

Abstract
Poly(dimethylsiloxane) (PDMS) is an attractive, versatile, and convenient material for use in biomedical devices that are in direct contact with the user. A crucial component in such a device is its surface in terms of antimicrobial properties preventing infection. Moreover, due to its inherent hydrophobicity, PDMS is rather prone to microbial colonization. Thus, developing an antimicrobial PDMS surface in a simple, large-scale, and applicable manner is an essential step in fully exploiting PDMS in the biomedical device industry. Current chemical modification methods for PDMS surfaces are limited; therefore, we present herein a new method for introducing an atom transfer radical polymerization (ATRP) initiator onto the PDMS surface via the base-catalyzed grafting of [(chloromethyl)phenylethyl]trimethoxysilane to the PDMS. The initiator surface was grafted with poly[2-(dimethylamino)ethyl methacrylate] (PDMAEMA) brushes via a surface-initiated supplemental activator and reducing agent ATRP (SI-SARA-ATRP). The use of sodium sulfite as a novel reducing agent in SI-SARA-ATRP allowed for polymerization during complete exposure to air. Moreover, a fast and linear growth was observed for the polymer over time, leading to a 400 nm thick polymer layer in a 120 min reaction time. Furthermore, the grafted PDMAEMA was quaternized, using various alkylhalides, in order to study the effect on surface antimicrobial properties. It was shown that antimicrobial activity not only depended highly on the charge density but also on the amphiphilicity of the surface. The fast reaction rate, high oxygen tolerance, increased antimicrobial activity, and the overall robustness and simplicity of the presented method collectively move PDMS closer to its full-scale exploitation in biomedical devices.
Introduction
Poly(dimethylsiloxane) (PDMS) elastomers have attracted considerable attention for their use in various biomedical devices. Products such as soft contact lenses,1,2 implantable cables,3 indwelling catheters,4 and many more5−8 can greatly benefit from the easy processing, excellent mechanical properties,8−10 biocompatibility,11 and other convenient properties of PDMS. Despite these many advantages, however, the hydrophobic nature of PDMS makes its surface biocompatible to cells11 and thus prone to microbial colonization, which may develop into an infection and result in severe complications in long-term users of such devices. The development of strategies to limit the growth of bacteria on PDMS surfaces is therefore necessary. Among several approaches to this problem on antimicrobial surfaces,12,13 the grafting of contact-killing cationic polymers containing quaternary amines has resulted in a great ability to kill bacteria.4,14,15 Moreover, the nonrelease nature of these surfaces may limit the development of resistance in bacteria, thus featuring permanent antimicrobial properties.16,17 In particular, quaternized poly(dimethylaminoethyl methacrylate) (PDMAEMA) has shown permanent bactericidal effects.18−20 In addition, when surface-grafted from a PDMS substrate, it has exhibited high protein-repelling capabilities, low bacterial attachment, and reduced cell adhesion compared to a native PDMS surface.21 However, due to the low reactivity of PDMS surfaces, there are only few—and complex—methods available for the fast and efficient modification of PDMS. Density, thickness, and surface coverage homogeneity are other essential factors when seeking to achieve excellent antimicrobial activity.19,22−24 Surface-initiated atom transfer radical polymerization (SI-ATRP) is one of the most popular and versatile techniques for surface modifications.25 Featuring good control over the abovementioned qualities, SI-ATRP serves as an invaluable tool offering precise surface manipulation suitable for antimicrobial studies.18−20,22,23 Indeed, several examples of DMAEMA surface modification by SI-ATRP have been reported.18−23 However, the requirement of an inert atmosphere in conventional SI-ATRP limits its application potential in scaled up production and hinders its commercial exploitation.
High oxygen sensitivity in conventional SI-ATRP has been tackled through different approaches, such as activator generated by electron transfer,26 activator regenerated by electron transfer (ARGET),27 and supplemental activator and reducing agent (SARA)28−30 SI-ATRP. The core idea behind these methods involves adding an amount of reducing agent to the polymerization mixture, which then helps maintain the metal–complex catalyst activity in the presence of oxygen through the continuous reduction of the catalyst. A notable example of an oxygen-tolerant, SI-ATRP of PDMAEMA was reported by Dong and Matyjaszewski.31 In their work, polymer brushes were grafted from an initiator-modified silicon wafer, using tin(II) 2-ethylhexanoate as a reducing agent. After an 18 h reaction time, the layer thickness of the modified surface was approximately 20 nm. Using this approach, a couple of reports showed how it was possible to transfer the conditions to other types of surfaces, whereby PDMAEMA was grafted from either filter paper32 or lignin nanofiber mats,33 for a number of different applications. In the latter case, ascorbic acid was used as a reducing agent, which resulted in a thickness of 72 nm after 6 h of polymerization. As another example, Dunderdale et al.34 showed how it was possible to graft DMAEMA from silicon wafer surfaces in an oxygen atmosphere (ARGET), leading to 2–300 nm thick grafts within 3 h. They even showed that simply painting the polymerization solution onto the wafer surface was sufficient to enable grafting to take place. This clearly demonstrates the great potential of using ARGET SI-ATRP for surface modification and tailoring the surface properties of solid substrates. However, to the best of our knowledge, no work on oxygen-tolerant ATRP of PDMAEMA grafted from a PDMS substrate has been reported to date. Thanks to their oxygen tolerance and simplicity, these methods offer great potential to achieve permanently antimicrobial PDMS surfaces on a commercial scale.
In this work, we present an oxygen-tolerant, SI-SARA-ATRP of PDMAEMA, using Na2SO3 as a novel reducing agent. This was validated by grafting DMAEMA from a silicon wafer to demonstrate the efficiency of the process, while the system was subsequently transferred to PDMS grafting in our open-to-air screening platform.35
Results and Discussion
A characteristic property of controlled radical polymerization is linear development in the polymer’s chain length as a function of reaction time, as well as the synthesized polymer having narrow dispersity. To confirm that the kinetics of the SI-SARA-ATRP system behaved in this manner, a model study was conducted on silicon wafers. The wafers were functionalized with a standard ATRP initiator (surface-anchored α-bromoisobutyryl bromide, APTES–BiBB) and were immersed into a glass vial holding an aqueous solution of H2O/MeOH (1:1, v/v) containing monomer (DMAEMA) and catalyst/ligand [Cu(II)Cl2/N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA)]. To enable the wafer to be gradually withdrawn from the polymerization mixture, a large headspace was used in the reaction. Depending on the size of the headspace, there would be a delay in the onset of polymerization to allow for the removal of excess oxygen, as also reported by Dunderdale et al.34 for their ARGET system. This delay in the onset of polymerization was prevented by replacing the air in the vial with nitrogen, but this was essentially not required for smaller amounts of oxygen in the system. The vial was sealed with a septum, and polymerization was initiated by the addition of Na2SO3. Through the use of ellipsometry, DMAEMA brush thickness could be measured at various time intervals of 10, 30, 60, 90, and 120 min (see Figure 1).
Figure 1.
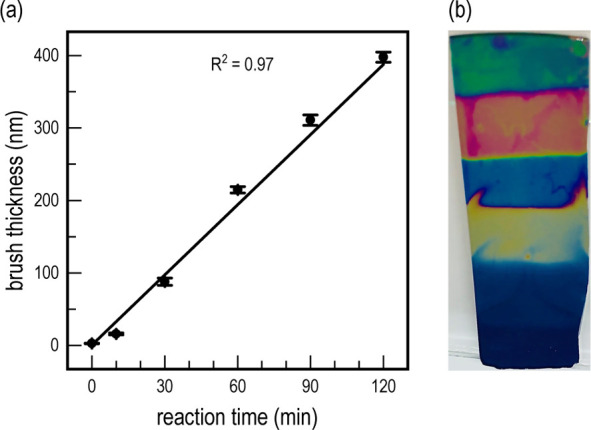
(a) Thickness of the grafted PDMAEMA layer over time. A clear linear trend in the growth of the polymer over time can be observed, yielding a very thick surface layer (400 nm) within a short reaction time (120 min). (b) Photographic image of the modified wafer, where the difference in the refractive index for the individual brush lengths can be observed as a change in wafer coloration. Polymerization time increases from the bottom to the top of the wafer.
Brush thickness propagation followed a clear linear trend with respect to reaction time. A common limitation for SI-ATRP is the long reaction times required to obtain sufficiently long brushes for effective surface modification. In this case, very large brush thicknesses were achieved within short polymerization times, with up to 400 nm being achieved after just 120 min. Interestingly, the difference in the refractive index of the various DMAEMA brush lengths could be directly observed on the silicon wafer—as shown in Figure 1.
In addition, this system was benchmarked against a more traditional SI-ARGET-ATRP system, in that Na2SO3 was replaced with sodium ascorbate as the reducing agent. However, reaction kinetics under these conditions was much slower and resulted in a brush thickness of only 34.15 ± 0.5 nm after 120 min (see Figure S1). This is attributed to the SARA mechanism, which generally exhibits a faster reaction rate, as compared to ARGET. In conclusion, employing Na2SO3 as the reducing agent facilitates the polymerization of DMAEMA at a much higher propagation rate, while still maintaining the linear brush length growth alongside relatively low standard deviation.
SI-SARA-ATRP Employed for Grafting PDMS on an Open Atmosphere Screening Platform
Inspired by the work of Brook et al.,36 in which thiols were introduced onto a PDMS surface via silylation with (3-mercaptopropyl)trimethoxysilane, we show herein how {[(chloromethyl)phenyl]ethyl}trimethoxysilane (CPTS) can be used in a similar fashion to graft an ATRP initiator to the PDMS surface. The presence of the ATRP initiator was confirmed by Fourier transform infrared (FT-IR) (Figure S2). The use of trimethoxysilane as a grafting unit resulted in the formation of a thin layer covering the entire surface, thereby providing an easy method for preparing a full ATRP initiator surface layer anchored to the PDMS—as illustrated in Figure 2a.
Figure 2.

(a) Reaction scheme of the PDMS surface modifications. First, activation of the PDMS surface via the base-catalyzed equilibration of CPTS onto the PDMS surface took place, followed by PDMAEMA grafting from the surface under SI-SARA-ATRP conditions. Finally, the surface was quaternized, using a direct reaction with alkyl halides. (b) Overview of the experimental setup of reactions conducted on the screening platform. PDMAEMA was grafted at three different polymerization times (10, 30, and 60 min), and each of these was then quaternized with a set of various alkyl halides.
This was then further utilized for grafting DMAEMA, using the same concentration of reagents as previously described, albeit now in a completely open atmosphere in a specially designed 96-well plate screening platform.35 The platform allowed for simultaneous brush length variation as well as quaternization of the side chain, using a series of alkylhalides, as illustrated in Figure 2b. Traditional ATRP experiments normally require completely inert conditions, which makes parallelized synthesis complicated and generally requires the use of a glovebox to allow for handling of multiple surfaces. ARGET- and SARA-ATRP, in this respect, have been proven to have advantages (as mentioned above). The use of a reducing agent enables both the reduction of the Cu(II) complex to Cu(I), as well as the removal of excess oxygen, thereby protecting the catalyst from unwanted oxidation. However, most experiments have been run under semi-open conditions with limited amounts of air, which still requires a closed environment.37−39 Herein, we show the possibility of conducting SI-SARA-ATRP in completely open conditions. The procedure allowed for the parallel synthesis of various DMAEMA brush lengths on a PDMS substrate, as well as post-modification via N-alkylation, as illustrated in Figure 2.
Initial analysis by FT-IR of the DMAEMA grafted surfaces showed a distinctive peak at 1728 cm–1 originating from the carbonyl ester of the methacrylate (see Figure 3a). The intensity of the peak increased in good correlation with polymerization time, and the high intensity corresponded well with the expected long brush length observed in the model study conducted on silicon wafers. Surface analysis by X-ray photoelectron spectroscopy (XPS) was also attempted, in order to confirm the presence of the DMAEMA polymer brush. Unfortunately, due to the inherent ability of PDMS to facilitate surface hydrophobic recovery,40 only the PDMS substrate was detected, even though the surface could clearly be observed visually to have been modified and wetted easily with water (see below). Therefore, to ensure that the grafted polymer was retained at the interface, the surface was stored under wet conditions prior to further use. By keeping the surface wet, a hydrophilic interaction with DMAEMA was favored, and the rearrangement could be suppressed before further modification. Because XPS is a high-vacuum technique, it is not possible to conduct the analysis in moist conditions to confirm the grafting chemically.
Figure 3.

(a) FT-IR spectra of pristine PDMS and PDMAEMA functionalized PDMS after 10, 30, and 60 min reaction times. The zoomed-in image shows the corresponding growth of a carbonyl stretch originating from PDMAEMA. (b) FT-IR spectra of pristine PDMS, PDMAEMA-modified PDMS at 30 min polymerization, and the quaternized PDMAEMA-modified PDMS with C1–C10 alkyl halides. The zoomed-in image shows the corresponding growth of C–H stretches originating from the alkyl halides.
To obtain the positively charged quaternary ammonium surfaces, N-alkylation was achieved by using a series of alkylhalides: iodomethane (C1), 1-bromobutane (C4), 1-bromooctane (C8), 1-bromodecane (C10), and 2-bromoethyl methylether [(CH2)2OCH3]. Again, the samples were stored in water immediately after reaction to prevent hydrophobic recovery. The surfaces were analyzed using FT-IR (see Figure 3b), whereby the addition of hydrocarbon chains to DMAEMA resulted in an increase in sp3 hybridization, as seen for all surfaces at 2928, 2905, and 2857 cm–1, respectively. As expected, DMAEMA had the lowest sp3 signal of all modifications due to the lack of a hydrocarbon chain at the side-chain amine. A significantly higher intensity was found for DMAEMA C1, DMAEMA C4, DMAEMA C8, and DMAEMA (CH2)2OCH3. The similar signal intensity between these modifications may be due to the extent of reaction. Iodomethane would be expected to be both more reactive and less sterically hindered than 1-bromobutane, leading to a higher sp3 signal. DMAEMA C8 had the second highest intensity, followed by DMAEMA C10, corresponding well with having the longest carbon chain of all the quarternizations. A similar trend was found at the 10 and 60 min polymerization times, although it was less evident at 60 min (see Figures S3 and S4). Due to the PDMS substrate having been stored under wet conditions, the surface was briefly washed with EtOH and dried in air before collecting the IR spectra. Nonetheless, there were clear signs of residual alcohol observed in the IR spectra at 1740–1570 and 3000–3650 cm–1, resulting from the OH stretches. Although the broad intensive peak at 1740–1570 cm–1 overlapped with the original carbonyl peak from the ester, the ability of PDMS to absorb water to such an extent strongly suggests the creation of a more hydrophilic surface.
Water contact angle (WCA) measurements were also conducted to investigate the change in hydrophilicity as a function of polymerization time and surface modification. Advancing contact angles did not initially reveal any significant differences compared to pristine PDMS or between individual surface modifications (see Figure 4a). On the other hand, a drastically more hydrophilic surface was observed from the receding WCA for all surface modifications, compared to PDMS (see Figure 4b).
Figure 4.

(a) Advancing WCA of the surface-functionalized PDMS. No significant differences are observed across the samples in comparison to the pristine PDMS surface. (b) Receding WCA of the surface-functionalized PDMS. A general decrease in the WCA can be observed when compared to the pristine surface. Moreover, a slight increase in the WCA can be observed via the increasing alkyl chain length in the quaternized surfaces (C1–C10).
The receding WCA for the pristine PDMS was 82°, while all the modified surfaces showed significantly lower receding contact angles. There is a clear trend of lower receding WCA’s for surface-grafted DMAEMA (hydrophilic) with increasing polymerization times (from 50 to 27°) which reflects the increased film thickness with increased polymerization time. For DMAEMA C1, which is also hydrophilic, a similar trend was observed in going from 47 to 37° at 10 and 60 min polymerization times, respectively. On the contrary, an increase in the contact angle was observed for the amphiphilic DMAEMA C4, which went from 37° at 10 min to 55° at 60 min. No significant differences were found for DMAEMA C8, DMAEMA C10, or DMAEMA (CH2)2OCH3 for any polymerization time. The discrepancy among the different modifications in terms of polymerization time is ascribed to the varying chain assembly due to nonelectrostatic effects, as recently described.41 Furthermore, WCA hysteresis, which was also calculated and can be found in Figure S5, shows a general increase after modification, further confirming the abovementioned conclusions. The IR and generally lower receding contact angles confirm a clear change in the surface chemistry of the PDMS substrate and corroborate the facilitation of the grafting and quaternization of DMAEMA.
Biofilm Formation
Contact-active antimicrobial surfaces are of great interest in the biomedical field as a means of creating constant sterile surfaces without the release of additives. These coatings, which can help protect against bacterial colonization and thereby prevent infection, contain polymeric quaternary ammonium and have been proven to induce a bactericidal effect through electrostatic interaction with the cell membrane of the bacteria. Although many studies have demonstrated this effect, our fundamental understanding of the structure–property relationship between the bactericidal properties and the chemical nature of the grafted polymer still requires further research. To aid in this investigation, a crystal violet assay was used as a way to quantify the biofilm inhibition properties of the modified surfaces against a Pseudomonas aeruginosa strain. The violet dye attaches to the DNA and proteins in cells, and the absorbance of the color can therefore be directly correlated with the number of viable bacteria found on the surface. The bacteriological results are shown in Figure 5.
Figure 5.
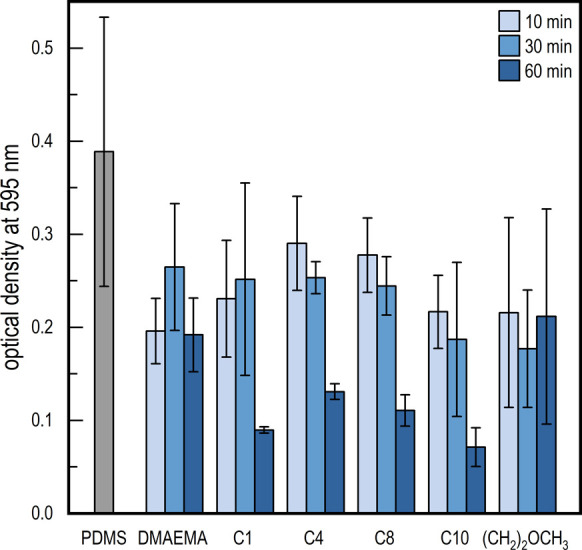
Crystal violet assay of PDMAEMA-functionalized PDMS. OD measured at 595 nm represents the growth of bacteria (P. aeruginosa) on the respective surfaces, where polymerization time can be confirmed as a key factor in reducing growth.
At first, no significant differences in optical density (OD) could be found when comparing the pristine PDMS with modified samples at either 10 or 30 min polymerization times. However, at 60 min, a significantly lower OD was observed for DMAEMA C1, DMAEMA C4, DMAEMA C8, and DMAEMA C10. Several studies have shown that the charge density for these types of surfaces needs to be above a certain threshold to engender antimicrobial activity.22−24 The longer polymer chains could similarly help increase both charge density as well as system mobility, thus increasing their bactericidal effect. On the other hand, this effect was not seen for DMAEMA, showing that positive charges are required for the bactericidal interaction with the cell membrane. DMAEMA (CH2)2OCH3, however, did not show any significant reduction in OD for any polymerization time, thereby indicating that a specific amphiphilicity needs to be present to facilitate proper integration with the membrane in order to induce the bactericidal effect.
Conclusions
Activating the PDMS surface, using direct silylation in alkaline conditions, was deemed a highly effective, simple, and efficient method. By employing this simple approach, further grafting of the surface using ATRP is an easy option for a broad range of products prepared in PDMS across the medico-industry. The grafting of extraordinarily long 400 nm DMAEMA brushes, within short reaction times (120 min), was successfully conducted through SI-SARA-ATRP by utilizing Na2SO3 as a novel reducing agent that effectively protected the Cu catalyst against oxidation, allowing the reaction to be conducted in a completely open atmosphere on a 96-well screening platform. Furthermore, parallel synthesis on the platform allowed for the investigation of the structure–property relationship between the antimicrobial activities of various brush lengths, as well as post-functionalization via N-alkylation, to create positively charged quaternary ammonium. The activity depended highly on the chain length of the polymer brush in order to obtain a sufficiently high charge density. Therefore, significantly lower numbers of viable bacteria were only seen for DMAEMA C1, C4, C8, and C10 at a 60 min polymerization time. No apparent reduction was observed for DMAEMA and DMAEMA (CH2)2OCH3, even at the longest grafting times, showing that not only charge density but also the amphiphilicity is important in facilitating optimal bactericidal properties.
Experimental Section
Materials
All chemicals and solvents were of analytical grade and used without further purification, unless stated otherwise. Inhibitors were removed from the monomers prior to use by passing them through a short plug flow column of basic alumina.
Characterization
WCA measurements were conducted on a DataPhysics OCA 20. Initially, a drop of 4 μL was placed on the surface with the needle inside, and the drop was then expanded and retracted. All WCAs reported are an average of three measurements. FT-IR spectroscopy was conducted on an attenuated total reflection (ATR)-FT-IR spectrometer (Thermo iS50 with a built-in diamond ATR with a resolution of 4 cm–1) to confirm the presence of functional groups. All spectra were normalized to a reference Si–O–Si stretch at 1007 cm–1. Absorbance measurements and UV–vis spectroscopy were carried out on a VICTOR X3 2030 multilabel reader (PerkinElmer). Brush thickness on the silicon wafers was measured using variable angle ellipsometry (M-2000, J.A. Woollam Co., Inc.) and conducted in air at room temperature over a wavelength range between 250 and 1000 nm and at six different angles of incidence (50, 55, 60, 65, 70, and 75°). Analysis of the ellipsometric data was done using the instrument software (CompleteEASE, J.A. Woollam Co., Inc.). The optical model comprised a thick Si substrate, an intermediate layer with a thickness of 1 nm, a SiO2 layer, and a transparent polymer layer. The thickness of the silicon oxide layer was determined in air before surface functionalization. The refractive index of the polymer layer is described using the Cauchy relation (n = A + B/λ2). Therefore, the model comprised a total of three free parameters (thickness, A, and B of the polymer layer). The average and standard deviations of the thickness were obtained by measuring at three different points of the sample.
Preparation of Silicon Wafers
Thermally oxidized silicon wafers with a SiO2 layer (WaferNet, USA) were used. Prior to functionalization, the wafer was rinsed with acetone, ethanol, and deionized water three times. The surface was subsequently plasma-cleaned (Harrick Plasma) for 3 min in 500 mTorr water vapor in order to produce hydroxyl groups. Thereafter, the wafer was left for 24 h in a desiccator together with a 4 mL solution of APTES in toluene (volume ratio: 1:1). The wafer was then rinsed with toluene, ethanol, and acetone and dried under a stream of argon. To graft the initiator onto the surface, the functionalized surfaces were placed into a mixture of dichloromethane (DCM) (20 mL) and triethylamine (1.3 mL) at 0 °C, following which 2-bromoisobutyrylbromide (1 mL) was added dropwise. The reaction mixture was stirred at room temperature for 12 h and rinsed with DCM, acetone, and EtOH three times and then dried under argon. The grafted initiator surfaces were used immediately after preparation for the polymerization stage.
SI-SARA-ATRP on Silicon Wafer
To a 40 mL vial, 2.6 mg Cu(II)Cl2, 8.14 μL PMDETA, 3.54 mL DMAEMA, and 9.45 mL MeOH/H2O (1:1, v/v) were added and mixed via magnetic stirring. A metal wire was fixed to a clamp, which was then fastened to the top of the silicon wafer and transferred to the vial. The metal wire was punched through a septum, the system was sealed, and the air evacuated and replaced with nitrogen three times. In a 4 mL vial, 9.45 mg of Na2SO3 was dissolved in 1 mL H2O and then 1 mL MeOH was added, following which the mixture was transferred to the reaction vial via a nitrogen-filled syringe to initiate polymerization. The total volume used was 15 mL with a final concentration of Cu(II)Cl2: 1.3 mM, PMDETA: 2.6 mM, Na2SO3: 5 mM, and DMAEMA: 1.4 M. To sample different DMAEMA brush length reaction times, the depth of the wafer was controlled by moving the sample out of the reaction mixture, using the attached metal wire, after 10, 30, 60, 90, and 120 min. The wafer was then briefly washed in pristine solvent (MeOH/H2O), water, and EtOH and dried in flowing air.
PDMS Preparation
An Elastollan RT 625 PDMS film was prepared in a mold consisting of an aluminum frame placed on top of a steel support. Approximately, 13 g of a 9:1 w/w part A to B mixture of Elastollan RT 625 was added to a mixing cup and placed in a speed mixer at 2700 rpm for 3 min before being poured into the mold. The PDMS was cured at 100 °C for 60 min and left to cool to room temperature.
Attachment of the ATRP Initiator
In a 250 mL beaker, KOH (1.700 g, 0.030 mol) was dissolved in MeOH (155 mL), following which CPTS (15.6 mL, 0.064 mol) was added. The solution was transferred to a Petri dish (d = 21 cm) along with the PDMS film and left for 6 h at 50 °C under magnetic stirring. After the reaction, the film was washed by being placed in a 250 mL beaker containing DCM for 10 min under magnetic stirring at room temperature. The film was then similarly washed in MeOH for 30 min under magnetic stirring.
SI-SARA-ATRP on the Screening Platform
The CPTS-modified PDMS film was mounted onto the screening platform as described in our previous work.35 To each well, 20 μL of solvent (MeOH/H2O, 1:1, v/v), 40 μL of DMAEMA (2.8 M in MeOH/H2O, 1:1, v/v), and 10 μL of Cu(II)Cl2/PMDETA (10.4 mM/20.8 mM in MeOH/H2O, 1:1, v/v) were added. To initiate polymerization, 10 μL of 40 mM Na2SO3 solution in MeOH/H2O, 1:1, v/v was transferred to each well. To obtain different chain lengths of DMAEMA, the reaction was terminated by removing the reaction mixture via a multi-pipette and by replacing it with a solvent. The wells were then filled with the solvent (MeOH/H2O, 1:1, v/v) and agitated at 200 rpm for 15 min, and the cleaning step was repeated with water. The setup was dried overnight in a vacuum oven at room temperature.
Quaternization of DMAEMA
To predetermined wells, 80 μL of 3 M stock solution of either iodomethane, 1-bromobutane, or 2-bromoethyl methyl ether in MeOH or 1-bromooctane or 1-bromodecane in MeOH/tetrahydrofuran (9:1, v/v) was added. The wells were covered with parafilm to avoid evaporation and were heated to 40 °C while being agitated overnight at 200 rpm. After the reaction was completed, the wells were emptied and subsequently filled with MeOH and agitated at 200 rpm for 15 min. The cleaning step was repeated with water, and the final modified PDMS was stored in wet conditions in a fridge at 5 °C.
Microtiter Dish Biofilm Formation Assay
P. aeruginosa ATCC 27853 was cultivated in 4 mL Luria Broth medium (Sigma-Aldrich, USA) at 37 °C in a cultivation incubator shaking overnight at 200 rpm. The P. aeruginosa culture suspension was adjusted to an OD OD600 of 0.05. A volume of 100 μL of the P. aeruginosa suspension was inoculated in the wells of the modified PDMS microplate system. The plates were then covered with an air-permeable sealing tape and placed in a plastic bag containing a moist napkin in order to maintain air humidity and avoid evaporation of the cultivation medium from the wells. The plates were then incubated at 37 °C for 20 h. Similar to the procedure described by O’Toole (2011),42 the planktonic P. aeruginosa cells were removed by discarding the culture, and the wells were washed with 120 μL of phosphate-buffered saline (PBS) buffer to remove any excess culture and loosely adhered cells. The plates were vigorously tapped on a tissue paper to remove excess buffer from the microtiter dish plates. A volume of 100 μL 0.01% crystal violet dye was added to each well, and the plates were incubated at room temperature for 15 min to stain the adherent bacteria. The crystal violet solution was discarded, and the wells were washed four times with 120 μL PBS buffer; excess liquid was removed each time by vigorously tapping the plate on tissue paper. A volume of 90 μL 99% ethanol was added to each well and mixed through pipetting to solubilize the crystal violet dye. Subsequently, 65 μL of the well content was transferred to a 96-well flat-bottom TPP plate (Techno Plastic Products AG, Switzerland), and absorbance was measured at 590 nm using a VICTOR X3 2030 multilabel reader (PerkinElmer). The assay was performed on duplicate PDMS plates, on which each modification was represented three times.
Acknowledgments
The work was financially supported by Innovation Fund Denmark and the Danish Council for Independent Research through grant no. DFF-7017-00109.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01611.
ARGET kinetics, FT-IR spectra, and WCA hysteresis (PDF)
Author Contributions
The manuscript was written through contributions by all authors, each of whom gave their approval to the final version. C.A. and L.Z. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Nicolson P. C.; Vogt J. Soft contact lens polymers: An evolution. Biomaterials 2001, 22, 3273–3283. 10.1016/S0142-9612(01)00165-X. [DOI] [PubMed] [Google Scholar]
- Lin C.-H.; Yeh Y.-H.; Lin W.-C.; Yang M.-C. Novel silicone hydrogel based on PDMS and PEGMA for contact lens application. Colloids Surf., B 2014, 123, 986–994. 10.1016/j.colsurfb.2014.10.053. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Moon J.-H.; Kim J. H.; Jeong S. M.; Lee S.-H. Flexible, stretchable and implantable PDMS encapsulated cable for implantable medical device. Biomed. Eng. Lett. 2011, 1, 199–203. 10.1007/s13534-011-0033-8. [DOI] [Google Scholar]
- Zhou C.; Wu Y.; Thappeta K. R. V.; Subramanian J. T. L.; Pranantyo D.; Kang E.-T.; et al. In Vivo Anti-Biofilm and Anti-Bacterial Non-Leachable Coating Thermally Polymerized on Cylindrical Catheter. ACS Appl. Mater. Interfaces 2017, 9, 36269–36280. 10.1021/acsami.7b07053. [DOI] [PubMed] [Google Scholar]
- Fujii T. PDMS-based microfluidic devices for biomedical applications. Microelectron. Eng. 2002, 61–62, 907–914. 10.1016/S0167-9317(02)00494-X. [DOI] [Google Scholar]
- Abbasi F.; Mirzadeh H.; Simjoo M. Hydrophilic interpenetrating polymer networks of poly(dimethyl siloxane) (PDMS) as biomaterial for cochlear implants. J. Biomater. Sci., Polym. Ed. 2006, 17, 341–355. 10.1163/156856206775997287. [DOI] [PubMed] [Google Scholar]
- Kawun P.; Leahy S.; Lai Y. A thin PDMS nozzle/diffuser micropump for biomedical applications. Sens. Actuators, A 2016, 249, 149–154. 10.1016/j.sna.2016.08.032. [DOI] [Google Scholar]
- Fan J.; Huang J.; Yan M.; Gong Z.; Cao L.; Chen Y. Thermoplastic multifunctional polysiloxane-based materials from broad gradient-transition multiphase separation. J. Mater. Chem. A 2020, 8, 16376–16384. 10.1039/d0ta05725h. [DOI] [Google Scholar]
- Döhler D.; Kang J.; Cooper C. B.; Tok J. B.-H.; Rupp H.; Binder W. H.; et al. Tuning the Self-Healing Response of Poly(dimethylsiloxane)-Based Elastomers. ACS Appl. Polym. Mater. 2020, 2, 4127–4139. 10.1021/acsapm.0c00755. [DOI] [Google Scholar]
- Fan J.; Huang J.; Gong Z.; Cao L.; Chen Y. Toward Robust, Tough, Self-Healable Supramolecular Elastomers for Potential Application in Flexible Substrates. ACS Appl. Mater. Interfaces 2021, 13, 1135–1144. 10.1021/acsami.0c15552. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhang X.; Chen Q.; Pan Y.; Liu C.; Shen C. A simple superhydrophobic/superhydrophilic Janus-paper with enhanced biocompatibility by PDMS and candle soot coating for actuator. Chem. Eng. J. 2021, 406, 126532. 10.1016/j.cej.2020.126532. [DOI] [Google Scholar]
- Tiller J. C.Antimicrobial Surfaces. Bioactive Surfaces; Springer, 2010; pp 193–217. [Google Scholar]
- Adlhart C.; Verran J.; Azevedo N. F.; Olmez H.; Keinänen-Toivola M. M.; Gouveia I.; et al. Surface modifications for antimicrobial effects in the healthcare setting: a critical overview. J. Hosp. Infect. 2018, 99, 239–249. 10.1016/j.jhin.2018.01.018. [DOI] [PubMed] [Google Scholar]
- Kurt P.; Wood L.; Ohman D. E.; Wynne K. J. Highly Effective Contact Antimicrobial Surfaces via Polymer Surface Modifiers. Langmuir 2007, 23, 4719–4723. 10.1021/la063718m. [DOI] [PubMed] [Google Scholar]
- Tiller J. C.; Liao C.-J.; Lewis K.; Klibanov A. M. Designing surfaces that kill bacteria on contact. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 5981–5985. 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.; Tiller J. C.; Lee S. B.; Lewis K.; Klibanov A. M. Insights into bactericidal action of surface-attached poly(vinyl-N-hexylpyridinium) chains. Biotechnol. Lett. 2002, 24, 801–805. 10.1023/A:1015584423358. [DOI] [Google Scholar]
- Lewis K.; Klibanov A. M. Surpassing nature: Rational design of sterile-surface materials. Trends Biotechnol. 2005, 23, 343–348. 10.1016/j.tibtech.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lee S. B.; Koepsel R. R.; Morley S. W.; Matyjaszewski K.; Sun Y.; Russell A. J. Permanent, nonleaching antibacterial surfaces, 1. Synthesis by atom transfer radical polymerization. Biomacromolecules 2004, 5, 877–882. 10.1021/bm034352k. [DOI] [PubMed] [Google Scholar]
- Huang J.; Murata H.; Koepsel R. R.; Russell A. J.; Matyjaszewski K. Antibacterial polypropylene via surface-initiated atom transfer radical polymerization. Biomacromolecules 2007, 8, 1396–1399. 10.1021/bm061236j. [DOI] [PubMed] [Google Scholar]
- Dong H.; Huang J.; Koepsel R. R.; Ye P.; Russell A. J.; Matyjaszewski K. Recyclable antibacterial magnetic nanoparticles grafted with quaternized poly(2-(dimethylamino)ethyl methacrylate) brushes. Biomacromolecules 2011, 12, 1305–1311. 10.1021/bm200031v. [DOI] [PubMed] [Google Scholar]
- Tu Q.; Wang J.-C.; Liu R.; He J.; Zhang Y.; Shen S.; et al. Antifouling properties of poly(dimethylsiloxane) surfaces modified with quaternized poly(dimethylaminoethyl methacrylate). Colloids Surf., B 2013, 102, 361–370. 10.1016/j.colsurfb.2012.08.033. [DOI] [PubMed] [Google Scholar]
- Murata H.; Koepsel R. R.; Matyjaszewski K.; Russell A. J. Permanent, non-leaching antibacterial surfaces-2: How high density cationic surfaces kill bacterial cells. Biomaterials 2007, 28, 4870–4879. 10.1016/j.biomaterials.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Huang J.; Koepsel R. R.; Murata H.; Wu W.; Lee S. B.; Kowalewski T.; et al. Nonleaching antibacterial glass surfaces via “grafting onto”: The effect of the number of quaternary ammonium groups on biocidal activity. Langmuir 2008, 24, 6785–6795. 10.1021/la8003933. [DOI] [PubMed] [Google Scholar]
- Kügler R.; Bouloussa O.; Rondelez F. Evidence of a charge-density threshold for optimum efficiency of biocidal cationic surfaces. Microbiology 2005, 151, 1341–1348. 10.1099/mic.0.27526-0. [DOI] [PubMed] [Google Scholar]
- Khabibullin A.; Mastan E.; Matyjaszewski K.; Zhu S.. Surface-Initiated Atom Transfer Radical Polymerization. Controlled Radical Polymerization at and from Solid Surfaces; Springer, 2015; pp 29–76. [Google Scholar]
- Dong L.; Liu X.; Xiong Z.; Sheng D.; Lin C.; Zhou Y.; et al. Preparation of UV-Blocking Poly(vinylidene fluoride) Films through SI-AGET ATRP Using a Colorless Polydopamine Initiator Layer. Ind. Eng. Chem. Res. 2018, 57, 12662–12669. 10.1021/acs.iecr.8b02373. [DOI] [Google Scholar]
- Matyjaszewski K.; Dong H.; Jakubowski W.; Pietrasik J.; Kusumo A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. 10.1021/la063402e. [DOI] [PubMed] [Google Scholar]
- Maaz M.; Elzein T.; Bejjani A.; Barroca-Aubry N.; Lepoittevin B.; Dragoe D.; et al. Surface initiated supplemental activator and reducing agent atom transfer radical polymerization (SI-SARA-ATRP) of 4-vinylpyridine on poly(ethylene terephthalate). J. Colloid Interface Sci. 2017, 500, 69–78. 10.1016/j.jcis.2017.03.115. [DOI] [PubMed] [Google Scholar]
- Konkolewicz D.; Krys P.; Góis J. R.; Mendonça P. V.; Zhong M.; Wang Y.; et al. Aqueous RDRP in the presence of Cu0: The exceptional activity of CuI confirms the SARA ATRP mechanism. Macromolecules 2014, 47, 560–570. 10.1021/ma4022983. [DOI] [Google Scholar]
- Konkolewicz D.; Wang Y.; Zhong M.; Krys P.; Isse A. A.; Gennaro A.; et al. Reversible-deactivation radical polymerization in the presence of metallic copper. A critical assessment of the SARA ATRP and SET-LRP mechanisms. Macromolecules 2013, 46, 8749–8772. 10.1021/ma401243k. [DOI] [Google Scholar]
- Dong H.; Matyjaszewski K. ARGET ATRP of 2-(Dimethylamino)ethyl Methacrylate as an Intrinsic Reducing Agent. Macromolecules 2008, 41, 6868–6870. 10.1021/ma8017553. [DOI] [Google Scholar]
- Laopa P. S.; Vilaivan T.; Hoven V. P. Positively charged polymer brush-functionalized filter paper for DNA sequence determination following Dot blot hybridization employing a pyrrolidinyl peptide nucleic acid probe. Analyst 2013, 138, 269–277. 10.1039/C2AN36133G. [DOI] [PubMed] [Google Scholar]
- Gao G.; Ko F.; Kadla J. F. Synthesis of Noble Monometal and Bimetal-Modified Lignin Nanofibers and Carbon Nanofibers Through Surface-Grafted Poly(2-(Dimethylamino)Ethyl Methacrylate) Brushes. Macromol. Mater. Eng. 2015, 300, 836–847. 10.1002/mame.201500006. [DOI] [Google Scholar]
- Dunderdale G. J.; Urata C.; Miranda D. F.; Hozumi A. Large-scale and environmentally friendly synthesis of pH-responsive oil-repellent polymer brush surfaces under ambient conditions. ACS Appl. Mater. Interfaces 2014, 6, 11864–11868. 10.1021/am503931j. [DOI] [PubMed] [Google Scholar]
- Andersen C.; Madsen N. J.; Daugaard A. E. Screening Platform for Identification of Suitable Monomer Mixtures Able To Form Thin-Film Coatings on Polyurethanes by UV-Initiated Free Radical Polymerization. ACS Appl. Polym. Mater. 2019, 1, 3295–3303. 10.1021/acsapm.9b00744. [DOI] [Google Scholar]
- Zhang J.; Chen Y.; Brook M. A. Facile Functionalization of PDMS Elastomer Surfaces Using Thiol–Ene Click Chemistry. Langmuir 2013, 29, 12432–12442. 10.1021/la403425d. [DOI] [PubMed] [Google Scholar]
- Ionov L.; Synytska A.; Diez S. Temperature-induced size-control of bioactive surface patterns. Adv. Funct. Mater. 2008, 18, 1501–1508. 10.1002/adfm.200800017. [DOI] [Google Scholar]
- Synytska A.; Svetushkina E.; Puretskiy N.; Stoychev G.; Berger S.; Ionov L.; et al. Biocompatible polymeric materials with switchable adhesion properties. Soft Matter 2010, 6, 5907–5914. 10.1039/c0sm00414f. [DOI] [Google Scholar]
- Bhut B. V.; Conrad K. A.; Husson S. M. Preparation of high-performance membrane adsorbers by surface-initiated AGET ATRP in the presence of dissolved oxygen and low catalyst concentration. J. Membr. Sci. 2012, 390–391, 43–47. 10.1016/j.memsci.2011.10.057. [DOI] [Google Scholar]
- Meincken M.; Berhane T. A.; Mallon P. E. Tracking the hydrophobicity recovery of PDMS compounds using the adhesive force determined by AFM force distance measurements. Polymer 2005, 46, 203–208. 10.1016/j.polymer.2004.11.012. [DOI] [Google Scholar]
- Ehtiati K.; Moghaddam S. Z.; Daugaard A. E.; Thormann E. Crucial Nonelectrostatic Effects on Polyelectrolyte Brush Behavior. Macromolecules 2021, 54, 3388–3394. 10.1021/acs.macromol.0c02526. [DOI] [Google Scholar]
- O’Toole G. A. Microtiter Dish Biofilm Formation Assay. J. Visualized Exp. 2011, 47, e2437. 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.