

Major depression affects millions of people and leads to debilitating symptoms. Although conventional antidepressants have been available and are often beneficial, they have a number of limitations including slow onset of action and inadequate response for a substantial fraction of patients. Recently, ketamine – primarily a noncompetitive N-Methyl-D-aspartic acid receptor (NMDAR) antagonist, among other actions – was approved as a novel treatment for treatment-resistant depression and suicidal ideation. This was an exciting development because ketamine can relieve depressive symptoms rapidly and with sustained effect. What is the biological basis for ketamine’s rapid antidepressant action? One framework gaining empirical support is that ketamine promotes neural plasticity. Specifically, ketamine appears to promote synaptogenesis in brain regions such as the medial frontal cortex and hippocampus, countering the dendritic atrophy and synapse loss associated with chronic stress and depression. This framework is supported by several studies that demonstrate a single dose of ketamine increases the number of dendritic spines (1) by elevating their formation rate in the frontal cortex (2, 3). Still unclear though, is when and how the plasticity is boosted. Specifically, when does ketamine enhance the propensity for neural plasticity – so far studies have looked only at synaptic connections, which is the final link in the chain of events. Moreover, how does ketamine enable neural plasticity – the full complement of molecular and cellular factors remains to be elucidated. Knowledge of both the timing and mechanisms underlying ketamine’s plasticity-promoting potential will be key to harnessing fast-acting antidepressants. In the current issue of Biological Psychiatry, Wu et al. (4) present compelling data to define a time window for ketamine’s plasticity potential, while uncovering dopamine as a crucial component of the mechanism.
Previous studies have provided clues into the time scale of ketamine’s effect on neural plasticity. In one longitudinal two-photon imaging study, a single dose of ketamine increased dendritic spine density in the medial frontal cortex within a day (2). Another recent study indicated ketamine begins to reverse stress-induced dendritic spine loss within 12 hours of drug administration but not before 6 hours (3). Intriguingly, blocking the plasticity actions of ketamine abolished its effect on motivated escape behavior in the mouse, suggesting the sustained antidepressant actions require neural plasticity in the medial frontal cortex (3). Together, these prior studies underscored the importance of dendritic structural remodeling for ketamine’s fast-acting antidepressant response. Still, a critical question remained: what is the time window in which ketamine engages neural plasticity to facilitate the behavioral improvements?
In the current study, Wu et al. developed an innovative method to probe a neuron’s likelihood to form new connections at the dendrites, termed plasticity potential, and applied it to investigate the timing of ketamine’s plasticity actions (4). The approach relies on the knowledge that a local burst of glutamate efflux is sufficient to produce a new dendritic spine in a cortical pyramidal neuron (5). Briefly, the authors used two-photon imaging to visualize dendrites of a layer 5 pyramidal neuron from brain slices of the medial frontal cortex, then performed two-photon uncaging of glutamate over a small volume near a dendritic branch to evoke spinogenesis. Using this approach, Wu et al. found that under control conditions new dendritic spines appeared 20-25% of the time. In the ketamine conditions, brain slices were collected at 2, 4, 12, 24, and 72 hours after administration. By the 2-hour time point, treatment significantly increased the probability of glutamate-evoked spinogenesis to ~50%. Notably, this time course corresponds to the time of a behavioral transition in ketamine-treated mice, where acute hyperlocomotor effects cease (6) and depressive-like behaviors begin to improve (3). The elevated plasticity potential was detected after 4 hours but dissipated by 12 hours, indicating a limited time window, which preceded the subsequent long-term increase in dendritic spine density. Figure 1 is an illustration of the time courses, plotted in the context of behavioral timing reported by previous studies.
Figure 1. A schematic of the reported time courses of ketamine’s neural and behavioral effects in mice and humans.
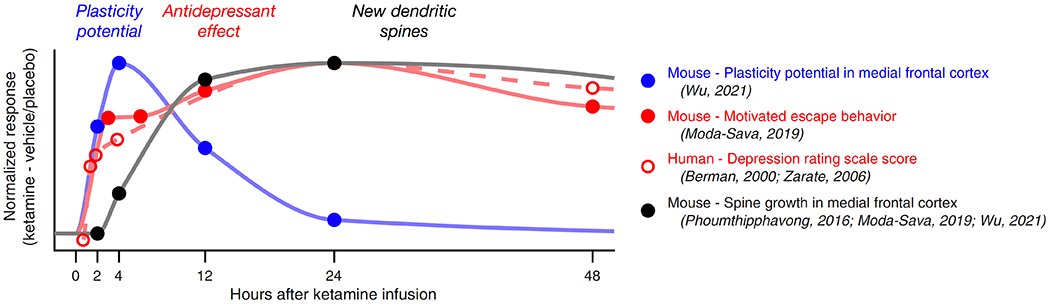
The circles denote the response, normalized by subtracting effects of ketamine from vehicle or placebo, at specific time points as reported in the current and previous studies.
Alongside our growing understanding of the time scale of ketamine’s effect, the underlying molecular and cellular mechanisms are also becoming more evident, including the role of dopamine. In one early study, subanesthetic ketamine increased dopamine level in the medial prefrontal cortex acutely for ~2 hours after drug injection (7). More recently, it was shown that photostimulation of neurons expressing the dopamine receptor Drd1 produced sustained antidepressant-like effects for up to 7 days, mimicking the effects of ketamine (8). By contrast, silencing Drd1-expressing neurons blocked the antidepressant action of ketamine (8). Overall, these results suggested Drd1-expressing neurons could be relevant to the antidepressant effects of ketamine, but whether it was the cell type or if there was a more specific role for the dopamine receptor remained unclear.
In light of the above, the study by Wu et al. provides mechanistic insights into the link between dopamine signaling, specifically protein kinase A (PKA) signal transduction through Drd1 receptors, and the actions of ketamine. To dive into the mechanism of the Drd1 receptor, Wu et al. started with three complementary experiments. First, they antagonized Drd1 receptors to block the increased spinogenesis evoked by ketamine, showing Drd1 receptors are necessary for the drug’s plasticity actions. Next, they used chemogenetic tools to inhibit dopaminergic neurons in the ventral tegmental area. Not surprisingly, the inhibition abolished the effect of ketamine on evoked spinogenesis as well as the delayed increase in spine density.
Conditionally knocking out Drd1 receptors in the medial prefrontal cortex similarly abolished ketamine’s enhancement of evoked spinogenesis. Intriguingly, direct activation of Drd1 receptors with the agonist SKF81297 mimicked ketamine’s impact on evoked spinogenesis. Taken together, these results demonstrated that signaling via Drd1 receptors mediates the increase in plasticity potential due to ketamine administration. Wu et al. then sought to uncover the signal transduction pathways that may underlie Drd1 receptor-dependent spinogenesis. One possibility is that the initiation of spinogenesis depends on glutamate but further enhancement requires the stimulatory actions of PKA downstream of Gαs- coupled Drd1 receptors. Consistent with this hypothesis, Wu et al. showed bath application of PKA suppressor H-89 or over-expression of endogenous PKA inhibitor (PKIα) blocked the expected increase in evoked spinogenesis by agonist activity at Drd1 receptors. Overall, these experiments underscore the essential role of Drd1-PKA signaling for the plasticity actions of ketamine on frontal cortical pyramidal neurons.
While the above findings provided insights into neural mechanisms, a key question remained: does Drd1 receptor-dependent spinogenesis contribute to the antidepressant action of ketamine? To examine this possibility Wu et al. used a learned helplessness model of depressive-like pathophysiology in mice. This paradigm is based on the finding that rodents exposed to an acute stressor, in this case inescapable foot shocks, have lasting deficits in motivated escape behavior. In the current study, mice exposed to inescapable foot shocks were less likely to escape subsequent avoidable foot shocks and showed a concomitant reduction in glutamate-evoked spinogenesis. Conversely, ketamine was protective against both the neural and behavioral deficits. Wu et al. went on to show the protective effects of ketamine on behavior may depend on dopamine release in frontal cortex. On one hand, in the absence of ketamine, optogenetically or chemogenetically-evoked activity of dopaminergic axon terminals in the medial frontal cortex was sufficient to rescue motivated escape behavior following learned helplessness. On the other hand, when dopamine release in the medial prefrontal cortex was chemogenetically inhibited, the protective effects of ketamine on behavior were abolished. Together, this exhaustive set of experiments implicate dopamine signaling in the antidepressant-like actions of ketamine.
The current study highlights dopaminergic transmission in the frontal cortex as a key ingredient in the plasticity actions of ketamine. Yet dopamine was studied in the context of glutamate-evoked spinogenesis, and additional glutamatergic mechanisms have been implicated in ketamine’s action. For instance, ketamine antagonizes NMDARs in dendrite-targeting GABAergic interneurons to increase dendritic excitability of pyramidal neurons (6). This cortical microcircuit mechanism may underlie a number of ketamine’s puzzling effects on neural activity and behavior, including the enhancement of synaptic plasticity (9). At a longer timescale, synaptic strength is maintained and stabilized through compensatory mechanisms. Ketamine may act to shift homeostatic set points to influence neural plasticity (10). How these different molecular and microcircuit mechanisms come together with dopamine signaling to exert antidepressant actions remains to be resolved. To this end, future studies will be needed to develop a complete picture of ketamine’s efficacy and biological action.
To summarize, at the beginning of this commentary, we talked about finding out when and how ketamine promotes structural plasticity. For the when, Wu et al. showed that the potential for glutamate-evoked spinogenesis rapidly increased 2 hours after ketamine administration. This effect curiously corresponds to the start of behavioral improvement and precedes the rise in dendritic spine density. For the how, dopamine, through Drd1-mediated activation of PKA, was demonstrated to be instrumental to the plasticity actions of ketamine in the frontal cortex. Knowing when ketamine promotes structural plasticity, we may leverage or potentially extend the time window of spinogenesis using interventions that augment the plasticity actions. Knowing how ketamine enhances structural plasticity may help uncover novel targets for developing better, more precise rapid-acting antidepressants.
Acknowledgements and Disclosures
This work was supported by the Yale Center for Psychedelic Science, NIH/NIMH grants R01MH112750 (A.C.K.) and R01MH121848 (A.C.K.), China Scholarship Council-Yale World Scholars Fellowship (H.W.), and NIH/NIGMS Medical Scientist Training grant T32GM007205 (N.K.S). The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. (2010): mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329:959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phoumthipphavong V, Barthas F, Hassett S, Kwan AC (2016): Longitudinal Effects of Ketamine on Dendritic Architecture In Vivo in the Mouse Medial Frontal Cortex. eNeuro. 3:ENEURO.0133-0115.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moda-Sava RN, Murdock MH, Parekh PK, Fetcho RN, Huang BS, Huynh TN, et al. (2019): Sustained rescue of prefrontal circuit dysfunction by antidepressant-induced spine formation. Science. 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu M, Minkowicz S, Dumrongprechachan V, Hamilton P, Kozorovitskiy Y (2021): Ketamine rapidly enhances glutamate-evoked dendritic spinogenesis in medial prefrontal cortex through dopaminergic mechanisms. Biological Psychiatry. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwon HB, Sabatini BL (2011): Glutamate induces de novo growth of functional spines in developing cortex. Nature. 474:100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali F, Gerhard DM, Sweasy K, Pothula S, Pittenger C, Duman RS, et al. (2020): Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines. Nat Commun. 11:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verma A, Moghaddam B (1996): NMDA receptor antagonists impair prefrontal cortex function as assessed via spatial delayed alternation performance in rats: modulation by dopamine. J Neurosci. 16:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hare BD, Shinohara R, Liu RJ, Pothula S, DiLeone RJ, Duman RS (2019): Optogenetic stimulation of medial prefrontal cortex Drd1 neurons produces rapid and long-lasting antidepressant effects. Nat Commun. 10:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Savalia NK, Shao L-X, Kwan AC (2020): A Dendrite-Focused Framework for Understanding the Actions of Ketamine and Psychedelics. Trends in Neurosciences. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kavalali ET, Monteggia LM (2020): Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron. 106:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]