonescu et al., 2014), we confirmed that expression of PICALM did not change APOE levels (Figure 4C,D).
Figure 4. Increasing PICALM expression rescues APOE4-induced endocytic defect in iPSC-derived astrocytes.
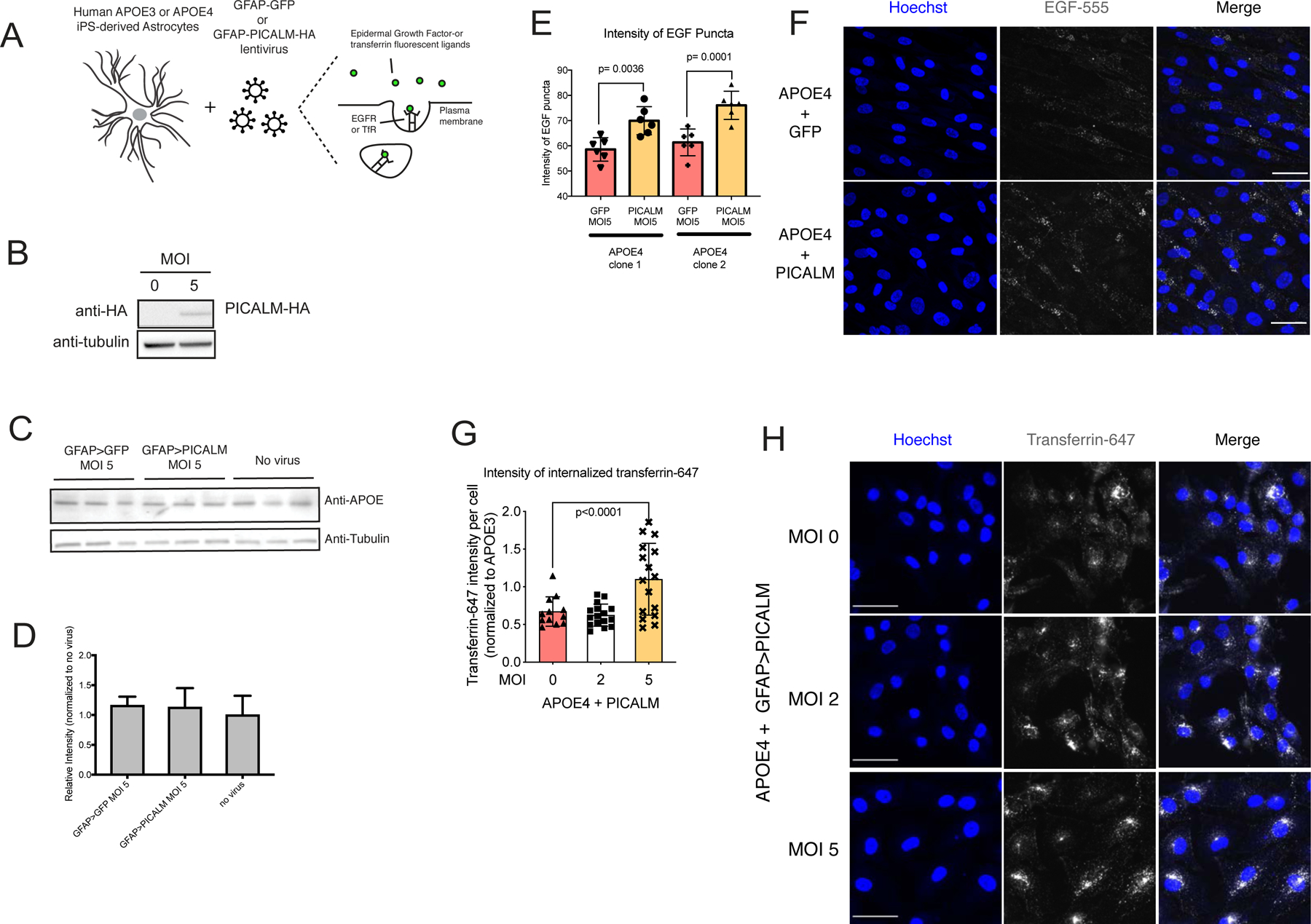
(A) A schematic assays fluorescent epidermal growth factor (EGF) or transferrin (Tf). (B) Western blot of PICALM-HA in iPSC-derived human astrocytes. (C) Western blot and (D) quantification of APOE levels in primary human astrocytes treated with PICALM and GFP viruses (MOI 5). Mean ± SD of n=3. Student’s t-test, **p<0.01, ***p<0.001, n=3 experiments. (E) Quantification of the intensity of EGF puncta per cell and (F) representative images. Mean ± SD of 80–100 cells per sample. Scale bar 50 μm. (G) Quantification of Tf internalization and (H) representative images with and without GFAP-PICALM-HA virus. Tukey’s multiple comparisons test. Mean ± SD of 80–100 cells per sample. See also Figure S4.
To examine the effect of PICALM overexpression on APOE4-induced endocytic disruptions, we again used the fluorescent ligands, EGF and Tf, as probes of the early endocytic process. We found that overexpression of PICALM increased EGF (Figure 4A,E,F) and Tf (Figure 4G,H) uptake suggesting a restoration of early endocytic capacity. Although not significant, we observed a trend towards increased EEA1 positive puncta volume and intensity in APOE4 astrocytes with increased expression of PICALM (Figure S4H,I). We observed that PICALM overexpression induced an endocytic defect in APOE3 astrocytes (Figure S4G). This concurs with previous work that noted overexpression of PICALM in a normal context may impair endocytosis (Tebar et al., 1999). These results suggest that the PICALM-mediated rescue observed for APOE4 is allele specific. Together, these results demonstrate that increasing cellular dose of PICALM uncouples the presence of APOE4 from its detrimental effects on endocytosis.
DISCUSSION
In this study, we used genetic and functional studies in iPSC-derived astrocytes and yeast both to characterize the conserved perturbation to endocytosis associated with APOE4, and to identify PICALM as a modifier of APOE4-associated defects. These findings align well with previous work showing that SNPs that lower expression of PICALM are associated with AD risk, while SNPs that increase PICALM expression decrease AD risk (Morgen et al., 2014; Parikh et al., 2014; Zhao et al., 2015).
Genetic links between APOE4 and PICALM have previously described (Harold et al., 2009; Lambert et al., 2009; Morgen et al., 2014). However, no clear functional connection has been reported between these risk factors. Given the shared action of these two risk factors on endocytosis, targeting of this cellular pathway could serve as a potential therapeutic avenue.
Prior studies examining APOE4’s role in trafficking have largely focused on the trafficking of specific Alzheimer’s disease-associated proteins like APP and its cleavage product, Aβ. In this study, we show that APOE4 can disrupt endocytosis independent of APP or Aβ. This finding suggests that disruptions to endocytosis could be a common defect among the many diseases for which APOE4 increases risk.
In neuronal and glial cell types where endocytic trafficking is critical to cellular function, even a mild disruption to endocytosis may profoundly change intracellular dynamics of many substrates. Chronic perturbation of trafficking pathways over the lifetime of an individual could lead to a disease-vulnerable cellular environment, which could be further exacerbated by other environmental or other genetic. Broad endocytic disruption may manifest as different phenotypic consequences in different cell types or brain regions, which may then lead to disease-specific consequences of APOE4.
This study examined the cellular consequences of the APOE4 allele in comparison to the APOE3 allele, considered the neutral allele with respect to AD risk. The APOE2 allele, which is considered protective with respect to AD risk, may act on similar or different pathways. We anticipate that future studies of APOE2, especially in the context of early endocytic disruptions will further expand our understanding of risk for or resilience to AD.
Our work has set up a paradigm to explore genotype-to-phenotype relationships. With advances in high-throughput sequencing, this type of investigation can be extended to understand particular combinations of polymorphisms present in various patient populations. Our study identified the interplay between two AD risk factors, APOE4 and PICALM. The techniques and systems used in this study can be used to systematically interrogate combinations of risk factors or protective factors to understand their impacts on cell biology. Given the genetic heterogeneity of AD and other late-onset neurodegenerative diseases, such studies will offer opportunities for genetic stratification as well as the identification of targeted therapies for such complex diseases.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Li-Huei Tsai (tsaiasst@mit.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact and may require completion of a Materials Transfer Agreement.
Data and Code availability
No custom code was used in this study.
Experimental model and subject details
iPSC-derived lines were generated from the parent lines Coriell #AG09173 and #AG10788 (both female). Line maintenance and derivation into various cell types was performed as previously described(Lin et al., 2018). After sorting, astrocytes derived from these iPSCs were maintained in 100mm dishes or T75 flasks at 37°C, 5% CO2 in Astrocyte Medium (ScienCell, Cat. #1801) with 2% fetal bovine serum (ScienCell Cat. #0010), 1% astrocyte growth supplement (ScienCell Cat. #1852), and 1% penicillin/streptomycin solution (ScienCell Cat. #0503). These were split (using TrypLE, Gibco) and plated at 1 million cells per T75 flask every 7–9 days. All experiments were performed on early passage numbered cells (<P8).
All yeast models described in this paper were constructed in a W303α background. For validation purposes, assays were performed in different backgrounds (BY4741, W303a) and all results were consistent across the different yeast backgrounds. Unless otherwise noted, yeast were grown at 30°C in minimal synthetic culture media (Sunrise Science Products) and supplemented with 2% glucose. For all experiments involving overexpression of Yap1802p or any other genetic modifiers, the yeast were switched into 2% (w/v) galactose media upon induction. Prior to switching the carbon source to galactose, the cultures were grown in 2% raffinose for a day to metabolically adjust to the non-fermentable carbon source. All cultures were grown at 30°C under shaking conditions in an incubator. Transformations of plasmids into yeast were carried out using a standard lithium acetate protocol (Gietz and Schiestl, 2007). All genetic modifiers tested were from the yeast ORF collection (FLEXgene, Dharmacon).
Inducible expression of APOE isoforms in S. cerevisiae
In order to generate APOE S. cerevisiae models where protein expression could be induced in a variety of carbon sources, we employed a two-plasmid system adapted from previous work (Aranda-Díaz et al., 2016). Independence of induction reagent from carbon source was especially crucial in the study of lipid homeostasis. The inducible expression system consists of a transcriptional regulator (ZEM) construct—a fusion of a the Zif268-DNA binding domain, the human estrogen receptor and the Msn2 activation domain. This ZEM was expressed in a constitutive manner from an S. paradoxus EIFα promoter. When estradiol is added, it binds the ZEM, the complex translocates to the nucleus where it binds to a synthetic promoter (pZ), which is a modified GAL1 promoter that drives the expression of APOE isoforms. To capture the trafficking of APOE through the secretory pathway, the APOE isoforms were expressed with a secretory signal sequence from the protein Kar2p. The ZEM construct was singly integrated at either the TRP or YBR032W locus. The pZ-Kar2pss-APOE construct was singly integrated at either the HIS or LEU loci.
For experiments that involved comparisons with other neurodegenerative disease proteins, similar models were constructed where the 42-amino acid Aβ peptide was expressed under the control of the pZ promoter and with a Kar2p signal sequence. The TDP-43 model referenced was expressed under the same inducible promoter but did not contain a secretory pathway signal sequence.
Yeast growth curves in liquid media and spotting assays
Yeast were grown overnight to saturation, diluted in the morning and grown to logarithmic phase. Cultures were then diluted to an OD600 of 0.05 in inducing media. The OD600 was monitored on a Multiskan GO Microplate Spectrophotometer (Thermo Fischer Scientific) taking readings every 15 minutes, at a temperature of 30°C with 15 seconds of shaking prior to every reading. The targeted screen of endocytic modulators was conducted using the centromeric plasmid yeast ORF library (Dharmacon).
For spotting assays, yeast were grown overnight to saturation, diluted in the morning and grown to logarithmic phase. They were then normalized to a starting OD600 of 0.5, diluted in five-fold serial dilution and spotted on agar plates with inducing and selective media. Spots were observed at 24h, 48h, and 72h after spotting.
Semi-denaturing oligomer blots
Yeast cultures were grown overnight to saturation, then diluted to an OD600 of 0.05 and grown for 8h at 30°C. Then 0.5 OD600 units of these cultures were plated on agar plates with inducing media and placed at 30°C for 15 hours. Induced yeast was then scraped from the plates and lysates were prepared using bead-beating for 10 minutes at 4°C in lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 1% (v/v) TritonX-100, 1 EDTA Free Mini Protease Cocktail Pellet per 5 mL of buffer, 50 mM N-ethyl-maleimide, 1 mM phenylmethylsulfonyl fluoride). Protein concentrations were measured using BCA assay and lysates were then resuspended in NativePAGE sample buffer (Thermo Fischer Scientific) and run on a 4–12% Bis-Tris Gel in MES running buffer at 90V, at 4°C. The gel was then transferred to a PVDF membrane using the iBLOT2 dry blotting system (Thermo Fischer Scientific). Blocking and antibody incubations were performed in 5% nonfat dry milk in PBS and washes were performed with PBS with 1% (v/v) Tween-20. Primary antibody incubations were performed overnight at 4°C. Secondary antibody incubations were performed at room temperature for 1h.
Western blots
Yeast cultures were grown to logarithmic phase and induced with estradiol for 6–12 hours. Cells were then harvested and the protein fraction was precipitated from 1.5 units of OD600 (optical density at 600 nm) culture using trichloroacetic acid. The resulting protein pellets were then resuspended in a urea-based sample buffer (2% SDS, 7M urea, 1 mM TCEP, 200 mM TRIS pH 6.8) and solubilized at 65 °C for 15 minutes. Bis-Tris acrylamide gels were run at 125V and the resulting gels were transferred to nitrocellulose membranes using the iBLOT2 dry blotting system (Thermo Fischer Scientific). Membranes were blocked with 5% dry milk in PBS, and incubated overnight with primary antibodies at 4°C. For APOE blots, the polyclonal goat AB947 antibody (Millipore Sigma) at 1:500 dilution was used. PGK1 was probed using rabbit polyclonal (ABIN568371) (1:5000 dilution) and amyloid-beta was probed using the 6E10 antibody (1:1000 dilution). Quantification of blot images from the Gel Doc imaging system (BioRad) was performed using ImageJ (NIH).
Astrocyte cultures were grown to confluence in a 6-well plate. 2 wells of a confluent 6-well plate were combined and used to create a single lysate. Cells were scraped off the plate in RIPA buffer with HALT protease inhibitors and PMSF. Cold pellets were spun down at 2000xg and supernatants were boiled with sample buffer (1:6) for western blotting. Blotting was carried out using similar protocol as for yeast, however, for astrocyte blots, a Transblot (BioRAD) transfer device was used.
Reporter assays
All fluorescent reporters described (except STE3) were tagged at their endogenous loci. Due to its low endogenous expression level, STE3 was expressed constitutively from the GPD promoter using an episomal plasmid. Tagged Rab constructs were N-terminally labelled with the mNeonGreen fluorophore, whereas STE3 and MUP1 were tagged C-terminally with mKate2 and mCherry, respectively. All yeast cells were imaged or analyzed in logarithmic growth phase. Images were acquired with a Nikon Plan Apo 100x oil objective (NA 1.4). For flow cytometry analysis yeast with fluorescent reporters were grown in inducing media to logarithmic phase and then incubated with propidium iodide (10 ng/mL, Sigma Aldrich) for 5 minutes. 15,000–20,000 cells were measured using the MACSquant VYB flow cytometer (Miltenyi Biotec). Gating and analysis was performed using FlowJo v8 (FlowJo LLC).
Invertase assay
The invertase construct (pHB4, Addgene # 53462) was inserted at the endogenous SUC2 locus (selection with nourseothricin) of all strains used. APOE-expressing and control yeast were inoculated into fructose-containing media (SF-CSM or –URA) (Sunrise Science Products) and grown to saturation. Cultures were then inoculated into galactose media (SG-CSM or –URA) and again grown to saturation to ensure complete metabolic switch and induction of modifier expression. Cultures were then inoculated into SG media at an optical density (OD600 nm) of 0.1 in the morning and grown for 4–6h until in log phase, then diluted for overnight induction of APOE or control genes at OD600 nm of 0.05 in SG-URA with 0nM or 1nM β-estradiol (Sigma-Aldrich E8875) in a 96-well format. Following 24h induction, cultures were briefly centrifuged and concentrated 2 to 20-fold to roughly equilibrate OD600 before invertase assay. The liquid glucose invertase assay to assess endocytic defects was performed according to previously published protocols (Dalton et al., 2015). Glucostat reagent was prepared fresh immediately prior to use (per 96 well plate: 13ml K2HPO4 0.1M pH7.0 (Sigma-Aldrich P3786), 26.7μl glucose oxidase 1000U/ml (Sigma-Aldrich G7141), 33.3μl HRP 1mg/ml (Sigma-Aldrich P6782), 66.7ul NEM 20mM (Sigma-Aldrich E1271), 200μl o-dianisidine 10mg/ml (Sigma-Aldrich F5803)). Following sucrose addition, the glucostat reagent was incubated 7–9 minutes to ensure a final OD540 reading between 0.05–0.3, after which the invertase reaction was stopped with 6N HCl. Sample readings were calibrated based on freshly prepared glucose standard curves run on the same assay plate. Three independent colonies were run per yeast genotype on each plate, and each plate was run in technical duplicate.
iPSC culture, derivation, and lentiviral production
Isogenic iPSC lines were created, maintained, derived and differentiated as described previously(Lin et al., 2018). All iPS cells displayed a normal karyotype. Cells were differentiated into astrocytes according to previously published protocols (Chen et al., 2014; Lin et al., 2018). Following derivation and FACS-based purification, astrocytes were maintained in Astrocyte medium (ScienCell) and passaged every 5–7 days and re-seeded at a density of 106- cells in a T-75 tissue culture flask. All cells were used within 8 passages of the second GLAST sorting.
Lentivirus encoding PICALM-HA under the control of a GFAP promoter was produced according to standard protocols using lipid-mediated transient transfection of 293T cells, and standard packaging plasmids (pMD2.G, and psPAX, Addgene 12259, 12260). Following supernatant collection, the virus was concentrated using LentiX-Concentrator (Clontech, 631231) according to manufacturer’s protocol. Viral titers were determined were determined using QuickTiter Lentivirus titer kit (Cell Biolabs) according to manufacturer’s protocol.
Immunostaining
All staining of astrocytes for markers of endocytosis were performed in the same manner. Cells were plated in 96-well glass bottom plates (IBIDI) at a density of 10,000 cells per well. First cells were fixed in 4% paraformaldehyde for 15 minutes at room temperature and then permeabilized using 0.2% TritonX-100 for 10 minutes at room temperature, blocked for 1 hour at room temperature using 1% normal goat serum, 1% normal donkey serum in 5% PBS. Primary antibodies were diluted according to supplier’s suggestions in blocking solution and incubated overnight at 4°C. The next day, cells were washed and incubated with secondary antibodies at 1:1000 dilution for an hour at room temperature and imaged using a Zeiss LSM 710 inverted microscope. Antibodies used were as follows: EEA1 (mouse BD biosciences 610457, 1:100), Rab5 (rabbit, CST, 1:400), Rab7 (rabbit, CST, 1:100), Rab11 (rabbit, CST, 1:50). Imaging and quantification criteria are described below.
Transferrin uptake, EGF uptake, and Lysotracker staining
APOE astrocytes were seeded at a density of 10,000 cells per well of a 96-well glass-bottom plate (IBIDI). Then cells were infected with lentiviruses encoding GFAP-PICALM-HA for 5 days. The cells were then placed on ice for 5 minutes to halt endocytic processes, and either EGF conjugated to AlexaFluor555 (Thermo Fischer Scientific E35350) or transferrin conjugated to AlexaFluor647 (Thermo Fischer Scientific T23366) was added to the media at a concentration of 100 ng/mL or 25 ug/mL and the cells were incubated at 37°C for 5 minutes. Cells were then washed three times with cold PBS and fixed with 4% paraformaldehyde for 20 minutes at room temperature. The cells were then washed with PBS three times and Hoechst 33342 was added at a final concentration of 2 μM to each well and imaged using a Ziess LSM 880 Laser Scanning Confocal Microscope taking 1 um sections of 10–15 um Z-stacks and analyzed using Imaris (Bitplane) and Fiji.
Lysotracker staining was performed according to manufacturer’s instructions. Lysotracker Red (Invitrogen) was used at a concentration of 50 nM with an incubation at 37°C for 30 minutes. Cells were then fixed and imaged as described above..
Quantification and statistical analysis
Data were processed using Microsoft Excel and statistical analysis was performed using GraphPad Prism. Image data were quantified using Imaris (Bitplane) and ImageJ (NIH). For each image quantification, the parameters analyzed (puncta number per cell, puncta intensity, volume, or overall staining intensity per cell) are specified in the graph y-axis. For measurements of puncta number, size, or intensity, the Imaris “spots” tool was used. For total intensity of staining, image J integrated density value was used. For “per cell” measurements, the data for the entire image were acquired and divided by the number of nuclei present in the image. Partial nuclei or cells were not counted as part of the image. All representative images are picked as the image with a quantification value closest to the mean of the distribution depicted in the graph. Details of statistical analysis for each experiment can be found in the figure legend (including test details, n, definition of significance, average, and precision metrics). Any specialized analysis software can be found in the Methods section.
Supplementary Material
ACKNOWLEDGEMENTS
We dedicate this work to the life and memory of our inspirational mentor, colleague, and friend Dr. Susan Lindquist. Without her guidance and support, this work would not have been possible. We would especially like to thank L.K. Clayton, D. Pincus, D. Jarosz, J. Rettenmaier, P. Tsvetkov, B. Bevis, S. Sarin, and members of the Tsai and Lindquist labs for helpful discussions and comments. We would like to thank the El-Samad lab (UCSF) for the plasmids used to construct our yeast models.
This work was supported by grants from the Whitehead Institute for Biomedical Research, the Picower Institute for Learning and Memory, The Neurodegeneration Consortium, the Belfer Family Foundation, the JPB foundation, the Edward N. and Della L. Thome Foundation, and the Howard Hughes Medical Institute. D.L. was supported by an American Parkinson’s Disease Foundation fellowship and the NIH (R21 NS087557). P.N. and G.S. were supported by fellowships from the Helen Hay Whitney Foundation. G.S. was supported by the EMBO Fellowship (ALTF 829-2015). P.N. was supported by a NIH K99 award (NIA AG055697-03) and, in part, by the intramural research program at NIDDK, NIH. This work is supported by an R01 from NIH (NIA RF1AG062377) awarded to L-H.T.
Footnotes
DECLARATION OF INTERESTS
P.N. and S.L. are inventors on US Patent US20160251665A. G.S. is an employee and shareholder of AstraZeneca. S.L. was a co-founder of Yumanity Therapeutics. L-H.T. is a member of the scientific advisory board of Yumanity Therapeutics.
REFERENCES
- Antonescu CN, McGraw TE, Klip A, 2014. Reciprocal regulation of endocytosis and metabolism. Cold Spring Harbor Perspectives in Biology 6, a016964–a016964. doi: 10.1101/cshperspect.a016964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Díaz A, Mace K, Zuleta I, Harrigan P, El-Samad H, 2016. Robust Synthetic Circuits for Two-Dimensional Control of Gene Expression in Yeast. ACS Synth. Biol 6, 545–554. doi: 10.1021/acssynbio.6b00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, Victor MB, Bonner JM, Mathys H, Lin Y-T, Ko T, Bennett DA, Cam HP, Kellis M, Tsai L-H, 2020. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nature Medicine 26, 952–963. doi: 10.1038/s41591-020-0886-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner DR, Chi RJ, Lemmon SK, 2012. Lessons from yeast for clathrin-mediated endocytosis. Nature Cell Biology 14, 2–10. doi: 10.1038/ncb2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burston HE, Maldonado-Báez L, Davey M, Montpetit B, Schluter C, Wendland B, Conibear E, 2009. Regulators of yeast endocytosis identified by systematic quantitative analysis. The Journal of Cell Biology 185, 1097–1110. doi: 10.1083/jcb.200811116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA, 2010. Endocytic Pathway Abnormalities Precede Amyloid β Deposition in Sporadic Alzheimer’s Disease and Down Syndrome. The American Journal of Pathology 157, 277–286. doi: 10.1016/S0002-9440(10)64538-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Jiang P, Xue H, Peterson SE, Tran HT, McCann AE, Parast MM, Li S, Pleasure DE, Laurent LC, Loring JF, Liu Y, Deng W, 2014. Role of astroglia in Down’s syndrome revealed by patient-derived human-induced pluripotent stem cells. Nature Communications 5. doi: 10.1038/ncomms5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small G, Roses AD, Haines JL, Pericak-Vance MA, 1993. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443 [DOI] [PubMed] [Google Scholar]
- Dalton L, Davey M, Conibear E, 2015. Large-Scale Analysis of Membrane Transport in Yeast Using Invertase Reporters, Methods in Molecular Biology 1270, 395–409. doi: 10.1007/978-1-4939-2309-0_27 [DOI] [PubMed] [Google Scholar]
- Darsow T, Odorizzi G, Emr SD, 2000. Invertase fusion proteins for analysis of protein trafficking in yeast. Meth. Enzymol 327, 95–106. doi: 10.1016/S0076-6879(00)27270-4 [DOI] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman J-W, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T, 2011. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nature Communications 2, 440–10. doi: 10.1038/ncomms1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z, 1999. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology 52, 244–248. doi: 10.1212/wnl.52.2.244 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, 2007. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2, 31–34. doi: 10.1038/nprot.2007.13 [DOI] [PubMed] [Google Scholar]
- Goode BL, Eskin JA, Wendland B, 2015. Actin and endocytosis in budding yeast. Genetics 199, 315–358. doi: 10.1534/genetics.112.145540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding C, Heuser J, Stahl P, 1983. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. The Journal of Cell Biology 97, 329–339. doi: 10.1083/jcb.97.2.329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JSK, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel K-H, Klopp N, Wichmann H-E, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J, 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics 41, 1088–1093. doi: 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlden H, 2006. Apolipoprotein E4 and traumatic brain injury. J Neurol Neurosurg Psychiatry 77, 1106–1107. doi: 10.1136/jnnp.2006.095513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, McCaffery JM, Lindquist S, Gitler AD, 2008. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A 105, 6439–6444. doi: 10.1073/pnas.0802082105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM, 2015. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348, 921–925. doi: 10.1126/science.aaa0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL, 1994. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. Journal of Biological Chemistry 269, 17386–17389. [PubMed] [Google Scholar]
- Lah JJ, Levey AI, 2000. Endogenous Presenilin-1 Targets to Endocytic Rather Than Biosynthetic Compartments. Molecular and Cellular Neuroscience 16, 111–126. doi: 10.1006/mcne.2000.0861 [DOI] [PubMed] [Google Scholar]
- Lambert J-C, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossù P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanché H, Dartigues J-F, Tzourio C, Gut I, Van Broeckhoven C, Alpérovitch A, Lathrop M, Amouyel P, 2009. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nature Genetics 41, 1094–1099. doi: 10.1038/ng.439 [DOI] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossù P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s Disease Initiative (EADI), Genetic and Environmental Risk in Alzheimer’s Disease, Alzheimer’s Disease Genetic Consortium, Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P, 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nature Genetics 45, 1452–1458. doi: 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-T, Seo J, Gao F, Feldman HM, Wen H-L, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai L-H, 2018. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1141–1154.e7. doi: 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C-C, Kanekiyo T, Xu H, Bu G, 2013. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 1–13. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Drubin DG, Sun Y, 2016. Clathrin-mediated endocytosis in budding yeast at a glance. J Cell Sci 129, 1531–1536. doi: 10.1242/jcs.182303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Rall SC, 2000. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 1, 507–537. doi: 10.1146/annurev.genom.1.1.507 [DOI] [PubMed] [Google Scholar]
- McIsaac RS, Oakes BL, Wang X, Dummit KA, Botstein D, Noyes MB, 2012. Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic Acids Research 41, e57–e57. doi: 10.1093/nar/gks1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgen K, Ramirez A, Frölich L, Tost H, Plichta MM, Kölsch H, Rakebrandt F, Rienhoff O, Jessen F, Peters O, Jahn H, Luckhaus C, Hüll M, Gertz H-J, Schröder J, Hampel H, Teipel SJ, Pantel J, Heuser I, Wiltfang J, Rüther E, Kornhuber J, Maier W, Meyer-Lindenberg A, 2014. Genetic interaction of PICALM and APOE is associated with brain atrophy and cognitive impairment in Alzheimer’s disease. Alzheimer’s & Dementia 10, S269–S276. doi: 10.1016/j.jalz.2013.11.001 [DOI] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JSK, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, George-Hyslop PS, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin L-W, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436–441. doi: 10.1038/ng.801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, van der Kant R, 2014. Stuck in traffic: an emerging theme in diseases of the nervous system. Trends in Neurosciences 37, 66–76. doi: 10.1016/j.tins.2013.11.006 [DOI] [PubMed] [Google Scholar]
- Nixon RA, 2005. Endosome function and dysfunction in Alzheimer’s disease and other neurodegenerative diseases. Neurobiology of Aging 26, 373–382. doi: 10.1016/j.neurobiolaging.2004.09.018 [DOI] [PubMed] [Google Scholar]
- Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, Ambat J, Levy E, Cookson MR, Mathews PM, Duff KE, 2017. The Endosomal–Lysosomal Pathway Is Dysregulated by APOE4 Expression in Vivo. Front. Neurosci 11, 263–12. doi: 10.3389/fnins.2017.00702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh I, Fardo DW, Estus S, 2014. Genetics of PICALM expression and Alzheimer’s disease. PLoS ONE 9, e91242. doi: 10.1371/journal.pone.0091242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I-H, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ, 2008. Disease-Specific Induced Pluripotent Stem Cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T, 2009. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature Genetics 41, 1308–1312. doi: 10.1038/ng.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD, 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences 90, 1977–1981. doi: 10.1073/pnas.90.5.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebar F, Bohlander SK, Sorkin A, 1999. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol. Biol. Cell 10, 2687–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The HD iPSC Consortium, 2012. Induced Pluripotent Stem Cells from Patients with Huntington’s Disease Show CAG-Repeat-Expansion-Associated Phenotypes. Stem Cell 11, 264–278. doi: 10.1016/j.stem.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH, 2008. Amyloid Precursor Protein Trafficking, Processing, and Function. Journal of Biological Chemistry 283, 29615–29619. doi: 10.1074/jbc.R800019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, Kramer P, Woltjer R, Trojanowski JQ, Weintraub D, Chen-Plotkin AS, Irwin DJ, Rick J, Schellenberg GD, Watson GS, Kukull W, Nelson PT, Jicha GA, Neltner JH, Galasko D, Masliah E, Quinn JF, Chung KA, Yearout D, Mata IF, Wan JY, Edwards KL, Montine TJ, Zabetian CP, 2013. APOE ϵ4 Increases Risk for Dementia in Pure Synucleinopathies. JAMA Neurology 70, 223–6. doi: 10.1001/jamaneurol.2013.600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, 1999. β-Secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. doi: 10.1126/science.286.5440.735 [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Zhao N, Caulfield TR, Liu C-C, Bu G, 2019. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol 1–18. doi: 10.1038/s41582-019-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J-T, Tan L, Hardy J, 2014. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci 37, 79–100. doi: 10.1146/annurev-neuro-071013-014300 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, Owens NC, Rege SV, Si G, Ahuja A, Zhu D, Miller CA, Schneider JA, Maeda M, Maeda T, Sugawara T, Ichida JK, Zlokovic BV, 2015. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nature Neuroscience 18, 978–987. doi: 10.1038/nn.4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 2013. Cerebrovascular Effects of Apolipoprotein E. JAMA Neurology 70, 440–12. doi: 10.1001/jamaneurol.2013.2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No custom code was used in this study.