

Abstract
The efficacy of all-trans retinoic acid (ATRA) for the treatment of chronic myeloid leukemia (CML) has been reported to be limited both as single-drug treatment or in combination with other drugs. Our previous study demonstrated that sphingosine 1-phosphate attenuated the effects of ATRA on human colon cancer cells by blocking the expression of retinoic acid receptor β. The aim of the present study was to investigate whether the ATRA-dependent proliferation inhibition of K562 cells was regulated by sphingosine kinases (SphKs). The results of cell proliferation assay and reverse transcription-PCR demonstrated that ATRA may exert synergistic effects with the SphK1 inhibitor SKI 5C or the pan-SphK inhibitor SKI II to inhibit the proliferation of K562 cells and upregulate the expression levels of the ATRA-inducible enzyme cytochrome P450 26A1 (CYP26A1). Knocking down the expression of SphK1 or SphK2 in K562 cells by small interfering RNA enhanced the inhibitory effects of ATRA and induced the expression of CYP26A1. Crude asterosaponins, which abrogated the expression of SphK2, also enhanced the effects of ATRA on K562 cells. In conclusion, the results of the present study demonstrated that SphKs may be involved in the regulation of the sensitivity of CML cells to ATRA.
Keywords: ATRA, K562 cells, SphK, SphK inhibitor, asterosaponins
Introduction
Chronic myeloid leukemia (CML) is characterized as a myeloproliferative disease (1). The majority of CML cases carry the Philadelphia chromosome, which is formed by a translocation between chromosomes 22 and 9, leading to the expression of the breakpoint cluster region protein (BCR)-ABL1 proto-oncogene (ABL) chimeric protein (2). The BCR-ABL fusion protein acts as a tyrosine kinase and drives the uncontrolled proliferation of CML cells (3). Tyrosine kinase inhibitors (TKIs) targeting the BCR-ABL protein have attracted attention as a treatment for CML; however, studies have reported that ~25% of patients with CML are resistant to the TKIs nilotinib and imatinib, and TKIs fail to eradicate the CML-initiating cells (4,5). Therefore, novel treatments for improving the therapeutic outcome of CML are urgently required.
As a natural and physiologically active metabolite of vitamin A, all-trans retinoic acid (ATRA) serves an important role in cell proliferation, differentiation and organogenesis (6). ATRA binds to and activates retinoic acid receptors (RARs). Retinoic acid X receptor (RXR) and RAR, both comprising three subtypes (α, β and γ), form heterodimers and regulate the transcription of certain genes such as cytochrome P450 26A1 (CYP26A1) and retinol-cellular retinol binding protein II (7). Retinoic acid response elements (RAREs) have been identified in the promoter region of these genes (7). The transcriptional activity of the RAR/RXR complex has been demonstrated to occur due to the binding of ATRA to the RAR receptor (8). Although ATRA also regulates gene expression independently of RAREs, this model is considered to be a typical pathway for retinoic acid to regulate cell differentiation, cell cycle and apoptosis (8). In addition, other intracellular signaling pathways, including the MAPK, VEGF, TGF-β, IFN-γ and NF-κB pathways, are also regulated by ATRA and its receptors (9). Due to the important role of ATRA in cell physiology, the antitumor application of retinoic acid has been considered (10–12).
ATRA has been successfully used as the main treatment modality for patients with acute promyelocytic leukemia (APL) (13). APL is a subtype of acute myeloid leukemia expressing the promyelocytic leukemia-retinoic acid receptor α (PML-RARα) tumor protein (13,14). ATRA serves an important role in inducing differentiation and promoting apoptosis of APL cells (13). The PML-RARα fusion protein is activated by pharmacological doses of ATRA, leading to cell cycle arrest and terminal differentiation of leukemia cells (15). Furthermore, ATRA-induced increase of p21/wild-type p53-activated fragment 1 gene expression levels may also lead to cell cycle arrest at the G1 phase (16). Due to its relatively low systemic toxicity, in addition to the treatment of leukemia, ATRA has also been studied in the therapy of cervical, lung and breast cancer as well as neuroblastoma (11,12,17,18). However, with the exception of APL, ATRA is has not been demonstrated to be effective in the treatment of other acute myeloid leukemia subtypes or CML. Identifying the mechanism underlying the development of drug resistance may not only contribute to the use of retinoic acid as a treatment for leukemia, but also provide strategies for optimizing single-agent or combined retinoic acid treatment regimens and overcoming drug resistance.
Sphingosine kinase (SphK) phosphorylates sphingosine to form sphingosine 1-phosphate (S1P) (19). Accumulating evidence has demonstrated that the SphK/S1P pathway is involved in several cancer processes, including cell proliferation, migration and angiogenesis (20). SphK has two subtypes, namely SphK1 and SphK2 (21). Previous studies have reported that SphK1 overexpression may serve an oncogenic role in several types of tumor such as leukemia and breast cancer (22,23). In contrast to SphK1, SphK2 is considered to be an apoptosis inducer (24,25). The sensitivity of tumor cells to retinoids may be regulated by the SphK/S1P pathway (26,27). SphK1 has been demonstrated to mediate the resistance of ovarian cancer cells to the synthetic retinoic N-(4-hydrooxyphenyl) retinamide (26). Our previous study demonstrated that S1P antagonized the inhibitory effects of ATRA on the HT-29 retinoic acid-sensitive human colon cancer cell line by blocking the expression of RARβ, suggesting that reducing the production of S1P or inhibiting the activity of SphKs may be an effective strategy for the treatment of tumors with retinoic acid (27). Other studies have reported that overexpression of BCR-ABL increases the expression levels of SphK1 in CML cells, and SphK1 improves the stability of the BCR-ABL protein (28,29).
The inefficiency of ATRA in the treatment of CML and the roles of SphKs in this process have not been studied in depth to date. Therefore, the present study was designed as a preliminary attempt to address this issue. Using the K562 human CML cell line, the present study aimed to determine whether inhibiting the activity or expression of SphKs may enhance the ATRA-dependent proliferation inhibition of K562 cells.
The present study aimed to determine the relationship between SphK1/2 and the sensitivity of K562 cells to ATRA using a pan-SphK inhibitor, a selective inhibitor of SphK1 and RNA interference. Since SphK1 has been demonstrated to be associated with the resistance of tumor to retinoids, SphK2 requires further study (26,27). Asterosaponins are a class of steroids isolated from starfish with diverse biological effects such as antitumor activity (30). In the present study, total saponins of starfish extracted from Asterias amurensis were tested to determine their ability to inhibit the expression of SphK2. As specific SphK2 inhibitors were not available, the total saponins were used to determine the role of Sphk2 in modulating the sensitivity of K562 cells to ATRA.
Materials and methods
Materials
ATRA was purchased from Sigma-Aldrich; Merck KGaA. SKI II, a pan-SphK inhibitor, and SKI 5C, a selective inhibitor of SphK1, were obtained from Santa Cruz Biotechnology, Inc. The RARE-tk-Luc plasmid was kindly provided by Professor Michael J. Spinella (Dartmouth Medical School, Lebanon, NH, USA) (31). The pCMV-Blank vector was purchased from the Beyotime Institute of Biotechnology.
Extraction of asterosaponins from starfish. Asterias amurensis were collected from the sea area of Yantai (Shandong, China) in January 2018, stored in seawater containing 3.75% (w/v) MgCl2 for 30 min and then frozen at −20°C. The frozen starfish were subsequently crushed, and extraction was performed with 65% (v/v) ethyl alcohol heat reflux; the extract was concentrated under low pressure. The concentrate was dispersed in water and extracted with petroleum ether three times. The aqueous phase was extracted with n-butanol three times, and the extract was concentrated under low pressure. The precipitate was dried to obtain a light brown powder. A small amount of the powder was examined and confirmed to be asterosaponins by a color reaction of concentrated sulfuric acid and hemolysis (32).
Cell line and culture
The K562 CML cell line was purchased from The Cell Bank of Type Culture Collection of The Chinese Academy of Sciences. The K562 cell line was selected due to the presence of the BCR-ABL fusion protein, which is associated with SphK1 expression (27,28). The cells were cultured in RPMI-1640 medium (HyClone; Cytiva) supplemented with 10% (v/v) fetal bovine serum (HyClone; Cytiva), 100 µg/ml streptomycin, 100 U/ml penicillin and 2 mM glutamine (Sangon Biotech Co., Ltd.) at 37°C in a 5% (v/v) CO2 humidified incubator.
Small interfering (si)RNA transfection
Negative control (siNC) and siRNAs targeting SphK1 or SphK2 were synthesized by Shanghai Gene Pharma, Co., Ltd. siRNAs were transfected into K562 cells (1×105 cells/ml) at 37°C using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) at a final concentration of 20 nM and incubated for 8 h. At 72 h post-transfection, the effects of the gene silencing were detected by western blotting. The siRNA sequences were as follows: SphK1 forward, 5′-GGCUGAAAUCUCCUUCACGTT-3′ and reverse, 5′-CGUGAAGGAGAUUUCAGCCTC-3′; SphK2 forward, 5′-GGGUAGUGCCUGAUCAAUGTT-3′ and reverse, 5′-CAUUGAUCAGGCACUACCCTC-3′; and siNC forward, 5′-UUCUCCGAACGGUCACGUTT-3′ and reverse, 5′-ACGUGACACGUUCGGAGAATT-3′.
Cell viability assay
K562 cells (2×103 cells per well) were cultured in a 96-well plate. ATRA (1 µM), SKI II (0.5 or 5 µM), SKI 5C (0.5 or 5 µM) or asterosaponins (10 or 50 µg/ml) were added and incubated for 72 h at 37°C as indicated. In order to study the association between the expression of SphKs and cell sensitivity to ATRA, the cells were seeded into a 96-well microplate at a density of 2×103 cells in 200 µl per well and treated with ATRA at 48 h post-transfection with SphK1/SphK2 siRNA. The control group was treated with 0.2% (v/v) DMSO. After 3 days, a Cell Counting Kit-8 (CCK-8) assay (Beijing Solarbio Science & Technology Co., Ltd.) was used to determine the effects of the drugs on cell viability. Three independent experiments were conducted in triplicate.
Reverse transcription-polymerase chain reaction (RT-PCR) analysis
RT-PCR analysis was performed to analyze the expression levels of CYP26A1, SphK1 and SphK2 in K562 cells. K562 cells were cultured in 6-well plate (6×105 cells/well) and were treated with ATRA (1 µM), SKI II (0.5 or 5 µM), SKI 5C (0.5 or 5 µM) or asterosaponins (10, 50 or 100 µg/ml) for 24 h at 37°C. Subsequently, total RNA was extracted from the cells by a Total RNA Extraction kit (Sangon Biotech Co., Ltd.) according to the manufacturer's instructions. For the cells transfected with siRNA, following transfection with SphK1/SphK2 siRNA or siNC, K562 cells (6×105 cells/well) were treated with ATRA (1 µM) for 24 h at 37°C prior to RNA extraction. RNA was reverse-transcribed into single-stranded cDNA using a ReverTra Ace® qPCR RT Kit (Toyobo Life Science). For conventional PCR, the RT product (1.5 µl) was used with the Quick Taq™ HS DyeMix (Toyobo Life Science). The PCR primer pairs were as follows: CYP26A1 forward, 5′-CCAAGGCAGCTCTACACTCAC-3′ and reverse, 5′-AGATGGCCAACATGAGCACA-3′; SphK1 forward, 5′-TCCTGGCACTGCTGCACTC-3′ and reverse, 5′-TAACCATCAATTCCCCATCCAC-3′; Sphk2 forward, 5′-CCAAGGCAGCTCTACACTCAC-3′, and reverse, 5′-AGATGGCCAACATGAGCACA-3′); and β-actin (reference gene) forward, 5′-GTCACCAACTGGGACGACA-3′ and reverse, 5′-TGGCCATCTCTTGCTCGAA-3′. The specific fragments amplified were as follows: 200 bp for CYP26A1, 260 bp for Sphk1, 178 bp for Sphk2 and 459 bp for β-actin. The PCR products were resolved on a 2% (w/v) agarose gel containing 0.1 µl/ml Gelview (BioTeke Corporation). Images of the gel were captured under an ultraviolet transmittance instrument. The relative light intensity was analyzed by AlphaEaseFC software (version 6.0.0; Alpha Innotech Corporation). Three experiments were carried out in three samples.
Luciferase reporter gene assay
Prior to transfection, K562 cells were cultured in a 6-well plate at 4×105 cells per well. The RARE-tk-Luc vector (1.2 µg) and β-galactosidase expression vector (80 ng) were transfected into the cells using Lipofectamine® 2000 at 37°C for 8 h. The total amount of DNA per well was adjusted to 1.5 µg using the pCMV-Blank empty vector (Beyotime Institute of Biotechnology). At 24 h post-transfection, ATRA (1 µM), SKI II (0.5 µM) and SKI 5C (0.5 µM) were added into the medium at the indicated concentrations at 37°C. After 24 h, the cells were centrifuged at 1,000 × g for 5 min at 4°C, washed twice with RPMI-1640 medium, resuspended in lysis buffer (BioVision, Inc.) and incubated at 4°C for 20 min. The cell lysate was centrifuged at 10,000 × g for 10 min at 4°C, and the luciferase activity of the supernatant was measured using a luciferase assay kit (BioVision, Inc.). The luciferase activity was normalized to that of β-galactosidase. Three experiments were performed in three samples.
Immunoblotting
K562 cells were centrifuged at 1,000 × g for 5 min at 4°C and lysed with RIPA buffer (Beyotime Institute of Biotechnology). The total lysates (20 µg) were then separated by 8% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were blocked for 10 h at 4°C in Tris-buffered saline with 0.05% (v/v) Tween-20 solution (Beyotime Institute of Biotechnology) containing 5% (w/v) non-fat milk. Subsequently, the membranes were incubated with primary antibodies against GAPDH (1:2,000; cat. no. 2118s; Cell Signaling Technology, Inc.), Sphk1 and Sphk2 (1:1,000) at 24°C for 1 h, followed by incubation with a goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:50; cat. no. a0208; Beyotime Institute of Biotechnology) for 1 h at room temperature. An enhanced chemiluminescence system (Cytiva) was used to visualize the blots according to the manufacturer's protocol. The polyclonal antibodies anti-SphK2 (cat. no. sc-366378) and anti-SphK1 (cat. no. sc-48825) were purchased from Santa Cruz Biotechnology.
Statistical analysis
SPSS version 17.0 (SPSS, Inc.) was used for statistical analysis. Data are presented as the mean ± SD of at least three independent experiments and were analyzed by one-way ANOVA. Multiple comparisons were performed using the Tukey's studentized range test. P<0.05 was considered to indicate a statistically significant difference.
Results
SphK inhibitors enhance the inhibitory effects of ATRA on K562 cell viability
In order to investigate the effects of SphK enzyme activity on the intracellular function of ATRA, K562 cell viability was analyzed following treatments with ATRA, SKI II and SKI 5C. The effects of ATRA, SKI II or SKI 5C alone on the viability of K562 cells were first determined (Fig. 1). ATRA (1 µM) reduced the viability of K562 cells by 14.4%. SKI II slightly inhibited the viability of K562 cells at 0.5 and 5 µM, with inhibition rates of 19.1 and 23.5%, respectively (P<0.01 vs. control). The inhibition rates of SKI 5C on K562 cells at 0.5 and 5 µM were 24.8 and 33.3%, respectively, compared with the control group (P<0.01).
Figure 1.
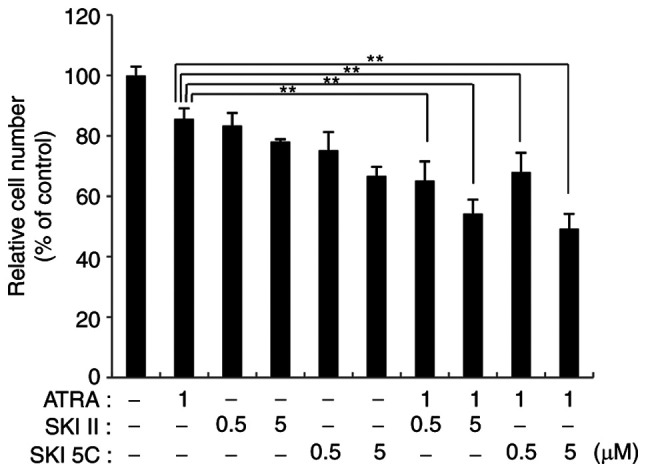
SphK inhibitors enhance the inhibitory effects of ATRA on K562 cell viability. Viable K562 cells treated with 1 µM ATRA, 0.5 or 5 µM SKI II (selective inhibitor of SphKs), or 0.5 or 5 µM SKI 5C (selective inhibitor of SphK1) alone or in combination for 72 h were evaluated by the Cell Counting Kit-8 assay. Data are presented as the mean ± SD of three independent replicates relative to the untreated control group. **P<0.01. ATRA, all-trans retinoic acid; SphK, sphingosine kinase; SKI, sphingosine kinase inhibitor.
When 1 µM ATRA was combined with 0.5 µM or 5 µM SKI II, the inhibition rate of K562 cells was 34.8 and 41.7%, respectively (P<0.01 vs. 1 µM ATRA alone). When ATRA was co-administered with SKI 5C, the inhibition rate of K562 cells significantly increased (1 µM ATRA + 0.5 µM SKI 5C, 32.1%; 1 µM ATRA + 5 µM SKI 5C, 47.5%; both P<0.01 vs. 1 µM ATRA alone). These results suggested that the combination of SphK inhibitors and ATRA effectively inhibited the viability of CML cells.
SKI II and SKI 5C increase ATRA-induced CYP26A1 transcription
In promyelocytic leukemia cells, CYP26 has been described as a retinoid-inducible gene (32). Therefore, the CYP26A1 expression levels were examined in K562 cells to determine the effects of SphK on ATRA activity. As presented in Fig. 2A, K562 cells expressed low levels of CYP26A1 mRNA, which significantly increased following ATRA treatment. Neither SKI II nor SKI 5C affected the expression levels of CYP26A1. When combined with SKI II or SKI 5C, the mRNA levels of CYP26A1 induced by ATRA were significantly higher compared with those observed in cells treated with ATRA alone (26 and 46%, respectively; P<0.01 vs. 1 µM ATRA). Thus, the increase in CYP26A1 transcription induced by SKI II and SKI 5C may be associated with the enhancement of ATRA-induced cytotoxicity in K562 cells.
Figure 2.

ATRA-induced CYP26A1 transcription in K562 cells is enhanced by sphingosine kinase inhibitors. (A) ATRA-induced CYP26A1 mRNA levels were enhanced by SKI II and SKI 5C. Reverse transcription-PCR assay was performed to analyze the mRNA levels of CYP26A1. (B) SKI II and SKI 5C increased the effects of ATRA on the activity of RARE. K562 cells were transfected with RARE-tk-Luc reporter plasmids and treated with either ATRA or a combination of ATRA with SKI II and SKI 5C. Data were normalized to β-galactosidase activity. Data are presented as the mean ± SD of three independent replicates. **P<0.01. ATRA, all-trans retinoic acid; CYP26A1, cytochrome P450 26A1; RARE, retinoic acid response element; SKI, sphingosine kinase inhibitor.
Luciferase reporter assay was used to test this hypothesis. The results revealed that the luciferase activity of K562 cells transfected with the RARE-tk-Luc reporter plasmid was 3-fold higher compared with that of the control cells following ATRA treatment (P<0.01 vs. control; Fig. 2B). When ATRA was combined with SphK inhibitors, the luciferase activity of K562 cells was further increased (by 45.7% with 0.5 µM SKI II and by 19.8% with 0.5 µM SKI 5C; both P<0.01 vs. ATRA alone). These results further suggested that the enzymatic activity of SphK may affect the role of ATRA in promoting the target gene transcription.
K562 cells are sensitive to ATRA following SphK knockdown
In order to determine the role of SphKs in the retinoic acid resistance of CML cells, siRNAs were used to inhibit the expression of SphK1 or SphK2 in K562 cells. The results demonstrated that K562 cells expressed high protein levels of SphK1 and SphK2 (Fig. 3A). The protein levels of SphKs in K562 cells significantly decreased (by 56.3% for SphK1 and by 43.6% for SphK2; P<0.01 vs. siNC) compared with those in the siNC group at 48 h post-transfection with the corresponding siRNA, and remained inhibited for ≥72 h (data not shown). Subsequently, the transfected K562 cells were treated with ATRA, and the levels of CYP26A1 mRNA were analyzed. ATRA increased the mRNA levels of CYP26A1 by 19.1% (P<0.05 vs. siNC) in siNC-transfected K562 cells (Fig. 3B). When cells transfected with siSphK1 or siSphK2 were treated with ATRA, the expression levels of CYP26A1 increased (by 55.1% for siSphK2 and by 31.1% for siSphK1; P<0.01 vs. siNC), suggesting that ATRA significantly enhanced the expression levels of CYP26A1 following knockdown of SphKs.
Figure 3.

K562 cells are sensitized to ATRA when SphK expression is knocked down. (A) SphK expression in K562 cells was downregulated by siRNA targeting SphK1 or SphK2. Knockdown efficiency was assessed by western blotting. (B) RT-PCR analysis was performed to detect the CYP26A1 mRNA levels in K562 cells transfected with siRNA targeting SphK1, SphK2 or siNC and subsequently treated with 1 µM ATRA. (C) The ATRA-dependent inhibitory effect in K562 cells was enhanced when SphK1 or SphK2 was knocked down. Viable cells were evaluated by a Cell Counting Kit-8 assay and presented as a percentage of the untreated control cells. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01. ATRA, all-trans retinoic acid; SphK, sphingosine kinase; CYP26A1, cytochrome P450 26A1; RT-PCR, reverse transcription-PCR.
The viability of the transfected cells was further analyzed by the CCK-8 assay. As presented in Fig. 3C, 1 µM ATRA slightly inhibited the proliferation of K562 cells transfected with siNC (by 14.4%; P<0.01 vs. vehicle control). Knockdown of SphK1 or SphK2 exerted no notable effects on the proliferation of K562 cells. When treated with ATRA, cells transfected with siSphK1 exhibited a higher rate of inhibition (19.1%) compared with the siNC group. Knockdown SphK2 also sensitized K562 cells to ATRA, and the inhibition rate of K562 cells treated with 1 µM ATRA was 24.8% (P<0.01 vs. siNC).
Asterosaponins abrogate SphK2 expression and enhance the effects of ATRA in K562 cells
Experiments were performed to determine the effects of starfish extracts on the activity of SphKs, and it was observed that the expression levels of SphK2 mRNA were downregulated by asterosaponins (10 µg/ml, 17.1%, P<0.05; 50 µg/ml, 52.7%, P<0.01; and 100 µg/ml, 57.7%, P<0.01, respectively, vs. control), whereas SphK1 mRNA levels were not significantly affected compared with those in the control group (Fig. 4A). These results were validated by western blotting (SphK2: 10 µg/ml, 15.1%, P<0.05; and 50 µg/ml, 55.2%, P<0.01, respectively, vs. control; Fig. 4B). Since asterosaponins were demonstrated to inhibit the expression of SphK2, it was hypothesized that they may enhance the inhibitory effects of ATRA on the proliferation of K562 cells, similar to SphK inhibitors. K562 cells were treated with ATRA and/or asterosaponins, and the cell viability was analyzed. K562 cell proliferation was not notably inhibited by ATRA, with an inhibition rate of 6.2% at 1 µM (P>0.05 vs. control) (Fig. 4C). Asterosaponins inhibited the proliferation of K562 cells (10 µg/ml, 18.1%, P<0.05; and 50 µg/ml, 39.7%, P<0.01, respectively, vs. control). When 1 µM ATRA was used together with asterosaponins, the viability rate of K562 cells was further reduced (10 µg/ml, 22.4%; and 50 µg/ml, 31.7%; both P<0.01 vs. asterosaponins alone).
Figure 4.

Asterosaponins inhibit SphK2 expression and enhance the ATRA-induced effects in K562 cells. (A) SphK2 mRNA levels were downregulated in K562 cells treated with asterosaponins (10, 50 or 100 µg/ml) for 24 h. SphK expression levels were determined by RT-PCR. (B) SphK2 protein expression levels were downregulated in K562 cells treated with asterosaponins (10 or 100 µg/ml) for 24 h. SphK protein levels determined by western blot assay. (C) Asterosaponins enhanced the ATRA-dependent inhibitory effect on the viability of K562 cells treated with ATRA (1 µM) and asterosaponins (10 or 100 µg/ml), alone or in combination, for 72 h. Viable cells were evaluated by a Cell Counting Kit-8 assay and presented as a percentage of the untreated control cells. (D) RT-PCR analysis was performed to detect the mRNA levels of CYP26A1 in K562 cells treated with ATRA (1 µM) and asterosaponins (100 µg/ml) for 24 h. Data are presented as the mean ± SD (n=3). *P<0.05 and **P<0.01. ATRA, all-trans retinoic acid; SphK, sphingosine kinase; CYP26A1, cytochrome P450 26A1; RT-PCR, reverse transcription-PCR.
The present study further investigated whether the effects of asterosaponins on ATRA activity occurred due to the enhanced expression of ATRA-targeted genes in cancer cells. The results of RT-PCR analysis demonstrated that K562 cells expressed CYP26A1 at low levels. Following treatment with ATRA, the levels of CYP26A1 in K562 cells significantly increased by 61.4% compared with those in the control group (P<0.01; Fig. 4D). The effects of 50 µg/ml asterosaponins alone on the expression levels of CYP26A1 were not significant; however, the mRNA levels of CYP26A1 in K562 cells were significantly enhanced when the cells were co-treated with asterosaponins and ATRA (increased by 27.9%; P<0.01 vs. 1 µM ATRA alone). Therefore, the increase in the transcription levels of CYP26A1 induced by asterosaponins may be the mechanism underlying the enhanced ATRA-induced cell proliferation inhibition in K562 cells.
Discussion
ATRA has been successfully used in the treatment of APL, but the effect of ATRA on CML has been unsatisfactory (8). In the present study, SphKs were demonstrated to be involved in the regulation of CML cell sensitivity to ATRA. SKI II and SKI 5C, two types of SphK inhibitors, enhanced the inhibitory effects of ATRA on the viability of K562 cells. When the expression of SphKs was knocked down by siRNA, the inhibitory effects of ATRA were also enhanced.
SphK1 is considered to be a potential target for drug intervention, particularly in leukemia cells, and its intracellular levels are associated with chemotherapy resistance (33). For example, the expression levels of SphK1 can predict the sensitivity of HL-60 acute myeloid leukemia cells to daunorubicin (34,35). Expression of SphK1 stimulated by BCR-ABL variant transcripts may be an important factor in the development of leukemia and drug resistance (36). In contrast to SphK1, SphK2 is considered to be an inducer of apoptosis (24,25); however, the experimental results of the present study demonstrated that SphK2 and SphK1 were similar in certain aspects. When the enzymatic activity was inhibited or the expression of SphK2 was downregulated, the cytotoxicity induced by ATRA in CML cells was enhanced compared with that induced by ATRA alone. Since no specific SphK2 inhibitor was available, the present study used SKI II, a pan-SphK inhibitor, and SKI 5C, a selective inhibitor of SphK1, to inhibit the activity of SphKs. The activity and phosphorylation levels of SphKs under various treatments were not detected in the present study, although phosphorylation is considered to serve an important role in the regulation of SphK activity (37). Despite these limitations, relevant experiments were performed by siRNA interference, and the results suggested that SphK1 and SphK2 may be involved in the ATRA resistance of K562 cells.
Retinoic acid induces the differentiation and/or apoptosis of tumor cells and has antioxidant properties; thus, it is considered to be a potential chemotherapeutic or chemopreventive drug (9). In addition to the successful use of ATRA in the treatment of patients with APL, other retinoic acids have been used in various cancer clinical trials, such as 13-cis-RA and bexarotene in patients with advanced non-small cell lung cancer (38,39). Retinoic acid is involved in cell differentiation via induction of target gene expression, such as MMP-9 and RARβ (40,41). Our previous study demonstrated that S1P promotes the ligand-dependent degradation of RARs and affects the transcription of RARβ (26). In the present study, the results demonstrated that treatment with ATRA induced the transcription of the target gene CYP26A1 in CML cells, which was also affected by SphKs. When SphK activity was inhibited or its expression was suppressed, ATRA strongly induced the expression levels of CYP26A1. However, the specific mechanism of ATRA action requires further research to be fully elucidated. Our previous and current studies suggested that the resistance of CML cells to ATRA may be associated with the interference with retinoic acid-induced target gene transcription by SphKs.
The BCR-ABL fusion protein is a marker of CML that is present in >98% of patients (42). The BCR-ABL fusion protein interacts with a variety of signaling proteins and activates a variety of signaling pathways through phosphorylation, including RAS, PI3K, protein kinase B, JNK, Src kinase and their downstream targets (42). The successful development of TKIs has improved the treatment outcomes of CML (43). Although TKIs are considered to be safe, they are also associated with a variety of adverse reactions, including gastrointestinal, cardiovascular, skin and liver toxicity (43). Notably, patients may develop resistance to TKIs (43). The BCR-ABL fusion protein has been reported induce TKI resistance by activating other non-receptor tyrosine kinases and their downstream signaling pathways (44). The BCR-ABL protein induces the expression of SphK1, which in turn improves the stability of BCR-ABL (28,29). The interaction between SphK1 and BCR-ABL may be implicated in the resistance of CML to retinoic acid, as the results of the present study demonstrated that SphK1 interferes with ATRA inhibiting cell proliferation and inducing gene expression. Additionally, similar to SphK1, SphK2 may also interfere with the action of ATRA. When the expression of SphK2 was downregulated by siRNA or asterosaponins in the present study, K562 cells were sensitized to the proliferation inhibition induced by ATRA, and the transcription levels of the ATRA target gene CYP26A1 were also enhanced compared with those in the siNC group. Thus, although the association between the BCR-ABL fusion protein and SphK2 is unclear, SphK2 may participate in the resistance of CML cells to ATRA. Asterosaponins have also been reported to have antitumor and anti-inflammatory properties (45,46). In the present study, starfish saponins were used as a tool to illustrate that the effects of ATRA on K562 cells were enhanced following specific inhibition of SphK2 expression as a supplement to the experiments of blocking SphK expression by siRNA. To the best of our knowledge, the present study was the first to demonstrate that asterosaponins specifically inhibited the expression of SphK2. However, the components of starfish extract that downregulate the expression of SphK2 must be isolated and identified, and the clinical value of the extract requires further investigation.
The results of the present study only demonstrated that SphKs were involved in the sensitivity of K562 cells to ATRA by determining cell viability, whereas the differentiation and apoptosis of K562 cells during this process were not analyzed. Induction of target gene transcription is considered to be an important pathway of ATRA-induced cell differentiation or apoptosis (9). Although the present study did not use flow cytometry to analyze the cell cycle and differentiation, the expression of SphKs was demonstrated to affect the ATRA-induced gene regulation by detecting the expression and transcriptional activity levels of the target genes. Thus, we hypothesized that the ATRA resistance of CML cells may be associated with the promotion of SphK expression by the BCR-ABL fusion protein in CML cells. Whether this phenomenon is observed in other CML cell lines requires further investigation. The mechanism through which SphKs affect the intracellular action of ATRA must also be further investigated. In addition, only the RAR agonist ATRA was assessed in the present study. It is not clear whether other retinoic acids, particularly specific RAR and RXR agonists, are also affected by SphKs. Therefore, further research, including animal studies, is required to determine the association between SphKs and CML resistance to retinoic acid and provide suggestions for clinical treatment.
In conclusion, the results of the present study demonstrated that SphKs were involved in the regulation of ATRA sensitivity in the K562 CML cell line. SphKs may be involved the low sensitivity of CML cells to ATRA. Therefore, ATRA combined with SphK inhibitors or drugs that reduce SphK expression levels may be an effective treatment for patients with CML.
Acknowledgements
The authors would like to thank Dr Michael J. Spinella (Dartmouth Medical School, Lebanon, NH, USA) for providing the RARE-tk-Luc plasmid..
Glossary
Abbreviations
- APL
acute promyelocytic leukemia
- ATRA
all-trans retinoic acid
- CML
chronic myeloid leukemia
- CYP26A1
cytochrome P450 26A1
- RAR
retinoic acid receptor
- RARE
retinoic acid response element
- RXR
retinoic acid X receptor
- SKI
sphingosine kinase inhibitor
- SphK
sphingosine kinase
- TKI
tyrosine kinase inhibitor
Funding Statement
The present study was supported by the Doctoral Research Fund of Yantai University (grant no. SM12B31).
Funding
The present study was supported by the Doctoral Research Fund of Yantai University (grant no. SM12B31).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
DS conceived and designed the study, and collected the data. DS and SW performed the experiments, analyzed the data and wrote the manuscript, discussed the results and revised the manuscript. DS and SW confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Laboratory Animal Ethics committee of Yantai University (approval no. 2019012007).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Schwartz SI. Myeloproliferative disorders. Ann Surg. 1975;182:464–471. doi: 10.1097/00000658-197510000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldman JM. Chronic myeloid leukemia: A historical perspective. Semin Hematol. 2010;47:302–311. doi: 10.1053/j.seminhematol.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96:3343–3356. doi: 10.1182/blood.V96.10.3343. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Peng C, Sullivan C, Li D, Li S. Critical molecular pathways in cancer stem cells of chronic myeloid leukemia. Leukemia. 2010;24:1545–1554. doi: 10.1038/leu.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang H, Li S. Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 2013;4:186–196. doi: 10.1007/s13238-013-2115-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kam RK, Deng Y, Chen Y, Zhao H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012;2:11. doi: 10.1186/2045-3701-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang R, Wang Y, Li R, Chen G. Transcriptional factors mediating retinoic acid signals in the control of energy metabolism. Int J Mol Sci. 2015;16:14210–14244. doi: 10.3390/ijms160614210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Theodosiou M, Laudet V, Schubert M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell Mol Life Sci. 2010;67:1423–1445. doi: 10.1007/s00018-010-0268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connolly RM, Nguyen NK, Sukumar S. Molecular pathways: Current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res. 2013;19:1651–1659. doi: 10.1158/1078-0432.CCR-12-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lotan R, Xu XC, Lippman SM, Ro JY, Lee JS, Lee JJ, Hong WK. Suppression of retinoic acid receptor-beta in premalignant oral lesions and its up-regulation by isotretinoin. N Engl J Med. 1995;332:1405–1410. doi: 10.1056/NEJM199505253322103. [DOI] [PubMed] [Google Scholar]
- 11.Arrieta O, Gonzalez-De la Rosa CH, Arechaga-Ocampo E, Villanueva-Rodriguez G, Ceron-Lizarraga TL, Martinez-Barrera L, Vázquez-Manríquez ME, Ríos-Trejo MA, Alvarez-Avitia MA, Hernández-Pedro N, et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:3463–3471. doi: 10.1200/JCO.2009.26.6452. [DOI] [PubMed] [Google Scholar]
- 12.Sutton LM, Warmuth MA, Petros WP, Winer EP. Pharmacokinetics and clinical impact of all-trans retinoic acid in metastatic breast cancer: A phase II trial. Cancer Chemother Pharmacol. 1997;40:335–341. doi: 10.1007/s002800050666. [DOI] [PubMed] [Google Scholar]
- 13.Degos L, Wang ZY. All trans retinoic acid in acute promyelocytic leukemia. Oncogene. 2001;20:7140–7145. doi: 10.1038/sj.onc.1204763. [DOI] [PubMed] [Google Scholar]
- 14.Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, Ferrara F, Fazi P, Cicconi L, Di Bona E, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 15.Borrow J, Goddard AD, Sheer D, Solomon E. Molecular analysis of acute promyelocytic leukemia breakpoint cluster region on chromosome 17. Science. 1990;249:1577–1580. doi: 10.1126/science.2218500. [DOI] [PubMed] [Google Scholar]
- 16.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer ME, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 17.Choi Y, Kim SY, Kim SH, Yang J, Park K, Byun Y. Inhibition of tumor growth by biodegradable microspheres containing all-trans-retinoic acid in a human head-and-neck cancer xenograft. Int J Cancer. 2003;107:145–148. doi: 10.1002/ijc.11354. [DOI] [PubMed] [Google Scholar]
- 18.Masetti R, Biagi C, Zama D, Vendemini F, Martoni A, Morello W, Gasperini P, Pession A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv Ther. 2012;29:747–762. doi: 10.1007/s12325-012-0047-3. [DOI] [PubMed] [Google Scholar]
- 19.Dayon A, Brizuela L, Martin C, Mazerolles C, Pirot N, Doumerc N, Nogueira L, Golzio M, Teissié J, Serre G, et al. Sphingosine kinase-1 is central to androgen-regulated prostate cancer growth and survival. PLoS One. 2009;4:e8048. doi: 10.1371/journal.pone.0008048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sukocheva O, Wang L, Verrier E, Vadas MA, Xia P. Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology. 2009;150:4484–4492. doi: 10.1210/en.2009-0391. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Toman RE, Goparaju SK, Maceyka M, Nava VE, Sankala H, Payne SG, Bektas M, Ishii I, Chun J, et al. Sphingosine kinase type 2 is a putative BH3-only protein that induces apoptosis. J Biol Chem. 2003;278:40330–40336. doi: 10.1074/jbc.M304455200. [DOI] [PubMed] [Google Scholar]
- 22.Sobue S, Iwasaki T, Sugisaki C, Nagata K, Kikuchi R, Murakami M, Takagi A, Kojima T, Banno Y, Akao Y, et al. Quantitative RT-PCR analysis of sphingolipid metabolic enzymes in acute leukemia and myelodysplastic syndromes. Leukemia. 2006;20:2042–2046. doi: 10.1038/sj.leu.2404386. [DOI] [PubMed] [Google Scholar]
- 23.Datta A, Loo SY, Huang B, Wong L, Tan SS, Tan TZ, Lee SC, Thiery JP, Lim YC, Yong WP, Lam Y, Kumar AP, Yap CT. SPHK1 regulates proliferation and survival responses in triple-negative breast cancer. Oncotarget. 2014;5:5920–5933. doi: 10.18632/oncotarget.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem. 2003;278:46832–46839. doi: 10.1074/jbc.M306577200. [DOI] [PubMed] [Google Scholar]
- 25.Illuzzi G, Bernacchioni C, Aureli M, Prioni S, Frera G, Donati C, Valsecchi M, Chigorno V, Bruni P, Sonnino S, Prinetti A. Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J Biol Chem. 2010;285:18594–18602. doi: 10.1074/jbc.M109.072801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun DF, Gao ZH, Liu HP, Yuan Y, Qu XJ. Sphingosine 1-phosphate antagonizes the effect of all-trans retinoic acid (ATRA) in a human colon cancer cell line by modulation of RARβ expression. Cancer Lett. 2012;319:182–189. doi: 10.1016/j.canlet.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 27.Li QF, Huang WR, Duan HF, Wang H, Wu CT, Wang LS. Sphingosine kinase-1 mediates BCR/ABL-induced upregulation of Mcl-1 in chronic myeloid leukemia cells. Oncogene. 2007;26:7904–7908. doi: 10.1038/sj.onc.1210587. [DOI] [PubMed] [Google Scholar]
- 28.Salas A, Ponnusamy S, Senkal CE, Meyers-Needham M, Selvam SP, Saddoughi SA, Apohan E, Sentelle RD, Smith C, Gault CR, et al. Sphingosine kinase-1 and sphingosine 1-phosphate receptor 2 mediate Bcr-Abl1 stability and drug resistance by modulation of protein phosphatase 2A. Blood. 2011;117:5941–5952. doi: 10.1182/blood-2010-08-300772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalle AM, Sachchidanand S, Pallu R. Bcr-Abl-independent mechanism of resistance to imatinib in K562 cells: Induction of cyclooxygenase-2 (COX-2) by histone deacetylases (HDACs) Leukemia Res. 2010;34:1132–1138. doi: 10.1016/j.leukres.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 30.Malyarenko TV, Kicha AA, Ivanchina NV, Kalinovsky AI, Popov RS, Vishchuk OS, Stonik VA. Asterosaponins from the Far Eastern starfish Leptasterias ochotensis and their anticancer activity. Steroids. 2014;87:119–127. doi: 10.1016/j.steroids.2014.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Kurie JM, Buck J, Eppinger TM, Moy D, Dmitrovsky E. 9-cis and all-trans retinoic acid induce a similar phenotype in human teratocarcinoma cells. Differentiation. 1993;54:123–129. doi: 10.1111/j.1432-0436.1993.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 32.Ozpolat B, Mehta K, Tari AM, Lopez-Berestein G. All-trans-Retinoic acid-induced expression and regulation of retinoic acid 4-hydroxylase (CYP26) in human promyelocytic leukemia. Am J Hematol. 2002;70:39–47. doi: 10.1002/ajh.10099. [DOI] [PubMed] [Google Scholar]
- 33.Pitson SM, Powell JA, Bonder CS. Regulation of sphingosine kinase in hematological malignancies and other cancers. Anticancer Agents Med Chem. 2011;11:799–809. doi: 10.2174/187152011797655078. [DOI] [PubMed] [Google Scholar]
- 34.Sobue S, Nemoto S, Murakami M, Ito H, Kimura A, Gao S, Furuhata A, Takagi A, Kojima T, Nakamura M, et al. Implications of sphingosine kinase 1 expression level for the cellular sphingolipid rheostat: Relevance as a marker for daunorubicin sensitivity of leukemia cells. Int J Hematol. 2008;87:266–275. doi: 10.1007/s12185-008-0052-0. [DOI] [PubMed] [Google Scholar]
- 35.Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, Cuvillier O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting shingosine kinase-1. Leukemia. 2005;20:95–102. doi: 10.1038/sj.leu.2404023. [DOI] [PubMed] [Google Scholar]
- 36.Powell JA, Lewis AC, Zhu W, Toubia J, Pitman MR, Wallington-Beddoe CT, Moretti PA, Iarossi D, Samaraweera SE, Cummings N, et al. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood. 2017;129:771–782. doi: 10.1182/blood-2016-06-720433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitson SM, Xia P, Leclercq TM, Moretti PA, Zebol JR, Lynn HE, Wattenberg BW, Vadas MA. Phosphorylation-dependent translocation of sphingosine kinase to the plasma membrane drives its oncogenic signalling. J Exp Med. 2005;201:49–54. doi: 10.1084/jem.20040559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Recchia F, Saggio G, Nuzzo A, Biondi E, Di Blasio A, Cesta A, Candeloro G, Alesse E, Rea S. Multicenter phase 2 study of interleukin-2 and 13-cis retinoic acid as maintenance therapy in advanced non-small-cell lung cancer. J Immunother. 2006;29:87–94. doi: 10.1097/01.cji.0000186244.85058.1b. [DOI] [PubMed] [Google Scholar]
- 39.Wakelee HA, Takimoto CH, Lopez-Anaya A, Chu Q, Middleton G, Dunlop D, Ramlau R, Leighl N, Rowinsky EK, Hao D, et al. The effect of bexarotene on atorvastatin pharmacokinetics: Results from a phase I trial of bexarotene plus chemotherapy in patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2012;69:563–571. doi: 10.1007/s00280-011-1772-z. [DOI] [PubMed] [Google Scholar]
- 40.Montesano R, Soulié P. Retinoids induce lumen morphogenesis in mammary epithelial cells. J Cell Sci. 2002;115((Pt 23)):4419–4431. doi: 10.1242/jcs.00164. [DOI] [PubMed] [Google Scholar]
- 41.de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A. Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature. 1990;343:177–180. doi: 10.1038/343177a0. [DOI] [PubMed] [Google Scholar]
- 42.Zhou T, Medeiros LJ, Hu S. Chronic myeloid leukemia: Beyond BCR-ABL1. Curr Hematol Malig Rep. 2018;13:435–445. doi: 10.1007/s11899-018-0474-6. [DOI] [PubMed] [Google Scholar]
- 43.Caldemeyer L, Dugan M, Edwards J, Akard L. Long-term side effects of tyrosine kinase inhibitors in chronic myeloid leukemia. Curr Hematol Malig Rep. 2016;11:71–79. doi: 10.1007/s11899-016-0309-2. [DOI] [PubMed] [Google Scholar]
- 44.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 45.Thao NP, Cuong NX, Luyen BT, Thanh NV, Nhiem NX, Koh YS, Ly BM, Nam NH, Kiem PV, Minh CV, Kim YH. Anti-inflammatory asterosaponins from the starfish Astropecten monacanthus. J Nat Prod. 2013;76:1764–1770. doi: 10.1021/np400492a. [DOI] [PubMed] [Google Scholar]
- 46.Ngoan BT, HanhT T, Vien le T, Diep CN, Thao NP, Thao do T, Thanh NV, Cuong NX, Nam NH, Thung do C, et al. Asterosaponins and glycosylated polyhydroxysteroids from the starfish Culcita novaeguineae and their cytotoxic activities. J Asian Nat Prod Res. 2015;17:1010–1017. doi: 10.1080/10286020.2015.1041930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.