

Abstract
It has become increasingly clear that the innate immune system plays an essential role in the generation of many types of neuropathic pain including that which accompanies cancer treatment. In this article we review current findings of the role of the innate immune system in contributing to cancer treatment pain at the distal endings of peripheral nerve, in the nerve trunk, in the dorsal root ganglion and in the spinal dorsal horn.
Keywords: paclitaxel, oxaliplatin, bortezomib, TLR4, macrophage
Introduction
Neurotoxicity is a primary concern for cancer treatments that rely on a range of chemotherapeutic agents, including taxanes (paclitaxel, docetaxel), platinum compounds (cisplatin, oxaliplatin, carboplatin), vinca alkaloids (vincristine), proteasome inhibitors (bortezomib, carfilzomib, ixazomib), angiogenesis inhibitors (thalidomide, lenalidomide, pomalidomide), and others (ifosfamide, pemtrexed). Patients commonly report sensory disturbances in their extremities that range from numbness and tingling to painful burning and electrical sensations in the hands and feet, termed chemotherapy-induced peripheral neuropathy (CIPN)[1–8]. The time course of this syndrome varies based on the type of chemotherapy drug, but symptoms tend to worsen as the cumulative dose increases. Worse yet, these symptoms can be severe and persist long after chemotherapy administration has ceased [3, 9]. Pain is still the number one dose-limiting side effect of chemotherapy, forcing patients to diminish the dose or discontinue treatment early [2, 10, 11]. Recent studies have focused on identifying methods to prevent or attenuate chemotherapy-induced neuropathy without impacting the cytotoxic activity of chemotherapy on cancer cells, since preserving peripheral nerve health during cancer treatment would ease patient burden and improve treatment outcomes. Investigations aimed at better understanding the mechanisms involved in the development and maintenance of CIPN have provided significant insight on this topic. One area that has come into focus is non-neuronal components of the nervous system that contribute to nociceptor sensitization and other aspects of CIPN, which may provide new targets for the development of adjuvant treatment for this syndrome [12–15]. In this paper, we review the innate immune mechanisms in the peripheral and central nervous system that contribute to CIPN.
Communication among neurons, resident immune cells, and immune cells recruited from the periphery plays an important role in the response to pathogens or tissue injury and is vital in the process of recovery, including tissue repair. Activation of immune cells promotes a state of neuroinflammation and can also trigger changes in the activity of nociceptors [16]. Persistence of these pathological changes can lead to debilitating chronic pain [16]. The complex interplay among neurons, glial cells and migratory immune cells has been examined extensively for its role in the development of neuropathy [17–21]. The immune system can be divided into two main aspects: innate and acquired immunity. Innate immunity includes both physical barriers to prevent infection, such as the skin, and chemical barriers that are activated when physical barriers are breached. Signaling among different immune components relies on specialized signaling molecules (chemokines and cytokines) released in the presence of injury or pathogens to recruit and activate immune cells and trigger an inflammatory response. In addition, antibody binding to antigens triggers the complement cascade, in which specialized plasma proteins assist in the identification and destruction of pathogens as well as recruitment of immune cells to the site. Inflammation triggers localized vasodilation, phagocytes (i.e., neutrophils, macrophages) are drawn in, and when activated, begin to release cytokines and chemokines that recruit additional immune cells to the site of inflammation. Many signaling molecules released by activated immune cells in an inflammatory state have been shown to directly sensitize nociceptors. Leukocytes that assist in the innate immune response includes mast cells, eosinophils, basophils, natural killer cells, phagocytes (neutrophils, macrophages, dendritic cells) and a subset of T cells (gamma/delta T cells). Innate immune activity connects to the acquired immune system when immune cells that express major histocompatibility complex (MHC) molecules (including activated macrophages) act as antigen-presenting cells (APCs) for lymphocytes responsible for the humoral (B cells) and cell-mediated (T cells) components of the acquired immune response.
Innate immune system in neuronal tissues
In the peripheral nervous system (PNS), immune-competent glial cells include Schwann cells and satellite glial cells. Schwann cells perform both cellular and humoral immune functions, including APC activity, phagocytosis, and inflammatory cytokine release [22]. Schwann cells can release IL-1β, IL-6, IL-8, MCP-1, interferon-α (IFN- α), and TNF-α, and are capable of signaling the immune complement cascade in response to cytokine binding [22–24]. IFN-α and TNF-α can also induce Schwann cells to upregulate the production of cytokine-inducible nitric oxide synthase, which can lead to an increase in NO production [22, 24]. Satellite glial cells, which are abundant in dorsal root ganglia (DRG), can also act as APCs and release pro-inflammatory cytokines [25, 26]. Activated satellite glial cells produce GFAP, release IL-1β, IL-6 and upregulate IL-6 receptor expression and pSTAT3 in lumbar DRG after chronic constriction injury (CCI)[27, 28].
In addition to glial cells, there are also phagocytes (i.e., macrophages and neutrophils) and granulocytes (neutrophils, mast cells, basophils, eosinophils) present in the PNS [29]. Macrophages are resident immune cells, but can also be recruited to the site of an inflammatory response [29]. Like glial cells, they can act as APCs and release and respond to pro-inflammatory cytokines, including IL-1, IL-6, and TNF-α [22]. One example of resident macrophages are the Langerhans cells (LC) in the skin, and studies have shown an increase in their numbers in inflammatory cutaneous diseases [30, 31]. Granulocytes can also be activated to release immune mediators. Neutrophils, which act as both phagocytes and granulocytes, release inflammatory cytokines and chemokines to recruit additional immune cells, and have been shown to invade affected nerve tissue and lumbar DRG following sciatic nerve injury [32–34]. Mast cells recruit macrophages and neutrophils to the site of nerve injury after sciatic nerve ligation [35]. Granulocytes also play a fundamental role in skin inflammation, especially during an allergic response [36]. Mast cells are found near peripheral nerves expressing substance P and CGRP in the skin and viscera and release a number of immune mediators, including histamine, serotonin, neuropeptides (i.e., substance P and CGRP), proteases, cytokines, chemokines, leukotrienes, prostaglandins, and growth factors [36]. Receptors for many of these effectors can be expressed on immune cells, peripheral nerve fibers, and other cells of the epithelium, skin, and blood vessels, and several have been shown to directly sensitize nociceptors [36–39]. Eosinophils, which are found primarily in mucosal tissues, release pro-inflammatory mediators as well as neurotrophins (including NGF)[40]. The cytotoxic compounds released by eosinophils are neurotoxic, and high eosinophil activity can lead to lesions of peripheral nerve tissue [40–43]. The immune complement system is also present in the PNS, and consists of proteins that, when triggered, form a complement activation cascade to stimulate phagocytosis, promote inflammation to recruit additional phagocytes, and activate cell-killing membrane attack complexes [44, 45].
In the central nervous system (CNS), the immune response to injury or pathogens is governed by immune-competent glial cells, which are divided into microglia and macroglia. Microglia act as the main immune cells of the CNS, and activation of microglia in the spinal cord plays a significant role in the development of neuropathic pain states [46]. At rest, microglia are resident macrophage-like cells that remain in a quiescent-like state and continually monitor the surrounding environment using branching projections that are capable of movement [47]. Microglia are highly sensitive to changes in the microenvironment of neurons, including potassium levels [48]. They express Ca2+-gated and inwardly rectifying K channels, which are increased under pathological conditions. Two-pore domain halothane-inhibited K+ channel type 1 (THIK-1), Cl− channels, and transient receptor potential cation channels (TRPV1 and TRPM7) are also present in microglia [49]. These ion channels contribute to the maintenance of the resting potential and/or depolarization, and could promote cellular functions such as surveillance and ramification and likely contribute to pathological functional phenotypes [49]. Microglia express toll-like receptors (TLRs) and play a key role in the innate and adaptive immune response of the CNS [50]. When changes in the environment occur, inflammatory stimuli produced by injury or disease are detected by receptors or other means (such as phagocytosis), and microglia undergo a number of changes. Upon activation, microglia shift to a rounded or ameboid shape and less ramified appearance, increase movement, and upregulate the expression of functional markers [51]. Activated microglia will begin to express surface molecules that are important for both innate and adaptive immune responses [52]. Activated microglia are capable of expressing signaling molecules, including pro-inflammatory cytokines and chemokines, which contribute to the recruitment and activation of additional immune cells [53]. Among the many signaling molecules released by activated microglia, including tumor necrosis factor-α (TNF-α), IL-6, IL-1β, nitric oxide (NO), bradykinins, prostaglandins, matrix metalloproteases (MMPs), and cathepsin S, several are known to sensitize nociceptive neurons [36–39, 50, 54–56].
Macroglia are comprised of oligodendrocytes and astrocytes. Oligodendrocytes produce myelin sheathing, while astrocytes play a role in development, maintain the blood brain barrier (BBB), and regulate the ionic microenvironment and concentration of neurotransmitters [46]. Both oligodendrocytes and astrocytes are immune-competent [46]. Astrocytes express receptors for pro-inflammatory cytokines in a resting state, including IL-1β, and will release IL-1β, IL-6, TNF-α, nitric oxide (NO), and pyruvate carboxylase (involved in glutamate synthesis) upon activation [46]. Like microglia, astrocytes are capable of phagocytosis and will undergo physical changes upon activation [46]. Astrocyte activation leads to proliferation, and the cells become enlarged and upregulate glial fibrillary acidic protein (GFAP) expression [46]. Under healthy conditions, astrocytes are very territorial, and this circumscribed area of action is lost in pathological states, in which their processes are enlarged and more ramified, making them overlap with adjacent and distant counterparts [51]. The expression of adhesion molecules is enhanced upon binding lipopolysaccharide (LPS), viral proteins, or inflammatory cytokines (including IL-1β, TNF-α, and interferon-γ), and activated astrocytes can act as antigen presenting cells (APCs)[52].
There are also meningeal, choroid plexus, and perivascular macrophages which derive from the same developmental pathway as microglia and monitor cerebrospinal fluid (CSF)[57]. In addition, blood-derived monocytes and granulocytes (mast cells, basophils, neutrophils), are able to infiltrate into the CNS in response to chemokine signals following tissue injury or pathogen detection [58]. Mast cells can be found in the meninges and release a number of pro-inflammatory immune mediators [59]. Mast cell recruitment has also been shown to be vital in the recovery from traumatic brain injury [60]. Basophils, like mast cells, are pro-inflammatory granulocytes, release immune mediators, including histamine and heparin [60]. Nerve growth factor (NGF) upregulates the release of these immune mediators from basophils and enhances the activity of eosinophils [61–63]. Neutrophils are rare in the CNS under normal basal conditions, but are recruited in high numbers in response to tissue injury or pathogens, where they perform phagocytosis, release reactive oxygen species, and degranulate to disperse proteolytic enzymes [64, 65].
Innate immune functions contributing to CIPN
Recruitment of immune cells to the PNS in response to chemotherapy administration
Along with resident immune cells, peripheral immune cells recruited to the PNS contribute to innate immune activation in response to chemotherapy administration. Naïve mouse DRGs have been shown to include a range of different immune cells representing less than 0.5% of the total number of cells [66]. After a single injection of paclitaxel (6 mg/kg, i.p.), large numbers of infiltrating innate immune cells, including macrophages, neutrophils, and monocytes, were found to have migrated into DRGs [66]. Transcriptomic analyses in mice have revealed upregulation of genes in DRG associated with inflammation and immune cell activation and recruitment following paclitaxel, vincristine and (to a lesser extent) cisplatin treatment [67, 68]. Increased macrophage activity in the DRG has been shown to contribute to CIPN induced by paclitaxel, oxaliplatin, vincristine, and bortezomib, indicating that macrophages play a fundamental role in the pathophysiology of this condition [69–72]. Macrophage infiltration also targets peripheral nerves after paclitaxel, vincristine, and bortezomib, but not oxaliplatin treatment [71, 73–75]. In rats, blocking the infiltration of macrophages into the DRG prevented the behavioral symptoms of CIPN and the loss of intraepidermal nerve fibers (IENFs) and DRG neurons associated with paclitaxel [72, 76, 77]. Likewise, intravenous immunoglobulin treatment that reduced peripheral nerve M1 macrophage infiltration also attenuated behavioral signs and IENF loss induced by bortezomib [74]. This pattern of DRG macrophage infiltration associated with neuronal damage and ENF loss has also been identified in Macaques with SIV-induced peripheral neuropathy, indicating that this mechanism is not limited to CIPN [78]. Macrophage infiltration to DRG and sciatic nerve has also been shown after CCI in rats, intrapancreatic nerves in patients with neuropathic pain due to pancreatic cancer, and is found in the peripheral nerves of rats with experimental diabetic neuropathy (streptozotocin), but not in a murine model of Type 2 diabetic neuropathy (high fat diet)[35, 79–81]. The relationship between macrophage infiltration, nociceptive behavior, and IENF loss may not hold for diabetic neuropathy since it also involves oxidative stress and cytokine release pathways that are triggered by hyperglycemia and features pathological changes to endothelial cells and capillaries in the endoneurium that do correlate with IENF loss in patients [82, 83].
Rats treated with paclitaxel and vincristine also showed an increase in the number and activation of Langerhans cells (LC) as APC in the epidermis of the paw, and this was followed by loss of IENF [84]. Activated LC can induce neuropathic pain symptoms through different mechanisms, including the release of NO, neurotrophic factors, and pro-inflammatory cytokines and chemokines, as seen in rats with CCI, where ENF loss and an increase in Langerhans cells were found in the skin and correlated with increased nociceptive behaviors [85–89]. Oxaliplatin treatment has also been shown to enhance the number of cutaneous mast cells and mast cell degranulation in the hind paw skin of mice [90]. Notably, mast cell deficient mice failed to develop mechanical allodynia after oxaliplatin injection [90]. Behavioral signs of paclitaxel and vincristine CIPN are also attenuated by mast cell stabilizers [91, 92].
Changes in number and activity of PNS glial cells and neurons
Nerve injury has been shown to induce a distinct pattern of neuroimmune activity, which initially triggers pro-inflammatory signaling associated with damage to the distal nerve and demyelination resulting in a state known as Wallerian degeneration (WD)[93]. The potent pro-inflammatory environment during WD persists during the nerve destruction and subsequent clean-up of debris as local immune cells activate, proliferate, and recruit additional immune cells to the site of injury [35, 93–96]. These changes begin rapidly after the initial injury, and within days to weeks, pro-inflammatory signals decline and are replaced by activity that acts to suppress the innate immune system and promote tissue repair [96–98]. Some of these signals are triggered early in the WD process, such as the promotion of an M2 macrophage response [98]. Compared to nerve injury models, chemotherapy drugs alter PNS immune activity in distinct ways, and not all types result in the same changes. The normal time course for nerve injury/repair is also altered given the chronic nature of the treatment and pathological damage that can impact immune cell functions.
Paclitaxel administration increased expression of activating transcription factor 3 (ATF3), a marker of nerve injury, and induced pathological changes in lumbar DRG neurons and satellite cells as well as sciatic nerve Schwann cells [75]. In addition, paclitaxel exposure resulted in cultured Schwann cells de-differentiating to an immature state with upregulated expression of p75 and galectin-3 [99]. Evidence of these changes were also identified in the sciatic nerve after in vivo paclitaxel administration [99]. After cisplatin administration, lumbar DRG were found to have pathological changes in satellite glial cells and their structural connections to neurons [100, 101]. In newborn rats, cisplatin administration was found to cause satellite glial and Schwann cell death in trigeminal ganglia [102]. Cisplatin and oxaliplatin exposure also led to dysfunction in cultured Schwann cell mitochondria [99]. Cultured satellite glial cells treated with oxaliplatin demonstrate changes in morphology and upregulate expression of the gap junction/hemichannel protein connexin-43, glial fibrillary acidic protein (GFAP), a marker of glial activation, and reactive oxygen species (ROS)[103]. Oxaliplatin administration has also been shown to cause satellite cell activation in the DRG [104]. Post-mortem examination of patient DRG after cisplatin or carboplatin showed evidence of satellite cell proliferation (nodes of Nageotte) and neuronal necrosis, but no axonal degeneration was identified [105]. In rats, bortezomib induced pathological changes in DRG satellite cells stemming from damage to mitochondria and the endoplasmic reticulum [106, 107].
Chemotherapy exposure can impact myelin formation, which may lead to axonal degeneration and impact nerve conduction. In cultured Schwann cells, exposure to paclitaxel or platinum chemotherapy compounds (cisplatin and oxaliplatin) decreased the expression of myelin basic protein (MBP)[99]. Disturbances in myelin formation were also detected in co-cultures of DRG neurons and Schwann cells at concentrations that were not neurotoxic [99]. Vincristine has been shown to damage DRG neurons and Schwann cells along with pathological changes to peripheral nerve axons [108, 109]. Bortezomib was shown to cause similar pathological changes to Schwann cells in the sciatic nerve, along with damage to myelin, axonal degeneration, and reduction in sciatic nerve conduction velocity [106, 107].
Changes in number and activity of CNS glial cells
Activation of glial cells in the spinal cord has been shown to contribute to the development of CIPN following paclitaxel, oxaliplatin, vincristine, and bortezomib administration, indicating that these cells play a fundamental role in CNS pathophysiology. Spinal glial cell activation has been identified in paclitaxel CIPN but reports of which cells types are affected have been inconsistent and may be related to differences in dose. In some studies, spinal microglia, but not astrocytes, were activated after paclitaxel treatment, and minocycline, an inhibitor of innate immune responses, blocked the development of hyperalgesia [15, 110]. Other studies show activation of astrocytes, but not microglia, in the dorsal horn of the spinal cord [111, 112]. Again, minocycline prevented the development of mechanical allodynia, but in this case it also reduced astrocyte activation [113]. In mice, paclitaxel treatment enhanced spinal microglia and astrocyte activity, though the extent of the response was greater in young mice (which had the highest response) and aged mice when compared to adult mice [114]. In addition, astrocyte activation has been identified in the anterior cingulate cortex following paclitaxel treatment [115].
Spinal microglia increase in number and are activated in parallel with the onset of behavioral symptoms of CIPN following oxaliplatin treatment, but return to basal levels within days [104]. In comparison, the number of astrocytes was increased in the spinal cord at the onset of behavioral symptoms and remained elevated for weeks afterwards. Astrocyte activation following oxaliplatin administration has also been identified in several brain areas involved in the processing of pain stimuli, including the anterior cingulate cortex, somatosensory cortex, ventrolateral periaqueductal gray, nucleus raphe magnus, and neostriatum [104]. The time course of these increases varied by brain region, with some increasing steadily week by week and others remaining elevated throughout the measurement period. Cisplatin is also associated with activation of spinal microglia through enhanced triggering receptor expressed on myeloid 2/DNAX-activating protein of 12 kDA (TREM2/DAP12) signaling, and intrathecal administration of minocycline or anti-TREM2 antibodies prevented microglia activation and attenuated the development of mechanical allodynia and loss of IENFs in mice [116]. In rat brain slices, cisplatin, applied in an ex vivo preparation, primarily localized to ribosomes of the endoplasmic reticulum in astroglial cells and the myelin sheathing of oligodendrocytes [117]. Intrathecal administration of minocycline (which is capable of modulating microglia) or fluorocitrate (presumably an astrocyte modulator) mitigated the development of mechanical and cold hypersensitivity as well as deficits in motor coordination associated with oxaliplatin [118]. The effect of fluorocitrate was greater than minocycline, in line with the observations that platinum chemotherapeutic agents accumulate primarily in astrocytes [117]. Notably, minocycline failed to prevent pathological changes to nuclei and nucleoli of DRG neurons [118]. Supporting the notion that astrocytes play a prominent role in platinum compound-induced neurotoxicity, oxaliplatin has been shown to upregulate spinal dorsal horn expression of astrocyte connexin-43 (or GJ1, a component of gap junctions), which contributes to the development of mechanical hypersensitivity and has been implicated in the enhanced release of cytokines and chemokines from astrocytes following nerve injury [119, 120]. Inhibition of connexin-43 prevented the development of hypersensitivity in oxaliplatin and bortezomib CIPN models [119, 121]. Activated astrocytes and microglia in the spinal dorsal horn also contribute to vincristine-induced CIPN in mice [122, 123]. In rats, vincristine robustly activated spinal astrocytes via oxidative stress (but not microglia), which induced the production of IL-1β and subsequent phosphorylation of NMDA receptors in spinal neurons [124]. Intrathecal administration of pentoxifylline (presumably astrocyte modulator) attenuated mechanical allodynia induced by vincristine, while minocycline was not effective [124].
Spinal and brain glial activation appear to follow distinct time courses and their contribution to CIPN development is influenced not only by the class of chemotherapy drug, but also on factors such as the dosing regimen and age of the subject. In addition, activated microglia, like macrophages, can take on both pro-inflammatory and anti-inflammatory roles depending on incoming signals [125]. Thus, the effectiveness of treatments aimed at the inhibition of microglia or astrocytes to attenuate CIPN could be limited, particularly where glial activation occurs only during the initial phase of treatment or where the pattern of activation is not consistent in the literature. Treatment efficacy would likely benefit from targeting specific mechanisms of glial activation (i.e., TREM2/DAP12 signaling, connexin-43) for drug classes where pro-inflammatory activation is consistent and remains elevated throughout the treatment period.
Upregulation of pro-inflammatory cytokine and chemokine signaling in DRG and spinal cord
Cytokines involved in innate immune signaling have also been identified as crucial participants in the pathophysiology driving CIPN [19]. Chemotherapeutic agents have been shown to promote the release of pro-inflammatory cytokines (such as IL-6 and TNF-α) and downregulate expression of anti-inflammatory cytokines IL-4 and IL-10 in DRG and spinal cord [14, 15, 126–130]. The activity of pro-inflammatory cytokines (including IL-1β and TNF-α) in DRG tissue appears to be of particular importance for paclitaxel CIPN. While blocking inflammatory cytokines in lumbar DRG attenuated paclitaxel-induced CIPN, no effect was seen when inflammatory cytokines were blocked in the spinal cord, sciatic nerve, or skin [127]. Paclitaxel also upregulates pro-inflammatory cytokines and chemokines in the spinal cord, including TNF-α, IFN-γ, IL-1β, IL-6, IL-12p70, CCL3 (along with CCR5 and P2Y7), CCL4, CCL11, and GM-CSF [15, 111, 131]. Upregulated cytokine expression in the DRG persisted for more than 5 weeks after paclitaxel administration, while spinal cord cytokine upregulation was present during (day 4) and after completion (day 8) of a 7d dosing period, but not at day 29 [111, 129]. Elevated plasma levels of several cytokines, including IL-1α, IL-1β, IL-6, TNF-α, IFN-γ, and CCL2, have also been found in paclitaxel-treated rats [132].
In mice, cisplatin treatment enhanced the release of both pro-inflammatory (IL-1β, IL-6, CCL2, TNF-α, LIF, PGE2, ATP) and anti-inflammatory (IL-10, erythropoietin) cytokines from Schwann cells [133]. Oxaliplatin was shown to increase the expression of IL-1β in the lumbar DRG of mice after a single dose [70]. In cultured satellite glial cells, oxaliplatin exposure increased pro-inflammatory cytokines IL-6 and TNF-α, while IL-1β release was decreased [103]. The functional consequences of the inflammatory effects induced by chemotherapeutics has been also demonstrated in vitro. For example, media conditioned by oxaliplatin-treated satellite glial cells was able to sensitize cultured DRG neurons [103].
Contrasting other chemotherapeutics, thalidomide and its analogues have been shown to induce changes in cytokine expression that produce an anti-inflammatory effect, including a decrease in TNF-α produced by macrophages and monocytes, although this is not always consistent [134–137]. Thalidomide reduced TNF-α production when PBMC were stimulated with LPS, an effect which was shown to be triggered by increased degradation of TNF-α mRNA [134]. Decreased TNF-α production associated with thalidomide treatment had beneficial clinical effects in patients with leprosy and tuberculosis [138, 139]. Thalidomide also modifies IL-12 production, though the direction of the change depends on the cell type and manner of cytokine stimulation. LPS-induced production of IL-12 is inhibited by thalidomide treatment in vitro, while thalidomide treatment in patients with HIV or tuberculosis increased circulating IL-12 levels [136, 140, 141]. In a mouse model of ischemia/reperfusion injury, thalidomide treatment decreased the area of infarct, reduced oxidative stress and pro-inflammatory cytokine release, and attenuated neurological deficits after cerebral middle artery occlusion [142]. Thalidomide also decreased the expression of adhesion molecules and phagocytosis by leukocytes [143]. Lenalidomide, which exhibits less neurotoxicity than its analogue thalidomide, increased expression of the anti-inflammatory cytokine IL-10 and decreased IL-1β, IL-6, IL-12, and the expression of adhesion molecules [144]. Conversely, thalidomide has been shown to increase production of cytokines in response to antigen or mitogen stimulation, and primarily those involved in adaptive immune system responses (T cell and B cell activity). Thalidomide enhanced production of pro-inflammatory cytokine IL-2 in human mononuclear cells, but only when stimulated with mitogens [145]. In antigen-stimulated PBMC, thalidomide upregulated IL-4 and IL-5 production, but also inhibited IFN-γ [146]. Taken together, these studies suggest that thalidomide and its analogues exhibit more of an anti-inflammatory profile with cytokines involved in innate immunity, which contrasts strongly with other chemotherapy drugs.
Multiple chemotherapeutic compounds induce the production of major pro-inflammatory cytokines, such as TNF-α, a molecule that plays a major role in the pathogenesis of multiple neuropathies and chronic pain. For example, TNF-α has been shown to upregulate the expression of voltage-gated sodium channels Nav1.3 and Nav1.8 in DRG neurons, increase the frequency of spontaneous EPSCs in spinal neurons, and inhibit spontaneous activity in GABAergic neurons [147, 148]. Notably, paclitaxel, when given in a regime that induces CIPN related behaviors, increases TNF-α expression in the DRG of rats [72]. In oxaliplatin-treated mice, increased expression of TNF-α and IL-1β led to upregulation of STAT3 in the DRG [149], a signaling pathway involved in the development of allodynia induced by nerve injury through the upregulation of DRG neuronal Nav1.6 [150]. Interestingly, a major source of TNF-α are cells of monocytic origin. In fact, monocytes treated with cisplatin rapidly increase the release of TNF-α in vitro [151]. In the CNS, TNF-α from activated microglia and astrocytes from the dorsal horn of the spinal cord increases in tandem with mechanical allodynia that arises from chronic vincristine treatment, a hypersensitive state that is reduced by intrathecal treatments that block TNF-α [108, 122]. Bortezomib also upregulates TNF-α expression in the DRG, leading to an increase in phosphorylated JNK1/2 mediated by TNF-α receptors 1 and 2 [152]. Knockout of these receptors attenuated the development of mechanical allodynia due to bortezomib administration [152].
Another major cytokine that plays important roles in cancer, neuropathies, and pain is IL-6, which possesses dual pro- and anti-nociceptive/inflammatory properties. Multiple experimental pain models display an upregulation of IL-6 and/or its receptor, and when administered together IL-6 and soluble IL-6 receptors (IL-6Rs) are capable of sensitizing nociceptors in the skin [153–155]. IL-6, along with NGF, has been shown to increase translation in sensory neurons through the eIF4F complex, leading to increased protein synthesis that contributes to nociceptive plasticity [156]. Increased IL-6 expression has also been observed in ovarian, breast, and colorectal cancer patients and was a predictor for poorer prognosis [157–159]. Higher levels of IL-6 and soluble IL-6Rs were also identified post-chemotherapeutic treatment in breast cancer patients with painful CIPN compared to those who did not develop painful CIPN [160]. After paclitaxel administration, IL-6 was upregulated in both DRG neurons and astrocytes, and there is evidence that satellite glial cells also contribute to IL-6 release [161]. Similarly, the release of IL-6 from peripheral macrophages that are recruited to nerve and DRG tissue plays an important role in the development of vincristine-induced mechanical allodynia [71]. It should be noted that concomitant treatment with IL-6 blocked development of behavioral signs and changes in sciatic nerve conduction velocity in rodents treated with cisplatin, paclitaxel, or vincristine, suggesting that systemic IL-6 has a neuroprotective effect in these models [162].
Changes in TLR signaling
Toll-like receptors are expressed by immune cells and are important regulators of cytokine and chemokine production in innate immunity through activation of the myeloid differentiation factor 88 (MyD88) signaling pathway [163, 164]. Several TLRs are also expressed by primary sensory neurons, including TLRs 3, 4, 7, and 9 [66, 163, 165–169]. TLRs in DRG and trigeminal ganglia are predominantly expressed by nociceptive neurons and contribute to pain and itch signaling [66, 163, 165–168, 170–172]. Administration of ligands for TLRs 3, 7, and 9 induced the expression of pro-inflammatory chemokines and cytokines from cultured mouse DRGs, including IL-1α, IL-1β, PGE2, CCL5, and CXCL10 [169].
TLR4 signaling plays a role in neuropathic pain that differs depending on sex [173]. In males, TLR4 activity in peripheral macrophages (but not nociceptors) contributes to mechanical hypersensitivity, while TLR4 activity in nociceptors (but not macrophages) is responsible for this effect in females [174]. TLR4 activates the MyD88 signaling pathway, which causes early activation of NF-κB, and TRIF, which causes late NF-κB activation and interferon-β production [66, 167, 175–179]. In addition, TLR4 activates MAPK signaling cascades, including ERK1/2, P38, and JNK [171]. Changes in the activation of various MAPKs in the DRG or spinal cord have previously been identified following oxaliplatin, cisplatin, paclitaxel, vincristine, and bortezomib. Increased levels of phosphorylated ERK (pERK) 1/2 have been reported in the lumbar DRG of rats with mechanical allodynia following oxaliplatin treatment [180]. Cisplatin and oxaliplatin were found to activate ERK 1/2 and p38 and reduce the activation of JNK/Sapk in cultured DRG neurons harvested from rat embryos [181, 182]. Paclitaxel treatment increased activation of ERK 1/2 and P38 as well as the expression of pERK 1/2, phosphorylated p38 (pP38), and NF-κB in the DRG [171]. Paclitaxel induced activation of MAPK and JNK as well as NF-κB nuclear translocation in mouse macrophages, an effect that was lost in TLR4-deficient macrophages but remained in MyD88-deficient macrophages [175]. Macrophages from MyD88-deficient mice also fail to produce TNF or NO in response to paclitaxel [175] Vincristine was shown to induce phosphorylation of ERK, p38, and JNK in the spinal cord [123]. Upregulation of phosphorylated JNK (pJNK) 1/2 was found in the DRG of bortezomib-treated rats [152].
Together, these pathways lead to phosphorylation and activation of multiple transcription factors, leading to the synthesis and release of pro-inflammatory cytokines and chemokines, including IFNα/γ, TNF-α, CCL-2, IL-1, and IL-6 [66, 167, 171, 175, 176]. TLR4 signaling is upregulated in the DRG following cisplatin or paclitaxel treatment [171, 172]. Paclitaxel also sensitized TRPV1 on DRG neurons through TLR4 signaling; in addition, the number of DRG neurons expressing TRPV1 increased after paclitaxel treatment and the expression of TRPV1 protein in DRG neurons increased after vincristine treatment [170, 183]. TLR4 activity was also found to be upregulated in dorsal horn astrocytes following paclitaxel treatment [168]. Like TLR4, the activation of TLRs 3,7, and 9 also upregulate expression of TRPV1 and sensitize TRPV1-expressing DRG neurons [169]. Macrophage TLR9 activity contributes to paclitaxel-induced CIPN in male mice only, suggesting that the effect of this receptor is also sex-dependent [184]. Blockade or knockout of TLR9 prevents paclitaxel-induced increases in TNF and CXCL1 in cultured macrophages and DRG of male mice only [184]. In contrast, bortezomib was shown to downregulate TLR4 signaling through MyD88 and block lipopolysaccharide (LPS) activation of MAPKs, NF-κB, and interferon regulatory transcription factor (IRF) 3 and 8 in dendritic cells [185].
Enhanced chemokine signaling
The chemokine CCL-2 and its receptor CCR2 play an important role in immune cell recruitment and activation in neuropathic pain. Increased spinal CCL2 signaling after nerve injury leads to the activation of spinal microglia, and blocking CCL2 prevents both microglial activation and nociceptive behavior after peripheral nerve injury [186]. Increased expression of CCL2 also occurs on nerve fibers following injury (CCI), attracting monocytes/macrophages to the injury site [187]. The development of neuropathic pain following CCI was dependent on this recruitment of monocytes/macrophages due to enhanced expression of CCL2 in sciatic nerve cells, as demonstrated when CCL2 knockout or CCR2 inhibition blocked both of these events [187]. Further, intraneural administration of CCL2 was sufficient to induce nociceptive behavior in mice, an effect also associated with monocyte/macrophage recruitment to the site of injection [187]. Upregulation of CCL-2/CCR-2 signaling in DRG neurons has also been shown to contribute to the development of CIPN following oxaliplatin, paclitaxel, and bortezomib treatment, and blocking CCL2/CCR2 signaling attenuated behavioral signs of CIPN [69, 110, 126, 132, 168, 188]. CCL2 upregulation in the DRG of rats treated with bortezomib was shown to be dependent on enhanced expression of the transcription factor c-Jun and activation transcription factor 3 (ATF3), where ATF3 increases recruitment of c-Jun to the ccl2 promotor [69]. CCL2/CCR2 signaling has previously been shown to alter the function of TTX-resistant sodium channel Nav1.8 in DRG neurons of naïve rats, and CCL2 administration led to depolarization and action potential generation in neurons [126, 189]. In DRG neurons from paclitaxel-treated rats, CCL-2 administration evoked calcium influx and generated action potentials in neurons that were previously not spontaneously active [161, 188]. CCL2 upregulation in the lumbar spinal cord also contributes to cold hyperalgesia in paclitaxel CIPN, since blocking spinal CCL2 prevents cold hyperalgesia and spinal microglial activation [110]. Likewise, vincristine treatment increases CCL2 expression; furthermore, interactions between CCR2 and CX3CR1 signaling in monocytes recruited to the sciatic nerve have been shown to contribute to vincristine-induced CIPN [190]. CX3CL1 also resulted in the activation of CX3CR1-expressing monocytes, inducing the release of ROS and sensitization of TRPA1 on sensory neurons in vincristine-treated mice [191]. CXCL1 is upregulated in primary sensory neurons (A fibers) after paclitaxel treatment, and blocking CXCL1 during paclitaxel administration inhibited the infiltration of macrophages into the DRG [76]. CXCL1 also appears to increase the activity of p38 mitogen-activated protein kinase (MAPK) in macrophages, and blocking this p38 MAPK activity inhibited macrophage recruitment to the DRG and prevented neuronal damage and mechanical allodynia during paclitaxel treatment [76]. Upregulation of CX3CL1 via NF-κB signaling in the DRG was also found to contribute to neuronal sensitization and thermal hyperalgesia in oxaliplatin-treated rats [192]. Similarly, increases in CXCL12 were found in the DRG of oxaliplatin-treated mice were inhibited by blocking STAT3, a transcription factor that is enhanced after nerve injury [149, 155].
Translational evidence of innate immunity in chemotherapy-induced peripheral neuropathy
Chemotherapeutic drugs might also induce systemic immunomodulatory effects. During early treatment, there is often a temporary immunosuppressive effect due to cytotoxic activity on immune cells, though evidence suggests that different chemotherapeutics can stimulate the human immune system in other ways as well [14, 193]. Currently, there are very few studies showing changes in plasma content of cytokines and chemokines in patients under chemotherapy. As mention elsewhere in this review, one study associated high levels of IL-6 and soluble receptors (sIL-6R) in women with breast cancer with CIPN symptoms induced by different chemotherapeutic agents [160]. Similarly, in advanced breast cancer patients, paclitaxel or docetaxel increased serum levels of pro-inflammatory factors such as IFN-γ, IL-2, IL-6, GM-CSF and augmented peripheral blood mononuclear natural killer cell activity. Interestingly, while both treatments led to a decrease of acute phase serum IL-1 and TNF-α levels, the effects of docetaxel are more pronounced than those of paclitaxel [194]. Oxaliplatin also is capable of inducing an acute immune response in colorectal cancer patients that displayed an increase in TNF-α and IL-6 [195]. Despite these studies showing the possible immunomodulatory effect of chemotherapy itself, the immune response induced by this type of drug varies greatly. Also, it is difficult to dissociate the effects of the cancer and the effect of chemotherapeutic agent used. However, an in vitro study suggests that docetaxel promotes the differentiation of primary human monocytes into pro-inflammatory macrophages with a M1 phenotype that displayed an elevated secretion of IL-8 and IL-1β, as well as the ability to present antigens to T cells [196]. Similarly, tumor-specific cytotoxic T cells or dendritic cells acquire a tolerogenic phenotype that allows tumor cells to thrive. However, it has been shown in vitro that low doses of paclitaxel can reverse this phenotype to a more reactive immune activity [197]. The immunomodulatory effects of chemotherapy could differ in combined therapy, for example it has been shown that metastatic breast cancer patients display immune cells with higher M2 (anti-inflammatory) markers (CD163, ARG1, and IL-10), and that the combination of bevacizumab and paclitaxel induced a reduction in IL-10, something that did not occur with paclitaxel alone [198]. Similar reductions in immunosuppressive monocytes in B-cell acute lymphoblastic leukemia pediatric patients has been documented following chemotherapeutic treatment [199].
In humans, there are limitations to evaluate the immune response under chemotherapy treatment as measurements of inflammatory mediators are mostly restricted to serum or plasma. Nonetheless, systemic changes in cytokines might be associated with tissue changes, as seen in a study with patients that received thalidomide who showed a dose-dependent sensorimotor length-dependent axonal peripheral neuropathy that was linked to Wallerian degeneration detected in sural nerve biopsy [200]. Similarly, in other conditions, higher expression of IL-6 and IL-8 was found in skin biopsies from patients with painful peripheral neuropathy [201], indicating that pro-inflammatory mediators are likely involved in the pathogenesis of clinical and pathological manifestations of CIPN. These findings are in line with the discussed observations in pre-clinical models of CIPN.
Conclusions
The interplay between the nervous system and components of the innate immune system adds to the complex nature of neurotoxicity during chemotherapeutic cancer treatments. While some mechanisms—macrophage infiltration, spinal glia activation—appear to play fundamental roles in the development of CIPN and can be observed after treatment with most neurotoxic chemotherapies, others are specific to each drug class, and some even differ between drugs of the same type. Patients, especially those with advanced cancer undergoing chemotherapy of multiple types (for example, combination therapy with platinum compounds and taxanes) together or consecutively, may have multiple distinct mechanisms contributing to CIPN that change based on the current regimen [202–204]. While prevention is the ultimate goal for adjuvant drug development, therapies providing neuroprotection may also confer greater resistance to chemotherapy in cancer cells or cause additional undesirable side effects [205, 206]. Thus, there is an imminent need for targeted therapies that bypass these shortcomings, and specific mechanisms of innate immune activity may provide innovative targets for drug development that will alleviate CIPN symptoms and allow patients to continue life-saving chemotherapy treatments. In addition to more targeted treatments for pain, greater understanding of the innate immune pathophysiology of CIPN could also help identify which patient populations are at a greater risk of developing symptoms. DNA analysis of blood samples from patients undergoing bortezomib treatment identified variations in genes associated with neuron and immune function, including Schwann cells, that were associated with a greater risk of developing peripheral neuropathy [207]. Distinct genes and single nucleotide polymorphisms (SNPs) in the gene expression profiles of myeloma plasma cells taken before treatment were found to contribute to the risk of developing early vs late onset peripheral neuropathy during bortezomib or vincristine treatments [208]. Among these were genes involved in the cell cycle and apoptosis, mitochondria dysfunction, and peripheral nervous system development and function. Similar analysis of DNA from peripheral blood samples in multiple myeloma patients identified distinct SNPs in genes associated with peripheral nervous system inflammation, growth, and repair that were associated with increased risk for developing peripheral neuropathy from vincristine or thalidomide treatment, including some that had been previously linked to peripheral neuropathy, such as SLC12A6 (rs7614902), which encodes the protein for a K-Cl cotransporter [209]. As the development of personalized cancer therapies progresses, treatments for the prevention or mitigation of chemotherapy neuropathy based on an individual’s genetic risk factors and specific innate immune targets could become customary in cancer treatment plans.
Fig. 1.
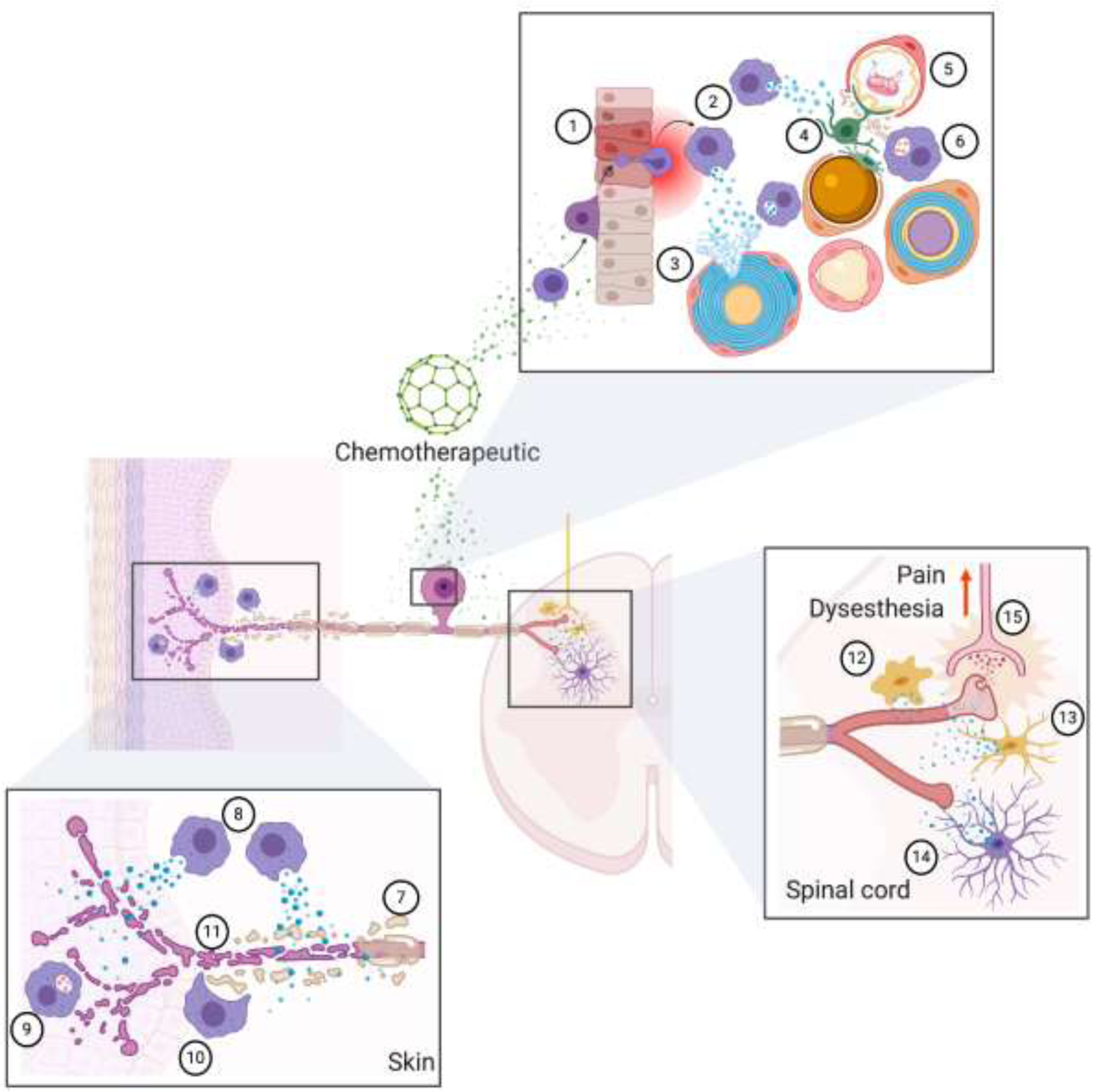
Chemotherapeutic agents directly act on peripheral nerves producing demyelination (1) and/or loss of intra epidermal nerve fibers (Skin). A cytotoxic or pro-inflammatory immune response is triggered due to direct actions of chemotherapeutics on leukocytes, or as a response of nerve fiber degeneration. Macrophages in particular migrate to the DRG, but also to the periphery secreting pro-inflammatory and pro-algesic molecules (2) and phagocyting degraded nerve fibers (3) or myelin (4). Chemotherapeutics also act in the CNS, and particularly in the spinal cord where microglial cells are activated, release pro-inflammatory and pro-nociceptive factors, and acquire an ameboid morphology (6) or a less ramified structure (7). Astrocytes are also affected, but they acquire a more ramified and elongated architecture (8). Both, microglia and astrocytes-derived factors sensitize primary sensory terminals, that increase their activity and release of neurotransmitters, resulting in an amplified nociceptive information in second order neurons (9) that ultimately is interpreted as pain. Created with BioRender.com.
Highlights.
Toll-like receptor 4 appears to be a common target for chemotherapy agents to induce inflammatory processes at peripheral nerve endings, in the peripheral axons, at the dorsal root ganglion cell bodies and in the spinal cord to promote treatment related pain.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Krarup-Hansen A, et al. , Neuronal involvement in cisplatin neuropathy: prospective clinical and neurophysiological studies. Brain, 2007. 130(4): p. 1076–1088. [DOI] [PubMed] [Google Scholar]
- [2].Bhagra A and Rao RD, Chemotherapy-induced neuropathy. Current oncology reports, 2007. 9(4): p. 290–299. [DOI] [PubMed] [Google Scholar]
- [3].Quasthoff S and Hartung HP, Chemotherapy-induced peripheral neuropathy. Journal of neurology, 2002. 249(1): p. 9–17. [DOI] [PubMed] [Google Scholar]
- [4].Hamers FPT, Gispen WH, and Neijt JP, Neurotoxic side-effects of cisplatin. European Journal of Cancer and Clinical Oncology, 1991. 27(3): p. 372–376. [DOI] [PubMed] [Google Scholar]
- [5].Kanbayashi Y, et al. , Statistical identification of predictors for peripheral neuropathy associated with administration of bortezomib, taxanes, oxaliplatin or vincristine using ordered logistic regression analysis. Anti-cancer drugs, 2010. 21(9): p. 877–881. [DOI] [PubMed] [Google Scholar]
- [6].Boyette-Davis JA, et al. , Persistent chemoneuropathy in patients receiving the plant alkaloids paclitaxel and vincristine. Cancer chemotherapy and pharmacology, 2013. 71(3): p. 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bastuji-Garin S, et al. , Incidence and risk factors for thalidomide neuropathy: a prospective study of 135 dermatologic patients. Journal of investigative dermatology, 2002. 119(5): p. 1020–1026. [DOI] [PubMed] [Google Scholar]
- [8].Staff NP, et al. , Chemotherapy‐induced peripheral neuropathy: a current review. Annals of neurology, 2017. 81(6): p. 772–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McWhinney SR, Goldberg RM, and McLeod HL, Platinum neurotoxicity pharmacogenetics. Molecular cancer therapeutics, 2009. 8(1): p. 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dworkin RH, et al. , Advances in neuropathic pain: diagnosis, mechanisms, and treatment recommendations. Archives of neurology, 2003. 60(11): p. 1524–1534. [DOI] [PubMed] [Google Scholar]
- [11].Richardson PG, et al. , A phase 2 study of bortezomib in relapsed, refractory myeloma. New England Journal of Medicine, 2003. 348(26): p. 2609–2617. [DOI] [PubMed] [Google Scholar]
- [12].Ji R-R, Chamessian A, and Zhang Y-Q, Pain regulation by non-neuronal cells and inflammation. Science, 2016. 354(6312): p. 572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Massey RL, Kim HK, and Abdi S, Brief review: chemotherapy-induced painful peripheral neuropathy (CIPPN): current status and future directions. Canadian Journal of Anesthesia/Journal canadien d’anesthésie, 2014. 61(8): p. 754–762. [DOI] [PubMed] [Google Scholar]
- [14].Lees JG, et al. , Immune-mediated processes implicated in chemotherapy-induced peripheral neuropathy. European Journal of Cancer, 2017. 73: p. 22–29. [DOI] [PubMed] [Google Scholar]
- [15].Makker PGS, et al. , Characterisation of Immune and Neuroinflammatory Changes Associated with Chemotherapy-Induced Peripheral Neuropathy. PLOS ONE, 2017. 12(1): p. e0170814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Watkins LR and Maier SF, Beyond Neurons: Evidence That Immune and Glial Cells Contribute to Pathological Pain States. Physiological Reviews, 2002. 82(4): p. 981–1011. [DOI] [PubMed] [Google Scholar]
- [17].Scholz J and Woolf CJ, The neuropathic pain triad: neurons, immune cells and glia. Nature Neuroscience, 2007. 10(11): p. 1361–1368. [DOI] [PubMed] [Google Scholar]
- [18].Calvo M, Dawes JM, and Bennett DLH, The role of the immune system in the generation of neuropathic pain. The Lancet Neurology, 2012. 11(7): p. 629–642. [DOI] [PubMed] [Google Scholar]
- [19].Wang X-M, et al. , Discovering cytokines as targets for chemotherapy-induced painful peripheral neuropathy. Cytokine, 2012. 59(1): p. 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Montague K and Malcangio M, The Therapeutic Potential of Monocyte/Macrophage Manipulation in the Treatment of Chemotherapy-Induced Painful Neuropathy. Frontiers in Molecular Neuroscience, 2017. 10(397). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ma J, et al. , Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: Targeting the source. Cancer, 2018. 124(11): p. 2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gold R, Archelos JJ, and Hartung H-P, Mechanisms of Immune Regulation in the Peripheral Nervous System. Brain Pathology, 1999. 9(2): p. 343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rutkowski JL, et al. , Signals for proinflammatory cytokine secretion by human Schwann cells. Journal of neuroimmunology, 1999. 101(1): p. 47–60. [DOI] [PubMed] [Google Scholar]
- [24].Tzekova N, Heinen A, and Küry P, Molecules involved in the crosstalk between immune-and peripheral nerve Schwann cells. Journal of clinical immunology, 2014. 34(1): p. 86–104. [DOI] [PubMed] [Google Scholar]
- [25].van Velzen M, et al. , Neuron-interacting satellite glial cells in human trigeminal ganglia have an APC phenotype. The Journal of Immunology, 2009. 183(4): p. 2456–2461. [DOI] [PubMed] [Google Scholar]
- [26].Afroz S, et al. , CGRP induces differential regulation of cytokines from satellite glial cells in trigeminal ganglia and orofacial nociception. International journal of molecular sciences, 2019. 20(3): p. 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dubový P, et al. , Satellite glial cells express IL-6 and corresponding signal-transducing receptors in the dorsal root ganglia of rat neuropathic pain model. Neuron Glia Biol, 2010. 6(1): p. 73–83. [DOI] [PubMed] [Google Scholar]
- [28].Takeda M, et al. , Enhanced excitability of nociceptive trigeminal ganglion neurons by satellite glial cytokine following peripheral inflammation. Pain, 2007. 129(1): p. 155–166. [DOI] [PubMed] [Google Scholar]
- [29].Thacker MA, et al. , Pathophysiology of Peripheral Neuropathic Pain: Immune Cells and Molecules. Anesthesia & Analgesia, 2007. 105(3): p. 838–847. [DOI] [PubMed] [Google Scholar]
- [30].Oaklander AL, The density of remaining nerve endings in human skin with and without postherpetic neuralgia after shingles. Pain, 2001. 92(1–2): p. 139–145. [DOI] [PubMed] [Google Scholar]
- [31].Calder J, Holten I, and McAllister R, Evidence for immune system involvement in reflex sympathetic dystrophy. Journal of hand surgery, 1998. 23(2): p. 147–150. [DOI] [PubMed] [Google Scholar]
- [32].Scapini P, et al. , The neutrophil as a cellular source of chemokines. Immunological Reviews, 2000. 177(1): p. 195–203. [DOI] [PubMed] [Google Scholar]
- [33].Perkins NM and Tracey DJ, Hyperalgesia due to nerve injury: role of neutrophils. Neuroscience, 2000. 101(3): p. 745–757. [DOI] [PubMed] [Google Scholar]
- [34].Morin N, et al. , Neutrophils invade lumbar dorsal root ganglia after chronic constriction injury of the sciatic nerve. Journal of Neuroimmunology, 2007. 184(1): p. 164–171. [DOI] [PubMed] [Google Scholar]
- [35].Zuo Y, et al. , Inflammation and hyperalgesia induced by nerve injury in the rat: a key role of mast cells. Pain, 2003. 105(3): p. 467–479. [DOI] [PubMed] [Google Scholar]
- [36].Nakashima C, et al. , Interaction of peripheral nerves and mast cells, eosinophils, and basophils in the development of pruritus. Experimental Dermatology, 2019. 28(12): p. 1405–1411. [DOI] [PubMed] [Google Scholar]
- [37].Vincent L, et al. , Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood, 2013. 122(11): p. 1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Schäfers M and Sorkin L, Effect of cytokines on neuronal excitability. Neuroscience letters, 2008. 437(3): p. 188–193. [DOI] [PubMed] [Google Scholar]
- [39].Binshtok AM, et al. , Nociceptors are interleukin-1β sensors. Journal of Neuroscience, 2008. 28(52): p. 14062–14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kobayashi H, et al. , Human eosinophils produce neurotrophins and secrete nerve growth factor on immunologic stimuli. Blood, 2002. 99(6): p. 2214–2220. [DOI] [PubMed] [Google Scholar]
- [41].Durack DT, Sumi SM, and Klebanoff SJ, Neurotoxicity of human eosinophils. Proceedings of the National Academy of Sciences, 1979. 76(3): p. 1443–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Weaver DF, et al. , Eosinophil‐induced neurotoxicity. Axonal neuropathy, cerebral infarction, and dementia, 1988. 38(1): p. 144–144. [DOI] [PubMed] [Google Scholar]
- [43].Sunohara N, et al. , Neurotoxicity of human eosinophils towards peripheral nerves. Journal of the Neurological Sciences, 1989. 92(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- [44].Morgan B, et al. , Role of complement in inflammation and injury in the nervous system. Experimental and clinical immunogenetics, 1997. 14(1): p. 19. [PubMed] [Google Scholar]
- [45].De Jonge RR, et al. , Expression of complement components in the peripheral nervous system. Human molecular genetics, 2004. 13(3): p. 295–302. [DOI] [PubMed] [Google Scholar]
- [46].Vallejo R, et al. , The Role of Glia and the Immune System in the Development and Maintenance of Neuropathic Pain. Pain Practice, 2010. 10(3): p. 167–184. [DOI] [PubMed] [Google Scholar]
- [47].Nimmerjahn A, Kirchhoff F, and Helmchen F, Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science, 2005. 308(5726): p. 1314–1318. [DOI] [PubMed] [Google Scholar]
- [48].Gehrmann J, Matsumoto Y, and Kreutzberg GW, Microglia: Intrinsic immuneffector cell of the brain. Brain Research Reviews, 1995. 20(3): p. 269–287. [DOI] [PubMed] [Google Scholar]
- [49].Izquierdo P, Attwell D, and Madry C, Ion Channels and Receptors as Determinants of Microglial Function. 2019. (1878–108X (Electronic)). [DOI] [PubMed]
- [50].Olson JK and Miller SD, Microglia Initiate Central Nervous System Innate and Adaptive Immune Responses through Multiple TLRs. The Journal of Immunology, 2004. 173(6): p. 3916–3924. [DOI] [PubMed] [Google Scholar]
- [51].Romero-Sandoval EA and Sweitzer S, Nonneuronal Central Mechanisms of Pain: Glia and Immune Response, in Progress in Molecular Biology and Translational Science, Price TJ and Dussor G, Editors. 2015, Academic Press: Burlington. p. 325–358. [DOI] [PubMed] [Google Scholar]
- [52].Shrikant P and Benveniste EN, The central nervous system as an immunocompetent organ: role of glial cells in antigen presentation. The Journal of Immunology, 1996. 157(5): p. 1819–1822. [PubMed] [Google Scholar]
- [53].Schomberg D and Olson JK, Immune responses of microglia in the spinal cord: Contribution to pain states. Experimental Neurology, 2012. 234(2): p. 262–270. [DOI] [PubMed] [Google Scholar]
- [54].Popovich PG, Wei P, and Stokes BT, Cellular inflammatory response after spinal cord injury in sprague-dawley and lewis rats. Journal of Comparative Neurology, 1997. 377(3): p. 443–464. [DOI] [PubMed] [Google Scholar]
- [55].Grace PM, Rolan PE, and Hutchinson MR, Peripheral immune contributions to the maintenance of central glial activation underlying neuropathic pain. Brain, Behavior, and Immunity, 2011. 25(7): p. 1322–1332. [DOI] [PubMed] [Google Scholar]
- [56].Khasabova IA, et al. , Sensitization of nociceptors by prostaglandin E2–glycerol contributes to hyperalgesia in mice with sickle cell disease. blood, 2019. 133(18): p. 1989–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Prinz M, Erny D, and Hagemeyer N, Ontogeny and homeostasis of CNS myeloid cells. Nature Immunology, 2017. 18(4): p. 385–392. [DOI] [PubMed] [Google Scholar]
- [58].Khorooshi R, et al. , Innate signaling within the central nervous system recruits protective neutrophils. Acta Neuropathologica Communications, 2020. 8(1): p. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mittal A, et al. , Mast Cell Neural Interactions in Health and Disease. Frontiers in Cellular Neuroscience, 2019. 13(110). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Geurts N, et al. , Basophils are dispensable for the recovery of gross locomotion after spinal cord hemisection injury. Journal of Leukocyte Biology, 2016. 99(4): p. 579–582. [DOI] [PubMed] [Google Scholar]
- [61].Bürgi B, et al. , Basophil priming by neurotrophic factors. Activation through the trk receptor. The Journal of Immunology, 1996. 157(12): p. 5582–5588. [PubMed] [Google Scholar]
- [62].Hamada A, et al. , Nerve growth factor enhances survival and cytotoxic activity of human eosinophils. British Journal of Haematology, 1996. 93(2): p. 299–302. [DOI] [PubMed] [Google Scholar]
- [63].Solomon A, et al. , Nerve growth factor is preformed in and activates human peripheral blood eosinophils. Journal of Allergy and Clinical Immunology, 1998. 102(3): p. 454–460. [DOI] [PubMed] [Google Scholar]
- [64].Mohanty T, et al. , Neutrophil extracellular traps in the central nervous system hinder bacterial clearance during pneumococcal meningitis. Nature Communications, 2019. 10(1): p. 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Manda-Handzlik A and Demkow U, The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells, 2019. 8(12): p. 1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Liu X-J, et al. , Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Research, 2014. 24(11): p. 1374–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Starobova H, et al. , Inflammatory and Neuropathic Gene Expression Signatures of Chemotherapy-Induced Neuropathy Induced by Vincristine, Cisplatin, and Oxaliplatin in C57BL/6J Mice. The Journal of Pain, 2019. [DOI] [PubMed]
- [68].Nishida K, et al. , Up‐regulation of matrix metalloproteinase‐3 in the dorsal root ganglion of rats with paclitaxel‐induced neuropathy. Cancer science, 2008. 99(8): p. 1618–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Liu C, et al. , Upregulation of CCL2 via ATF3/c-Jun interaction mediated the Bortezomib-induced peripheral neuropathy. Brain, behavior, and immunity, 2016. 53: p. 96–104. [DOI] [PubMed] [Google Scholar]
- [70].Li D, et al. , Preventive effects of bee venom derived phospholipase A2 on oxaliplatin-induced neuropathic pain in mice. Toxins, 2016. 8(1): p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kiguchi N, et al. , The critical role of invading peripheral macrophage-derived interleukin-6 in vincristine-induced mechanical allodynia in mice. European journal of pharmacology, 2008. 592(1–3): p. 87–92. [DOI] [PubMed] [Google Scholar]
- [72].Zhang H, et al. , Dorsal Root Ganglion Infiltration by Macrophages Contributes to Paclitaxel Chemotherapy-Induced Peripheral Neuropathy. The Journal of Pain, 2016. 17(7): p. 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Tsubota M, et al. , Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: negative impact of anticoagulants. Journal of neuroinflammation, 2019. 16(1): p. 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Meregalli C, et al. , High-dose intravenous immunoglobulins reduce nerve macrophage infiltration and the severity of bortezomib-induced peripheral neurotoxicity in rats. Journal of neuroinflammation, 2018. 15(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Peters CM, et al. , Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Experimental neurology, 2007. 203(1): p. 42–54. [DOI] [PubMed] [Google Scholar]
- [76].Huang Z-Z, et al. , CX3CL1-mediated macrophage activation contributed to paclitaxel-induced DRG neuronal apoptosis and painful peripheral neuropathy. Brain, Behavior, and Immunity, 2014. 40: p. 155–165. [DOI] [PubMed] [Google Scholar]
- [77].Liu C-C, et al. , Prevention of paclitaxel-induced allodynia by minocycline: effect on loss of peripheral nerve fibers and infiltration of macrophages in rats. Molecular pain, 2010. 6: p. 1744-8069–6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Laast VA, et al. , Macrophage-mediated dorsal root ganglion damage precedes altered nerve conduction in SIV-infected macaques. The American journal of pathology, 2011. 179(5): p. 2337–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Conti G, et al. , Macrophage infiltration and death in the nerve during the early phases of experimental diabetic neuropathy: a process concomitant with endoneurial induction of IL-1β and p75NTR. Journal of the neurological sciences, 2002. 195(1): p. 35–40. [DOI] [PubMed] [Google Scholar]
- [80].Gonçalves NP, et al. , Schwann cell p75 neurotrophin receptor modulates small fiber degeneration in diabetic neuropathy. Glia, 2020. 68(12): p. 2725–2743. [DOI] [PubMed] [Google Scholar]
- [81].Demir IE, et al. , Perineural mast cells are specifically enriched in pancreatic neuritis and neuropathic pain in pancreatic cancer and chronic pancreatitis. PloS one, 2013. 8(3): p. e60529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Dyck PJ and Giannini C, Pathologic alterations in the diabetic neuropathies of humans: a review. Journal of Neuropathology & Experimental Neurology, 1996. 55(12): p. 1181–1193. [DOI] [PubMed] [Google Scholar]
- [83].Yagihashi S, Yamagishi S-I, and Wada R, Pathology and pathogenetic mechanisms of diabetic neuropathy: correlation with clinical signs and symptoms. Diabetes research and clinical practice, 2007. 77(3): p. S184–S189. [DOI] [PubMed] [Google Scholar]
- [84].Siau C, Xiao W, and Bennett GJ, Paclitaxel-and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Experimental neurology, 2006. 201(2): p. 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Qureshi AA, et al. , Langerhans cells express inducible nitric oxide synthase and produce nitric oxide. Journal of investigative dermatology, 1996. 107(6): p. 815–821. [DOI] [PubMed] [Google Scholar]
- [86].Torii H, et al. , Expression of neurotrophic factors and neuropeptide receptors by Langerhans cells and the Langerhans cell-like cell line XS52: further support for a functional relationship between Langerhans cells and epidermal nerves. Journal of investigative dermatology, 1997. 109(4): p. 586–591. [DOI] [PubMed] [Google Scholar]
- [87].Deng L, Ding W, and Granstein RD, Thalidomide Inhibits Tumor Necrosis Factor-α Production and Antigen Presentation by Langerhans Cells. Journal of Investigative Dermatology, 2003. 121(5). [DOI] [PubMed] [Google Scholar]
- [88].Berthier-Vergnes O, et al. , TNF-α enhances phenotypic and functional maturation of human epidermal Langerhans cells and induces IL-12 p40 and IP-10/CXCL-10 production. FEBS letters, 2005. 579(17): p. 3660–3668. [DOI] [PubMed] [Google Scholar]
- [89].Lindenlaub T and Sommer C, Epidermal innervation density after partial sciatic nerve lesion and pain-related behavior in the rat. Acta neuropathologica, 2002. 104(2): p. 137–143. [DOI] [PubMed] [Google Scholar]
- [90].Sakamoto A, Andoh T, and Kuraishi Y, Involvement of mast cells and proteinase-activated receptor 2 in oxaliplatin-induced mechanical allodynia in mice. Pharmacological Research, 2016. 105: p. 84–92. [DOI] [PubMed] [Google Scholar]
- [91].Gao W, et al. , Quercetin ameliorates paclitaxel-induced neuropathic pain by stabilizing mast cells, and subsequently blocking PKCε-dependent activation of TRPV1. Acta Pharmacologica Sinica, 2016. 37(9): p. 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jaggi AS, et al. , Pharmacological investigations on mast cell stabilizer and histamine receptor antagonists in vincristine-induced neuropathic pain. Naunyn-Schmiedeberg’s archives of pharmacology, 2017. 390(11): p. 1087–1096. [DOI] [PubMed] [Google Scholar]
- [93].Saxena S and Caroni P, Mechanisms of axon degeneration: from development to disease. Progress in neurobiology, 2007. 83(3): p. 174–191. [DOI] [PubMed] [Google Scholar]
- [94].Shamash S, Reichert F, and Rotshenker S, The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. Journal of Neuroscience, 2002. 22(8): p. 3052–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Perrin FE, et al. , Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1α and interleukin-1β in Wallerian degeneration. Brain, 2005. 128(4): p. 854–866. [DOI] [PubMed] [Google Scholar]
- [96].Mueller M, et al. , Macrophage response to peripheral nerve injury: the quantitative contribution of resident and hematogenous macrophages. Laboratory investigation, 2003. 83(2): p. 175–185. [DOI] [PubMed] [Google Scholar]
- [97].Omura T, et al. , Spatiotemporal quantification of recruit and resident macrophages after crush nerve injury utilizing immunohistochemistry. Brain research, 2005. 1057(1–2): p. 29–36. [DOI] [PubMed] [Google Scholar]
- [98].Ydens E, et al. , Acute injury in the peripheral nervous system triggers an alternative macrophage response. Journal of neuroinflammation, 2012. 9(1): p. 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Imai S, et al. , Taxanes and platinum derivatives impair Schwann cells via distinct mechanisms. Scientific reports, 2017. 7(1): p. 5947–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Cece R, et al. , Ultrastructural aspects of DRG satellite cell involvement in experimental cisplatin neuronopathy. Journal of submicroscopic cytology and pathology, 1995. 27(4): p. 417–425. [PubMed] [Google Scholar]
- [101].Corsetti G, et al. , Cytoplasmic Changes in Satellite Cells of Spinal Ganglia Induced by Cisplatin Treatment in Rats. Ultrastructural Pathology, 2000. 24(4): p. 259–265. [DOI] [PubMed] [Google Scholar]
- [102].Sugimoto T, et al. , Peripheral neuroglial death induced by cisplatin administration in newborn rats. NeuroReport, 2001. 12(1): p. 137–140. [DOI] [PubMed] [Google Scholar]
- [103].Schmitt L-I, et al. , Activation and functional modulation of satellite glial cells by oxaliplatin lead to hyperexcitability of sensory neurons in vitro. Molecular and Cellular Neuroscience, 2020: p. 103499. [DOI] [PubMed]
- [104].Di Cesare Mannelli L, et al. , Morphologic Features and Glial Activation in Rat Oxaliplatin-Dependent Neuropathic Pain. The Journal of Pain, 2013. 14(12): p. 1585–1600. [DOI] [PubMed] [Google Scholar]
- [105].Krarup-Hansen A, et al. , Histology and platinum content of sensory ganglia and sural nerves in patients treated with cisplatin and carboplatin: an autopsy study. Neuropathology and applied neurobiology, 1999. 25(1): p. 29–40. [DOI] [PubMed] [Google Scholar]
- [106].Meregalli C, et al. , Bortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in rats. European Journal of Pain, 2010. 14(4): p. 343–350. [DOI] [PubMed] [Google Scholar]
- [107].Cavaletti G, et al. , Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Experimental neurology, 2007. 204(1): p. 317–325. [DOI] [PubMed] [Google Scholar]
- [108].Kiguchi N, et al. , Involvement of inflammatory mediators in neuropathic pain caused by vincristine. International review of neurobiology, 2009. 85: p. 179–190. [DOI] [PubMed] [Google Scholar]
- [109].Topp KS, Tanner KD, and Levine JD, Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine‐induced painful peripheral neuropathy in the rat. Journal of Comparative Neurology, 2000. 424(4): p. 563–576. [PubMed] [Google Scholar]
- [110].Pevida M, et al. , Spinal CCL2 and microglial activation are involved in paclitaxel-evoked cold hyperalgesia. Brain research bulletin, 2013. 95: p. 21–27. [DOI] [PubMed] [Google Scholar]
- [111].Burgos E, et al. , Cannabinoid agonist WIN 55,212–2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. European journal of pharmacology, 2012. 682(1–3): p. 62–72. [DOI] [PubMed] [Google Scholar]
- [112].Zhang H, et al. , Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of Paclitaxel-induced painful neuropathy. The Journal of Pain, 2012. 13(3): p. 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Boyette-Davis J, et al. , Intraepidermal nerve fiber loss corresponds to the development of taxol-induced hyperalgesia and can be prevented by treatment with minocycline. PAIN®, 2011. 152(2): p. 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Ruiz‐Medina J, et al. , Paclitaxel‐induced neuropathic pain is age dependent and devolves on glial response. European journal of pain, 2013. 17(1): p. 75–85. [DOI] [PubMed] [Google Scholar]
- [115].Masocha W, Astrocyte activation in the anterior cingulate cortex and altered glutamatergic gene expression during paclitaxel-induced neuropathic pain in mice. PeerJ, 2015. 3: p. e1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Hu L-Y, et al. , Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain, behavior, and immunity, 2018. 68: p. 132–145. [DOI] [PubMed] [Google Scholar]
- [117].Zueva L, et al. , Electron Microscopy in Rat Brain Slices Reveals Rapid Accumulation of Cisplatin on Ribosomes and Other Cellular Components Only in Glia. Chemotherapy Research and Practice, 2014. 2014: p. 174039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Di Cesare Mannelli L, et al. , Glial role in oxaliplatin-induced neuropathic pain. Experimental Neurology, 2014. 261: p. 22–33. [DOI] [PubMed] [Google Scholar]
- [119].Yoon S-Y, et al. , Spinal astrocyte gap junctions contribute to oxaliplatin-induced mechanical hypersensitivity. The Journal of Pain, 2013. 14(2): p. 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Chen G, et al. , Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain, 2014. 137(8): p. 2193–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Robinson CR and Dougherty PM, Spinal astrocyte gap junction and glutamate transporter expression contributes to a rat model of bortezomib-induced peripheral neuropathy. Neuroscience, 2015. 285: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kiguchi N, et al. , Up-regulation of tumor necrosis factor-alpha in spinal cord contributes to vincristine-induced mechanical allodynia in mice. Neuroscience letters, 2008. 445(2): p. 140–143. [DOI] [PubMed] [Google Scholar]
- [123].Shen Y, et al. , Exogenous induction of HO-1 alleviates vincristine-induced neuropathic pain by reducing spinal glial activation in mice. Neurobiology of Disease, 2015. 79: p. 100–110. [DOI] [PubMed] [Google Scholar]
- [124].Ji X-T, et al. , Spinal astrocytic activation contributes to mechanical allodynia in a rat chemotherapy-induced neuropathic pain model. PloS one, 2013. 8(4): p. e60733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Jha MK, Lee W-H, and Suk K, Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochemical pharmacology, 2016. 103: p. 1–16. [DOI] [PubMed] [Google Scholar]
- [126].Zhang H, et al. , Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. The Journal of Pain, 2013. 14(10): p. 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Kim HK, et al. , Rolipram, a Selective Phosphodiesterase 4 Inhibitor, Ameliorates Mechanical Hyperalgesia in a Rat Model of Chemotherapy-Induced Neuropathic Pain through Inhibition of Inflammatory Cytokines in the Dorsal Root Ganglion. Frontiers in Pharmacology, 2017. 8(885). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Janes K, et al. , Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain, behavior, and immunity, 2015. 44: p. 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Ledeboer A, et al. , Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain, behavior, and immunity, 2007. 21(5): p. 686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Doyle T, et al. , Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. Journal of Neuroscience, 2012. 32(18): p. 6149–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ochi-Ishi R, et al. , Involvement of the chemokine CCL3 and the purinoceptor p2× 7 in the spinal cord in paclitaxel-induced mechanical allodynia. Molecular pain, 2014. 10: p. 1744–8069-10–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Al‐Mazidi S, et al. , Blocking of cytokines signalling attenuates evoked and spontaneous neuropathic pain behaviours in the paclitaxel rat model of chemotherapy‐induced neuropathy. European Journal of Pain, 2018. 22(4): p. 810–821. [DOI] [PubMed] [Google Scholar]
- [133].Öztürk G, et al. , Effect of leukemia inhibitory factor in experimental cisplatin neuropathy in mice. Cytokine, 2005. 29(1): p. 31–41. [DOI] [PubMed] [Google Scholar]
- [134].Moreira A, et al. , Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. The Journal of experimental medicine, 1993. 177(6): p. 1675–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Muller GW, et al. , Amino-substituted thalidomide analogs: potent inhibitors of TNF-α production. Bioorganic & medicinal chemistry letters, 1999. 9(11): p. 1625–1630. [DOI] [PubMed] [Google Scholar]
- [136].Corral LG, et al. , Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-α. The Journal of Immunology, 1999. 163(1): p. 380–386. [PubMed] [Google Scholar]
- [137].Jacobson JM, et al. , Thalidomide for the treatment of oral aphthous ulcers in patients with human immunodeficiency virus infection. New England Journal of Medicine, 1997. 336(21): p. 1487–1493. [DOI] [PubMed] [Google Scholar]
- [138].Sampaio EP, et al. , The influence of thalidomide on the clinical and immunologic manifestation of erythema nodosum leprosum. Journal of Infectious Diseases, 1993. 168(2): p. 408–414. [DOI] [PubMed] [Google Scholar]
- [139].Tramontana JM, et al. , Thalidomide treatment reduces tumor necrosis factor α production and enhances weight gain in patients with pulmonary tuberculosis. Molecular Medicine, 1995. 1(4): p. 384–397. [PMC free article] [PubMed] [Google Scholar]
- [140].Moller DR, et al. , Inhibition of IL-12 production by thalidomide. The Journal of Immunology, 1997. 159(10): p. 5157–5161. [PubMed] [Google Scholar]
- [141].Haslett PA, et al. , Thalidomide stimulates T cell responses and interleukin 12 production in HIV-infected patients. AIDS research and human retroviruses, 1999. 15(13): p. 1169–1179. [DOI] [PubMed] [Google Scholar]
- [142].Palencia G, et al. , Anti-apoptotic, anti-oxidant, and anti-inflammatory effects of thalidomide on cerebral ischemia/reperfusion injury in rats. Journal of the neurological sciences, 2015. 351(1–2): p. 78–87. [DOI] [PubMed] [Google Scholar]
- [143].Barnhill RL, et al. , Studies on the anti-inflammatory properties of thalidomide: effects on polymorphonuclear leukocytes and monocytes. Journal of the American Academy of Dermatology, 1984. 11(5): p. 814–819. [DOI] [PubMed] [Google Scholar]
- [144].Kotla V, et al. , Mechanism of action of lenalidomide in hematological malignancies. Journal of hematology & oncology, 2009. 2(1): p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Shannon E and Sandoval F, Thalidomide increases the synthesis of IL-2 in cultures of human mononuclear cells stimulated with Concanavalin-A, Staphylococcal enterotoxin A, and purified protein derivative. Immunopharmacology, 1995. 31(1): p. 109–116. [DOI] [PubMed] [Google Scholar]
- [146].McHugh S, et al. , The immunosuppressive drug thalidomide induces T helper cell type 2 (Th2) and concomitantly inhibits Th1 cytokine production in mitogen‐and antigen‐stimulated human peripheral blood mononuclear cell cultures. Clinical & Experimental Immunology, 1995. 99(2): p. 160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Zhang H, Nei H, and Dougherty PM, A p38 mitogen-activated protein kinase-dependent mechanism of disinhibition in spinal synaptic transmission induced by tumor necrosis factor-α. Journal of Neuroscience, 2010. 30(38): p. 12844–12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].He X-H, et al. , TNF-α contributes to up-regulation of Nav1. 3 and Nav1. 8 in DRG neurons following motor fiber injury. PAIN®, 2010. 151(2): p. 266–279. [DOI] [PubMed] [Google Scholar]
- [149].Li Y-Y, et al. , Activation of STAT3-mediated CXCL12 up-regulation in the dorsal root ganglion contributes to oxaliplatin-induced chronic pain. Molecular pain, 2017. 13: p. 1744806917747425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ding H-H, et al. , TNF-α/STAT3 pathway epigenetically upregulates Nav1. 6 expression in DRG and contributes to neuropathic pain induced by L5-VRT. Journal of neuroinflammation, 2019. 16(1): p. 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Gan X, Jewett A, and Bonavida B, Activation of human peripheral-blood-derived monocytes by cis-diamminedichloroplatinum: enhanced tumoricidal activity and secretion of tumor necrosis factor-alpha. Natural immunity, 1992. 11(3): p. 144–155. [PubMed] [Google Scholar]
- [152].Zhang J, et al. , TNF-α-mediated JNK activation in the dorsal root ganglion neurons contributes to Bortezomib-induced peripheral neuropathy. Brain, Behavior, and Immunity, 2014. 38: p. 185–191. [DOI] [PubMed] [Google Scholar]
- [153].Obreja O, et al. , Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. Pain, 2002. 96(1–2): p. 57–62. [DOI] [PubMed] [Google Scholar]
- [154].De Jongh RF, et al. , The role of interleukin-6 in nociception and pain. Anesthesia & Analgesia, 2003. 96(4): p. 1096–1103. [DOI] [PubMed] [Google Scholar]
- [155].Yamauchi K, et al. , Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. Journal of neurochemistry, 2006. 96(4): p. 1060–1070. [DOI] [PubMed] [Google Scholar]
- [156].Melemedjian OK, et al. , IL-6-and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. Journal of Neuroscience, 2010. 30(45): p. 15113–15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Penson RT, et al. , Cytokines IL‐1β, IL‐2, IL‐6, IL‐8, MCP‐1, GM‐CSF and TNFα in patients with epithelial ovarian cancer and their relationship to treatment with paclitaxel. International journal of gynecological cancer, 2000. 10(1): p. 33–41. [DOI] [PubMed] [Google Scholar]
- [158].Zhang G-J and Adachi I, Serum interleukin-6 levels correlate to tumor progression and prognosis in metastatic breast carcinoma. Anticancer research, 1999. 19(2): p. 1427–1432. [PubMed] [Google Scholar]
- [159].Shimazaki J, et al. , In patients with colorectal cancer, preoperative serum interleukin-6 level and granulocyte/lymphocyte ratio are clinically relevant biomarkers of long-term cancer progression. Oncology, 2013. 84(6): p. 356–361. [DOI] [PubMed] [Google Scholar]
- [160].Starkweather A, Increased interleukin-6 activity associated with painful chemotherapy-induced peripheral neuropathy in women after breast cancer treatment. Nursing research and practice, 2010. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Li Y, et al. , Chemotherapy induced peripheral neuropathy in a dish: dorsal root ganglion cells treated in vitro with paclitaxel show biochemical and physiological responses parallel to that seen in vivo. Pain, 2020. [DOI] [PMC free article] [PubMed]
- [162].Callizot N, et al. , Interleukin-6 protects against paclitaxel, cisplatin and vincristine-induced neuropathies without impairing chemotherapeutic activity. Cancer chemotherapy and pharmacology, 2008. 62(6): p. 995–1007. [DOI] [PubMed] [Google Scholar]
- [163].Liu T, Gao Y-J, and Ji R-R, Emerging role of Toll-like receptors in the control of pain and itch. Neuroscience bulletin, 2012. 28(2): p. 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Mitterreiter JG, et al. , Satellite glial cells in human trigeminal ganglia have a broad expression of functional Toll‐like receptors. European Journal of Immunology, 2017. 47(7): p. 1181–1187. [DOI] [PubMed] [Google Scholar]
- [165].Liu T, et al. , TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. The Journal of clinical investigation, 2012. 122(6): p. 2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Liu T, et al. , Toll-like receptor 7 mediates pruritus. Nature neuroscience, 2010. 13(12): p. 1460–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Liu X-J, et al. , TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Scientific Reports, 2016. 6: p. 28188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Li Y, et al. , Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. The Journal of Pain, 2014. 15(7): p. 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Qi J, et al. , Painful Pathways Induced by TLR Stimulation of Dorsal Root Ganglion Neurons. The Journal of Immunology, 2011. 186(11): p. 6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Li Y, et al. , The Cancer Chemotherapeutic Paclitaxel Increases Human and Rodent Sensory Neuron Responses to TRPV1 by Activation of TLR4. The Journal of Neuroscience, 2015. 35(39): p. 13487–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Li Y, et al. , MAPK signaling downstream to TLR4 contributes to paclitaxel-induced peripheral neuropathy. Brain, Behavior, and Immunity, 2015. 49: p. 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Park HJ, et al. , Toll-like receptor signaling regulates cisplatin-induced mechanical allodynia in mice. Cancer chemotherapy and pharmacology, 2014. 73(1): p. 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Sorge RE, et al. , Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. Journal of Neuroscience, 2011. 31(43): p. 15450–15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Burton M, et al. , Uncovering Cell-Specific Mechanisms in Sex Differences in TLR4-Dependent Pain. The Journal of Pain, 2019. 20(4, Supplement): p. S1. [Google Scholar]
- [175].Byrd‐Leifer CA, et al. , The role of MyD88 and TLR4 in the LPS‐mimetic activity of Taxol. European journal of immunology, 2001. 31(8): p. 2448–2457. [DOI] [PubMed] [Google Scholar]
- [176].O’Brien J, et al. , Taxol and colchicine increase LPS-induced pro-IL-1 beta production, but do not increase IL-1 beta secretion. A role for microtubules in the regulation of IL-1 beta production. The Journal of Immunology, 1995. 154(8): p. 4113–4122. [PubMed] [Google Scholar]
- [177].Zaks-Zilberman M, Zaks TZ, and Vogel SN, Induction of proinflammatory and chemokine genes by lipopolysaccharide and paclitaxel (Taxol™) in murine and human breast cancer cell lines. Cytokine, 2001. 15(3): p. 156–165. [DOI] [PubMed] [Google Scholar]
- [178].O’Neill LA and Bowie AG, The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Reviews Immunology, 2007. 7(5): p. 353–364. [DOI] [PubMed] [Google Scholar]
- [179].Palsson-McDermott E and O’Neill L, Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4 Immunology 113: 153–162. Find this article online, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Maruta T, et al. , Upregulation of ERK phosphorylation in rat dorsal root ganglion neurons contributes to oxaliplatin-induced chronic neuropathic pain. PloS one, 2019. 14(11): p. e0225586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Scuteri A, et al. , Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology, 2009. 30(2): p. 312–319. [DOI] [PubMed] [Google Scholar]
- [182].Scuteri A, et al. , NGF protects dorsal root ganglion neurons from oxaliplatin by modulating JNK/Sapk and ERK1/2. Neuroscience letters, 2010. 486(3): p. 141–145. [DOI] [PubMed] [Google Scholar]
- [183].Chiba T, et al. , Vincristine-induced peripheral neuropathic pain and expression of transient receptor potential vanilloid 1 in rat. Journal of pharmacological sciences, 2017. 133(4): p. 254–260. [DOI] [PubMed] [Google Scholar]
- [184].Luo X, et al. , Macrophage Toll-like Receptor 9 Contributes to Chemotherapy-Induced Neuropathic Pain in Male Mice. The Journal of Neuroscience, 2019. 39(35): p. 6848–6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Nencioni A, et al. , Proteasome inhibitor bortezomib modulates TLR4-induced dendritic cell activation. Blood, 2006. 108(2): p. 551–558. [DOI] [PubMed] [Google Scholar]
- [186].Thacker MA, et al. , CCL2 is a key mediator of microglia activation in neuropathic pain states. European journal of pain, 2009. 13(3): p. 263–272. [DOI] [PubMed] [Google Scholar]
- [187].Van Steenwinckel J, et al. , Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain, behavior, and immunity, 2015. 45: p. 198–210. [DOI] [PubMed] [Google Scholar]
- [188].Illias AM, et al. , Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain, 2018. 159(7): p. 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Belkouch M, et al. , The chemokine CCL2 increases Nav1. 8 sodium channel activity in primary sensory neurons through a Gβγ-dependent mechanism. Journal of Neuroscience, 2011. 31(50): p. 18381–18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Montague K, et al. , A novel interaction between CX 3 CR 1 and CCR 2 signalling in monocytes constitutes an underlying mechanism for persistent vincristine-induced pain. Journal of neuroinflammation, 2018. 15(1): p. 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Old EA, et al. , Monocytes expressing CX3CR1 orchestrate the development of vincristine-induced pain. The Journal of clinical investigation, 2014. 124(5): p. 2023–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Wang J, et al. , Upregulation of CX3CL1 mediated by NF-κB activation in dorsal root ganglion contributes to peripheral sensitization and chronic pain induced by oxaliplatin administration. Molecular pain, 2017. 13: p. 1744806917726256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Zitvogel L, et al. , Immunological aspects of cancer chemotherapy. Nature reviews immunology, 2008. 8(1): p. 59–73. [DOI] [PubMed] [Google Scholar]
- [194].Tsavaris N, et al. , Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. British journal of cancer, 2002. 87(1): p. 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Tonini G, et al. , Oxaliplatin may induce cytokine-release syndrome in colorectal cancer patients. Journal of biological regulators and homeostatic agents, 2002. 16(2): p. 105. [PubMed] [Google Scholar]
- [196].Millrud CR, Mehmeti M, and Leandersson K, Docetaxel promotes the generation of anti-tumorigenic human macrophages. Experimental cell research, 2018. 362(2): p. 525–531. [DOI] [PubMed] [Google Scholar]
- [197].Matsuhashi T, et al. , A low, non-toxic dose of paclitaxel can prevent dendritic cell-precursors from becoming tolerogenic dendritic cells with impaired functions. Biomedical Research, 2014. 35(6): p. 369–380. [DOI] [PubMed] [Google Scholar]
- [198].Cattin S, et al. , Bevacizumab specifically decreases elevated levels of circulating KIT+ CD11b+ cells and IL-10 in metastatic breast cancer patients. Oncotarget, 2016. 7(10): p. 11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Lima D, Lemes R, and Matos D, Immunosuppressive monocytes (CD14+/HLA-DR low/−) increase in childhood precursor B-cell acute lymphoblastic leukemia after induction chemotherapy. Medical Oncology, 2018. 35(3): p. 36. [DOI] [PubMed] [Google Scholar]
- [200].Chaudhry V, et al. , Thalidomide-induced neuropathy. Neurology, 2002. 59(12): p. 1872–1875. [DOI] [PubMed] [Google Scholar]
- [201].Üçeyler N, et al. , Elevated proinflammatory cytokine expression in affected skin in small fiber neuropathy. Neurology, 2010. 74(22): p. 1806–1813. [DOI] [PubMed] [Google Scholar]
- [202].Delaloge S, et al. , Pilot study of the paclitaxel, oxaliplatin, and cisplatin combination in patients with advanced/recurrent ovarian cancer. American journal of clinical oncology, 2000. 23(6): p. 569–574. [DOI] [PubMed] [Google Scholar]
- [203].Rowinsky EK, et al. , Sequence-dependent cytotoxic effects due to combinations of cisplatin and the antimicrotubule agents taxol and vincristine. Journal of cancer research and clinical oncology, 1993. 119(12): p. 727–733. [DOI] [PubMed] [Google Scholar]
- [204].Golub DV, Civelek AC, and Sharma VR, A regimen of taxol, Ifosfamide, and platinum for recurrent advanced squamous cell cancer of the anal canal. Chemotherapy research and practice, 2011. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Bairati I, et al. , Antioxidant vitamins supplementation and mortality: a randomized trial in head and neck cancer patients. International journal of cancer, 2006. 119(9): p. 2221–2224. [DOI] [PubMed] [Google Scholar]
- [206].Park S, et al. , Mechanisms underlying chemotherapy-induced neurotoxicity and the potential for neuroprotective strategies. Current medicinal chemistry, 2008. 15(29): p. 3081–3094. [DOI] [PubMed] [Google Scholar]
- [207].Favis R, et al. , Genetic variation associated with bortezomib-induced peripheral neuropathy. Pharmacogenetics and genomics, 2011. 21(3): p. 121–129. [DOI] [PubMed] [Google Scholar]
- [208].Broyl A, et al. , Mechanisms of peripheral neuropathy associated with bortezomib and vincristine in patients with newly diagnosed multiple myeloma: a prospective analysis of data from the HOVON-65/GMMG-HD4 trial. The lancet oncology, 2010. 11(11): p. 1057–1065. [DOI] [PubMed] [Google Scholar]
- [209].Johnson DC, et al. , Genetic factors underlying the risk of thalidomide-related neuropathy in patients with multiple myeloma. Journal of Clinical Oncology, 2011. 29(7): p. 797–804. [DOI] [PubMed] [Google Scholar]