

Graphical Abstract
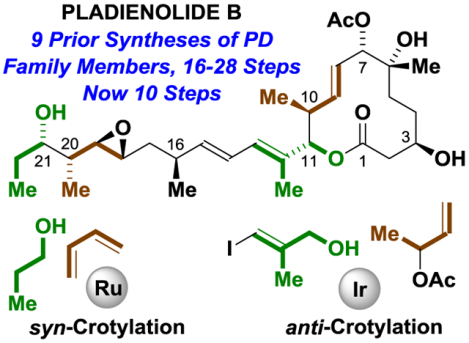
Total Synthesis Fueled by Alcohol. The potent spliceosome modulator pladienolide B, which bears 10 stereogenic centers, is prepared in 10 steps (LLS). Asymmetric alcohol-mediated carbonyl crotylations catalyzed by ruthenium and iridium that occur with syn- and anti-diastereoselectivity, respectively, were used to form the C20-C21 and C10-C11 C-C bonds.
Keywords: Enantioselective, Hydrogen Transfer, Iridium, Ruthenium, Cancer
The pladienolides are a family of 12-membered macrolides isolated in 2004 by researchers at Eisai from the culture broth of Mer-11107, an engineered strain of Streptomyces platensis (Figure 1).[1] Earlier in 1994, a related polyketide, FD-895, was isolated from the Okinawan soil bacteria Streptomyces hygroscopicus A-9561.[2] The pladienolides were identified in a cell-based reporter assay for hypoxia-induced gene expression controlled by human VEGF promoter[1a] and were shown to inhibit proliferation of multiple drug resistant human cancer cells at low nanomolar IC50 values.[3] In 2007, researchers at Eisai determined that the pladienolides bind splicing factor 3b (SF3b),[4,5] inducing cell cycle arrest at both G1 and G2/M phase. In 2018, researchers at the Max Planck Institute for Biophysical Chemistry and H3 Biomedicine, Inc., obtained a crystal structure of SF3b bound by pladienolide B, providing precise insight into its mode of action.[6] Eisai launched two phase I clinical trials of pladienolide analogue E7107,[7] which were discontinued due to side-effects involving vision loss. Interest in anti-cancer drugs that target the spliceosome persisted, and in 2017 the pladienolide derivative H3B-8800 developed by Eisai and H3 Biomedicine, Inc. was granted orphan drug status by the FDA for treatment of myelogenous and chronic myelomonocytic leukemia.[8]
Figure 1.
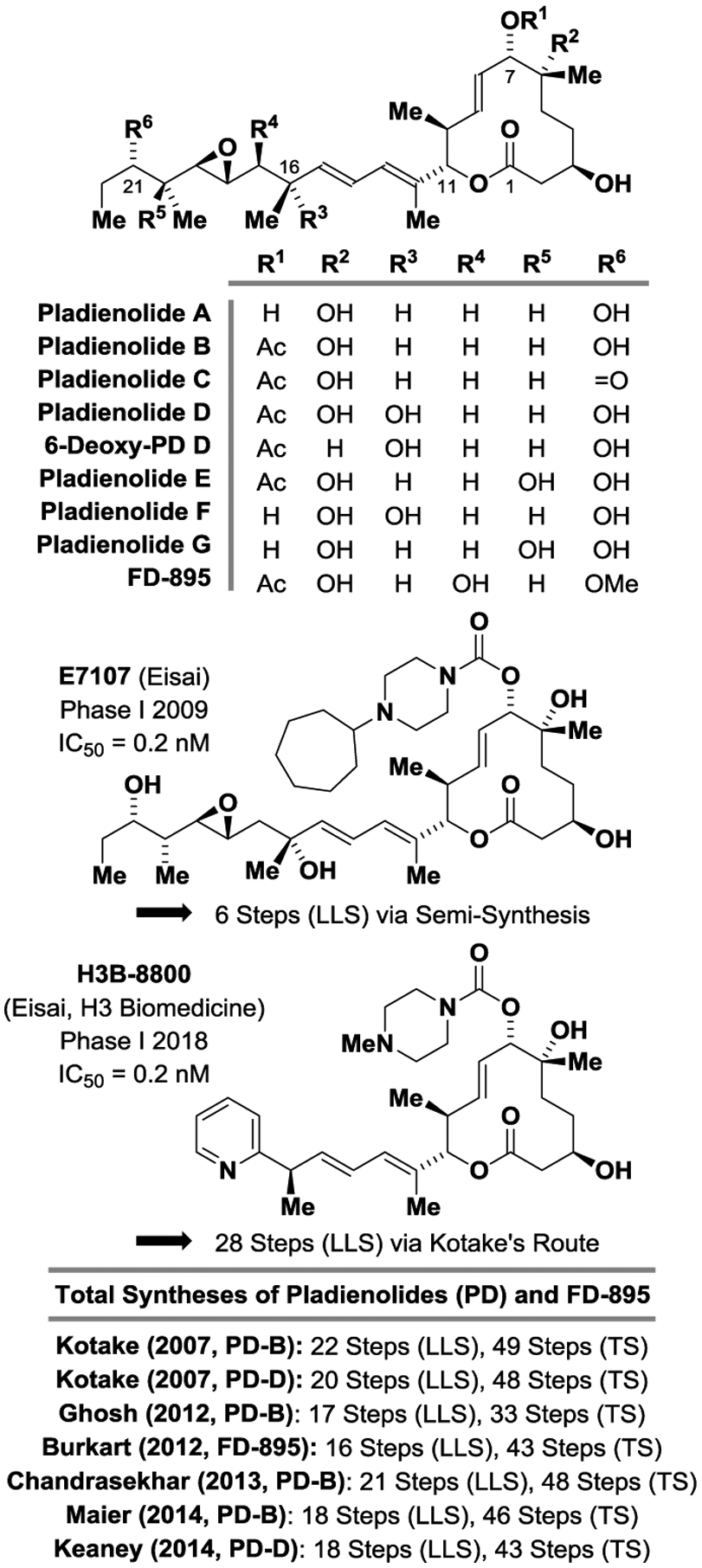
Pladienolides A-G, FD-895 and related clinical candidates E7107 and H3B-8800. LLS = Longest Linear Sequence. TS = Total Steps.
Although pladienolide and its derivatives show great promise vis-á-vis cancer treatment, concise routes to compounds of this class remain elusive. To date, four total syntheses of pladienolide B (and its C7 epimer) and have been reported,[9] along with the total synthesis of FD-895 (and its C17 epimer).[10] These routes range in length between 16–22 steps (LLS) (Figure 1). The manufacturing routes to the clinical candidate E7107 involves a 6 step (LLS) semi-synthesis from Pladienolide D[11a] and, according the patent literature, H3B-8800 is prepared in 8 steps (LLS) from synthetic pladienolide D prepared using Kotake’s route,[9a] giving a total of 28 steps (LLS).[11b] Finally, the syntheses of pladienolide and FD-895 substructures also have been disclosed.[12] As documented in the review literature,[13] catalytic methods for the direct enantioselective conversion of lower alcohols to higher alcohols developed in our laboratory have the potential to streamline type 1 polyketide total synthesis by circumventing discrete alcohol-to-carbonyl redox reactions and operations associated with the installation and removal of chiral auxiliaries and protecting groups.[14] To enable more concise entry to the pladienolides and related clinical candidates, the total synthesis of pladienolide B, the most potent member of this class, was undertaken. Here, using asymmetric alcohol-mediated carbonyl crotylations catalyzed by ruthenium[15] and iridium[16] that occur with syn- and anti-diastereoselectivity, we report the total synthesis of pladienolide B in 10 steps (LLS).
Our retrosynthesis of pladienolide B maximizes convergency for optimal step-economy (Scheme 1). Specifically, it was anticipated pladienolide B could be formed via Suzuki cross-coupling of Fragments A and B, as similar Stille cross-couplings were used to in the total syntheses of pladienolide B reported by Chandrasekhar[9c] and Maier,[9d] and Burkart’s synthesis of FD-895.[10a] Also, a related Suzuki coupling was used in the total synthesis of 6-deoxypladienolide D reported by Keaney.[9e] Fragment A was envisioned to arise from Fragments C and D via Yamaguchi esterification[17] followed by Grubbs’ ring-closing metathesis, as practiced in all prior total syntheses of the pladienolides and FD-895 with the exception of Maier’s synthesis.[9d] Fragment C is accessible via dienolate-mediated epoxide ring opening. A related ring-opening of an epoxide lacking the C6 methyl group was used by Ghosh in the total syntheses of pladienolide B.[9b] Fragment D is accessible via asymmetric iridium-catalyzed anti-crotylation of alcohol 10 using α-methyl allyl acetate 11 as the crotyl donor.[16] Fragment B could be obtained from Fragments E and F through a sequence involving Grubbs’ cross-metathesis, Shi epoxidation[19] and Cu-catalyzed alkyne hydroboration. Fragment E is accessible via asymmetric ruthenium-catalyzed syn-crotylation of n-propanol using butadiene as the crotyl donor.[15] Finally, fragment F can be prepared via asymmetric iridium-catalyzed reductive allylation of the acetylenic aldehyde 12 mediated by 2-propanol using allyl acetate as pronucleophile,[18] followed by propargyl substitution with inversion using the Normant reagent.[20] Thus, a total of three C-C bonds are formed via asymmetric alcohol-mediated carbonyl allylation (C16-C17) and crotylation (C20-C21 and C10-C11), the latter with both syn- and anti-diastereoselectivity.
Scheme 1.
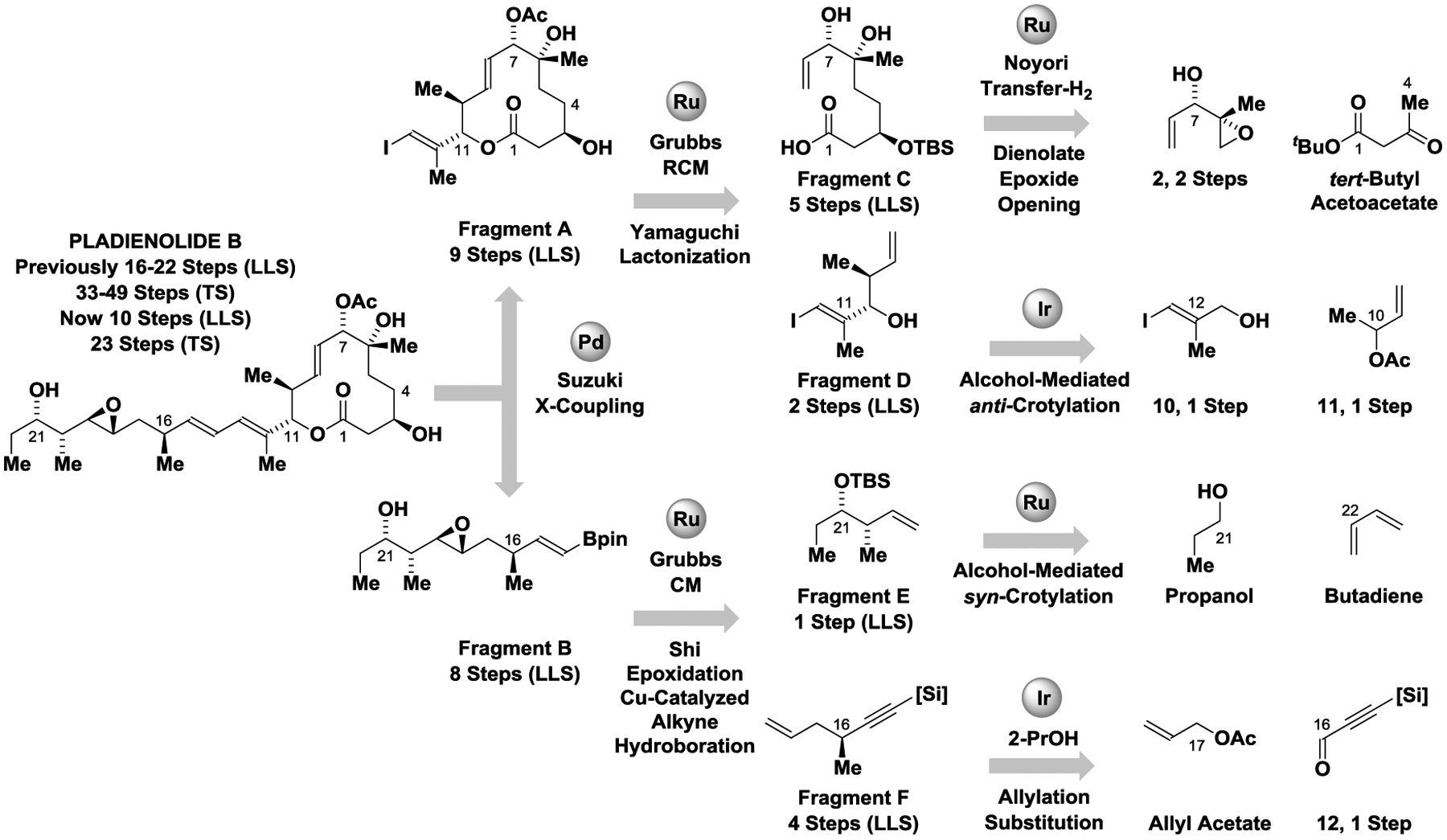
Retrosynthetic analysis of pladienolide b: maximizing convergency for step-economy.
The synthesis of Fragment A begins with the kinetic resolution of the doubly allylic alcohol 1, which is prepared from methacrolein and the vinyl Grignard reagent,[21] using the Sharpless asymmetric epoxidation (Scheme 2).[22] Exclusive epoxidation of the more substituted alkene was observed, providing glycidol 2 in highly enantiomerically enriched form. The unprotected epoxide was exposed to the dienolate derived from tert-butyl acetoacetate followed by acetic anhydride to form the product of epoxide ring opening 3 in 86% yield.[23] Compound exists as a dynamic mixture of hydroxy ketone 3 and, predominantly, the 5-membered lactol. If the free hydroxyl is present at C7 (rather than the acetate), a six-membered lactol is formed that does not participate in the subsequent ketone reduction (not shown). Ruthenium-catalyzed transfer hydrogenation of 3 mediated by formic acid enables access to the β-hydroxy ester 4 as a 4:1 mixture of diastereomers.[24] Acidic cleavage of the tert-butyl ester gave the carboxylic acid, which was isolated as a single stereoisomer. Treatment of the hydroxy acid with tert-butyldimethylsilyl chloride results in silylation of both the C1 carboxylic acid and C3 hydroxyl groups. Addition of methanolic K2CO3 to the reaction mixture results in concomitant cleavage of the silyl ester and the C7 acetate[25] to furnish the C6-C7 diol, Fragment C. Under Yamaguchi conditions, Fragment C and Fragment D undergo esterification to form compound 5 despite the presence of the unprotected C7 hydroxyl.[17,26] The formation of Fragment D is accomplished via asymmetric iridium-catalyzed alcohol-mediated carbonyl anti-crotylation[16] of the allylic alcohol 10, which is prepared via zirconium-catalyzed carboalumination of progaryl alcohol (Scheme 3).[27] Finally, ring-closing metathesis of 5 followed by acetylation of the C7 hydroxyl with acidic workup provides Fragment A. It is worth noting that although allyl acetates are well-established participants in diverse metathetic processes, attempted metathesis of the C7 allyl acetates 6 and 7 resulted in low conversion accompanied by formation of the unanticipated C-C bond cleavage side-products, methyl ketones 8 and 9.[28]
Scheme 2.
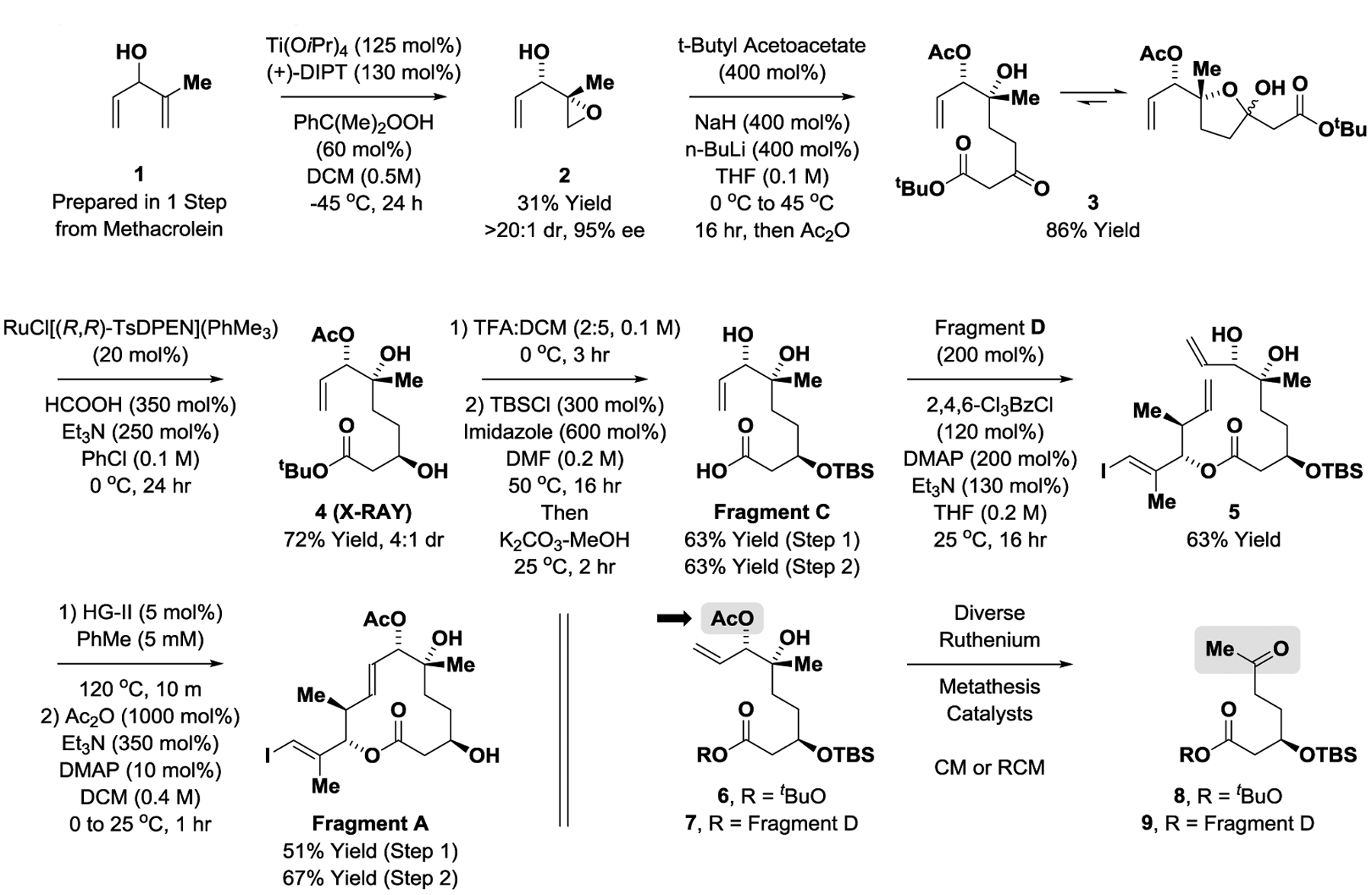
Synthesis of Fragment A.a
aYields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Scheme 3.
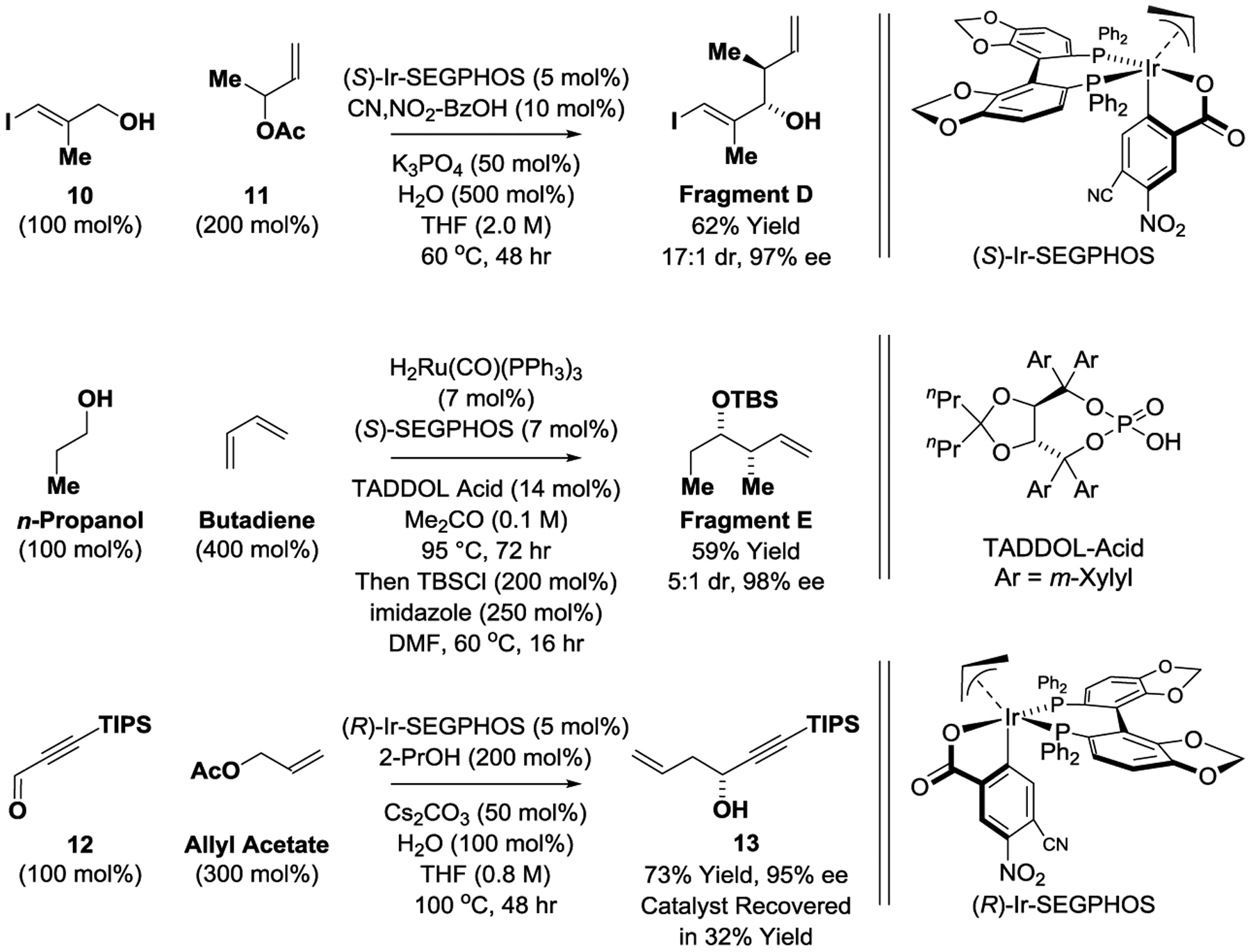
Catalytic enantioselective anti- and syn-crotylations of allylic alcohol 10 and propanol to form Fragments D and E, respectively, and the catalytic enantioselective allylation of acetylenic aldehyde 12 mediated by 2-propanol to form propargyl alcohol 13.
aYields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
With Fragment A in hand, the preparation of Fragment B, and therefrom, the total synthesis of pladienolide B could be achieved (Scheme 4). The formation of Fragment B begins with conversion of propargyl alcohol 13 to Fragment F via acylation with methyl chloroformate followed by methyl-substitution of the mixed carbonate using the Normant reagent.[20] Propargyl alcohol 13 is prepared via asymmetric iridium-catalyzed reductive coupling of acetylenic aldehyde 12 and allyl acetate mediated by 2-propanol (Scheme 3).[18] Although a slight erosion in enantiomeric enrichment was observed in each step of the conversion of propargyl alcohol 13 to Fragment F, the latter could be formed in 91% enantiomeric excess. Cross-metathesis of Fragments E and F catalyzed by the second-generation Grubbs catalyst was followed by concomitant fluoride-mediated removal of the silyl protecting groups to provide compound 15, which was isolated as a single stereoisomer. In the cross-metathesis, an excess of Fragment E was required to suppress homo-coupling of Fragment F. Excess Fragment E could be recovered in 46% yield and recycled. The 1,5-acetylenic olefin 14 readily underwent highly chemo- and diastereoselective Shi epoxidation[19] and alkyne hydroboration[29] to deliver Fragment B. Finally, Suzuki cross-coupling under conditions reported by Keaney[9e] provided pladienolide B in a total of 10 steps (LLS), the shortest route to any pladienolide family member reported to date.[30]
Scheme 4.
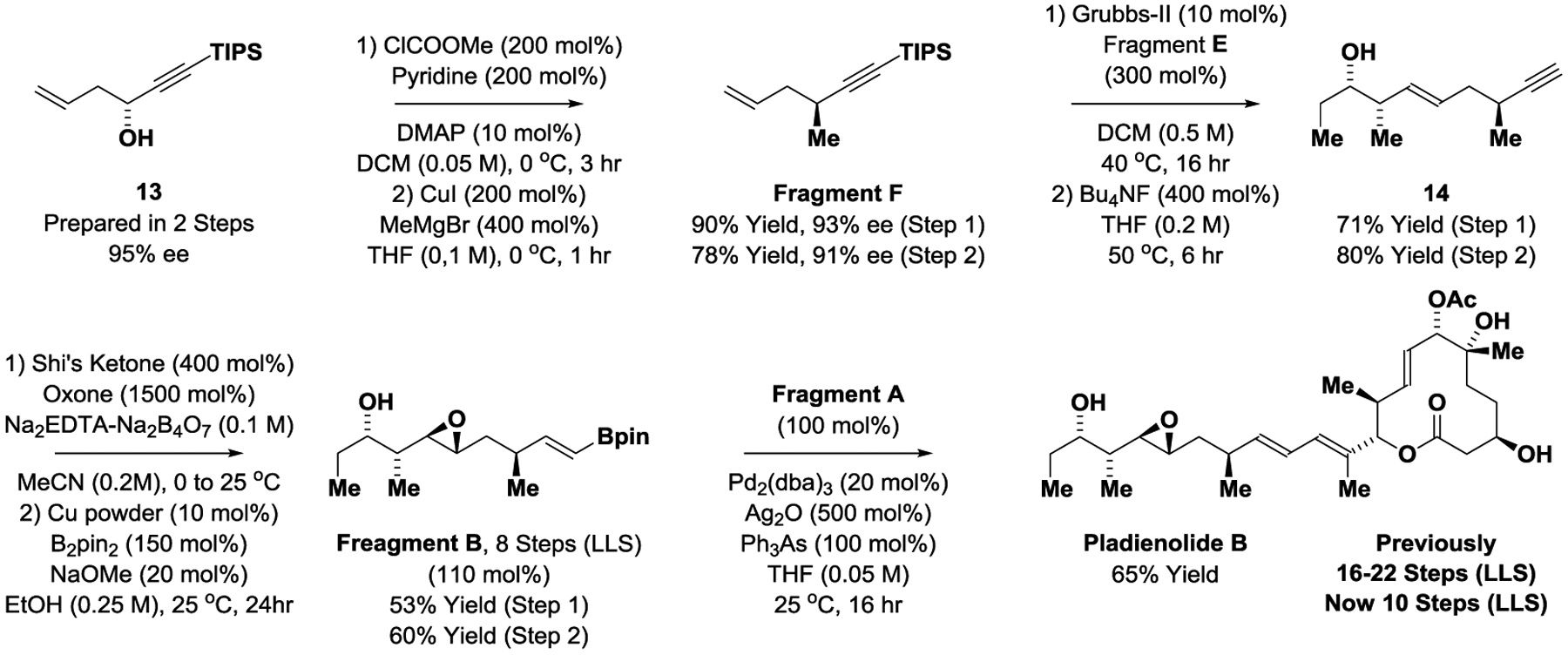
Synthesis of Fragment B and total synthesis of pladienolide B.a
aYields are of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
In conclusion, by maximizing convergency and exploiting diverse metal-mediated methods for C-C coupling, a concise total synthesis of the spliceosome modulator pladienolide B was achieved. Among the 10 stereogenic centers found in pladienolide B, half are formed using catalytic asymmetric alcohol-mediated carbonyl additions. Specifically, enantioselective alcohol-mediated carbonyl crotylations catalyzed by ruthenium and iridium that occur with syn- and anti-diastereoselectivity, respectively, were used to forge the C20-C21 and C10-C11 C-C bonds. Additionally, an enantioselective asymmetric iridium-catalyzed reductive coupling of acetylenic aldehyde 12 with allyl acetate mediated by 2-propanol was used to form forge the C16-C17 bond. It is our hope that the present synthetic route will broader access to pladienolide and related clinical candidates for cancer chemotherapy that target the spliceosome.
Supplementary Material
Acknowledgments
The Welch Foundation (F-0038) and the NIH (RO1-GM093905) are acknowledged for partial support of this research. Mr. Haifeng Chen is acknowledged for skillful technical assistance.
References
- [1].For isolation and structure determination of pladienolides A-G, see:; a) Sakai T, Sameshima T, Matsufuji M, Kawamura N, Dobashi K, Mizui Y, J. Antibiot 2004, 57, 173–179; [DOI] [PubMed] [Google Scholar]; b) Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y, J. Antibiot 2004, 57, 180–187; [DOI] [PubMed] [Google Scholar]; c) Asai N, Kotake Y, Niijima J, Fukuda Y, Uehara T, Sakai T, J. Antibiot 2007, 60, 364–369. [DOI] [PubMed] [Google Scholar]
- [2].For isolation and structure determination (via total synthesis) of FD-895, see:; a) Seki-Asano M, Okazaki T, Yamagishi M, Sakai N, Takayama Y, Hanada K, Morimoto S, Takatsuki A, Mizoue K, J. Antibiot 1994, 47, 1395–1401; [DOI] [PubMed] [Google Scholar]; b) Villa R, Mandel AL, Jones BD, LaClair JJ, Burkart MD, Org. Lett 2012, 14, 5396–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizui Y; Sakai T; Iwata M; Uenaka T; Okamoto K; Shimizu H; Yamori T; Yoshimatsu K; Asada MJ Antibiot. 2004, 57, 188–196. [DOI] [PubMed] [Google Scholar]
- [4].Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y, Shimizu H, Uesugi M, Ishihama Y, Iwata M, Mizui Y, Nat. Chem. Biol 2007, 3, 570–575. [DOI] [PubMed] [Google Scholar]
- [5].For a review of pladienolide and related natural products that inhibit splicing factor 3B subunit 1, see:; Pham D, Koide K, Nat. Prod. Rep 2016, 33, 637–647. [DOI] [PubMed] [Google Scholar]
- [6].Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Lührmann R, Larsen N, Buonamici S, Pena V, Mol. Cell 2018, 70, 265–273. [DOI] [PubMed] [Google Scholar]
- [7].ClinicalTrials.gov Identifier: NCT00499499;; b) Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O’Brien J, LoRusso P, Invest New Drugs. 2014,. 32, 436–444; [DOI] [PubMed] [Google Scholar]; c) Dehm SM, Clin Cancer Res. 2013,. 19, 6064–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].ClinicalTrials.gov Identifier: NCT02841540;; Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, Fekkes P, Karr C, Klimek V, Lai G, Lee L, Kumar P, Lee SC-W, Liu X, Mackenzie C, Meeske C, Mizui Y, Padron E, Park E, Pazolli E, Peng S, Prajapati S, Taylor J, Teng T, Wang J, Warmuth M, Yao H, Yu L, Zhu P, Abdel-Wahab O, Smith PG, Buonamici S, Nature Med. 2018, 24, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].For prior total syntheses of pladienolide B and 6-deoxypladienolide D, see:; a) Kanada RG, Itoh D, Nagai M, Niijima J, Asai N, Mizui Y, Abe S, Kotake Y, Angew. Chem. Int. Ed 2007, 46, 4350–4355; [DOI] [PubMed] [Google Scholar]; b) Ghosh AK, Anderson DD, Org. Lett 2012, 14, 4730–4733; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kumar VP, Chandrasekhar S Org. Lett 2013, 15, 3610–3613; [DOI] [PubMed] [Google Scholar]; d) Müller S, Sasse F, Maier ME, Eur. J. Org. Chem 2014, 1025–1036; [Google Scholar]; e) Arai K, Buonamici S, Chan B, Corson L, Endo A, Gerard B, Hao M-H, Karr C, Kira K, Lee L, Liu X, Lowe JT, Luo T, Marcaurelle LA, Mizui Y, Nevalainen M, O’Shea M. Welzel, Park ES, Perino SA, Prajapati S, Shan M, Smith PG, Tivitmahaisoon P, Wang JY, Warmuth M, Wu K-M, Yu L, Zhang H, Zheng G-Z, Keaney GF, Org. Lett 2014, 16, 5560–5563. [DOI] [PubMed] [Google Scholar]
- [10].For total syntheses of FD-895 and 17S-FD-895, see:; a) Villa R, Mandel AL, Jones BD, La Clair JJ, Burkart MD, Org. Lett 2012, 14, 5396–5399; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chan WC, La Clair JJ, León B, Trieger KA, Slagt MQ, Verhaar MT, Bachera DU, Rispens MT, Hofman RM, de Boer VL, van der Hulst R, Bus R, Hiemstra P, Neville ML, Mandla KA, Figueroa JS, Jamieson C, Burkart MD, Cell Rep. Phy. Sci 2020, 1, 100277. [Google Scholar]
- [11].For the semi-synthesis of E7107, see:; a) Kotake Y, Niijima J, Fukuda Y, Kanada RM, Nakashima T, Yoshida M, Tsuchida T, inventors; Eisai R&D Management Co., Ltd., Merican Corporation, assignee. Physiologically Active Substances. United States patent US 7,550,503. 2009. June 23. [Google Scholar]; For the synthesis of H3B-8800, see:; b) Keaney GF, Wang J, Gerard B, Arai K, Liu X, Zheng GZ, Kira K, Tibitma-Haisoon P, Prajapati S, Kotake Y, Kanada MR, inventors; Eisai R&D Management Co., Ltd., assignee. A Solid State Form of Pladienolide Pyridine Compounds and Methods of Use. International patent WO 2017/087667. 2017. May 26. [Google Scholar]
- [12].For syntheses of pladienolide and FD-895 substructures, see:; a) Mandel AL, Jones BD, La Clair JJ, Burkhart MD, Bioorg. Med. Chem. Lett 2007, 17, 5159–5164; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Skaanderup PR, Jensen T, Org. Lett 2008, 10, 2821–2824; [DOI] [PubMed] [Google Scholar]; c) Müller S, Mayer T, Sasse F, Maier ME, Org. Lett 2011, 13, 3940–3943. [DOI] [PubMed] [Google Scholar]
- [13].For selected reviews highlighting recent advances in type I polyketide total synthesis, see:; a) Dechert-Schmitt A-MR, Schmitt DC, Gao X, Itoh T, Krische MJ, Nat. Prod. Rep 2014, 31, 504–513; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Feng J, Kasun ZA, Krische MJ, J. Am. Chem. Soc 2016, 138, 5467–5478; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schwan J, Christmann M, Chem. Soc. Rev 2018, 47, 7985–7995; [DOI] [PubMed] [Google Scholar]; d) Liu H, Lin S, Jacobsen KM, Poulsen TB, Angew. Chem., Int. Ed 2019, 58, 13630–13642; [DOI] [PubMed] [Google Scholar]; e) Friedrich RM, Friestad GK, Nat. Prod. Rep 2020, 37, 1229–1261. [DOI] [PubMed] [Google Scholar]
- [14].For selected reviews on alcohol-mediated carbonyl allylation and propargylation via metal-catalyzed hydrogen auto-transfer, see:; a) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371–2380; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ambler BR, Woo SK, Krische MJ, ChemCatChem 2019, 11, 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].For ruthenium-catalyzed syn-crotylation mediated by butadiene, see:; McInturff EL, Yamaguchi E, Krische MJ, J. Am. Chem. Soc 2012, 134, 20628–20631; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gao X, Woo SK, Krische MJ, J. Am. Chem. Soc 2013, 135, 4223–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].For iridium-catalyzed anti-crotylation mediated by α-methyl allyl acetate, see:; a) Kim IS, Han SB, Krische MJ, J. Am. Chem. Soc 2009, 131, 2514–2520; [DOI] [PMC free article] [PubMed] [Google Scholar]; Gao X, Townsend IA, Krische MJ, J. Org. Chem 2011, 76, 2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Inanaga J; Hirata K; Saeki H; Katsuki T; Yamaguchi M Bull. Chem. Soc. Jpn 1979, 52, 1989–1993. [Google Scholar]
- [18].Brito GA, Della-Felice F, Luo G, Burns AS, Pilli R, Rychnovsky SD, Krische MJ, Org. Lett 2018, 20, 4144–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang Z-X, Tu Y, Frohn M, Zhang J-R, Shi Y, J. Am. Chem. Soc 1997, 119, 11224–11235. [Google Scholar]; Formation of the C18–19 epoxide was accomplished using the Shi epoxidation in all prior pladienolide syntheses.
- [20].a) Brinkmeyer RS, Macdonald TL, J. Chem. Soc., Chem. Commun 1978, 876–877; [Google Scholar]; b) Macdonald TL, Reagan DR, Brinkmeyer RS, J. Org. Chem 1980, 45, 4740–4747; [Google Scholar]; c) Overman LE, Ball KL, J. Am. Chem. Soc 1981, 103, 1853–1855. [Google Scholar]
- [21].Bertus P, Drouin L, . Szymoniak Laroche, J., Tetrahedron. 2004, 60, 1375–1383. [Google Scholar]
- [22].Ogura Y, Sato H, Kuwahara S, Org. Lett 2016, 18, 2399–2402. [DOI] [PubMed] [Google Scholar]
- [23].Lygo B, O’Connor N, Synlett, 1992, 6, 529–530. [Google Scholar]
- [24].Fujii A, Hashiguchi S, Uematsu N, Ikariya T, Noyori R, J. Am. Chem. Soc 1996, 118, 2521–2522. [Google Scholar]
- [25].For a related 2-step process, see:; Law C, Meng Y, Koo SM, Morken JP, Angew. Chem. Int. Ed 2019, 58, 6654–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Related esterifications en route to pladienolide B have involved compounds in which the C7 hydroxyl is protected (see references 9b and 9c).
- [27].a) Rand CL, Van Horn DE, Moore MW, Negishi E, J. Org. Chem 1981, 46, 4093–4096; [Google Scholar]; b) Lölsberg W, Werle S, Neudörfl JM, Schmalz HG, Org. Lett 2012, 14, 5996–5999. [DOI] [PubMed] [Google Scholar]
- [28].As revealed in a study on allylic substituent effects in the ring-closing metathesis, allylic hydroxyl groups exert a large, favorable effect on reaction rates:; Hoye TR, Zhao H Org. Lett 1999, 1, 1123–1125. [DOI] [PubMed] [Google Scholar]
- [29].Zhao J, Niu Z, Fu H, Li Y, Chem. Commun 2014, 50, 2058–2060. [DOI] [PubMed] [Google Scholar]
- [30].While our manuscript was under review, Rhoades et al. reported total syntheses of pladienolide B and H3B-8800 using our method for enantioselective iridium-catalyzed anti-crotylation (DOI: 10.1021/jacs.1c01135). As their aldehyde starting material is not commercially available and requires a 2 step synthesis, their routes to pladienolide B and H3B-8800 are each 10 steps (10 LLS; 25 TS and 10 LLS; 21 TS, respectively), making their longest linear sequence to pladienolide B equivalent in length to own. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.