

Abstract
There is an increasing evidence that inflammation contributes to clinical and functional outcomes in traumatic brain injury (TBI). Many successful target-engaging, lesion-reducing, symptom-alleviating, and function-improving interventions in animal models of TBI have failed to show efficacy in clinical trials. Timing and immunological context are paramount for the direction, quality, and intensity of immune responses to TBI and the resulting neuroanatomical, clinical, and functional course. We present components of the immune system implicated in TBI, potential immune targets, and target-engaging interventions. The main objective of our article is to point toward modifiable molecular and cellular mechanisms that may modify the outcomes in TBI, and contribute to increasing the translational value of interventions that have been identified in animal models of TBI.
Keywords: Depression, glia, immune challenge, immunomodulation, inflammation, priming, probiotic, traumatic brain injury
Traumatic brain injury (TBI) is an all-encompassing term to describe an intracranial injury after an external mechanical force results in damage to the brain structure or alteration in its functioning [1]. Both the causes and courses of TBI are heterogenous. Mechanisms of injury resulting in TBI are varied, including blunt forces (from bumps to blows), inertial loading, penetrating wounds, and blast injuries [1]. Just as the causes of TBI vary, the extent of damage also varies. Immediate injury after TBI can result in complications ranging from an unnoticed bruise with transient deficits to bleeding, or brain dysfunction resulting in permanent disability, or even death [2]. In evaluation of potential TBI patients, it is important to gain an accurate history of injury and complications during injury (including loss of consciousness), evaluate and treat symptoms [2], and to perform serial assessments in the post-injury period [3].
Etiological and pathophysiological heterogeneity of TBI are two of the major factors that produce inconsistent results between animal research and randomized clinical trials. It is essential to identify precise mechanisms for the primary [1] and secondary injuries associated with TBI to adequately target specific subgroups that are characterized by shared pathophysiology. After TBI, the cellular cascade of inflammation could potentially have beneficial effects [4–7]. However, if inflammation is too intense, prolonged, and unremitting, it may be harmful. Secondary injury has a slower progression ranging from minutes to years following the mechanical insult and is driven by several pathways, including: 1) free radical generation causing damage to molecular structures of cellular membranes [8], 2) excitotoxicity secondary to excess of glutamate [9, 10], and 3) systemic and central nervous system (CNS) immune activation [11]. It has been hypothesized that immune activation and modulation can significantly change the clinical and functional course in TBI [12]. Within minutes of impact, glia and recruited immune cells generate signals that initiate an inflammatory cascade [13]. The activation of the immune system has a vital role in cleaning the areas of the primary injury and preventing subsequent infections. However, to this day, targeting immune activation has been unsuccessful in improving clinical outcome in patients with TBI [14, 15], perhaps because of the greater heterogeneity of the causes of inflammation and their interactions in clinical TBI as compared to animal models, as well as genetic, environmental, pharmacological, and cultural differences between participants. The complex immunological mechanisms, including the influence of interacting immune-modifying factors involved in human TBI, presents challenges in developing an adequate animal model.
Interventional paradigms may be designed to:
Target early inflammatory processes that stimulate beneficial effects and inhibit detrimental ones.
Modulate overly prolonged or intense immune activation.
Clarify how immune challenges before or following TBI (e.g., reactivating infections, allergen exposure in atopic individuals, or exacerbation of autoimmune disorders) may affect its course and outcomes.
A better understanding of the complex molecular cascade of inflammation and immune function post-TBI is vital for both patient-specific diagnostic interventions and development of novel treatments, as well as in developing coupling between animal models and clinical interventional studies.
SEVERITY OF TBI
The methods of assessing the severity of TBIs in a variety of clinical settings include the mechanism [closed versus (vs.) penetrating], the clinical severity, and the type and degree of structural damage [16]. The most common tool used to evaluate TBI severity in the clinical setting is the Glasgow Coma Scale (GCS). The GCS includes assessment of best verbal response, best eye-opening response, and best motor response, with an overall score range of 3–15, with 3 being the most severe and 15 the least severe [1].
The three major categories of TBI severity are mild, moderate, and severe, often dichotomized into mild vs. moderate/severe.
EPIDEMIOLOGY OF TRAUMATIC BRAIN INJURY
TBI is a significant public health problem. It has been referred to as the “silent epidemic”. Yet, even if underestimated, the epidemiological characteristics of TBI are staggering. The lifetime prevalence of TBI is up to 40% of adults [17]. The global estimate of TBI indicates an incidence of 64 million and 74 million new cases yearly worldwide, with the highest incidence in USA, Canada, and Europe [18]. Overall, the most common causes of TBI are falls and motor vehicle accidents [19]. TBI incidence peaks three times during the lifespan: childhood, adolescence, and older adulthood [17]. TBI is the leading cause of death in children over 1 year old worldwide [20]. The estimated incidence of TBI in children aged 0–14 years old is about 475,000 in the United States [21]. The geriatric population incurs the most TBI-related hospital visits and deaths. Given the aging population, the number of older adults afflicted by TBI is likely to continue to increase [17]. Military personnel are also at particularly high risk due to combat, accidents, and activities during training [22].
ANIMAL MODELS OF TRAUMATIC BRAIN INJURY
Presenting the immune changes induced by TBI, common for all models and specific to each animal model in detail, is beyond the scope of the current article. Yet, we will briefly describe specific models with some of the inflammation findings uncovered using those models. For a presentation in depth, the reader is directed to a recent review of the topic [23].
Given the complexity of TBI, it is not surprising to see a variety of approaches that aim at modeling it in animals [24–26]. Animal models of TBI are commonly designed and classified by how the head injury is induced in the animal subject, rather than by its severity [25, 26]. Severity of injury in a particular animal model depends on the specific mechanical impact parameters that are used in the experiment. Therefore, in most cases, the same animal model can be used to represent both severe and milder forms of TBI, depending on the particular parameters used in the study [27]. At minimum, animal models of TBI can be classified by a single distinguishing factor, namely, closed vs. open head models. In the closed-head injury models, the animal’s skull is left intact, while in open-head models, craniotomy or craniectomy is performed to open a window on the skull to reach the brain for induction of TBI. Neuroinflammation is a key feature of all animal TBI models. Proliferation of reactive astrocytes and microglia, which is a hallmark cellular response to TBI and is often evidenced by upregulation of certain proteins, such as glial fibrillary acidic protein (GFAP) and ionized calcium-binding adaptor molecule 1 (Iba1), along with increased levels of proinflammatory cytokines and chemokines, has been consistently observed across different types of animal models of TBI [23, 28, 29]. Another common finding is that repeated injuries exacerbate the neuroinflammatory response across different animal models [1, 30].
Closed-head models
Most closed-head animal TBI models require a blunt impact to the head for the induction of TBI. Some models only mimic the rotational forces that the brain is subjected to during TBI but not the mechanical impact [31–35]. Blast-injury models can be considered impactless since brain injury is achieved by the shock wave from the blast rather than a mechanical blow to the head [36]. The critical factor in interpreting the results of these animal models of TBI is the possibility of contusion, hematoma, and skull fractures. Presence of hematoma or contusion can change the course of the immune response and should be taken into account when the results are interpreted [37, 38].
In a weight drop model of TBI, a solid object falls freely in a guiding tube from a predetermined height onto a subject’s head, which is usually anesthetized during the procedure to induce TBI [39–42]. Enhanced reactive gliosis and astrogliosis have been shown with several variations of the weight drop model in both mouse and rat subjects [43–46], in addition to increased proinflammatory cytokine [e.g., interleukin (IL)-1, tumor necrosis factor-alpha (TNF-α)] levels and their gene expression [41, 47]. These neuroinflammatory responses are enhanced with repeated injuries [1, 44]. Another method to induce closed-head TBI is the utilization of pneumatically or electronically controlled piston as the impactor instead of a free-falling weight [48–52]. Similar to the findings in the weight drop models, drastically increased GFAP and Iba1 levels have been reported in this TBI model [53–55]. However, elevation of proinflammatory cytokines has not been consistently observed with this model [53, 54, 56].
An alternative to the weight drop- and piston-based animal models of TBI are the projectile impactor models. These models use accelerated projectiles as impactors and have the ability to model the head impacts from high-speed small mass objects [57–61]. Using this approach, it has been demonstrated that repeated impacts to the head causes extensive increases in neuroinflammatory markers and microgliosis, including increased M1-type microglia [61, 62].
The blast-induced TBI model is another example of a closed-head TBI model. In blast-induced TBI models, the animal is secured in a blast tube where a shock wave travels and reaches the animal’s head to create TBI [36]. An acute neuroinflammatory response occurs acutely in blast-induced TBI models and levels of various chemokines and cytokines [e.g., IL-1α, IL-6, IL-10, interferon-gamma (IFN-γ), monocyte chemoattractant protein-1] increase in a short time frame (2–4 h after blast TBI) [63, 64].
Open-head models
The controlled cortical impact (CCI) model is one of the most utilized animal models of TBI that was originally developed in ferrets and then applied predominantly to rodents [65, 66]. In CCI, anesthetized animals undergo a craniectomy or craniotomy procedure, depending on the experimental design, and the dura is exposed [65, 66]. During the injury, a piston impactor moving at a predetermined speed hits the exposed dura to create TBI. Modified versions of CCI in which the skull is kept intact and the piston impacts the skull directly should be considered a separate model [67]. CCI was originally thought to produce focal injury around the impact site, but later studies showed that it also produces more diffuse injury and neurodegeneration throughout the brain distant from the cortical impact site [68]. CCI has been shown to induce acute increases in chemokines (i.e., CXCL1) and cytokines (i.e., IFN-γ, TNF-α) and also certain anti-inflammatory cytokines (IL-4, IL-13) along with activated microglia [69, 70]. Fluid percussion injury (FPI) is another commonly used open-head TBI animal model [71–74]. FPI and CCI are similar in terms of their application of craniectomy to open a window on the skull. Through this opening, however, instead of blunt force impact from a piston, rapid and transient hydraulic pressure is applied through the opening in the skull to create TBI [75]. FPI induces both focal and diffuse injury as well [25, 75]. FPI increases activated microglia and macrophages in the brain [76]. In a more recent study, FPI has been shown to initiate a neuroinflammatory response acutely and increase levels of various chemokines and cytokines in the brain [77].
Even though relatively uncommon among civilian TBI patients, penetrating injury to the brain from a ballistic high-speed object (i.e., a bullet) is a particular concern for the military. The penetrating ballistic-like brain injury (PBBI) model was developed to address this particular type of injury [78]. In PBBI, similar to CCI and FPI, a skull window is opened through craniectomy. After that, a probe with an inflatable tip is inserted into the brain. To create injury, the probe tip is inflated very rapidly. This mimics effects of a bullet wound in the brain [78]. PBBI has been shown to induce loss of brain tissue, extensive intracranial hemorrhages and edema [78–80]. Neuroinflammation after PBBI is widespread and occurs rapidly, and given the major disruption of the blood-brain barrier (BBB), the central events are rapidly mirrored in the periphery. Levels of proinflammatory cytokines (IL-1β, IL-6, IFN-γ, TNF-α) and markers of microglial reactivity increase acutely after PBBI [81, 82].
THE IMMUNE SYSTEM: A BRIEF SUMMARY
The immune system includes structures, cells, and molecules that together are charged with defending the self against pathogens, malignant cells, transplanted tissue and, most commonly, microorganisms, as well as with eliminating damaged tissue that consumes resources and represents a risk of systemic toxicity, and ultimately sepsis. The innate immune system is characterized by rapid and non-specific responses and limited capacity to cascade the response based on the memory of previous encounters with antigens. It plays an essential role in preventing or limiting the invasion by microorganisms, and in the removal of debris, wound healing, and tissue remodeling. The innate immune system includes anatomical barriers (e.g., skin, mucosa) and physiological gradient barriers (e.g., pH, temperature), antimicrobial peptides, cellular effectors such as innate lymphoid cells (e.g., natural killer cells), phagocytes (e.g., neutrophils, macrophages, microglia, and dendritic cells), and granulocytes (e.g., eosinophils, basophils, mast cells, and neutrophils) that mainly release immune molecular mediators from their granules [83, 84]. To sense danger signals or pathogens and to trigger immune responses, these martial cells use “pattern recognition receptors”, including nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), Toll-like receptors (TLRs), and scavenger receptors [83, 85]. Chemokines and cytokines are released to amplify and initiate a cascade of immune responses and recruit other immune cells. Distal signaling via the vascular system and neural input to the brain may affect behavior, ultimately centralizing behavior towards the bare essentials necessary to survive the threat (i.e., “sickness behavior”, involving an overall retreat from the environment, a decrease in energy and purposeful activity, an increase in sleepiness and propensity to sleep, and decrease in appetite and food intake).
Adaptive immunity involves the cascading activation and expansion of T and/or B lymphocytes targeting specific antigens, and a memory of previous antigen exposure allowing a targeted, intense, and rapid response to future antigen challenges [86, 87]. T lymphocytes (CD4+ and CD8+ T cells) have cytotoxic, helper, or regulatory functions, and are activated and reactivated by recognizing antigens displayed in major histocompatibility complexes. B lymphocytes produce immunoglobulins and have reciprocal interactions with T cells and cells of the innate immunity [88]. Adaptive immunity is also involved in autoimmune responses, as well as atopy.
SPECIFIC COMPONENTS OF THE IMMUNE RESPONSE TO TBI
The timeline of immune response to TBI, reflecting the elevation of various components of the immune system, is summarized in Fig. 1. Several methods, including pharmacological probing, targeting with antibodies, and utilizing genetically modified animals, have been used to infer the role of molecular signaling pathways and cellular immunity implicated in TBI (described in Tables 1A and 1B). Molecular pathways and cell types involved in the inflammatory response to TBI are well-described in other mechanism-oriented reviews [11, 30, 89, 90]. Below, we list some of the components of this immune response.
Fig. 1.
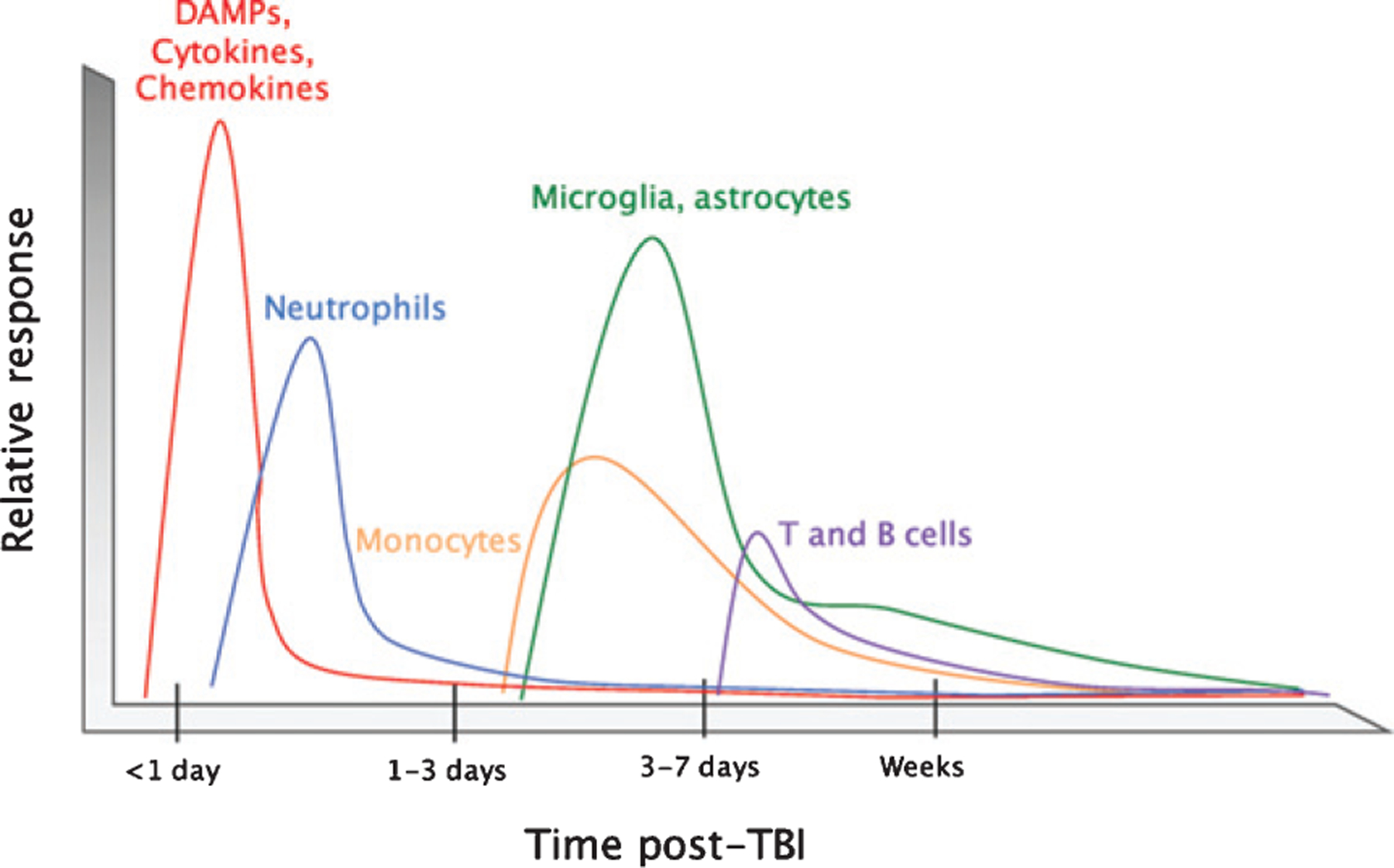
Timeline of the immune response to TBI. After impact, rapid tissue release of damage-associated molecular patterns (DAMPs) prompts resident cells to secrete chemokines and cytokines. These molecules attract neutrophils, which contribute to circumscribing the injury site and promoting the removal of damaged and dead cells and debris. Infiltrating monocytes and activated glia begin to predominate 3–5 days post-injury to defend against infection and perform reparative functions. T and B cells can also be recruited to sites of brain pathology at later time points in the response (3–7 days post-injury). The timeline is modeled to human TBI across modalities, and is likely to correspond to the great majority of animal models of TBI (Reproduced from McKee CA, Lukens JR (2016) Emerging roles for the immune system in traumatic brain injury. Front Immunol 7, 556).
Table 1.
Rodent models used to characterize the role of signaling pathways and immune cell types in TBI. Table 1A describes molecular signaling pathways and Table 1B describes the cellular components of the immune system in TBI, illustrating approaches based on antibody targeting, as well as genetically modified mice (Reproduced from McKee CA, Lukens JR (2016) Emerging roles for the immune system in traumatic brain injury. Front Immunol 7, 556)
| Table 1A. Molecular signaling pathways | ||||
|---|---|---|---|---|
| Inflammatory mediator | Animal line/model | Purpose | Major findings in animals exposed to TBI | Reference |
| IL-1 | Anti-IL-1β antibody | Blockade of IL-1β signaling | Reductions in macrophages/microglia, neutrophils, and T cell numbers in the brain, improvement in learning tasks, and decreased tissue loss | [289, 290] |
| IL-1 R antagonist | Neutralize IL-1 | Higher expression of proinflammatory cytokines in macrophages | [291] | |
| ASC | Anti-ASC | Limit inflammasome assembly | Reduced caspase-1 activation and IL-1β production, decreased lesion volume | [292] |
| ASC knockout | Abrogate inflammasome assembly | No improvements in lesion volume, histopathology, cell death, or motor function | [293] | |
| NLRP1 | NLRP1 knockout | Prevent NLRP1 inflammasome assembly | No improvements in lesion volume, histopathology, cell death, or motor function | [293] |
| IL-6 | IL-6 knockout | Ablation of IL-6 signaling | Fewer reactive astrocytes and macrophages, increased neuronal death | [294] |
| IL-6 knockout | Ablation of IL-6 signaling | Poor behavioral performance, higher IL-1β levels in the cortex | [295] | |
| GFAP-IL-6 overexpression | Increase IL-6 expression in astrocytes | Greater recruitment of glia and immune cells to the lesion, decreased oxidative stress and neuronal death | [296] | |
| Anti-IL-6 antibody | Neutralize IL-6 | Reduced some inflammatory and behavioral effects of post-injury hypoxia | [297] | |
| TNF-α | TNF-α inhibitor post-TBI | Inhibit TNF-α signaling | Early administration improved cognitive performance, and decreased neuronal apoptosis and astrogliosis | [298] |
| TNFR1 knockout | Disrupt TNF-α signaling through TNFR1 | Improved neurological function and neuronal survival/lesion volume, decreased numbers of CD11b+cells in the brain | [299] | |
| TNFR2 knockout | Reduce TNFR2 signaling | Worsened neurological function and no protection from tissue loss | [299] | |
| TNFR2/Fas knockout | Abrogate TNF-α signaling through TNFR2 | Impaired motor and cognitive performance | [142] | |
| G-CSF | G-CSF injection post-TBI | Enhance G-CSF signalling | Improved cognitive performance and increased hippocampal neurogenesis, higher glial activation and production of BDNF and GDNF | [300] |
| GM-CSF | GM-CSF knockout | Disrupt GM-CSF signalling | Worsened cognitive deficits as well as cell and tissue loss, reduced astrogliosis | [301] |
| Type 1 IFN | IFNAR knockout or IFNAR blocking antibody | Block type 1 IFN signalling | Reduced lesion volume, more anti-inflammatory cytokine signaling, increased glial activation, these effects were hematopoietic cell-dependent | [302] |
| IL-10 | IL-10 knockout, IL-10 injection | Modulate IL-10 signaling | Diminished protective effects of hyperbaric oxygen treatment, including lesion volume, edema, cognitive improvement, and decreased cytokine production in IL-10 knockout mice, while IL-10 injection improved these outcomes | [303] |
| TGF-β | TAK1 inhibition | Disrupt signaling downstream of TGF-β | Improved neuronal survival and motor function, decreased NF-κB signaling and inflammatory cytokine production | [304] |
| TGIF shRNAv knockdown | Ablation of downstream TGF-β signalling | Decreased infarct volume and microglia numbers, improved motor function | [305] | |
| APOE | APOEϵ4 overexpression | APOEϵ4 overexpression | Worsened brain pathology, BBB breakdown, and neurological impairments | [306, 307] |
| TREM2 | TREM2 knockout | Abrogate TREM2 signaling | Altered macrophage distribution, hippocampal neuroprotection, and fewer cognitive deficits | [308] |
| Table 1B. Cellular mediators | ||||
| Cell type | Animal line/model | Purpose | Major findings in animals exposed to TBI | Reference |
| Neutrophils | IgM RP-3 | Neutrophil depletion | No significant decrease in BBB permeability | [151] |
| Anti-Gr1 antibody | Neutrophil depletion | Decreased edema, apoptosis, and microglia/macrophage activation, no significant changes in BBB integrity | [153] | |
| CXCR2 knockout | Reduce neutrophil infiltration | Decreased cell death, no significant changes in BBB permeability or behavior | [148] | |
| Neutrophil elastase knockout | Reduce neutrophil effector functions | Decreased edema and apoptotic neurons, but no decrease in tissue volume loss or behavioral improvement | [154] | |
| Macrophages and microglia | CD11b-TK | Deplete CD11b-expressing cells | Reductions in microglia numbers in the brain, no improvement in axonal injury, treatment toxic at high dosage | [309] |
| CD11b-DTR | Deplete CD11b-expressing cells | No change in lesion size, treatment caused inflammatory response without injury | [310] | |
| CCX872 (CCR2 antagonist) | Reduce CCR2 signaling functions | Reduced macrophages in the brain, altered pro- and anti-inflammatory cytokine expression, less cognitive dysfunction | [162] | |
| CCR2 knockout | Limit CCR2-mediated recruitment of monocytes | Reduced numbers of infiltrating monocytes, improved learning and memory | [161] | |
| CCR2RFP/RFP | Disrupt recruitment of monocytes | Reduced monocyte recruitment, cavity volume, and axonal pathology | [160] | |
| CX3CR1 knockout | Abrogate CX3CR1 signaling functions in macrophages and microglia | Short-term neuroprotection and lower inflammatory response, long-term functional impairments and elevated myeloid cell activation | [311] | |
| T cells | Rag1 knockout | Genetic ablation of B and T cells | No changes in neurological outcome, BBB integrity, pro- or anti-apoptotic mediators, hippocampal architecture, or astroglial activation | [312] |
| FTY720 | Sequester lymphocytes and reduce their migration to the brain | Decreased circulating lymphocytes, decreased neutrophils and macrophages/microglia in ipsilateral hemisphere | [313] | |
Early immune “danger” signaling in TBI
In the damaged CNS, the initiation of immune signaling is started by recognition of damage-associated molecular patterns (DAMPS), while microorganism invasion in the CNS is signaled by pathogen-associated molecular patterns (PAMPS) [91]. These molecules interact with receptors, such as TLRs, NLRs, and scavenger receptors that serve as “danger” sensors and help to initiate the inflammatory cascade [85, 92–94].
The DAMPS’ recognition leads to the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB)-inducing kinase [94] and elevation of proinflammatory cytokines, such as TNF-α and IL-6 [95]. A reduced expression of inflammatory markers and smaller brain lesions after impact were observed in TLR4-deficient mice relative to wild-type controls [96], and less edema was observed after TBI after administration of VGX-1027, a selective TLR4 blocker [97]. Brain injury in humans and experimental animals induces expression of High Mobility Group Box 1 protein (HMGB1) [98], a protein binding to TLR4 and initiating inflammatory cascades [99–101], and its inhibition diminishes both inflammation as well as BBB breakdown [102]. Other proteins triggering inflammation after TBI are the protein chaperones involved in tissue reorganization and wound healing [103, 104], and have immunomodulatory and neuroprotective effects [105–108]. Other inflammation triggering and perpetuating mechanisms include the activation of the purinergic receptors by adenosine tri-phosphate (ATP) released in the extracellular space secondary to tissue damage, leading to activation of inflammatory cascades [109, 110] and promotion of neutrophil recruitment [111, 112], as well as the formation of inflammasomes in the cytosol [113, 114]. Multiple cells in the CNS—such as neurons, astrocytes, microglia, macrophages, and astrocytes—can contain assembled inflammasomes [115, 116], and levels of inflammasome proteins in the cerebrospinal fluid (CSF) generally predict a poorer outcome in TBI [117].
Inflammatory gene expression
After TBI, a program of immune gene expression is induced that determines the progression of the subsequent inflammatory cascade [118]. Qualitatively, genes implicated in antigen presentation [CD74, CD86, major histocompatibility complex (MHC) II], phagocytosis [C3, C4, Fc gamma receptor (FcgR) and FCGR4], astrocyte activation [Aquaporin (AQP)4 and GFAP), chemotaxis [chemokine ligand (CCL)2, CCL4, chemokine (C-X-C motif) ligand (CXCL)1 and CXCL4], cytokine signaling [IL-1β, IFN-γ, IL-6, IL-12, IL-10 and transforming growth factor-beta (TGF-β)] are, surprisingly, similarly upregulated in both mild and severe TBI [118], and thus, are unlikely to be key mediators of severity of TBI outcome.
Peripheral innate immune activation after TBI
It is known that the body responds to TBI by engineering both central and peripheral inflammatory responses [119–122], and that a single brain injury can elicit complex changes in the systemic immune system. In fact, the immunologic complex known as the systemic inflammatory response syndrome characterized by alterations in circulating leukocyte numbers, complement proteins, coagulants, and inflammatory cytokines [120, 122], seems to result from post-TBI disruption of the BBB and CNS vasculature along with consequent leakage of central inflammatory mediators into the periphery [119–122].
The complement system
Various complement components have been found in the CSF of TBI patients [123, 124] and correlate with the severity of BBB dysfunction [124], and interrupting the complement cascade in various animal models of TBI has been consistently found to be neuroprotective [125–132].
Cytokines
Cytokines are cellular messengers that are secreted by multiple cells implicated in TBI that can convey proinflammatory or anti-inflammatory signals to other immune and nonimmune cells [133]. Often produced during sterile injury immune responses, IL-1β is elevated in the CSF of patients with severe TBI for at least 24 h, with levels that are linked with a less favorable prognosis [134, 135]. This signal is not just a marker of severity, but likely a component of a mediating adaptive mechanism, as administering an IL-1β antagonist in animal models reduces neurological symptoms, improves neurological functioning, and reduces the lesion volume [136].
Similarly, IL-18 is detected early post-TBI [137] and administering IL-18 antagonists in rodents one hour post-brain injury led to improved symptomatic recovery [137]. Thus, IL-1β and IL-18 have been associated with negative outcomes post-TBI. In contrast, other cytokines have been associated with either a good or a bad prognosis. For instance, a classical proinflammatory cytokine, TNF-α, shows heterogenous links with outcome in TBI. On one hand, it is elevated in TBI patients and, moreover, neuroprotective effects can be elicited by its blockade in rodent TBI models [138, 139]. However, while less severe neurological symptoms and functional deficits occur one week after TBI in mice that are genetically deficient in TNF-α, greater deficits and motor symptoms were seen at 2–4 weeks after injury with larger lesions at later time points [140]. It also appears that the experimental model of TBI plays a strong moderating effect on the functional outcome of TNF-α inhibition. In particular, concomitant Fas receptor and TNF-α inhibition appears to lead to worse functional outcomes in a mouse concussion model with no structural brain damage based on impact and head acceleration, while in models of cerebral contusion, the Fas receptor and TNF-α inhibition improved functional outcome and reduced tissue damage [141, 142]. This suggests that TNF-α has a dual role in contributing to both tissue repair and recovery, as well as to neuroinflammation and excitotoxicity.
Anti-inflammatory cytokines, such as IL-10 and TGF-β, counteract the effects of proinflammatory cytokines and reduce neuroinflammation, and overall, improve the functional and symptomatic outcomes and lesion dimension. For instance, the cytokine IL-10 appears to be neuroprotective if administered early and peripherally (but not intrathecally) in a fluid percussion concussion [143], suggesting that its anti-inflammatory effect has to act, at least initially, on the peripheral immune system for beneficial effects to occur. Similarly, TGF-β1 appears to be neuroprotective in a weight drop model in rats, where the cytokine is expressed in perilesional neurons and astrocytes, and consistently, knockout of the TGF-β1 results in increased neuronal death, larger lesion size, and worse neurological deficits [16].
Phagocytes/glial cells
Post-TBI, myeloid cells in the CNS are first to be activated followed by neutrophils, which are usually among the first peripheral immune cells to be mobilized towards the site of injury within just a few hours (h) [111, 144–146]. There is a significant surge in the number of peripheral neutrophils in the early hours after TBI that lasts until 48 h post-injury [147]. Perivascular spaces and meninges seem particularly vulnerable to alignment of neutrophils after cortical injury [37, 111]. Promoting complexity, there are data to define neutrophils as neuroprotective [111, 148], actively involved in healing after neurological injury [149, 150], neutral [151, 152], or alternatively, neurotoxic [121, 151–154]. Similarly, monocytes and macrophages are directed by temporal and biological contextual factors towards either tissue damage or repair [155, 156].
The perivascular spaces and brain parenchyma become hosts for monocytes within the first two days after injury. These monocytes then differentiate into macrophages and remain in the same location for weeks after the original injury [157]. The chemokine CCL2 signals the deployment of monocytes [158, 159] after TBI and elicits CCR2 + monocyte recruitment. Some studies suggest that interfering with this pathway decreases the lesion size and may lead towards improved neurological outcomes in animal models of TBI [158, 160–162]. Yet, other studies demonstrate the repair capacity of macrophages after CNS injury [163–165]. For microglial priming, the reader is directed to discussion later in the article at the section on “Age effects/microglial priming.”
Astrocytes
Astrocytes respond directly and indirectly to TBI via HMGB1/Receptor for advanced glycation end products (RAGE) signaling and activation of the NF-kB pathway [98], leading to secretion of chemokines/cytokines and enhanced astrocyte phagocytic activity. Yet, post-TBI reactive astrogliosis limits the spread of damage and inflammatory response to the unaffected CNS area, with astrocytes also providing metabolic support for neurons and their synapses [166, 167]. Astrocytes interact with other immune cells, such as microglia, in releasing various growth factors like insulin like growth factor 1 (IGF1) and nerve growth factor (NGF) that promote post-TBI healing [4].
T cells
Brain injury results in a cell-mediated immune activation [168, 169] with recruitment of T cells in the brain [170] independent of their antigenic specificity. Detailing the adaptive immunity activation in detail is beyond the intended scope of this review, However, it is important to mention that while overreactive adaptive immunity has been thought to represent a negative prognostic factor, the CD4+ T cells autoreactive to CNS antigens appear to be mainly neuroprotective, in particular for injured axons [5–7].
LIPIDS, IMMUNE MODULATION, AND TBI
Polyunsaturated fatty acids (PUFA)
Lipids are a diverse molecular class, important for signaling and immunomodulation. A number of studies reported increases in levels of free fatty acids (FFA), including polyunsaturated fatty acids (PUFA), in tissues, CSF, and serum in both human TBI and experimental models of TBI [171, 172]. Notably, two independent mass spectroscopy imaging studies of TBI reported PUFA-containing lipid signatures within the injury site in rat models, including multiple phosphatidylcholines [173], diacylglycerols, and sphingolipids [174].
PUFAs are important substrates for the cyclooxygenase (COX), lipoxygenase (LOX), and cytochrome P450 (CYP) pathways, ultimately yielding a range of immunomodulatory molecules, such as prostaglandins, thromboxanes, leukotrienes, and resolvins that mediate signaling through G-protein coupled receptors [175, 176]. Two classes of PUFAs, ω-3 (i.e., docosahexaenoic acid, DHA; eicosapentaenoic acid, EPA) and ω-6 (arachidonic acid) are generally implicated in opposing anti- and proinflammatory functions, respectively, that have been extensively studied [177–179].
Multiple regimens for supplementation with ω-3 s have been implemented to limit putative immune-mediated pathology in various rodent models of TBI [180–183]. Furthermore, in rodent models of mild to severe TBI, treatment with DHA or fish oil (containing DHA and other ω-3 s) showed significant improvements in cognitive function and mitigation of neuronal damage [181–184]. In humans, there are no adequate data to make a determination about efficacy and safety of ω-3 s treatment in TBI, and despite anecdotal evidence, pathophysiological targeting, as well as advocacy [185], well-designed human clinical trials are necessary. Additionally, a theoretical possibility of impaired initiation of clotting, and thus, increased risk of bleeding due to decreased eicosanoid synthesis exists [186], even if this is disputed based on analogical reasoning [185], and to our knowledge, is unsupported by large-scale safety data.
Statins
Cholesterol-lowering drugs, in particular, statins, have also been implicated in reduction of TBI pathogenesis, as well as in anti-inflammatory effects. The primary effect of statins is inhibition of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase (a rate-limiting enzyme in the mevalonate pathway), but there are well-recognized pleiotropic effects of statins, including direct immunomodulation [187].
The positive significant immune effects of statins in animal models are reductions of IL-6, TNF-α, and intracellular adhesion molecule 1 (ICAM-1), attenuation of cerebral edema and glial cell activation, and reduction of the permeability of the BBB [188, 189]. The upstream immunomodulatory mechanism of statins is the inhibition of TLR4 and NF-κB [188]. It has been shown that statins have an anti-apoptotic effect via downregulating the caspase pathways, which can be overly activated after TBI [190]. In animal models of TBI, statins have resulted in reduction of parenchymal hemorrhage [191], improved cerebral blood flow [192], decreased cerebral edema [193], improved neuronal survival and regenerative angiogenesis and synaptogenesis [194], improved neurological outcome [195], reduced intravascular thrombosis, improved spatial learning [196], and reduced cognitive deficits [197], including in severe forms of TBI [198]. In a small double-blind randomized trial in patients with moderate-severe TBI, 72 h after TBI, rosuvastatin significantly decreased the level of TNF-α but not of IL-1β, IL-6, or IL-10, and led to a reduction in the disability score [199]. Preinjury atorvastatin use leads to a reduction in mortality and improved functional outcomes in elderly exposed to TBI [200].
Consistent with the findings described above, a retrospective analysis found a significant reduction of hospital mortality in statin-using patients admitted for TBI, as compared to nonusers [201]. In contrast, a recent observational study did not confirm any benefit of statins in TBI [202]. Meanwhile, another study in Asia [203] did not find any benefit of a statin pretreated status in patients with moderate-severe TBI—a difference that could be attributed to: 1) using multiple statins rather than purely atorvastatin; 2) a predominantly Asian population; 3) potential pharmacokinetic differences; or, finally, 4) withholding the statin medications during the inpatient hospitalizations. In a randomized clinical trial of atorvastatin 20 mg as compared to placebo in patients after moderate-severe TBI (32 participants in each group), no statin–placebo difference in the volume of the contusion and its expansion was found at any time point, and yet, significantly superior scores on the Glasgow Outcome Scale and the Disability Rating Scale were observed in the statin group at the 3-month follow-up period, with no differences at baseline [204]. An important area of research is to evaluate the differential effects of statins that more easily penetrate the BBB (lipophilic ones) vs. those that act mainly at the periphery on neuronal cholesterol levels, neuroprotection, neuroproliferation, neuroinflammation, and oxidative stress. This will guide the selection of specific statins for specific molecular pathways implicated in TBI.
OTHER IMMUNE-TARGETING INTERVENTIONS IN ANIMAL MODELS OF TBI
Metabotropic glutamate receptor 5 agonists
Metabotropic glutamate receptor 5 (MGluR5) agonists, such as (RS)-2-chloro-5-hydroxyphenylglycine (CHPG), attenuate microglial activation by actions at MGlurR5 on microglia and neurons and inhibit caspase-dependent apoptosis. CHPG attenuates microglial activation through NADPH oxidase [205] with a potent neuroprotective effect observed early in treatment [206]. Moreover, a delayed administration reduced neurodegeneration and long-term inflammation. Specifically, if administered after 1 month, it lowered subsequent activation of microglia expressing NADPH oxidase subunits, inhibited hippocampal neuronal loss, and lesion progression measured by T2-weighted MRI (at one, two, and three months), as well as white matter loss measured by diffusion tensor imaging at four months post-TBI [207].
Minocycline
Minocycline, a common antibiotic, has been explored for its ability to attenuate inflammation after brain injury and improve prognosis. Minocycline is a lipophilic, tetracycline-derivative antibiotic that is most commonly associated with treatment of acne and various infections. Its anti-inflammatory and anti-edematous properties at higher concentrations were investigated in the treatment of TBI, given its favorable safety profile and ability to cross the BBB [2, 12, 208]. The drug decreases secretion of cytokines and chemokines that promote inflammation. Minocycline results in decreased downstream nitric oxide production and decreases the activation of macrophages and microglia, and decreases expression of multiple cytokines (TNF-α, IL-1β, IL-6, and MMP-9) [2, 90, 209]. Additionally, minocycline promotes neuroprotective responses after brain injury in mice [210]. Several experimental studies have shown promise for use of minocycline as an intervention after TBI. These results indicated that minocycline decreased cerebral edema lasting up to 24 h post-injury by decreasing immediate release of inflammatory markers like IL-1β and MMP-9, but with no difference in oxidative stress as measured by glutathione levels [208]. On a macroscopic level, minocycline has been shown to decrease gray and white matter changes after TBI in mice [209]. Lesions after TBI are tempered, as evidenced by decreased focal brain lesion volume and decreased olfactory bulb atrophy in treated mice [209, 211]. Minocycline was able to attenuate memory deficits, an effect that was sustained until at least 13 weeks post-injury [212]. Other studies have indicated mice with minocycline treatment after TBI perform better on the tasks of spatial learning and memory [12].
Considering the difficulty in extrapolating the results from animal studies into practice, there are a number of guidelines for future immune targeting interventions to be developed. Studies should be conducted across models and species to provide evidence of molecular or cellular target engagement, and should add histological (whenever possible) or imaging outcomes to the behavioral ones. Factors such as brain penetration as well as pharmacokinetic and pharmacodynamic measurements with optimal dosage should be identified [213] and the therapeutic window for any intervention intended to become a treatment should not be less than 6 h [213].
THE TIME COURSE OF TBI-INDUCED INFLAMMATION
As previously discussed, mechanical forces leading to TBI lead to direct, primary damage that results in axonal injury, contusion, or hemorrhage [16]. The secondary damage occurs hours to days after the initial insult [16] as a result of widespread neuroinflammation [11], oxidative stress [214], and excitotoxicity [9]. Within the inflammatory milieu, TBI is not an isolated event. Subsequent, co-occurring, and especially previous immune challenge and inflammation-mediated medical conditions interact with TBI with synergistic, antagonistic, or additive effects.
Both central and peripheral nervous systems are involved in the complex and dynamic inflammatory response induced by TBI, with genetics, location and severity of the trauma, secondary injury cascades, sex, and age as important moderators [11]. It has been proposed that modulation of the immune response could be central in identifying clinically relevant and effective therapeutic interventions for TBI, as all brain injuries trigger inflammatory responses [12].
A common immune phenomenon in the long-term recovery after TBI is the tardive enhanced inflammatory response that occurs post-injury. Experimental TBI results in changes that are usually coupled with prolonged elevation in glial/macrophage reactivity as a component of neuroinflammation [215–218], which is consistent with studies in humans that identified increased binding of PK-[11C](R)PK11195 (expressed by activated microglia) ligand on imaging between 11 months and 17 years post-injury [219], and higher mRNA expression of microglial markers CD68 and OX-6 at 1-year post-injury [220]. Many age-related neurodegenerative disorders, including Alzheimer’s disease, also have cell-mediated neuroinflammation as a prominent feature [221].
Age effects/microglial priming
Whereas microglia display an amplified proinflammatory response with aging that is referred to as “microglial priming” (first described in a model of prion disease) [222], macrophages display an age-related reduction in the proinflammatory response to an immune challenge [223]. Characteristics of a primed profile of microglia, in addition to morphological changes, include: 1) reduced activation threshold to express and release proinflammatory molecules; 2) exaggerated inflammatory response to immune challenge; and 3) increased basal expression of inflammatory markers and mediators [224]. Additionally, in older rodents, in contrast to younger rodents, although white matter demyelination injury induces a reduced macrophage/microglia response [225], a gray matter injury provokes an enhanced macrophage/microglia response [70, 226] (Fig. 2).
Fig. 2.

Traumatic brain injury-induced macrophage response varies in reaction to immune stressors that occur before, with, and after the injury. A solid black line and gray shading depicts normal, age-related health burden. A) Traumatic brain injury (TBI) occurring in the absence of Aβ (dotted black line) or tau results in acute macrophage-related neuroinflammation that subsides over time. TBI in the presence of Aβ (solid red line) results in an acute blunted macrophage response that increases at chronic post-injury time points. TBI in the presence of pathological tau (solid blue line) results in an enhanced macrophage response to TBI that remains elevated at chronic post-injury time points. B) Over time, macrophage-related neuroinflammation increases with normal health burden. Single TBI (dotted black line) results in acute macrophage-related neuroinflammation that subsides over time. Pre-injury peripheral immune challenge at sub-threshold levels (red line) attenuates the post-injury macrophage-related inflammatory response to TBI. Post-injury peripheral immune challenge (solid blue line) causes a hyper-active macrophage response correlating with behavioral dysfunction. Repetitive post-injury immune challenge (dotted blue line), similar to what is observed in repetitive TBI, increases macrophage-related neuroinflammation and correlates with the advanced neuropathology. The figure represents a model of what occurs in human TBI, translating specific findings in rodent TBI studies. (Reproduced from Kokiko-Cochran ON, Godbout JP (2018) The inflammatory continuum of traumatic brain injury and Alzheimer’s disease. Front Immunol 9, 672.)
PERSISTENT PROINFLAMMATORY PROFILES AFTER TBI
Reactive microglia
Brain trauma induces a persistent proinflammatory profile with increased inflammatory cytokines TNF-α and IL-6 (detectable in the CSF for up to 12 months and directly associated with measures of disinhibition and functional impairment [227, 228]) and priming in microglia [229–231] (Fig. 3). Increased CR3/43+ (MHC-II+) and CD68+ reactive microglia have been reported several months after brain injury in an immunohistochemical analysis of autopsied brains [232]. Especially, white matter in these autopsied brains had an increased immune load of microglial reactivity in the presence of traumatic axonal injury [232]. An association of CR3/43+ cells with increased myelin basic protein long-term after brain trauma (2–8 years) has also been reported in other studies [220].
Fig. 3.
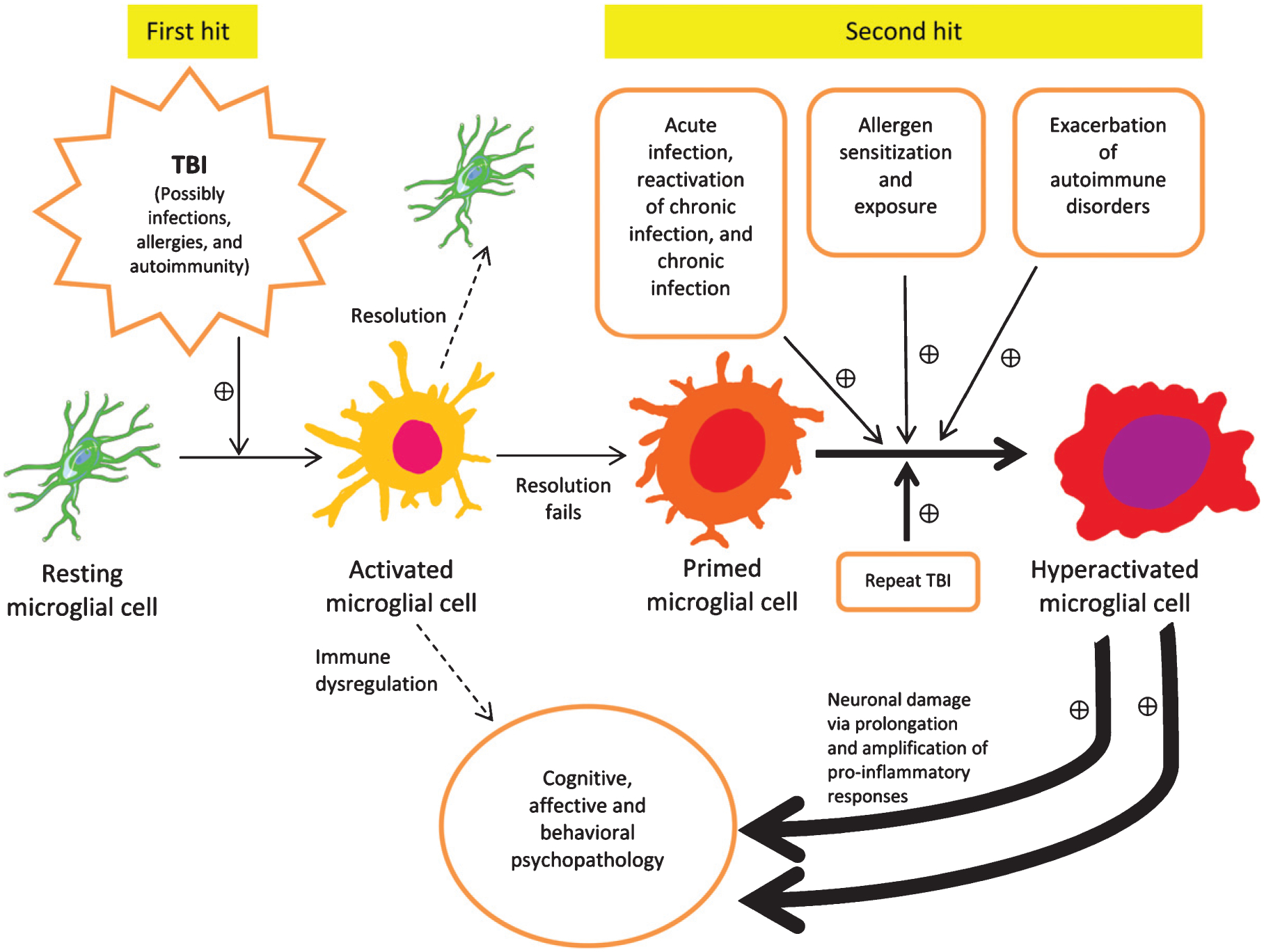
Double hit model illustrates how during the first hit microglia become activated, and then potentially primed. A second hit may include any source of inflammation, such as intense stress, autoimmune disease reactivation, allergen exposure in atopic individuals, and most commonly, infections (acute, chronic, chronic reactivated) and/or repeat TBI. The primed microglia convey a risk for developing behavioral, affective, and cognitive disorders, and exposure to subsequent inflammation driven “hits” exponentially increases that probability, as well as symptomatic intensity and functional deterioration. The double hit model is expected to fully represent all TBI models in rodents, as well as its translation to clinical TBI, with the difference that in human TBI, a multitude of individual immune stressors interact with TBI in contrast to experimental animals living in conditions characterized by limited variation between individuals in exposure to various immune modifiers. This is a top-of-the-list possible explanation of why immune targeting interventions for TBI that are beneficial in rodents do not translate in humans, with few exceptions (e.g., aerobic exercise).
Multiple Immune Challenges and Multi-hit Models
Pre-injury peripheral mild immune challenges may improve recovery following TBI:
Experimental models of TBI have a reported improved behavioral recovery and reduced lesion volume after pretreatment with low doses of LPS [233]. Additionally, attenuation of IL-1β and TNF-α overexpression and neuronal loss was found in the hippocampus when LPS treatment was administered before TBI [234]. Even a higher dose of LPS administered over shorter duration (1.0 mg/kg, single intraperitoneal dose for 4 days) prior to a lateral cryogenic brain injury has been demonstrated to decrease lesion volume and neuronal death [235]. In sum, a pre-treatment inflammatory stimulus may help protect, in part, the CNS from a secondary, generally more potent, stimulus, such as TBI, likely via pre-conditioning leading to the induction of neuroprotective responses (see Fig. 2, upper graph).
Immune challenges after TBI
A primed microglial phenotype, defined by increased expression of MHC-II and CD68 and altered morphology is consistently induced by experimental models of TBI [231, 236]. Primed microglia show some increased reactivity at baseline, but a strongly increased reactivity after an immune trigger [30].
In rats that sustained TBI via impact acceleration, an exaggerated inflammatory response associated with cognitive impairments and depressive-like behavior 3 months post-injury has been reported to be induced by administering LPS, as early as 5 days after the primary injury [237]. Findings from these studies substantiate the fact that post-TBI immune challenges negatively impact symptoms and functional outcomes and lead to increased deficits, as well as highlight the long period of increased vulnerability to subsequent immune challenges. However, a decreased production of inflammatory cytokines and reduced neuronal injury were reported if LPS treatment was given 1 day after repetitive mild TBI (3 TBIs, 5 days apart) [43]. In contrast, if the administration of LPS was delayed to 5 days after repetitive TBI, worsening of neuronal damage characterized by aggregation and phosphorylation of tau, impaired behavioral recovery, and elevation of inflammatory cytokines was observed [43]. Together, these findings emphasize the substantial temporality of the immune response to TBI and its interactions with other immune triggers.
Common clinically related immune challenges, such as allergies [238, 239], autoimmune disorders [240, 241] and/or infections [242, 243], produce a proinflammatory response and lower the threshold for priming of microglia [244, 245]. Thus, further clinical studies should investigate the role of these immune-mediated conditions and their treatment in altering the course of TBI. Clinically, there is potential benefit to be uncovered from assertively diagnosing and aggressively treating these conditions when co-occurring with TBI.
Psychological stress, a common occurrence in reaction to TBI and events that contributed to it and surrounding it, has been consistently demonstrated to upregulate immune responses acutely and chronically, with secondary loops related to hypothalamic-pituitary-adrenal (HPA) axis dysregulation (with both suppression and hyperactivity) contributing to dynamic sequential changes in immune function [246] and progression of symptomatic and functional deficits. HPA axis suppression and the diminished modulatory role of HPA axis on immune alterations post-TBI, and vulnerability to stress, sleep deprivation, and circadian dysregulation, represent important domains that may provide translational value for minimizing early symptoms and long-term consequences after TBI [246].
Immune challenges in aged- and TBI-exposed mice lead to an exaggerated immune response
Microglial priming, characterized by altered morphology and higher expression of complement receptor-3 (CD11b) and MHC-II, has been reported in aged rodents [247, 248]. As compared to young adult mice being challenged with LPS, aged animals have an increased microglial expression of IL-1β upon induction of TBI, leading to a depressive-like phenotype [249] and prolongation of sickness behavior [250], as well as greater spatial memory impairments in parallel to the higher expression of IL-6, TNF-α, and IL-1β in the hippocampus [251, 252]. As compared to TBI-exposed mice that were given saline, TBI-exposed mice that received a peripheral LPS challenge 30 days after brain injury were reported to have an exaggerated microglial response, which was depicted by higher depressive-like behaviors and increased TNF-α, MHC-II, and IL-1β expression [231]. Moreover, memory recall deficits in TBI mice are exaggerated by administration of LPS 30 days after brain injury [236].
CHRONIC NEURODEGENERATIVE CONSEQUENCES OF TBI
History of TBI is predictively associated with future onset and severity of all forms of neurodegenerative diseases, including chronic traumatic encephalopathy (CTE), Alzheimer’s, Parkinson’s, Huntington’s, and vascular dementias [253], as well as psychiatric sequelae such as depression [254] believed to be, at least in part, consequences of microglial priming and perpetuation of neuroinflammation. However, complete replacement of primed microglia with nonreactive microglia failed to reduce astrocyte-mediated inflammation, and more importantly, markers of neuronal death (reviewed by Tapp et al. [246]). A single TBI produces long-term plastic changes in multiple aspects of the immune function (i.e., acute phase response, TLR signaling, NF-κB signaling, complement system) that take part in reciprocally activating and perpetuating immune molecular networks [255]. In repetitive TBI, the first TBI is a priming event, which creates the foundation for an elevated inflammatory response that occurs upon subsequent exposure to more brain injuries. It is important to note that TBI has persistent and profound effects on systemic immunity that show similarities to changes linked with aging [256].
Chronic traumatic encephalopathy
Development of CTE, a neurodegenerative disease characterized by abnormal tau accumulation in the sulci of the cortex, is an example of chronic cognitive impairment associated with repetitive TBI via participation in contact sports [257]. Even though some cases of CTE may have presence of amyloid-β (Aβ) plaques, their location and distribution are different from those of Alzheimer’s disease subjects [258–260]. A common feature of CTE is glial reactivity, including microglial reactivity as well as astrocytic accumulation of abnormal tau [257, 261]. In a study utilizing PET imaging, retired National Football League players were reported to have increased translocator protein binding in the entorhinal cortex, supramarginal gyrus, hippocampus, and the parahippocampal cortex, as compared to sex- and age-matched control subjects who did not have a history of repetitive head trauma [262]. In parallel to these findings, other studies on American football players showed increased CD68+ microglia in their brains, which could have partially mediated the deposition of abnormal tau [263].
A recent review describes the animal models of repetitive mild TBI and includes the discussion on available evidence related to the development of tau and Aβ pathology after the brain injury [264]. The number of injuries in repetitive TBI varies from 2–10 over a period of several days or weeks, but its severity is usually referred to as “mild.” Recently, a highly repetitive mouse model of TBI has been described that includes 30 injuries [265]. Generally, on one injury day, administration of 1 or 2 brain injuries is done. At chronic post-injury time points, consistent presence of increased behavioral impairment, amyloid-β protein precursor, and phosphorylated tau have been reported in non-transgenic mice exposed to repetitive mild TBI [265].
Alzheimer’s disease
Dementia risk later in life is elevated by history of TBI [266, 267]. Chronic inflammation induced by TBI has been suggested to be a major contributor to the development of neurodegenerative disease [220, 268]. Indeed, many studies have documented an association between TBI and vulnerability to dementia, with underlying mechanisms involving damage to white matter tracts and neuronal networks, impaired cognitive resources, and deposition of a Aβ and tau [266, 269], with neurotoxic forms of tau and amyloid leading to neuroinflammation and perpetuating a cascading vicious circle [270]. The interruption of this vicious circle by targeting persistent posttraumatic immune responses is a logical aim of future clinical trials in TBI [271]. The recent confirmation of long-term dysregulation of immunoregulatory genes provides optimism through targetable molecular networks that can be modified during a much longer therapeutic window, e.g., months after brain injury [255]. Future interventional studies in animals should consider strong interactive factors that can robustly modify the immunological consequences of TBI (e.g., dementia continuum), and the application in humans should account for immunological triggers and precipitants (by design, inclusion, and adjustment) that originate in the environment, lifestyle (e.g., exercise, sleep patterns), and the temporal dynamic of immune-mediated medical conditions [272].
MICROBIOME AND PROBIOTIC INTERVENTIONS
One factor contributing to increases in chronic inflammatory disorders in Western countries is thought to be failing immunoregulation, attributable to reduced exposure to the microbial environment within which the mammalian immune system co-evolved [273]. Our understanding of the “gut-brain axis” [274] is steeply increasing. The three proposed mechanisms bidirectionally linking the gut with the brain are: 1) changes in sympathetic sensorimotor function or vagal tone, 2) alterations in neuroendocrine signaling, and 3) alterations in immune signaling and gut, as well as BBB permeability [275]. The metabolites of the microbiome’s colonizing bacteria can be both anti-inflammatory and neuroprotective [274] and the gut microbiome has a prominent role in maintaining a delicate homeostasis of the immune system [276], and integrity of barrier functions. Rodents with germ-free gastrointestinal systems have decreased tight junction integrity (and increased BBB permeability) likely contributing to increased microglial activation [274].
Changes in gut microbiome are associated with increased neuroinflammatory and autoimmune reactions in multiple sclerosis patients [277] and have been implicated in other autoimmune-mediated diseases mediated by T cells including rheumatoid arthritis, inflammatory bowel disease, and type 1 diabetes [274]. Krezalek and colleagues coined the term “pathobiome” to describe changes in the microbiome after TBI [276]. There is a proposed feedback loop in which TBI and subsequent induction of the “pathobiome” result in a neuroinflammatory cascade, thus, further augmenting the effects of brain injury by cell death, BBB disruption, edema, and hemorrhage [276]. Changes in the gut microbiome occur as soon as 2 h after a TBI in rodent studies [276], with subsequent increases in permeability in the intestinal wall making TBI recipients susceptible to translocation of bacterial pathogens, resulting in exponentially increased local, systemic, and CNS inflammation [276].
Certain studies on probiotic administration have reported increases in anti-inflammatory cytokines such as IL-10 and decreases in inflammatory cytokine production in the intestine, and decreases in stressrelated hormone production induced by brain injury [276]. A 2017 review by Brenner identifies three studies indicating a benefit for the treatment of TBI [278]. Two randomized clinical trials in TBI patients in the ICU showed patients who received probiotics had decreased ICU length of stay, decreased infections, infection with fewer number of pathogens, treatment with fewer types of antibiotics, and decreased duration (in days) on artificial ventilation. As initial studies are promising in the use of probiotics in TBI patients immediately following their injury during hospital stay [279], further research on long-term effects and benefits across various severities and types of TBI is much needed.
In sum, in TBI, in contrast to many other neuropsychiatric domains, there is a disquieting disconnect between bench and bedside, with successful preclinical studies failing to translate [255]. It is commonly stated that the lack of efficacy demonstrated in human clinical trials of very promising interventions developed in animal models of TBI, including molecules directed at immune targets, is due to the incomplete representation of human TBI by the specific animal models used, specifically the much higher pathogenetic heterogeneity of clinical TBI. Yet, we see two factors that are equally important. First, human TBI does not occur in isolation—concurrent stressors, levels of immunomodulatory hormones and vitamins, variability in fitness and common endocrine dysregulation and metabolic syndrome, common infections, allergies and exposure to allergens, past TBIs, including barely noticed concussive or subconcussive events, exposure to heat and cold, and photoperiodic and circadian factors, are all known to modify immune response, and some are known to modify neuroprotection and neurogenesis. Second, many immune factors are highly context-dependent, with a good number of them playing either a protective or degenerative role, dependent on the time sequence, dose, and interaction with other immune stressors or immune-mediated clinical conditions, either as a trait or state. Reciprocal cascading interactions exist—while there is long-term immune activation in many TBI patients, TBI has an immediate peripheral immunosuppressive effect that is similar to advanced aging [256], and that can lead to vulnerability to acute infections and reactivation of chronic infections that lead to immediate increased mortality [280], and long-term negative outcomes in TBI [281, 282]. Prior brain trauma, prior extracranial trauma, polytrauma, and their interactions with infections [283], in particular nosocomial infections, may result in a different course of brain injury and immune contribution to brain injury than if it would happen in isolation. Sleep abnormalities are often present in TBI patients [284] and in animal models of TBI [285], and are known to affect immune function, functional changes of HPA axis, and stress vulnerability [246]. These have not yet been studied in interventional animal studies of TBI that could mimic more closely the human experience of TBI. These processes are currently oversimplified in animal models that generate treatment candidates but are not formally tested before being transposed to humans. Ideally, these factors would be analytically studied in animals, determining the specific association of immune-modifying factors in interaction, considering simultaneity, precedence, sequence, repetitiveness, and dose-response [272]. While life in experimental animals is forcefully homogenized, with identical genetic background, similar food, and sleep-wake and rest-activity opportunities, it is not the case in humans. Sources of heterogeneity include genetic variation, exposure to activities that modify the response of the immune system to TBI, such as acute, latent, and chronic infections, atopy with allergen exposure, autoimmune conditions, exercise, degree of general as well as context-specific psychological stress (e.g., realization of personal loss of physical, social and economical abilities via TBI and potential loss through demise or severe sequelae in others who have been co-exposed to the same trauma as the TBI patient). Additionally, regular or recent intake of common medications for common conditions that have immunomodulatory properties (such as statins, metformin and psychotropic medications) can markedly increase immune response heterogeneity in clinical samples. Additive or multiplicative interactions between these immune-modifying factors make the specific manipulation of candidate therapeutic molecules, identified in animal research and utilized in humans, to be overly simplified, and call for concentric interventions addressing individual combinations of interacting sources of immune dysregulation in animal models. Their translation to human studies requires large multicenter collaborations allowing the adequate number of participants to select specific interacting immune-modifying factors to be addressed by specific immune targeting interventions.
TAKE HOME MESSAGE
Attenuation of secondary injury has been a major goal of neuroprotective treatment, and despite strong experimental data, many trials for human TBI have failed to produce results [213]. In our evaluation, the complexity and interindividual differences of interactive factors that up or downregulate immune function prior to TBI in humans relative to the homogenous condition of experimental animals is the main cause of poor animal to human translation of significant interventional results with beneficial outcomes in animal models. The failure of the human trials to show consistency have also been attributed to: 1) considering secondary mechanisms in isolation rather than in interaction; 2) inadequate attention to animal model species, strains, age, gender, and timing; 3) the major difference of anesthetics being used in animal models; 4) the relative impermeability of the BBB; 5) the definition of meaningful translational behavioral outcomes; 6) epigenetic variability in humans resulting in pharmacogenetic heterogeneity; 7) not considering predicting biomarkers that would justify physiologically the need of the intervention in a particular participant; 8) inadequate sample size [213]; and 9) less diffuse axonal injury because of much smaller brains in rodents. Animal studies generally do not make pharmacokinetic and pharmacodynamic measurements, and do not try to optimize dosage to tissue concentration.
While we are noticing the disturbing lack of translation between targeting the immune system in animal models and clinical TBI, there is, however, one element of congruence—aerobic exercise that has shown significant benefits (cognitive performance) in patients with mild and moderate TBI [286], as it adequately translates the rodent study on pre-conditioning with exercise (daily running for four weeks) leading to reduced increases in IL-1β, TNF-α, and markers of BBB permeability and lesser decreases in IL-10 post-brain injury, as well as less motor function impairment after fluid percussion injury in rats [287]. Similar results have been found in a mouse study suggesting that there is a therapeutic window, specifically late after TBI (5 weeks after brain trauma) for reduction of longer-term inflammation and cognitive impairment [288]. This late administration of exercise is important given that there are concerns regarding aerobic exercise early after TBI, considering that, as shown previously in this article, immune activation appears to be beneficial in the early phase of TBI for cleaning of the impacted tissue, as well as for protection of infections, in addition to increasing brain oxygen demand and generation of additional free radicals with exercise. Furthermore, the later administration of an immunomodulating intervention is consistent with literature suggesting that targetable molecular networks can be modified during a much longer therapeutic window than initially thought, e.g., months after brain injury [255], although a more exact translation of the therapeutic window in humans is highly desirable. Finding the temporal sweet-spot for switching from immunotherapies geared toward clearance of debris, reparative and neuroprotective effects of inflammation, and those geared toward decreasing chronic unremitting inflammation [11], should represent a central topic for future research.
ACKNOWLEDGMENTS
This work was supported by the Rocky Mountain MIRECC for Suicide Prevention, Aurora CO, USA, and the Military and Veteran Microbiome Consortium for Research and Education, and in part by the Distinguished Investigator Award from the American Foundation for Suicide Prevention (Postolache, PI, Rujescu, Co-I, DIG 1-162-12). This study also was supported by the VA Merit Review CSR&D grant 1I01CX001310-01A1 (Postolache, PI). Additional support for the writing of this manuscript was provided by Saint Elizabeths Hospital Psychiatry Residency training program (DC Department of Behavioral Health, Washington, DC, USA). The authors thank Po Yu Yen, Anna Spector and Nasrollah Navid for their help in editing the final version of the manuscript; Aline Dagdag for the coordination of our team; and to Lisa A. Brenner, Ph.D., for her support and encouragement throughout the project. The opinions expressed in this article belong to the authors and should not be construed as the official statements of the Veterans Affairs Administration or the American Foundation of Suicide Prevention.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1150r1).
REFERENCES
- [1].Fehily B, Fitzgerald M (2017) Repeated mild traumatic brain injury: potential mechanisms of damage. Cell Transplant 26, 1131–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV (2015) Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat 11, 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Silver JM, McAllister TW, Arciniegas DB (2019) Textbook of traumatic brain injury. American Psychiatric Publishing. [Google Scholar]
- [4].Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999) Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5, 49–55. [DOI] [PubMed] [Google Scholar]
- [6].Schwartz M (2000) Beneficial autoimmune T cells and posttraumatic neuroprotection. Ann N Y Acad Sci 917, 341–347. [DOI] [PubMed] [Google Scholar]
- [7].Schwartz M, Raposo C (2014) Protective autoimmunity: a unifying model for the immune network involved in CNS repair. Neuroscientist 20, 343–358. [DOI] [PubMed] [Google Scholar]
- [8].Anthonymuthu TS, Kenny EM, Bayir H (2016) Therapies targeting lipid peroxidation in traumatic brain injury. Brain Res 1640, 57–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dorsett CR, McGuire JL, DePasquale EA, Gardner AE, Floyd CL, McCullumsmith RE (2017) Glutamate neuro-transmission in rodent models of traumatic brain injury. J Neurotrauma 34, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Faden AI, Demediuk P, Panter SS, Vink R (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244, 798–800. [DOI] [PubMed] [Google Scholar]
- [11].Simon DW, McGeachy MJ, Bayir H, Clark RSB, Loane DJ, Kochanek PM (2017) The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol 13, 572. [DOI] [PubMed] [Google Scholar]
- [12].Bergold PJ (2016) Treatment of traumatic brain injury with anti-inflammatory drugs. Exp Neurol 275 Pt 3, 367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Corps KN, Roth TL, McGavern DB (2015) Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, Fernandes J, Gogichaisvili T, Golden N, Hartzenberg B, Husain M, Ulloa MI, Jerbi Z, Khamis H, Komolafe E, Laloe V, Lomas G, Ludwig S, Mazairac G, Munoz Sanchez Mde L, Nasi L, Olldashi F, Plunkett P, Roberts I, Sandercock P, Shakur H, Soler C, Stocker R, Svoboda P, Trenkler S, Venkataramana NK, Wasserberg J, Yates D, Yutthakasemsunt S (2005) Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 365, 1957–1959. [DOI] [PubMed] [Google Scholar]
- [15].Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, Laloe V, Munoz-Sanchez A, Arango M, Hartzenberg B, Khamis H, Yutthakasemsunt S, Komolafe E, Olldashi F, Yadav Y, Murillo-Cabezas F, Shakur H, Edwards P (2004) Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet 364, 1321–1328. [DOI] [PubMed] [Google Scholar]
- [16].Maas AI, Stocchetti N, Bullock R (2008) Moderate and severe traumatic brain injury in adults. Lancet Neurol 7, 728–741. [DOI] [PubMed] [Google Scholar]
- [17].Gardner RC, Dams-O’Connor K, Morrissey MR, Manley GT (2018) Geriatric traumatic brain injury: epidemiology, outcomes, knowledge gaps, and future directions. J Neurotrauma 35, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung Y-C, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM (2018) Estimating the global incidence of traumatic brain injury. J Neurosurg 1, 1–18. [DOI] [PubMed] [Google Scholar]
- [19].Nguyen R, Fiest KM, McChesney J, Kwon C-S, Jette N, Frolkis AD, Atta C, Mah S, Dhaliwal H, Reid A (2016) The international incidence of traumatic brain injury: a systematic review and meta-analysis. Can J Neurol Sci 43, 774–785. [DOI] [PubMed] [Google Scholar]
- [20].Murphy S, Duhaime A-C (2016) The role of neuroimaging in the diagnosis, treatment, and prognosis of traumatic brain injury in children. J Pediatr Neuroradiol 5, 002–006. [Google Scholar]
- [21].Dewan MC, Mummareddy N, Wellons JC III, Bonfield CM (2016) Epidemiology of global pediatric traumatic brain injury: qualitative review. World Neurosurg 91, 497–509. e491. [DOI] [PubMed] [Google Scholar]
- [22].Helmick KM, Spells CA, Malik SZ, Davies CA, Marion DW, Hinds SR (2015) Traumatic brain injury in the US military: epidemiology and key clinical and research programs. Brain Imaging Behav 9, 358–366. [DOI] [PubMed] [Google Scholar]
- [23].Chiu C-C, Liao Y-E, Yang L-Y, Wang J-Y, Tweedie D, Karnati HK, Greig NH, Wang J-Y (2016) Neuroinflammation in animal models of traumatic brain injury. J Neurosci Methods 272, 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shultz SR, McDonald SJ, Vonder Haar C, Meconi A, Vink R, van Donkelaar P, Taneja C, Iverson GL, Christie BR (2017) The potential for animal models to provide insight into mild traumatic brain injury: Translational challenges and strategies. Neurosci Biobehav Rev 76, 396–414. [DOI] [PubMed] [Google Scholar]
- [25].Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev Neurosci 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cernak I (2005) Animal models of head trauma. NeuroRx 2, 410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ma X, Aravind A, Pfister BJ, Chandra N, Haorah J (2019) Animal models of traumatic brain injury and assessment of injury severity. Mol Neurobiol 56, 5332–5345. [DOI] [PubMed] [Google Scholar]
- [28].Pekny M, Pekna M (2016) Reactive gliosis in the pathogenesis of CNS diseases. Biochim Biophys Acta 1862, 483–491. [DOI] [PubMed] [Google Scholar]
- [29].Donat CK, Scott G, Gentleman SM, Sastre M (2017) Microglial activation in traumatic brain injury. Front Aging Neurosci 9, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Witcher KG, Eiferman DS, Godbout JP (2015) Priming the inflammatory pump of the CNS after traumatic brain injury. Trends Neurosci 38, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Raghupathi R, Mehr MF, Helfaer MA, Margulies SS (2004) Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J Neurotrauma 21, 307–316. [DOI] [PubMed] [Google Scholar]
- [32].Krave U, Al-Olama M, Hansson H-A (2011) Rotational acceleration closed head flexion trauma generates more extensive diffuse brain injury than extension trauma. J Neurotrauma 28, 57–70. [DOI] [PubMed] [Google Scholar]
- [33].Fijalkowski RJ, Stemper BD, Pintar FA, Yoganandan N, Crowe MJ, Gennarelli TA (2007) New rat model for diffuse brain injury using coronal plane angular acceleration. J Neurotrauma 24, 1387–1398. [DOI] [PubMed] [Google Scholar]
- [34].Friess SH, Ichord RN, Ralston J, Ryall K, Helfaer MA, Smith C, Margulies SS (2009) Repeated traumatic brain injury affects composite cognitive function in piglets. J Neurotrauma 26, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gao W, Xu H, Liang M, Huang JH, He X (2013) Association between reduced expression of hippocampal glucocorticoid receptors and cognitive dysfunction in a rat model of traumatic brain injury due to lateral head acceleration. Neurosci Lett 533, 50–54. [DOI] [PubMed] [Google Scholar]
- [36].Long JB, Bentley TL, Wessner KA, Cerone C, Sweeney S, Bauman RA (2009) Blast overpressure in rats: recreating a battlefield injury in the laboratory. J Neurotrauma 26, 827–840. [DOI] [PubMed] [Google Scholar]
- [37].Holmin S, Soderlund J, Biberfeld P, Mathiesen T (1998) Intracerebral inflammation after human brain contusion. Neurosurgery 42, 291–298; discussion 298–299. [DOI] [PubMed] [Google Scholar]
- [38].Harish G, Mahadevan A, Pruthi N, Sreenivasamurthy SK, Puttamallesh VN, Keshava Prasad TS, Shankar SK, Srinivas Bharath MM (2015) Characterization of traumatic brain injury in human brains reveals distinct cellular and molecular changes in contusion and pericontusion. J Neurochem 134, 156–172. [DOI] [PubMed] [Google Scholar]
- [39].Foda MA, Marmarou A (1994) A new model of diffuse brain injury in rats: Part II: Morphological characterization. J Neurosurg 80, 301–313. [DOI] [PubMed] [Google Scholar]
- [40].Marmarou A, Foda MAA-E, Brink Wvd, Campbell J, Kita H, Demetriadou K (1994) A new model of diffuse brain injury in rats: Part I: Pathophysiology and biomechanics. J Neurosurg 80, 291–300. [DOI] [PubMed] [Google Scholar]
- [41].Khuman J, Meehan III WP, Zhu X, Qiu J, Hoffmann U, Zhang J, Giovannone E, Lo EH, Whalen MJ (2011) Tumor necrosis factor alpha and Fas receptor contribute to cognitive deficits independent of cell death after concussive traumatic brain injury in mice. J Cereb Blood Flow Metab 31, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kane MJ, Angoa-Pérez M, Briggs DI, Viano DC, Kreipke CW, Kuhn DM (2012) A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods 203, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Corrigan F, Arulsamy A, Collins-Praino LE, Holmes JL, Vink R (2017) Toll like receptor 4 activation can be either detrimental or beneficial following mild repetitive traumatic brain injury depending on timing of activation. Brain Behav Immun 64, 124–139. [DOI] [PubMed] [Google Scholar]
- [44].McAteer KM, Corrigan F, Thornton E, Turner RJ, Vink R (2016) Short and long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLoS One 11, e0160220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Acabchuk R, Briggs DI, Angoa-Pérez M, Powers M, Wolferz R Jr, Soloway M, Stern M, Talbot LR, Kuhn DM, Conover JC (2016) Repeated mild traumatic brain injury causes focal response in lateral septum and hippocampus. Concussion 1, CNC13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Albayram O, Kondo A, Mannix R, Smith C, Tsai C-Y, Li C, Herbert MK, Qiu J, Monuteaux M, Driver J (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Weil ZM, Gaier KR, Karelina K (2014) Injury timing alters metabolic, inflammatory and functional outcomes following repeated mild traumatic brain injury. Neurobiol Dis 70, 108–116. [DOI] [PubMed] [Google Scholar]
- [48].Uryu K, Laurer H, McIntosh T, Praticò D, Martinez D, Leight S, Lee VM-Y, Trojanowski JQ (2002) Repetitive mild brain trauma accelerates Aβ deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci 22, 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Huh JW, Widing AG, Raghupathi R (2007) Repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: a preliminary report. J Neurotrauma 24, 15–27. [DOI] [PubMed] [Google Scholar]
- [50].Raghupathi R, Huh JW (2007) Diffuse brain injury in the immature rat: evidence for an age-at-injury effect on cognitive function and histopathologic damage. J Neurotrauma 24, 1596–1608. [DOI] [PubMed] [Google Scholar]
- [51].Cernak I, Vink R, Zapple DN, Cruz MI, Ahmed F, Chang T, Fricke ST, Faden AI (2004) The pathobiology of moderate diffuse traumatic brain injury as identified using a new experimental model of injury in rats. Neurobiol Dis 17, 29–43. [DOI] [PubMed] [Google Scholar]
- [52].Namjoshi DR, Cheng WH, McInnes KA, Martens KM, Carr M, Wilkinson A, Fan J, Robert J, Hayat A, Cripton PA (2014) Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): a novel, surgery-free model of traumatic brain injury. Mol Neurodegener 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Haber M, Hutchinson E, Sadeghi N, Cheng W, Namjoshi D, Cripton P, Irfanoglu M, Wellington C, Diaz-Arrastia R, Pierpaoli C (2017) Defining an analytic framework to evaluate quantitative MRI markers of traumatic axonal injury: preliminary results in a mouse closed head injury model. eNeuro 4, ENEURO.0164–17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Namjoshi DR, Cheng WH, Bashir A, Wilkinson A, Stukas S, Martens KM, Whyte T, Abebe ZA, McInnes KA, Cripton PA (2017) Defining the biomechanical and biological threshold of murine mild traumatic brain injury using CHIMERA (Closed Head Impact Model of Engineered Rotational Acceleration). Exp Neurol 292, 80–91. [DOI] [PubMed] [Google Scholar]
- [55].Cheng WH, Stukas S, Martens KM, Namjoshi DR, Button EB, Wilkinson A, Bashir A, Robert J, Cripton PA, Wellington CL (2018) Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp Neurol 301, 26–38. [DOI] [PubMed] [Google Scholar]
- [56].Haar CV, Martens KM, Bashir A, McInnes KA, Cheng WH, Cheung H, Stukas S, Barron C, Ladner T, Welch KA (2019) Repetitive closed-head impact model of engineered rotational acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic innervation in the olfactory tubercle and generates white matter inflammation, tau phosphorylation and degeneration. Exp Neurol 317, 87–99. [DOI] [PubMed] [Google Scholar]
- [57].Viano DC, Hamberger A, Bolouri H, Säljö A (2009) Concussion in professional football: animal model of brain injury—part 15. Neurosurgery 64, 1162–1173. [DOI] [PubMed] [Google Scholar]
- [58].Mychasiuk R, Hehar H, Candy S, Ma I, Esser MJ (2016) The direction of the acceleration and rotational forces associated with mild traumatic brain injury in rodents effect behavioural and molecular outcomes. J Neurosci Methods 257, 168–178. [DOI] [PubMed] [Google Scholar]
- [59].Leung LY, Larimore Z, Holmes L, Cartagena C, Mountney A, Deng-Bryant Y, Schmid K, Shear D, Tortella F (2014) The WRAIR projectile concussive impact model of mild traumatic brain injury: re-design, testing and preclinical validation. Ann Biomed Eng 42, 1618–1630. [DOI] [PubMed] [Google Scholar]
- [60].Browning JR, Whiteman AC, Leung LY, Lu X-CM, Shear DA (2017) Air-puff induced vocalizations: A novel approach to detecting negative affective state following concussion in rats. J Neurosci Methods 275, 45–49. [DOI] [PubMed] [Google Scholar]
- [61].Mountney A, Boutté AM, Cartagena CM, Flerlage WF, Johnson WD, Rho C, Lu X-C, Yarnell A, Marcsisin S, Sousa J (2017) Functional and molecular correlates after single and repeated rat closed-head concussion: indices of vulnerability after brain injury. J Neurotrauma 34, 2768–2789. [DOI] [PubMed] [Google Scholar]
- [62].Madathil SK, Wilfred BS, Urankar SE, Yang W, Leung LY, Gilsdorf J, Shear D (2018) Early microglial activation following closed-head concussive injury is dominated by pro-inflammatory M-1 type. Front Neurol 9, 964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cho HJ, Sajja VSSS, VandeVord PJ, Lee YW (2013) Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience 253, 9–20. [DOI] [PubMed] [Google Scholar]
- [64].Kochanek PM, Dixon CE, Shellington DK, Shin SS, Bayír H, Jackson EK, Kagan VE, Yan HQ, Swauger PV, Parks SA (2013) Screening of biochemical and molecular mechanisms of secondary injury and repair in the brain after experimental blast-induced traumatic brain injury in rats. J Neurotrauma 30, 920–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lighthall JW (1988) Controlled cortical impact: a new experimental brain injury model. J Neurotrauma 5, 1–15. [DOI] [PubMed] [Google Scholar]
- [66].Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39, 253–262. [DOI] [PubMed] [Google Scholar]
- [67].Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, Saatman KE, Raghupathi R, Hoshino S, Grady MS (2001) Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg 95, 859–870. [DOI] [PubMed] [Google Scholar]
- [68].Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW (2005) Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma 22, 252–265. [DOI] [PubMed] [Google Scholar]
- [69].Dalgard C, Cole J, Kean W, Lucky J, Sukumar G, McMullen D, Pollard H, Watson W (2012) The cytokine temporal profile in rat cortex after controlled cortical impact. Front Mol Neurosci 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Sandhir R, Onyszchuk G, Berman NEJ (2008) Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol 213, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Dixon CE, Lyeth BG, Povlishock JT, Findling RL, Hamm RJ, Marmarou A, Young HF, Hayes RL (1987) A fluid percussion model of experimental brain injury in the rat. J Neurosurg 67, 110–119. [DOI] [PubMed] [Google Scholar]
- [72].Sullivan HG, Martinez J, Becker DP, Miller JD, Griffith R, Wist AO (1976) Fluid-percussion model of mechanical brain injury in the cat. J Neurosurg 45, 520. [PubMed] [Google Scholar]
- [73].Stalhammar D, Galinat BJ, Allen AM, Becker DP, Stonnington HH, Hayes RL (1987) A new model of concussive brain injury in the cat produced by extradural fluid volume loading: I. Biomechanical properties. Brain Inj 1, 73–91. [DOI] [PubMed] [Google Scholar]
- [74].Lyeth BG (2016) Historical review of the fluid-percussion TBI model. Front Neurol 7, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kabadi SV, Hilton GD, Stoica BA, Zapple DN, Faden AI (2010) Fluid-percussion–induced traumatic brain injury model in rats. Nat Protoc 5, 1552–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Shultz SR, Bao F, Omana V, Chiu C, Brown A, Cain DP (2012) Repeated mild lateral fluid percussion brain injury in the rat causes cumulative long-term behavioral impairments, neuroinflammation, and cortical loss in an animal model of repeated concussion. J Neurotrauma 29, 281–294. [DOI] [PubMed] [Google Scholar]
- [77].Clausen F, Marklund N, Hillered L (2018) Acute inflammatory biomarker responses to diffuse traumatic brain injury in the rat monitored by a novel microdialysis technique. J Neurotrauma 36, 201–211. [DOI] [PubMed] [Google Scholar]
- [78].Williams AJ, Hartings JA, Lu X-CM, Rolli ML, Dave JR, Tortella FC (2005) Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma 22, 313–331. [DOI] [PubMed] [Google Scholar]
- [79].Williams AJ, Ling GSF, Tortella FC (2006) Severity level and injury track determine outcome following a penetrating ballistic-like brain injury in the rat. Neurosci Lett 408, 183–188. [DOI] [PubMed] [Google Scholar]
- [80].Shear DA, Lu X-CM, Pedersen R, Wei G, Chen Z, Davis A, Yao C, Dave J, Tortella FC (2011) Severity profile of penetrating ballistic-like brain injury on neurofunctional outcome, blood–brain barrier permeability, and brain edema formation. J Neurotrauma 28, 2185–2195. [DOI] [PubMed] [Google Scholar]
- [81].Williams AJ, Wei HH, Dave JR, Tortella FC (2007) Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J Neuroinflammation 4, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wei HH, Lu X-CM, Shear DA, Waghray A, Yao C, Tortella FC, Dave JR (2009) NNZ-2566 treatment inhibits neuroinflammation and pro-inflammatory cytokine expression induced by experimental penetrating ballistic-like brain injury in rats. J Neuroinflammation 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Chaplin DD (2010) Overview of the immune response. J Allergy Clin Immunol 125, S3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sonnenberg GF, Artis D (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].PrabhuDas MR, Baldwin CL, Bollyky PL, Bowdish DME, Drickamer K, Febbraio M, Herz J, Kobzik L, Krieger M, Loike J, McVicker B, Means TK, Moestrup SK, Post SR, Sawamura T, Silverstein S, Speth RC, Telfer JC, Thiele GM, Wang XY, Wright SD, El Khoury J (2017) A consensus definitive classification of scavenger receptors and their roles in health and disease. J Immunol 198, 3775–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Schenkel JM, Masopust D (2014) Tissue-resident memory T cells. Immunity 41, 886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Weisel F, Shlomchik M (2017) Memory B cells of mice and humans. Annu Rev Immunol 35, 255–284. [DOI] [PubMed] [Google Scholar]
- [88].Warrington R, Watson W, Kim HL, Antonetti FR (2011) An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol 7 Suppl 1, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Gyoneva S, Ransohoff RM (2015) Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci 36, 471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jassam YN, Izzy S, Whalen M, McGavern DB, El Khoury J (2017) Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron 95, 1246–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81, 1–5. [DOI] [PubMed] [Google Scholar]
- [92].Jounai N, Kobiyama K, Takeshita F, Ishii KJ (2012) Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front Cell Infect Microbiol 2, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Matzinger P (1994) Tolerance, danger, and the extended family. Annu Rev Immunol 12, 991–1045. [DOI] [PubMed] [Google Scholar]
- [94].Lee H, Lee S, Cho IH, Lee SJ (2013) Toll-like receptors: sensor molecules for detecting damage to the nervous system. Curr Protein Pept Sci 14, 33–42. [DOI] [PubMed] [Google Scholar]
- [95].Hang CH, Shi JX, Li JS, Wu W, Yin HX (2005) Concomitant upregulation of nuclear factor-kB activity, proinflammatory cytokines and ICAM-1 in the injured brain after cortical contusion trauma in a rat model. Neurol India 53, 312–317. [DOI] [PubMed] [Google Scholar]
- [96].Ahmad A, Crupi R, Campolo M, Genovese T, Esposito E, Cuzzocrea S (2013) Absence of TLR4 reduces neurovascular unit and secondary inflammatory process after traumatic brain injury in mice. PLoS One 8, e57208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, Dhandapani KM (2014) High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 62, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Gao TL, Yuan XT, Yang D, Dai HL, Wang WJ, Peng X, Shao HJ, Jin ZF, Fu ZJ (2012) Expression of HMGB1 and RAGE in rat and human brains after traumatic brain injury. J Trauma Acute Care Surg 72, 643–649. [DOI] [PubMed] [Google Scholar]
- [99].Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A (2008) HMGB1: endogenous danger signaling. Mol Med 14, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Parker TM, Nguyen AH, Rabang JR, Patil AA, Agrawal DK (2017) The danger zone: Systematic review of the role of HMGB1 danger signalling in traumatic brain injury. Brain Inj 31, 2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. [DOI] [PubMed] [Google Scholar]
- [102].Okuma Y, Liu K, Wake H, Zhang J, Maruo T, Date I, Yoshino T, Ohtsuka A, Otani N, Tomura S, Shima K, Yamamoto Y, Yamamoto H, Takahashi HK, Mori S, Nishibori M (2012) Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann Neurol 72, 373–384. [DOI] [PubMed] [Google Scholar]
- [103].Binder RJ (2014) Functions of heat shock proteins in pathways of the innate and adaptive immune system. J Immunol 193, 5765–5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zuo D, Subjeck J, Wang XY (2016) Unfolding the role of large heat shock proteins: new insights and therapeutic implications. Front Immunol 7, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Van Molle W, Wielockx B, Mahieu T, Takada M, Taniguchi T, Sekikawa K, Libert C (2002) HSP70 protects against TNF-induced lethal inflammatory shock. Immunity 16, 685–695. [DOI] [PubMed] [Google Scholar]
- [106].Kim JY, Kim N, Zheng Z, Lee JE, Yenari MA (2013) The 70 kDa heat shock protein protects against experimental traumatic brain injury. Neurobiol Dis 58, 289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Eroglu B, Kimbler DE, Pang J, Choi J, Moskophidis D, Yanasak N, Dhandapani KM, Mivechi NF (2014) Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J Neurochem 130, 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kim N, Kim JY, Yenari MA (2015) Pharmacological induction of the 70-kDa heat shock protein protects against brain injury. Neuroscience 284, 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Eltzschig HK, Sitkovsky MV, Robson SC (2012) Purinergic signaling during inflammation. N Engl J Med 367, 2322–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Junger WG (2011) Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol 11, 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P (2010) Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366. [DOI] [PubMed] [Google Scholar]
- [113].Gold M, El Khoury J (2015) beta-amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin Immunopathol 37, 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- [115].Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH (2013) Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38, 2072–2083. [DOI] [PubMed] [Google Scholar]
- [116].Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15, 84–97. [DOI] [PubMed] [Google Scholar]
- [117].Adamczak S, Dale G, de Rivero Vaccari JP, Bullock MR, Dietrich WD, Keane RW (2012) Inflammasome proteins in cerebrospinal fluid of brain-injured patients as biomarkers of functional outcome: clinical article. J Neurosurg 117, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lagraoui M, Latoche JR, Cartwright NG, Sukumar G, Dalgard CL, Schaefer BC (2012) Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Front Neurol 3, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Keel M, Trentz O (2005) Pathophysiology of polytrauma. Injury 36, 691–709. [DOI] [PubMed] [Google Scholar]
- [120].Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S (2009) Systemic inflammatory response following acute traumatic brain injury. Front Biosci (Landmark Ed) 14, 3795–3813. [DOI] [PubMed] [Google Scholar]
- [121].Weaver LC, Bao F, Dekaban GA, Hryciw T, Shultz SR, Cain DP, Brown A (2015) CD11d integrin blockade reduces the systemic inflammatory response syndrome after traumatic brain injury in rats. Exp Neurol 271, 409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Wilcockson DC, Campbell SJ, Anthony DC, Perry VH (2002) The systemic and local acute phase response following acute brain injury. J Cereb Blood Flow Metab 22, 318–326. [DOI] [PubMed] [Google Scholar]
- [123].Kossmann T, Stahel PF, Morganti-Kossmann MC, Jones JL, Barnum SR (1997) Elevated levels of the complement components C3 and factor B in ventricular cerebrospinal fluid of patients with traumatic brain injury. J Neuroimmunol 73, 63–69. [DOI] [PubMed] [Google Scholar]
- [124].Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T (2001) Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma 18, 773–781. [DOI] [PubMed] [Google Scholar]
- [125].Fluiter K, Opperhuizen AL, Morgan BP, Baas F, Ramaglia V (2014) Inhibition of the membrane attack complex of the complement system reduces secondary neuroaxonal loss and promotes neurologic recovery after traumatic brain injury in mice. J Immunol 192, 2339–2348. [DOI] [PubMed] [Google Scholar]
- [126].Leinhase I, Holers VM, Thurman JM, Harhausen D, Schmidt OI, Pietzcker M, Taha ME, Rittirsch D, Huber-Lang M, Smith WR, Ward PA, Stahel PF (2006) Reduced neuronal cell death after experimental brain injury in mice lacking a functional alternative pathway of complement activation. BMC Neurosci 7, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Leinhase I, Schmidt OI, Thurman JM, Hossini AM, Rozanski M, Taha ME, Scheffler A, John T, Smith WR, Holers VM, Stahel PF (2006) Pharmacological complement inhibition at the C3 convertase level promotes neuronal survival, neuroprotective intracerebral gene expression, and neurological outcome after traumatic brain injury. Exp Neurol 199, 454–464. [DOI] [PubMed] [Google Scholar]
- [128].Leinhase I, Rozanski M, Harhausen D, Thurman JM, Schmidt OI, Hossini AM, Taha ME, Rittirsch D, Ward PA, Holers VM, Ertel W, Stahel PF (2007) Inhibition of the alternative complement activation pathway in traumatic brain injury by a monoclonal anti-factor B antibody: a randomized placebo-controlled study in mice. J Neuroinflammation 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Longhi L, Perego C, Ortolano F, Zanier ER, Bianchi P, Stocchetti N, McIntosh TK, De Simoni MG (2009) C1-inhibitor attenuates neurobehavioral deficits and reduces contusion volume after controlled cortical impact brain injury in mice. Crit Care Med 37, 659–665. [DOI] [PubMed] [Google Scholar]
- [130].Rich MC, Keene CN, Neher MD, Johnson K, Yu ZX, Ganivet A, Holers VM, Stahel PF (2016) Site-targeted complement inhibition by a complement receptor 2-conjugated inhibitor (mTT30) ameliorates post-injury neuropathology in mouse brains. Neurosci Lett 617, 188–194. [DOI] [PubMed] [Google Scholar]
- [131].Stahel PF, Flierl MA, Morgan BP, Persigehl I, Stoll C, Conrad C, Touban BM, Smith WR, Beauchamp K, Schmidt OI, Ertel W, Leinhase I (2009) Absence of the complement regulatory molecule CD59a leads to exacerbated neuropathology after traumatic brain injury in mice. J Neuroinflammation 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].You Z, Yang J, Takahashi K, Yager PH, Kim HH, Qin T, Stahl GL, Ezekowitz RA, Carroll MC, Whalen MJ (2007) Reduced tissue damage and improved recovery of motor function after traumatic brain injury in mice deficient in complement component C4. J Cereb Blood Flow Metab 27, 1954–1964. [DOI] [PubMed] [Google Scholar]
- [133].Ziebell JM, Morganti-Kossmann MC (2010) Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 7, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hayakata T, Shiozaki T, Tasaki O, Ikegawa H, Inoue Y, Toshiyuki F, Hosotubo H, Kieko F, Yamashita T, Tanaka H, Shimazu T, Sugimoto H (2004) Changes in CSF S100B and cytokine concentrations in early-phase severe traumatic brain injury. Shock 22, 102–107. [DOI] [PubMed] [Google Scholar]
- [135].Shiozaki T, Hayakata T, Tasaki O, Hosotubo H, Fuijita K, Mouri T, Tajima G, Kajino K, Nakae H, Tanaka H, Shimazu T, Sugimoto H (2005) Cerebrospinal fluid concentrations of anti-inflammatory mediators in early-phase severe traumatic brain injury. Shock 23, 406–410. [DOI] [PubMed] [Google Scholar]
- [136].Jones NC, Prior MJ, Burden-Teh E, Marsden CA, Morris PG, Murphy S (2005) Antagonism of the interleukin-1 receptor following traumatic brain injury in the mouse reduces the number of nitric oxide synthase-2-positive cells and improves anatomical and functional outcomes. Eur J Neurosci 22, 72–78. [DOI] [PubMed] [Google Scholar]
- [137].Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, Otto VI, Rancan M, Kossmann T, Redaelli CA, Trentz O, Shohami E, Stahel PF (2002) Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab 22, 971–978. [DOI] [PubMed] [Google Scholar]
- [138].Chio CC, Lin JW, Chang MW, Wang CC, Kuo JR, Yang CZ, Chang CP (2010) Therapeutic evaluation of etanercept in a model of traumatic brain injury. J Neurochem 115, 921–929. [DOI] [PubMed] [Google Scholar]
- [139].Shohami E, Bass R, Wallach D, Yamin A, Gallily R (1996) Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab 16, 378–384. [DOI] [PubMed] [Google Scholar]
- [140].Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK (1999) Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A 96, 8721–8726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Bermpohl D, You Z, Lo EH, Kim HH, Whalen MJ (2007) TNF alpha and Fas mediate tissue damage and functional outcome after traumatic brain injury in mice. J Cereb Blood Flow Metab 27, 1806–1818. [DOI] [PubMed] [Google Scholar]
- [142].Yang J, You Z, Kim HH, Hwang SK, Khuman J, Guo S, Lo EH, Whalen MJ (2010) Genetic analysis of the role of tumor necrosis factor receptors in functional outcome after traumatic brain injury in mice. J Neurotrauma 27, 1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Knoblach SM, Faden AI (1998) Interleukin-10 improves outcome and alters proinflammatory cytokine expression after experimental traumatic brain injury. Exp Neurol 153, 143–151. [DOI] [PubMed] [Google Scholar]
- [144].Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM (1997) Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol 61, 279–285. [DOI] [PubMed] [Google Scholar]
- [145].Clark RS, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM (1994) Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma 11, 499–506. [DOI] [PubMed] [Google Scholar]
- [146].Szmydynger-Chodobska J, Strazielle N, Zink BJ, Ghersi-Egea JF, Chodobski A (2009) The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J Cereb Blood Flow Metab 29, 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Rhind SG, Crnko NT, Baker AJ, Morrison LJ, Shek PN, Scarpelini S, Rizoli SB (2010) Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J Neuroinflammation 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Semple BD, Bye N, Ziebell JM, Morganti-Kossmann MC (2010) Deficiency of the chemokine receptor CXCR2 attenuates neutrophil infiltration and cortical damage following closed head injury. Neurobiol Dis 40, 394–403. [DOI] [PubMed] [Google Scholar]
- [149].Lammermann T, Afonso PV, Angermann BR, Wang JM, Kastenmuller W, Parent CA, Germain RN (2013) Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 498, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Stirling DP, Liu S, Kubes P, Yong VW (2009) Depletion of Ly6 G/Gr-1 leukocytes after spinal cord injury in mice alters wound healing and worsens neurological outcome. J Neurosci 29, 753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Whalen MJ, Carlos TM, Kochanek PM, Clark RS, Heineman S, Schiding JK, Franicola D, Memarzadeh F, Lo W, Marion DW, Dekosky ST (1999) Neutrophils do not mediate blood-brain barrier permeability early after controlled cortical impact in rats. J Neurotrauma 16, 583–594. [DOI] [PubMed] [Google Scholar]
- [152].Whalen MJ, Carlos TM, Wisniewski SR, Clark RS, Mellick JA, Marion DW, Kochanek PM (2000) Effect of neutropenia and granulocyte colony stimulating factor-induced neutrophilia on blood-brain barrier permeability and brain edema after traumatic brain injury in rats. Crit Care Med 28, 3710–3717. [DOI] [PubMed] [Google Scholar]
- [153].Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F (2012) Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Semple BD, Trivedi A, Gimlin K, Noble-Haeusslein LJ (2015) Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol Dis 74, 263–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Rua R, McGavern DB (2015) Elucidation of monocyte/macrophage dynamics and function by intravital imaging. J Leukoc Biol 98, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44, 450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Beschorner R, Nguyen TD, Gozalan F, Pedal I, Mattern R, Schluesener HJ, Meyermann R, Schwab JM (2002) CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol 103, 541–549. [DOI] [PubMed] [Google Scholar]
- [158].Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC (2010) Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2−/− mice. J Cereb Blood Flow Metab 30, 769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, Chodobski A (2012) Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab 32, 93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Gyoneva S, Kim D, Katsumoto A, Kokiko-Cochran ON, Lamb BT, Ransohoff RM (2015) Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J Neuroinflammation 12, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, Cozen ML, Charo I, Huang EJ, Liu J, Nakamura MC (2014) CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma 31, 1677–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S (2015) CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT (2017) Triggering receptor expressed on myeloid cells 2 deficiency alters acute macrophage distribution and improves recovery after traumatic brain injury. J Neurotrauma 34, 423–435. [DOI] [PubMed] [Google Scholar]
- [164].Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med 6, e1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O, Schwartz M, Kokaia Z (2016) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36, 4182–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Burda JE, Bernstein AM, Sofroniew MV (2016) Astrocyte roles in traumatic brain injury. Exp Neurol 275 Pt 3, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sofroniew MV (2015) Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci 16, 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Clausen F, Lorant T, Lewen A, Hillered L (2007) T lymphocyte trafficking: a novel target for neuroprotection in traumatic brain injury. J Neurotrauma 24, 1295–1307. [DOI] [PubMed] [Google Scholar]
- [169].Lenzlinger PM, Hans VH, Joller-Jemelka HI, Trentz O, Morganti-Kossmann MC, Kossmann T (2001) Markers for cell-mediated immune response are elevated in cerebrospinal fluid and serum after severe traumatic brain injury in humans. J Neurotrauma 18, 479–489. [DOI] [PubMed] [Google Scholar]
- [170].Hirschberg DL, Moalem G, He J, Mor F, Cohen IR, Schwartz M (1998) Accumulation of passively transferred primed T cells independently of their antigen specificity following central nervous system trauma. J Neuroimmunol 89, 88–96. [DOI] [PubMed] [Google Scholar]
- [171].Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW (2003) Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci Lett 349, 136–138. [DOI] [PubMed] [Google Scholar]
- [172].Sparvero LJ, Amoscato AA, Kochanek PM, Pitt BR, Kagan VE, Bayir H (2010) Mass-spectrometry based oxidative lipidomics and lipid imaging: applications in traumatic brain injury. J Neurochem 115, 1322–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Mallah K, Quanico J, Trede D, Kobeissy F, Zibara K, Salzet M, Fournier I (2018) Lipid changes associated with traumatic brain injury revealed by 3D MALDI-MSI. Anal Chem 90, 10568–10576. [DOI] [PubMed] [Google Scholar]
- [174].Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, Balaban CD, Barbacci D, Schultz JA, Gouty S, Cox BM, Woods AS (2016) Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J Neurosci Methods 272, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Ariel A, Serhan CN (2007) Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 28, 176–183. [DOI] [PubMed] [Google Scholar]
- [176].Serhan CN, Haeggstrom J (2010) Lipid mediators in acute inflammation and resolution: Eicosanoids, PAF, resolvins, and protectins. In Fundamentals of Inflammation, Serhan CN, Ward PA, Gilroy DW. Cambridge University Press, pp. 153–174. [Google Scholar]
- [177].Calder PC (2006) n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr 83, 1505s–1519 s. [DOI] [PubMed] [Google Scholar]
- [178].Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8, 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Innes JK, Calder PC (2018) Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids 132, 41–48. [DOI] [PubMed] [Google Scholar]
- [180].Mills JD, Bailes JE, Sedney CL, Hutchins H, Sears B (2011) Omega-3 fatty acid supplementation and reduction of traumatic axonal injury in a rodent head injury model. J Neurosurg 114, 77–84. [DOI] [PubMed] [Google Scholar]
- [181].Chen X, Pan Z, Fang Z, Lin W, Wu S, Yang F, Li Y, Fu H, Gao H, Li S (2018) Omega-3 polyunsaturated fatty acid attenuates traumatic brain injury-induced neuronal apoptosis by inducing autophagy through the upregulation of SIRT1-mediated deacetylation of Beclin-1. J Neuroinflammation 15, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Zhu W, Chi N, Zou P, Chen H, Tang G, Zhao W (2017) Effect of docosahexaenoic acid on traumatic brain injury in rats. Exp Ther Med 14, 4411–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Pu H, Jiang X, Wei Z, Hong D, Hassan S, Zhang W, Liu J, Meng H, Shi Y, Chen L, Chen J (2017) Repetitive and prolonged omega-3 fatty acid treatment after traumatic brain injury enhances long-term tissue restoration and cognitive recovery. Cell Transplant 26, 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D (2014) Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 34, 3743–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Lewis MD (2016) Concussions, traumatic brain injury, and the innovative use of omega-3 s. J Am Coll Nutr 35, 469–475. [DOI] [PubMed] [Google Scholar]
- [186].Kramer HJ, Stevens J, Grimminger F, Seeger W (1996) Fish oil fatty acids and human platelets: dose-dependent decrease in dienoic and increase in trienoic thromboxane generation. Biochem Pharmacol 52, 1211–1217. [DOI] [PubMed] [Google Scholar]
- [187].York AG, Williams KJ, Argus JP, Zhou QD, Brar G, Vergnes L, Gray EE, Zhen A, Wu NC, Yamada DH, Cunningham CR, Tarling EJ, Wilks MQ, Casero D, Gray DH, Yu AK, Wang ES, Brooks DG, Sun R, Kitchen SG, Wu TT, Reue K, Stetson DB, Bensinger SJ (2015) Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163, 1716–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Chen G, Zhang S, Shi J, Ai J, Qi M, Hang C (2009) Simvastatin reduces secondary brain injury caused by cortical contusion in rats: possible involvement of TLR4/NF-kappaB pathway. Exp Neurol 216, 398–406. [DOI] [PubMed] [Google Scholar]
- [189].Chen SF, Hung TH, Chen CC, Lin KH, Huang YN, Tsai HC, Wang JY (2007) Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci 81, 288–298. [DOI] [PubMed] [Google Scholar]
- [190].Cheng G, Wei L, Zhi-Dan S, Shi-Guang Z, Xiang-Zhen L (2009) Atorvastatin ameliorates cerebral vasospasm and early brain injury after subarachnoid hemorrhage and inhibits caspase-dependent apoptosis pathway. BMC Neurosci 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lu D, Mahmood A, Qu C, Goussev A, Lu M, Chopp M (2004) Atorvastatin reduction of intracranial hematoma volume in rats subjected to controlled cortical impact. J Neurosurg 101, 822–825. [DOI] [PubMed] [Google Scholar]
- [192].Wang H, Lynch JR, Song P, Yang HJ, Yates RB, Mace B, Warner DS, Guyton JR, Laskowitz DT (2007) Simvastatin and atorvastatin improve behavioral outcome, reduce hippocampal degeneration, and improve cerebral blood flow after experimental traumatic brain injury. Exp Neurol 206, 59–69. [DOI] [PubMed] [Google Scholar]
- [193].Turkoglu OF, Eroglu H, Okutan O, Gurcan O, Bodur E, Sargon MF, Öner L, Beskonaklí E (2009) Atorvastatin efficiency after traumatic brain injury in rats. Surg Neurol 72, 146–152. [DOI] [PubMed] [Google Scholar]
- [194].Lu D, Goussev A, Chen J, Pannu P, Li Y, Mahmood A, Chopp M (2004) Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J Neurotrauma 21, 21–32. [DOI] [PubMed] [Google Scholar]
- [195].Qu C, Lu D, Goussev A, Schallert T, Mahmood A, Chopp M (2005) Effect of atorvastatin on spatial memory, neuronal survival, and vascular density in female rats after traumatic brain injury. J Neurosurg 103, 695–701. [DOI] [PubMed] [Google Scholar]
- [196].Lu D, Mahmood A, Goussev A, Schallert T, Qu C, Zhang ZG, Li Y, Lu M, Chopp M (2004) Atorvastatin reduction of intravascular thrombosis, increase in cerebral microvascular patency and integrity, and enhancement of spatial learning in rats subjected to traumatic brain injury. J Neurosurg 101, 813–821. [DOI] [PubMed] [Google Scholar]
- [197].Peng W, Yang J, Yang B, Wang L, Xiong XG, Liang Q (2014) Impact of statins on cognitive deficits in adult male rodents after traumatic brain injury: a systematic review. Biomed Res Int 2014, 261409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Mountney A, Boutté AM, Gilsdorf J, Lu X-C, Tortella FC, Shear DA (2016) Intravenous administration of simvastatin improves cognitive outcome following severe traumatic brain injury in rats. J Neurotrauma 33, 1492–1500. [DOI] [PubMed] [Google Scholar]
- [199].Sanchez-Aguilar M, Tapia-Perez JH, Sanchez-Rodriguez JJ, Vinas-Rios JM, Martinez-Perez P, de la Cruz-Mendoza E, Sanchez-Reyna M, Torres-Corzo JG, Gordillo-Moscoso A (2013) Effect of rosuvastatin on cytokines after traumatic head injury. J Neurosurg 118, 669–675. [DOI] [PubMed] [Google Scholar]
- [200].Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, Gordillo-Moscoso A, Martinez-Perez P, Madeville P, de la Cruz-Mendoza E, Chalita-Williams J (2008) Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758). J Neurotrauma 25, 1011–1017. [DOI] [PubMed] [Google Scholar]
- [201].Khokhar B, Simoni-Wastila L, Slejko JF, Perfetto E, Zhan M, Smith GS (2017) In-hospital mortality following traumatic brain injury among older medicare beneficiaries, comparing statin users with nonusers. J Pharm Technol 33, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Whyte J, Ketchum JM, Bogner J, Brunner RC, Hammond FM, Zafonte R, Whiteneck GG, Weintraub A (2018) Effects of statin treatment on outcomes after traumatic brain injury. J Neurotrauma, doi: 10.1089/neu.2017.5545. [DOI] [PubMed] [Google Scholar]
- [203].Neilson SJ, See AA, King NK (2016) Effect of prior statin use on outcome after severe traumatic brain injury in a South-East Asian population. Brain Inj 30, 993–998. [DOI] [PubMed] [Google Scholar]
- [204].Farzanegan GR, Derakhshan N, Khalili H, Ghaffarpasand F, Paydar S (2017) Effects of atorvastatin on brain contusion volume and functional outcome of patients with moderate and severe traumatic brain injury; a randomized double-blind placebo-controlled clinical trial. J Clin Neurosci 44, 143–147. [DOI] [PubMed] [Google Scholar]
- [205].Loane DJ, Stoica BA, Pajoohesh-Ganji A, Byrnes KR, Faden AI (2009) Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. J Biol Chem 284, 15629–15639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Loane DJ, Stoica BA, Byrnes KR, Jeong W, Faden AI (2013) Activation of mGluR5 and inhibition of NADPH oxidase improves functional recovery after traumatic brain injury. J Neurotrauma 30, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Byrnes KR, Loane DJ, Stoica BA, Zhang J, Faden AI (2012) Delayed mGluR5 activation limits neuroinflammation and neurodegeneration after traumatic brain injury. J Neuroinflammation 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Homsi S, Federico F, Croci N, Palmier B, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2009) Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res 1291, 122–132. [DOI] [PubMed] [Google Scholar]
- [209].Homsi S, Piaggio T, Croci N, Noble F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2010) Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma 27, 911–921. [DOI] [PubMed] [Google Scholar]
- [210].Siopi E, Cho AH, Homsi S, Croci N, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2011) Minocycline restores sAPPα levels and reduces the late histopathological consequences of traumatic brain injury in mice. J Neurotrauma 28, 2135–2143. [DOI] [PubMed] [Google Scholar]
- [211].Siopi E, Calabria S, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2012) Minocycline restores olfactory bulb volume and olfactory behavior after traumatic brain injury in mice. J Neurotrauma 29, 354–361. [DOI] [PubMed] [Google Scholar]
- [212].Siopi E, Llufriu-Dabén G, Fanucchi F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2012) Evaluation of late cognitive impairment and anxiety states following traumatic brain injury in mice: the effect of minocycline. Neurosci Lett 511, 110–115. [DOI] [PubMed] [Google Scholar]
- [213].Kabadi SV, Faden AI (2014) Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci 15, 1216–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Hall ED, Vaishnav RA, Mustafa AG (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Pierce JE, Smith DH, Trojanowski JQ, McIntosh TK (1998) Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 87, 359–369. [DOI] [PubMed] [Google Scholar]
- [216].Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK (1997) Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma 14, 715–727. [DOI] [PubMed] [Google Scholar]
- [217].Carbonell WS, Grady MS (1999) Regional and temporal characterization of neuronal, glial, and axonal response after traumatic brain injury in the mouse. Acta Neuropathol 98, 396–406. [DOI] [PubMed] [Google Scholar]
- [218].Carbonell WS, Maris DO, McCall T, Grady MS (1998) Adaptation of the fluid percussion injury model to the mouse. J Neurotrauma 15, 217–229. [DOI] [PubMed] [Google Scholar]
- [219].Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ (2011) Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 70, 374–383. [DOI] [PubMed] [Google Scholar]
- [220].Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W (2013) Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 136, 28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW (2016) Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139, 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25, 9275–9284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Shaw AC, Goldstein DR, Montgomery RR (2013) Age-dependent dysregulation of innate immunity. Nat Rev Immunol 13, 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Norden DM, Muccigrosso MM, Godbout JP (2015) Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 96, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Zhao C, Li WW, Franklin RJ (2006) Differences in the early inflammatory responses to toxin-induced demyelination are associated with the age-related decline in CNS remyelination. Neurobiol Aging 27, 1298–1307. [DOI] [PubMed] [Google Scholar]
- [226].Kumar A, Stoica BA, Sabirzhanov B, Burns MP, Faden AI, Loane DJ (2013) Traumatic brain injury in aged animals increases lesion size and chronically alters microglial/macrophage classical and alternative activation states. Neurobiol Aging 34, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Juengst SB, Kumar RG, Arenth PM, Wagner AK (2014) Exploratory associations with tumor necrosis factor-alpha, disinhibition and suicidal endorsement after traumatic brain injury. Brain Behav Immun 41, 134–143. [DOI] [PubMed] [Google Scholar]
- [228].Kumar RG, Diamond ML, Boles JA, Berger RP, Tisherman SA, Kochanek PM, Wagner AK (2015) Acute CSF interleukin-6 trajectories after TBI: associations with neuroinflammation, polytrauma, and outcome. Brain Behav Immun 45, 253–262. [DOI] [PubMed] [Google Scholar]
- [229].Loane DJ, Kumar A, Stoica BA, Cabatbat R, Faden AI (2014) Progressive neurodegeneration after experimental brain trauma: association with chronic microglial activation. J Neuropathol Exp Neurol 73, 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Ziebell JM, Taylor SE, Cao T, Harrison JL, Lifshitz J (2012) Rod microglia: elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J Neuroinflammation 9, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014) Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biol Psychiatry 76, 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Smith C, Gentleman SM, Leclercq PD, Murray LS, Griffin WS, Graham DI, Nicoll JA (2013) The neuroinflammatory response in humans after traumatic brain injury. Neuropathol Appl Neurobiol 39, 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Longhi L, Gesuete R, Perego C, Ortolano F, Sacchi N, Villa P, Stocchetti N, De Simoni MG (2011) Long-lasting protection in brain trauma by endotoxin preconditioning. J Cereb Blood Flow Metab 31, 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Eslami M, Sayyah M, Soleimani M, Alizadeh L, Hadjighassem M (2015) Lipopolysaccharide preconditioning prevents acceleration of kindling epileptogenesis induced by traumatic brain injury. J Neuroimmunol 289, 143–151. [DOI] [PubMed] [Google Scholar]
- [235].Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD (2012) Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32, 11706–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Muccigrosso MM, Ford J, Benner B, Moussa D, Burnsides C, Fenn AM, Popovich PG, Lifshitz J, Walker FR, Eiferman DS, Godbout JP (2016) Cognitive deficits develop 1month after diffuse brain injury and are exaggerated by microglia-associated reactivity to peripheral immune challenge. Brain Behav Immun 54, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Collins-Praino LE, Arulsamy A, Katharesan V, Corrigan F (2018) The effect of an acute systemic inflammatory insult on the chronic effects of a single mild traumatic brain injury. Behav Brain Res 336, 22–31. [DOI] [PubMed] [Google Scholar]
- [238].Mirotti L, Castro J, Costa-Pinto F, Russo M (2010) Neural pathways in allergic inflammation. J Allergy (Cairo) 2010, 491928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Zhou L, Chen L, Li X, Li T, Dong Z, Wang YT (2018) Food allergy induces alteration in brain inflammatory status and cognitive impairments. Behav Brain Res 364, 374–382. [DOI] [PubMed] [Google Scholar]
- [240].Wang J, Yang C, Zhao Q, Zhu Z, Li Y, Yang P (2017) Microglia activation induced by serum of SLE patients. J Neuroimmunol 310, 135–142. [DOI] [PubMed] [Google Scholar]
- [241].Talebi F, Ghorbani S, Chan WF, Boghozian R, Masoumi F, Ghasemi S, Vojgani M, Power C, Noorbakhsh F (2017) MicroRNA-142 regulates inflammation and T cell differentiation in an animal model of multiple sclerosis. J Neuroinflammation 14, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Perry VH, Cunningham C, Holmes C (2007) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7, 161–167. [DOI] [PubMed] [Google Scholar]
- [243].Fischer HG, Reichmann G (2001) Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol 166, 2717–2726. [DOI] [PubMed] [Google Scholar]
- [244].Streit WJ, Mrak RE, Griffin WST (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Norden DM, Godbout JP (2013) Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39, 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Tapp ZM, Godbout JP, Kokiko-Cochran ON (2019) A tilted axis: maladaptive inflammation and HPA axis dysfunction contribute to consequences of TBI. Front Neurol 10, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].VanGuilder HD, Bixler GV, Brucklacher RM, Farley JA, Yan H, Warrington JP, Sonntag WE, Freeman WM (2011) Concurrent hippocampal induction of MHC II pathway components and glial activation with advanced aging is not correlated with cognitive impairment. J Neuroinflammation 8, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Frank MG, Barrientos RM, Biedenkapp JC, Rudy JW, Watkins LR, Maier SF (2006) mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging 27, 717–722. [DOI] [PubMed] [Google Scholar]
- [249].Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, Castanon N, Kelley KW, Dantzer R, Johnson RW (2008) Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology 33, 2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW (2005) Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 19, 1329–1331. [DOI] [PubMed] [Google Scholar]
- [251].Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW (2008) Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun 22, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF (2009) Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun 23, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [253].Chauhan NB (2014) Chronic neurodegenerative consequences of traumatic brain injury. Restor Neurol Neurosci 32, 337–365. [DOI] [PubMed] [Google Scholar]
- [254].Rapoport MJ (2012) Depression following traumatic brain injury: epidemiology, risk factors and management. CNS Drugs 26, 111–121. [DOI] [PubMed] [Google Scholar]
- [255].Boone DR, Weisz HA, Willey HE, Torres KEO, Falduto MT, Sinha M, Spratt H, Bolding IJ, Johnson KM, Parsley MA, DeWitt DS, Prough DS, Hellmich HL (2019) Traumatic brain injury induces long-lasting changes in immune and regenerative signaling. PLoS One 14, e0214741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Ritzel RM, Doran SJ, Barrett JP, Henry RJ, Ma EL, Faden AI, Loane DJ (2018) Chronic alterations in systemic immune function after traumatic brain injury. J Neurotrauma 35, 1419–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I, Perl DP, Stein TD, Vonsattel JP, Stewart W, Tripodis Y, Crary JF, Bieniek KF, Dams-O’Connor K, Alvarez VE, Gordon WA (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA (2009) Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68, 709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Tartaglia MC, Hazrati LN, Davis KD, Green RE, Wennberg R, Mikulis D, Ezerins LJ, Keightley M, Tator C (2014) Chronic traumatic encephalopathy and other neurodegenerative proteinopathies. Front Hum Neurosci 8, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Gavett BE, Cantu RC, Shenton M, Lin AP, Nowinski CJ, McKee AC, Stern RA (2011) Clinical appraisal of chronic traumatic encephalopathy: current perspectives and future directions. Curr Opin Neurol 24, 525–531. [DOI] [PubMed] [Google Scholar]
- [261].Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, Goletiani CJ, Maglakelidze GM, Casey N, Moncaster JA, Minaeva O, Moir RD, Nowinski CJ, Stern RA, Cantu RC, Geiling J, Blusztajn JK, Wolozin BL, Ikezu T, Stein TD, Budson AE, Kowall NW, Chargin D, Sharon A, Saman S, Hall GF, Moss WC, Cleveland RO, Tanzi RE, Stanton PK, McKee AC (2012) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Coughlin JM, Wang Y, Minn I, Bienko N, Ambinder EB, Xu X, Peters ME, Dougherty JW, Vranesic M, Koo SM, Ahn HH, Lee M, Cottrell C, Sair HI, Sawa A, Munro CA, Nowinski CJ, Dannals RF, Lyketsos CG, Kassiou M, Smith G, Caffo B, Mori S, Guilarte TR, Pomper MG (2017) Imaging of glial cell activation and white matter integrity in brains of active and recently retired National Football League players. JAMA Neurol 74, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH, Mez J, Montenigro PH, Solomon TM, Alosco ML, Stern RA, McKee AC, Stein TD (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Edwards G 3rd, Moreno-Gonzalez I, Soto C (2017) Amyloid-beta and tau pathology following repetitive mild traumatic brain injury. Biochem Biophys Res Commun 483, 1137–1142. [DOI] [PubMed] [Google Scholar]
- [265].Winston CN, Noel A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, Wilkins TE, Alikhani AD, Zapple DN, Villapol S, Planel E, Burns MP (2016) Dendritic spine loss and chronic white matter inflammation in a mouse model of highly repetitive head trauma. Am J Pathol 186, 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Mendez MF (2017) What is the relationship of traumatic brain injury to dementia? J Alzheimers Dis 57, 667–681. [DOI] [PubMed] [Google Scholar]
- [267].Nordstrom A, Nordstrom P (2018) Traumatic brain injury and the risk of dementia diagnosis: A nationwide cohort study. PLoS Med 15, e1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Aungst SL, Kabadi SV, Thompson SM, Stoica BA, Faden AI (2014) Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J Cereb Blood Flow Metab 34, 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Bloom GS (2014) Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71, 505–508. [DOI] [PubMed] [Google Scholar]
- [270].Heneka MT, Golenbock DT, Latz E (2015) Innate immunity in Alzheimer’s disease. Nat Immunol 16, 229–236. [DOI] [PubMed] [Google Scholar]
- [271].Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI, Rebeck GW, Burns MP (2009) Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat Med 15, 377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Kokiko-Cochran ON, Godbout JP (2018) The inflammatory continuum of traumatic brain injury and Alzheimer’s disease. Front Immunol 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Lowry CA, Smith DG, Siebler PH, Schmidt D, Stamper CE, Hassell JE Jr., Yamashita PS, Fox JH, Reber SO, Brenner LA, Hoisington AJ, Postolache TT, Kinney KA, Marciani D, Hernandez M, Hemmings SM, Malan-Muller S, Wright KP, Knight R, Raison CL, Rook GA (2016) The microbiota, immunoregulation, and mental health: implications for public health. Curr Environ Health Rep 3, 270–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [274].Logsdon AF, Erickson MA, Rhea EM, Salameh TS, Banks WA (2018) Gut reactions: How the blood–brain barrier connects the microbiome and the brain. Exp Biol Med 243, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [275].Pärtty A, Kalliomäki M, Wacklin P, Salminen S, Isolauri E (2015) A possible link between early probiotic intervention and the risk of neuropsychiatric disorders later in childhood: a randomized trial. Pediatr Res 77, 823. [DOI] [PubMed] [Google Scholar]
- [276].Zhu C, Grandhi R, Patterson T, Nicholson S (2018) A review of traumatic brain injury and the gut microbiome: insights into novel mechanisms of secondary brain injury and promising targets for neuroprotection. Brain Sci 8, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Rodriguez M, Wootla B, Anderson G (2016) Multiple sclerosis, gut microbiota and permeability: role of tryptophan catabolites, depression and the driving down of local melatonin. Curr Pharm Des 22, 6134–6141. [DOI] [PubMed] [Google Scholar]
- [278].Brenner LA, Stearns-Yoder KA, Hoffberg AS, Penzenik ME, Starosta AJ, Hernandez TD, Hadidi DA, Lowry CA (2017) Growing literature but limited evidence: A systematic review regarding prebiotic and probiotic interventions for those with traumatic brain injury and/or posttraumatic stress disorder. Brain Behav Immun 65, 57–67. [DOI] [PubMed] [Google Scholar]
- [279].Painter TJ, Rickerds J, Alban RF (2015) Immune enhancing nutrition in traumatic brain injury - A preliminary study. Int J Surg 21, 70–74. [DOI] [PubMed] [Google Scholar]
- [280].Selassie AW, Fakhry SM, Ford DW (2011) Population-based study of the risk of in-hospital death after traumatic brain injury: the role of sepsis. J Trauma 71, 1226–1234. [DOI] [PubMed] [Google Scholar]
- [281].Kesinger MR, Kumar RG, Wagner AK, Puyana JC, Peitzman AP, Billiar TR, Sperry JL (2015) Hospital-acquired pneumonia is an independent predictor of poor global outcome in severe traumatic brain injury up to 5 years after discharge. J Trauma Acute Care Surg 78, 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Suz P, Vavilala MS, Souter M, Muangman S, Lam AM (2006) Clinical features of fever associated with poor outcome in severe pediatric traumatic brain injury. J Neurosurg Anesthesiol 18, 5–10. [DOI] [PubMed] [Google Scholar]
- [283].Sun M, McDonald SJ, Brady RD, O’Brien TJ, Shultz SR (2018) The influence of immunological stressors on traumatic brain injury. Brain Behav Immun 69, 618–628. [DOI] [PubMed] [Google Scholar]
- [284].Viola-Saltzman M, Musleh C (2016) Traumatic brain injury-induced sleep disorders. Neuropsychiatr Dis Treat 12, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [285].Rowe RK, Harrison JL, Morrison HW, Subbian V, Murphy SM, Lifshitz J (2019) Acute post-traumatic sleep may define vulnerability to a second traumatic brain injury in mice. J Neurotrauma 36, 1318–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Chin LM, Keyser RE, Dsurney J, Chan L (2015) Improved cognitive performance following aerobic exercise training in people with traumatic brain injury. Arch Phys Med Rehabil 96, 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].de Castro MRT, Ferreira APO, Busanello GL, da Silva LRH, da Silveira Junior MEP, Fiorin FDS, Arrifano G, Crespo-López ME, Barcelos RP, Cuevas MJ, Bresciani G, González-Gallego J, Fighera MR, Royes LFF (2017) Previous physical exercise alters the hepatic profile of oxidative-inflammatory status and limits the secondary brain damage induced by severe traumatic brain injury in rats. J Physiol 595, 6023–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Piao CS, Stoica BA, Wu J, Sabirzhanov B, Zhao Z, Cabatbat R, Loane DJ, Faden AI (2013) Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol Dis 54, 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [289].Clausen F, Hånell A, Israelsson C, Hedin J, Ebendal T, Mir AK, Gram H, Marklund N (2011) Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 34, 110–123. [DOI] [PubMed] [Google Scholar]
- [290].Clausen F, Hånell A, Björk M, Hillered L, Mir AK, Gram H, Marklund N (2009) Neutralization of interleukin-1β modifies the inflammatory response and improves histological and cognitive outcome following traumatic brain injury in mice. Eur J Neurosci 30, 385–396. [DOI] [PubMed] [Google Scholar]
- [291].Helmy A, Guilfoyle MR, Carpenter KL, Pickard JD, Menon DK, Hutchinson PJ (2016) Recombinant human interleukin-1 receptor antagonist promotes M1 microglia biased cytokines and chemokines following human traumatic brain injury. J Cereb Blood Flow Metab 36, 1434–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [292].de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29, 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Brickler T, Gresham K, Meza A, Coutermarsh-Ott S, Williams TM, Rothschild DE, Allen IC, Theus MH (2016) Nonessential role for the NLRP1 inflammasome complex in a murine model of traumatic brain injury. Mediators Inflamm 2016, 6373506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [294].Penkowa M, Giralt M, Carrasco J, Hadberg H, Hidalgo J (2000) Impaired inflammatory response and increased oxidative stress and neurodegeneration after brain injury in interleukin-6-deficient mice. Glia 32, 271–285. [DOI] [PubMed] [Google Scholar]
- [295].Ley EJ, Clond MA, Singer MB, Shouhed D, Salim A (2011) IL6 deficiency affects function after traumatic brain injury. J Surg Res 170, 253–256. [DOI] [PubMed] [Google Scholar]
- [296].Penkowa M, Giralt M, Lago N, Camats J, Carrasco J, Hernandez J, Molinero A, Campbell IL, Hidalgo J (2003) Astrocyte-targeted expression of IL-6 protects the CNS against a focal brain injury. Exp Neurol 181, 130–148. [DOI] [PubMed] [Google Scholar]
- [297].Yang SH, Gangidine M, Pritts TA, Goodman MD, Lentsch AB (2013) Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 40, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Baratz R, Tweedie D, Wang J-Y, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG (2015) Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J Neuroinflammation 12, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [299].Longhi L, Perego C, Ortolano F, Aresi S, Fumagalli S, Zanier ER, Stocchetti N, De Simoni MG (2013) Tumor necrosis factor in traumatic brain injury: effects of genetic deletion of p55 or p75 receptor. J Cereb Blood Flow Metab 33, 1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Song S, Kong X, Acosta S, Sava V, Borlongan C, Sanchez-Ramos J (2016) Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J Neurosci Res 94, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Shultz SR, Tan XL, Wright DK, Liu SJ, Semple BD, Johnston L, Jones NC, Cook AD, Hamilton JA, O’Brien TJ (2014) Granulocyte-macrophage colony-stimulating factor is neuroprotective in experimental traumatic brain injury. J Neurotrauma 31, 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Karve IP, Zhang M, Habgood M, Frugier T, Brody KM, Sashindranath M, Ek CJ, Chappaz S, Kile BT, Wright D, Wang H, Johnston L, Daglas M, Ates RC, Medcalf RL, Taylor JM, Crack PJ (2016) Ablation of type-1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro 3, ENEURO.0128–15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Chen X, Duan XS, Xu LJ, Zhao JJ, She ZF, Chen WW, Zheng ZJ, Jiang GD (2014) Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience 266, 235–243. [DOI] [PubMed] [Google Scholar]
- [304].Zhang D, Hu Y, Sun Q, Zhao J, Cong Z, Liu H, Zhou M, Li K, Hang C (2013) Inhibition of transforming growth factor beta-activated kinase 1 confers neuroprotection after traumatic brain injury in rats. Neuroscience 238, 209–217. [DOI] [PubMed] [Google Scholar]
- [305].Chio CC, Chang CP, Lin MT, Su FC, Yang CZ, Tseng HY, Liu ZM, Huang HS (2014) Involvement of TG-interacting factor in microglial activation during experimental traumatic brain injury. J Neurochem 131, 816–824. [DOI] [PubMed] [Google Scholar]
- [306].Johnson VE, Stewart W, Smith DH (2010) Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 11, 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Saber M, Kokiko-Cochran O, Puntambekar SS, Lathia JD, Lamb BT (2017) Triggering receptor expressed on myeloid cells 2 deficiency alters acute macrophage distribution and improves recovery after traumatic brain injury. J Neurotrauma 34, 423–435. [DOI] [PubMed] [Google Scholar]
- [309].Bennett RE, Brody DL (2014) Acute reduction of microglia does not alter axonal injury in a mouse model of repetitive concussive traumatic brain injury. J Neurotrauma 31, 1647–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Frieler RA, Nadimpalli S, Boland LK, Xie A, Kooistra LJ, Song J, Chung Y, Cho KW, Lumeng CN, Wang MM, Mortensen RM (2015) Depletion of macrophages in CD11b diphtheria toxin receptor mice induces brain inflammation and enhances inflammatory signaling during traumatic brain injury. Brain Res 1624, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Zanier ER, Marchesi F, Ortolano F, Perego C, Arabian M, Zoerle T, Sammali E, Pischiutta F, De Simoni M-G (2016) Fractalkine receptor deficiency is associated with early protection but late worsening of outcome following brain trauma in mice. J Neurotrauma 33, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Weckbach S, Neher M, Losacco JT, Bolden AL, Kulik L, Flierl MA, Bell SE, Holers VM, Stahel PF (2012) Challenging the role of adaptive immunity in neurotrauma: Rag1(−/−) mice lacking mature B and T cells do not show neuroprotection after closed head injury. J Neurotrauma 29, 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Mencl S, Hennig N, Hopp S, Schuhmann MK, Albert-Weissenberger C, Siren AL, Kleinschnitz C (2014) FTY720 does not protect from traumatic brain injury in mice despite reducing posttraumatic inflammation. J Neuroimmunol 274, 125–131. [DOI] [PubMed] [Google Scholar]