

Abstract
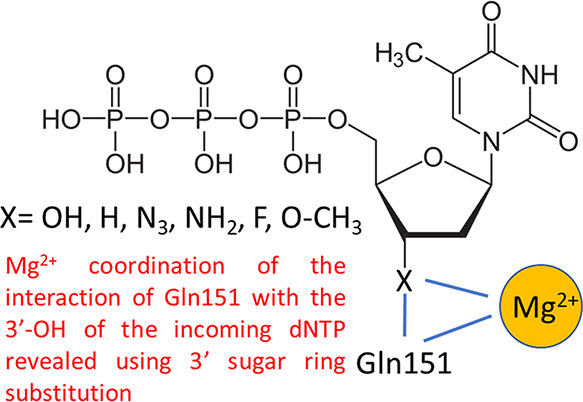
Deoxythymidine triphosphate analogues with various 3′ substituents in the sugar ring (−OH (dTTP)), −H, −N3, −NH2, −F, −O–CH3, no group (2′,3′-didehydro-2′,3′-dideoxythymidine triphosphate (d4TTP)), and those retaining the 3′-OH but with 4′ additions (4′-C-methyl, 4′-C-ethyl) or sugar ring modifications (d-carba dTTP) were evaluated using pre-steady-state kinetics in low (0.5 mM) and high (6 mM) Mg2+ with HIV reverse transcriptase (RT). Analogues showed diminished observed incorporation rate constants (kobs) compared to dTTP ranging from about 2-fold (3′-H, −N3, and d4TTP with high Mg2+) to >10-fold (3′-NH2 and 3′-F with low Mg2+), while 3′-O-CH3 dTTP incorporated much slower than other analogues. Illustrating the importance of interactions between Mg2+ and the 3′-OH, kobs using 5 μM dTTP and 0.5 mM Mg2+ was only modestly slower (1.6-fold) than with 6 mM Mg2+, while analogues with 3′ alterations incorporated 2.8–5.1-fold slower in 0.5 mM Mg2+. In contrast, 4′-C-methyl and d-carba dTTP, which retain the 3′-OH, were not significantly affected by Mg2+. Consistent with these results, analogues with 3′ modifications were better inhibitors in 6 versus 0.5 mM Mg2+. Equilibrium dissociation constant (KD) and maximum incorporation rate (kpol) determinations for dTTP and analogues lacking a 3′-OH indicated that low Mg2+ caused a several-fold greater reduction in kpol with the analogues but did not significantly affect KD, results consistent with a role for 3′-OH/Mg2+ interactions in catalysis rather than nucleotide binding. Overall, results emphasize the importance of previously unreported interactions between Mg2+ and the 3′-OH of the incoming nucleotide and suggest that inhibitors with 3′-OH groups may have advantages in low free Mg2+ in physiological settings.
Introduction
Nucleoside reverse transcriptase inhibitors (NRTIs) are a hallmark of antiretroviral therapy (ART). All NRTIs currently approved for ART are “chain terminators” and either lack a hydroxyl group at the 3′ ribose position or have a substituted sugar or other group replacing ribose. In all cases, there is no group present for addition of the next base by reverse transcriptase (RT) (reviewed in refs (1−3)). A second class of emerging NRTIs referred to as “translocation inhibitors” retain the 3′-OH but contain modifications at other positions that dramatically slow the addition of the next base.4−7 The first approved HIV drug, azidothymidine (also referred to as zidovudine, AZT, or ZDV), is essentially dTTP with the 3′-OH replaced by an azido group. Dideoxy drugs replace the 3′-OH with H including dideoxyinosine (also referred to as didanosine or ddI) and dideoxycytidine (a formerly approved HIV drug also referred to as zalcitabine or ddC). Other drugs remove both the 3′-OH and a hydrogen atom from the 3′ position by addition of a carbon double bond to the ribose ring (2′,3′-didehydro-3′-deoxythimidine) (also referred to as stavudine or d4T) and the guanosine analogue abacavir (ABC). Finally, some approved drugs substantially modify or remove the ribose sugar including lamivudine (3TC), emtricitabine (FTC), and tenofovir disoproxil fumarate (TDF). Despite the success of drugs lacking a 3′-OH, for some translocation inhibitors, this group is pivotal in promoting the phosphorylation of the prodrug in cells.8
Others have examined the effect of different groups at the 3′ positions of thymidine and adenosine analogues for inhibition of HIV RT.9,10 For thymidine, inhibition assays using steady-state kinetics and measuring incorporation on poly(rA)-oligo(dT10) indicated that substitutions of fluorine (F), amino (NH2), azido (N3), and hydrogen (H) at the 3′ position were all effective inhibitors of dTTP incorporation with modest overall differences, with 3′-F and 3′-N3 being the most and least effective, respectively.9 Despite this, only the 3′-N3 derivative (marketed as AZT) has been an effective HIV drug. Drug effectiveness is complicated by several factors including stability, uptake, cellular phosphorylation, target and off-target utilization, and resistance profiles, among others. Even small modification could potentially affect any of these steps. The F and H 3′ substitutions (as well as several other changes at other positions) on dATP were also tested for anti-HIV activity and cell toxicity.10 Both analogues had lower cytotoxicity than AZT; however, they also had much lower potencies. Still, the selectivity index for 3′-H (ddATP) was only about 4.5-fold lower than that for AZT suggesting that it could be an effective drug.
Previous results indicated that the level of Mg2+ in in vitro reactions can affect the fidelity of HIV RT, efficiency of reverse transcription, and the potency of both NRTIs and nonnucleoside reverse transcriptase inhibitors (NNRTIs). Fidelity increased significantly11,12 in reactions performed in low Mg2+ (0.25–0.5 mM) that more closely mimics cellular free Mg2+ conditions,13−17 in comparison to the high Mg2+ (∼6 mM) typically used in in vitro reactions with RT. The efficiency of reverse transcription, as judged by the length of products produced in primer extension reactions, was greater with physiological Mg2+ concentrations.18 The effect on NRTIs and NNRTIs was opposite, with the former being less effective in low Mg2+ and the latter being more effective.18,19
In this report, we used pre-steady-state kinetic analysis at a fixed nucleotide concentration (5 μM) that approximated cellular dNTP concentrations20−22 and high (6 mM) or low (0.5 mM) Mg2+ concentrations to examine the incorporation rates of dTTP and several dTTP analogues with substitutions at the 3′ position. This included 3′-OH (dTTP), −N3 (AZT), −NH2, −F, −O–CH3, −H (ddTTP), and d4T (no group at this position). Three translocation-type dTTP analogue inhibitors (4′-C-methyl, 4′-C-ethyl, and d-carba dTTP) that retained the 3′-OH were also tested. Determinations of kpol and KD were also performed for some analogues under pre-steady-state conditions. Overall, the results illustrate a potential benefit for NRTI drugs with 3′-OH groups, especially under physiological Mg2+ concentrations, while also uncovering a previously unreported direct or indirect interaction between the 3′-OH of incoming nucleotides and Mg2+.
Results
Substitution of the 3′-OH Differentially Affects the Incorporation Rates of dTTP Analogues
All the analogues tested, including chain terminators and translocation inhibitors, were incorporated more slowly than dTTP in both 0.5 and 6 mM Mg2+, in pre-steady-state assays with 5 μM nucleotide (Table 1, see Figure 1 for an example of an analysis). This amount was chosen as it approximately mimics the level of nucleotides in dividing T cells.20−22 The incorporation rate ordered from highest to lowest in 6 mM Mg2+ was as follows: 3′-OH > (−N3, no group (d4TTP), −H) > (4′-C-methyl, 4′-C-ethyl, −NH2) > (−F, d-carba) ≫ −O–CH3. In 0.5 mM Mg2+, the order was as follows: 3′-OH > (4′-C-methyl) > (−N3, no group, −H, 4′-C-ethyl, d-carba, −NH2) > −F ≫ −O–CH3. The 3′-O–CH3 analogue was by far the slowest, and no data could be obtained using pre-steady-state conditions. However, steady-state analysis showed that the analogue could be incorporated by RT (Figure 2). Among the chain terminating analogues, at 6 mM Mg2+, a −N3, −H, or no group at the 3′ position was most tolerated. The 3′-F was modestly slower than 3′-NH2, and the incorporation rate of 3′-O-CH3 was negligible in comparison (see above). Note that these results agree with a previous steady-state analysis of some of these analogues indicating that they were efficiently incorporated by HIV RT (see ref (9) and Introduction). However, the relative potency of the various inhibitors did not agree with our pre-steady-state analysis.
Table 1. Comparison of Pre-Steady-State Incorporation of dTTP Analogues.
| nucleotide ([Mg2+])a | kobs (s–1) ± standard deviationb | relative rate vs dTTP same [Mg2+] | 6/0.5 mM Mg2+ with the same nucleotide |
|---|---|---|---|
| dTTP (0.5 mM) | 8.6 ± 1.3 | 1 | 1.6 |
| dTTP (6 mM) | 14.1 ± 1.2 | 1 | |
| analogues with substitutions at the 3′ position (chain terminators) | |||
| ddTTP (0.5 mM) | 1.4 ± 0.7 | 0.16 | 5.1 |
| ddTTP (6 mM) | 7.2 ± 1.0 | 0.51 | |
| AZT (0.5 mM) | 2.0 ± 0.5 | 0.23 | 4.5 |
| AZT (6 mM) | 8.9 ± 1.9 | 0.63 | |
| D4T (0.5 mM) | 1.8 ± 0.7 | 0.21 | 4.6 |
| D4T (6 mM) | 8.3 ± 0.8 | 0.59 | |
| 3′-amino dTTP (0.5 mM) | 0.81 ± 0.13 | 0.094 | 3.8 |
| 3′-amino dTTP (6 mM) | 3.1 ± 0.1 | 0.22 | |
| 3′-fluoro dTTP (0.5 mM) | 0.54 ± 0.13 | 0.063 | 2.8 |
| 3′-fluoro dTTP (6 mM) | 1.5 ± 0.1 | 0.11 | |
| 3′-O-methyl dTTP (0.5 mM)c | 3.3 × 10–5± 0.6 × 10–5c | NA | 5.4 |
| 3′-O-methyl dTTP (6 mM)c | 1.8 × 10–4± 0.2 × 10–4c | NA | |
| analogues with a 3′ hydroxyl (translocation inhibitors) | |||
| 4′-C-methyl dTTP (0.5 mM) | 4.5 ± 1.4 | 0.52 | 0.88 |
| 4′-C-methyl dTTP (6 mM) | 3.9 ± 2.8 | 0.28 | |
| 4′-C-ethyl dTTP (0.5 mM) | 1.3 ± 0.1 | 0.15 | 3.1 |
| 4′-C-ethyl dTTP (6 mM) | 3.9 ± 0.4 | 0.28 | |
| d-carba dTTP (0.5 mM) | 1.1 ± 0.5 | 0.13 | 1.2 |
| d-carba dTTP (6 mM) | 1.3 ± 0.1 | 0.092 | |
All nucleotides were tested at 5 μM final concentration. The free Mg2+ concentration in reactions is listed (see Materials and Methods). The nucleic acid primer–template used was: 20-nt primer, 5′-TTCCCGTCAGCCTAGCTGAG-3′, and 36-nucleotide template, 5′-ATCGTCGTACTCAGCTAGGCTGACGGGAAATTGTAT-3′.
kobs is the observed rate constant for nucleotide addition. Results are averages from three or more independent experiments ± standard deviations. See Figure 1 for an example of an experiment.
Values for 3′-O-methyl dTTP are from steady-state experiments as the rate was too low to reliably measure by pre-steady-state methods. Units are nM/min. They cannot be directly compared to other values for kobs. Conditions were 20 nM primer–template, 20 nM RT, and 100 μM 3′-O-methyl dTTP in the same buffer as pre-steady-state reactions, and reactions were measured over 30 min.
Figure 1.

Example of pre-steady-state determination of the observed rate constant (kobs) of nucleotide addition with 5 μM dTTP or AZTTP and 0.5 or 6 mM Mg2+. (A) Assays were conducted as described in Materials and Methods with a 20-nt 5′-32P end-labeled DNA primer bound to a 36-nt DNA template using rapid quench analysis. Time points in these assays were 10, 20, 40, 80, 100, 200, and 500 ms and were conducted in duplicate. For nucleotide analogues with slower incorporation rates, longer time points were also used. Positions of the primer (P) and primer + 1 nucleotide (P+1) are shown for assays with dTTP and AZTTP at 0.5 or 6 mM Mg2+. FE, full extension; −E, no enzyme added. (B) The data was fitted to an exponential equation as described in Materials and Methods to yield kobs (s–1) used to produce the data in Table 1.
Figure 2.
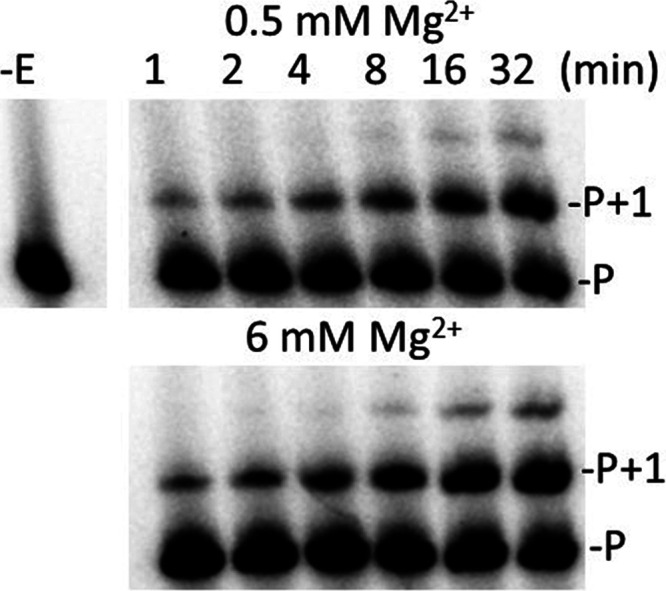
Addition of 3′-O–CH3 dTTP in steady-state assays with HIV RT with 0.5 or 6 mM Mg2+. Reactions were carried out as described in Materials and Methods using a 20-nt 5′-32P end-labeled DNA primer bound to a 36-nt DNA template. All reactions contained 100 μM 3′-O-CH3 dTTP with either 0.5 or 6 mM Mg2+. Reactions were carried out for the indicated times and resolved on a 20% denaturing gel. The proportion of the extended primer was quantified on a phosphorimager and used to determine the observed rate constant (kobs) of nucleotide addition. −E, no enzyme added; P, 5′-32P end-labeled DNA primer; P + 1, primer extended with 3′-O-CH3 dTTP.
Incorporation rates for translocation inhibitors were less dependent on the Mg2+ conditions. Rates for the 4′-C-methyl and 4′-C-ethyl derivatives were similar in 6 mM Mg2+, and both were about 3-fold faster than d-carba dTTP. In 0.5 mM Mg2+, only the incorporation rate for 4′-C-ethyl dTTP declined significantly.
Effect of Lower Mg2+ on Incorporation Is Largely Independent of the Type of 3′ Substitution
All analogues that did not have a 3′-OH were incorporated more slowly in 0.5 than 6 mM Mg2+. The magnitudes of the decreases were in a range between 2.8-fold and 5.1-fold (Table 1), with 3′-F and 3′-H (ddTTP) being the lowest and highest, respectively. However, the difference between the various 3′ analogues was small and, in most cases, not significant. This indicates that the low Mg2+ effect results more from the loss of the 3′-OH than a property of the substitution. The 4′-C-methyl and d-carba translocation inhibitor analogues were not strongly affected by lower Mg2+, while dTTP showed just a 1.6-fold decrease in the incorporation rate with low Mg2+ (Table 1). Curiously, 4′-C-ethyl dTTP behaved more like the chain terminators and was incorporated about 3 times more slowly in 0.5 mM Mg2+. It is notable, however, that despite possessing a 3′-OH, 4′-C-ethyl dTTP actually behaves like a chain terminator as unlike 4′-C-methyl and d-carb dTTP, a nucleotide cannot be added to this moiety (see ref (23) and see Discussion).
Analogues without a 3′-OH Demonstrate a More Profound Loss of Catalytic Efficiency in Lower Mg2+
The observed incorporation rate of a nucleotide or analogue at a particular concentration is dependent on its affinity for the enzyme and the overall rate of the catalytic steps. Several reports have shown that 3′-OH substitutions may alter both the binding affinity (i.e., equilibrium dissociation constant (KD)) for HIV RT and the maximum incorporation rate (kpol).24−28 We choose three dNTPs/analogues, dTTP, AZTTP, and d4TTP, to evaluate more thoroughly and determine both pre-steady-state KD and kpol values at 0.5 and 6 mM Mg2+. Although it would have been more complete to test all 10 analogues, a combination of limiting amounts of material and time made it more feasible to test a smaller representative set. The decision to focus on AZTTP and d4TTP was made because there is extensive literature on these compounds, and they are approved HIV drugs. The KD values for all three compounds were similar with d4TTP showing a modestly higher KD than the others at both Mg2+ concentrations (Figure 3 and Table 2). The level of Mg2+ did not strongly affect the KD value for any of the nucleotides. Values for KD at 6 mM obtained here were comparable to previous results with high Mg2+.28 As expected from the results in Table 1, changes in kpol were more pronounced, especially for the nucleotides lacking a 3′-OH (AZTTP and d4TTP). The decline in catalytic efficiency (kpol/KD), with 0.5 versus 6 mM Mg2+, was also more pronounced for these analogues. For example, AZTTP was incorporated only 1.5-fold less efficiently than dTTP in 6 mM Mg2+ but 7.2-fold less efficiently in 0.5 mM Mg2+ (Table 2). The data again illustrates that the lack of a 3′-OH has a more pronounced negative effect on incorporation in low, more physiological Mg2+, and a decrease in the observed incorporation rate (kobs) as opposed to changes in affinity (KD) for the analogues is the predominant factor.
Figure 3.

Pre-steady-state determination of the equilibrium dissociation constants (KD) and the maximum incorporation rates (kpol) for dTTP, AZTTP, and d4TTP. Assays were conducted as described in Materials and Methods with a 20-nt 5′-32P end-labeled DNA primer bound to a 36-nt DNA template using rapid quench analysis. Data points are from independent experiments at various concentrations of dTTP (A), AZTTP (B), or d4TTP (C) (see Figure 1 for an example). Points from several experiments (typically 2–3, while only 1 point was used for the highest d4TTP concentration) were plotted and fit to a hyperbolic equation for ligand binding with single site saturation to determine values as described in Materials and Methods. The indicated values for KD and kpol are derived from the curve fit program. Values are ±standard errors as determined by the program.
Table 2. Pre-Steady State Kinetic Data for HIV RT Incorporation of TTP, AZTTP, and D4TTP.
| nucleotide | [Mg2+] (mM) | kpol (s–1)a | Kd (μM)a | kpol/Kd | efficiency declineb |
|---|---|---|---|---|---|
| dTTP | 0.5 | 14.3 ± 0.8 | 3.3 ± 0.6 | 4.3 | 1 |
| AZTTP | 0.5 | 3.3 ± 0.3 | 5.5 ± 1.7 | 0.6 | 7.2 |
| d4TTP | 0.5 | 4.9 ± 0.5 | 7.6 ± 2.2 | 0.64 | 6.7 |
| dTTP | 6 | 25.0 ± 1.3 | 4.2 ± 0.7 | 6.0 | 1 |
| AZTTP | 6 | 17.0 ± 0.6 | 3.9 ± 0.6 | 3.9 | 1.5 |
| d4TTP | 6 | 22.3 ± 1.7 | 11.6 ± 2.3 | 1.9 | 3.2 |
Values were derived from graphs shown in Figure 2 and are predicted values based on curve fitting ± standard error (as determined by the fitting program). See Table 1 and Materials and Methods for a description of the primer template used.
Numbers represent the fold decline in enzyme efficiency relative to dTTP at the same Mg2+ concentration. Enzyme catalytic efficiency was taken as kpol/Kd.
Inhibition Assays on a Heteropolymeric DNA Template Are Generally Consistent with the Kinetic Analysis
To evaluate the effectiveness of the various analogues at terminating DNA synthesis, a primer extension assay was performed on a 100-nt DNA template primed with a 20-nt 5′-32P-labeled DNA primer (Figure 4). Assays contained 5 μM dCTP, dGTP, dATP, and dTTP. Analogues were included at 3 μM when present. Consistent with the 6/0.5 mM Mg2+ ratios shown in Table 1, all chain terminators and 4′-C-ethyl dTTP were less effective (as judged by a lower level of paused (terminated) products and an increase in fully extended products) in 0.5 mM Mg2+ than in 6 mM Mg2+. Among translocation inhibitors, 4′-C-methyl dTTP was essentially equivalent at both 0.5 and 6 mM Mg2+ while d-carba was a weak inhibitor under both conditions. The 3′-F analogue was the weakest inhibitor among the chain terminators and was also incorporated the slowest in pre-steady-state reactions (Table 1, note that 3′-O-CH3 produced no inhibition in this assay (data not shown) and its incorporation was not measurable in pre-steady-state reactions (see above)). However, it is important to note that the inhibition observed in these reactions is a function of both incorporation kinetics and binding affinity of the analogue for RT versus binding affinity for dTTP. Therefore, a direct correlation between the results in Table 1 and those in the inhibition assay would not necessarily be expected. Finally, results in Table 1 were performed using a single sequence position on a primer-template while the experiments in Figure 4 use a long template with positions for thymidine incorporation presented in several sequence contexts.
Figure 4.

Primer extension on a 100-nt DNA template with HIV RT. Reactions were carried out as described in Materials and Methods using a 20-nt 5′-32P end-labeled DNA primer bound to a 100-nt DNA template. All reactions contained 5 μM dNTPs and 3 μM of the indicated dTTP analogue with either 0.5 or 6 mM Mg2+. Reactions were carried out for 30 min and resolved on an 8% denaturing gel. −E, no enzyme added. The *d4TTP lane came from another gel and a separate experiment.
Discussion
In this report, several chain-terminating 3′-OH substitutions of dTTP were tested for incorporation by HIV RT. All the analogues with 3′ alterations were incorporated, and several (e.g., AZT, ddTTP, and d4TTP) were incorporated with only modestly slower rates (∼2-fold) than dTTP in 6 mM Mg2+ (Table 1). In contrast, the rate of incorporation compared to dTTP dropped to ∼4.5–16-fold slower (excluding 3′-O-CH3, which was much slower at both high and low Mg2+) in 0.5 mM Mg2+. This is consistent with previous experiments showing that NRTIs are better inhibitors with higher Mg2+ concentrations in vitro,19 an observation that was also shown for analogues tested here (Figure 4). Overall, the results support a previously unreported interaction, either direct or indirect, between Mg2+ and the 3′-OH of incoming nucleotides. Further, the results suggest that this interaction plays a role in catalysis rather than nucleotide binding affinity (see below).
The importance of the 3′-OH was supported by experiments with translocation-type inhibitors (4′-C-methyl and d-carba dTTP), which retain the 3′-OH and behaved more like dTTP in that their rates of incorporation were less affected by low Mg2+ (Table 1). However, 4′-C-ethyl dTTP was an exception, showing a 3.1-fold lower rate of incorporation in low Mg2+ than in high Mg2+. As was noted, this analogue terminates further extension by RT, in contrast to 4′-C-methyl and d-carba dTTP, which delay extension.23 This suggests that the 3′-OH may be in the wrong position for catalysis, and this position may also weaken the proposed interactions of the 3′-OH with Mg2+. The translocation inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine triphosphate (EFdATP) is currently in clinical trials. It also binds strongly to HIV RT as the ethynyl group can access a hydrophobic pocket in RT, which may stabilize binding.7,29 This and favorable kinetics lead to EFdATP showing a competitive advantage over the natural dATP nucleotide.30 EFdATP inhibition is also insensitive to Mg2+ concentration,19 and incorporated EFdA can accept additional nucleotides at a highly reduced rate.30 This suggests that translocation inhibitors with the 3′-OH positioned for nucleotide addition behave more like natural nucleotides with respect to Mg2+ interactions.
It was interesting that the 4′-C-ethyl dTTP was clearly a more effective inhibitor than 4′-C-methyl or d-carba dTTP at 6 mM Mg2+ during extension on the long template (Figure 4). Even at 0.5 mM Mg2+, where 4′-C-ethyl dTTP was incorporated much more slowly than 4′-C-methyl dTTP and at a rate similar to d-carba dTTP (Table 1), it still was modestly more effective than 4′-C-methyl dTTP and much more effective than d-carba dTTP in the template inhibition assay. This may be due to two factors. First, 4′-C-ethyl dTTP, as stated above, behaves like a chain terminator and does not allow the addition of other nucleotides after incorporation.23 Additional nucleotides can be added with delayed kinetics with 4′-C-methyl and d-carba dTTP,23,31 and for these analogues, extended products in the reactions in Figure 4 may include DNA chains containing more than one inhibitor nucleoside. Second, the KD values for 4′-C-ethyl, 4′-C-methyl, and d-carba dTTP binding to HIV RT were measured at 6 mM Mg2+ in a separate analysis on a different template (manuscript in preparation). The KD value for d-carba dTTP was ∼4–8-fold higher than the others. This helps explain why d-carba dTTP is by far the weakest inhibitor among both translocation and chain terminating inhibitors in the template extension inhibition assay (Figure 4). It binds more weakly to RT than the other translocation inhibitors, and this, coupled with a relatively low incorporation rate (Table 1), weakens inhibition.
Although positioning of the 3′-OH of an incoming nucleotide is coordinated through many interactions with HIV RT, glutamine 151 (Q151) presumably interacts directly with this group through a proposed hydrogen bond between the amide oxygen from Q151 and the 3′-OH hydrogen atom. Mutations in Q151 like Q151N (glutamine to asparagine) that disrupt this interaction weaken dNTP binding but do not significantly affect kpol.24 This indicates that Q151 likely plays a key role in stabilizing dNTP binding. Dideoxy dTTP, which lacks a 3′-OH, demonstrated weaker binding to HIV RT than dTTP. However, binding relative to dTTP improves with Q151N, which has weakened association with the 3′-OH. This again predicts a role for Q151 in stabilizing binding of dNTPs through interactions with the 3′-OH, as the advantage of a 3′-OH for binding is mitigated when Q151 is altered to a noninteracting amino acid.
High divalent cation concentrations can also improve incorporation of analogues without 3′-OH groups with other polymerases, similar to what was found here. The Klenow fragment of E. coli polymerase I (KF) discriminates strongly against ddNTPs relative to dNTPs. However, ddTTP incorporation improves in high Mg2+ showing an optimum of 25 mM, which is several-fold greater than the optimal concentration for incorporation of dTTP (∼2 mM).32 Like Q151N for HIV RT, an E710A (glutamic acid to alanine) KF mutation reduced ddNTP/dNTP discrimination, consistent with a role for this amino acid in interactions with the 3′-OH. The authors hypothesize that Mg2+ may bridge an interaction between KF E710 and the 3′-OH group of incoming nucleotides, although more complex explanations where E710 interacts with Mg2+via repositioning of other active site residues could not be ruled out by the data. It was also not clear if the Mg2+ ion in question was metal ion A and/or B, the putative metal ions involved in nucleotide catalysis at polymerase active sites, or another, as yet undescribed metal ion. In this regard, it is interesting that a third metal ion has been proposed to be involved in polymerase nucleotide catalysis and may play a role in these interactions.33−37 Interestingly, unlike Q151N, E710A had a larger effect on kcat rather than on binding, leading the authors to postulate that its interaction with the 3′-OH does not contribute to ground-state binding. Instead, the data was more consistent for E710–Mg2+ interactions having a role in the transition state occurring after nucleotide binding but before catalysis.32 Our work indicates that lowering the concentration of Mg2+ disproportionately depresses the rate of catalysis for all thymidine analogues lacking a 3′-OH without strongly affecting affinity (based on KD values) for a limited set (dTTP, AZTTP, and d4TTP) of tested nucleotides (Figure 3 and Tables 1 and 2). This is consistent with the observations with the KF and suggests that Mg2+ may interact with the 3′-OH, either directly or indirectly, in a step after nucleotide binding during the transition state or the chemistry step (the latter was ruled for the KF by demonstrating a lack of a sulfur elemental effect,32 but we did not test this possibility).
It was noteworthy that HIV RT was able to incorporate all nine analogues that were tested. Only 3′-O-CH3 dTTP showed very poor incorporation. For chain terminating analogues, there was not a clear consistent chemical property that explained the different rates of incorporation of the analogues. However, electronegative groups that, unlike a 3′-OH, could not donate a H atom for hydrogen bonding (e.g., 3′-F and 3′-O-CH3) were incorporated the slowest. Groups with potential for positive charge or hydrogen bond formation (e.g., 3′-NH2, and 3′-N3) and small groups (e.g., 3′-H (ddTTP) or d4TTP (no group)) were better substrates, although all were less efficiently incorporated than dTTP, especially in low Mg2+ (Tables 1 and 2). It is possible that 3′-NH2 and 3′-N3 groups may interact with Q151 through charge interactions or H-bonding, and this may help compensate for the loss of the 3′-OH. Both ddTTP and d4TTP could not establish these interactions, but the small size of the 3′ substitution and the noncharge nature could minimize repulsive interactions. Also, d4TTP is different from the other tested chain terminators as the 2′,3′-didehydro configuration of the sugar moiety alters the structure of the sugar. This makes it more difficult to directly compare it to the other 3′-modified analogues.
Conclusions
In conclusion, our results support a role for interactions between Mg2+ and the 3′-OH of the incoming nucleotide that enhances the rate of catalysis. Interactions between Mg2+ and the 3′-OH terminus of the growing DNA chain and the incoming nucleotide phosphate backbone have been well established and are part of polymerase dogma. In these instances, the divalent cation is directly interacting with the reacting moieties of nucleotide catalysis. Presumably, the 3′-OH of the incoming nucleotide is spatially removed from the catalytic center. This suggests that an interaction with it that enhances catalysis would likely have a stabilizing (e.g., stabilization of a transition state) rather than a chemical role. An example could be that Mg2+ effects established interactions between Q151 and the 3′-OH. This could occur directly or indirectly through positioning of the nucleotide in a manner that promotes the Q151–3′-OH interaction. Further experiments, including those with Q151 mutants and mutations at other positions that effect Mg2+ binding, will be required to better understand the mechanism behind these observations. Finally, the results demonstrate that inhibitors without a 3′-OH are likely to suffer a competitive disadvantage in the cellular environment where the concentration of free Mg2+ is relatively low. Since there are many factors determining inhibitor efficacy (see Introduction), this weakness can be overcome by other properties. This has been the case thus far for essentially all approved NRTIs. However, our results suggest that further improvements may be possible with inhibitors that retain the 3′-OH and achieve inhibition through limiting RT translocation.
Materials and Methods
Materials
3′-Fluor (F) and 3′-O-methyl (O–CH3) dTTP were synthesized by AX Molecules Inc. (San Marcos, CA). 3′-Amino (NH2) dTTP and dideoxy TTP (ddTTP) were from TriLink Bio Technologies (San Diego, CA). AZTTP and d4TTP were from Sierra Bioresearch (Tucson, AZ). 4′-C-Methyl, 4′-C-ethyl, and d-carba dTTP23,31 were a generous gift from Dr. Stephen Hughes (National Institutes of Health). All other nucleotides were from Roche Diagnostics Corporation (Indianapolis, IN). All nucleotides had purity levels >95% (as determined by mass spectroscopy and TLC analysis). T4 polynucleotide kinase (PNK) was from New England Biolabs (Ipswich, MA). Radiolabeled compounds were from PerkinElmer (Waltham, MA). DNA oligonucleotides were from Integrated DNA Technologies (IDT) (Coralville, IA). G-25 spin columns were from Harvard Apparatus (Holliston, MA). All other chemicals were obtained from Thermo Fisher Scientific (Waltham, MA), VWR International (Radnor, PA), or Sigma-Aldrich (St. Louis, MO).
Preparation of HIV Reverse Transcriptase (HIV RT)
The expression plasmids (pRT66 and pRT51) for HIV-1 RT (HXB2 sequence) were used to produce HIV RT.38 The enzyme, which is a nontagged heterodimer consisting of a 1:1 molar ratio of p66 and p51, was prepared as described.39
Preparation of Primer-Templates for Pre-Steady-State Kinetics and Inhibitor Analysis on the 100-nt Template
For pre-steady-state kinetics, the 5′-32P-labeled 20-nt primer (5′-TTCCCGTCAGCCTAGCTGAG-3′) and 36-nt template (5′-ATCGTCGTACTCAGCTAGGCTGACGGGAAATTGTAT-3′) were mixed at a molar ratio of 1:1.25 (primer/template) in a buffer containing 50 mM Tris-HCl (pH 8), 80 mM KCl, and 1 mM DTT. For inhibitor analysis, during primer extension on a long template, the 5′-32P-labeled 20-nt primer (5′-TTGTTGTCTCTTCCCCAAAC-3′) and 100-nt template (5′-TGGCCTTCCCACAAGGGAAGGCCAGGGAATTTTCTTCAGAGCAGACCAGAGCCAACAGCCCCACCAGAAGAGAGCTTCAGGTTTGGGGAAGAGACAACAA-3′) were mixed at a molar ratio of 1:1.5 (primer/template). Hybrids were formed by heating to 80 °C for 5 min and then slow cooling in a PCR machine to 4 °C.
Pre-Steady-State Kinetic Parameters for dTTP or dTTP Analogue Incorporation
Experiments were performed using a QFM-4000 from Bio-Logic Scientific Instruments. All experiments were carried out at 37 °C. For each reaction, 20 μL of solution containing 50 nM (in primer) primer–template prepared as described above (final concentration 25 nM in reactions) and 50 nM HIV-1 RT (HXB2) (final concentration 25 nM in reactions) in buffer 1 (50 mM Tris–HCl (pH 8), 80 mM KCl, 1 mM DTT, and 0.5 or 6 mM MgCl2) were loaded into one loop of the apparatus. A second loop contained 20 μL of a solution with various concentrations of dTTP or specific dTTP analogues in the same buffer. Concentrations used ranged from 1 to 100 μM (final concentrations in reaction) for experiments to determine KD and kpol (depending on the nucleotide) and were 5 μM for pre-steady-state rate constant (kobs) determinations with various analogues. For reactions with higher nucleotide concentrations, the starting Mg2+ concentration in the 0.5 mM Mg2+ reactions was adjusted to keep the free Mg2+ concentration at 0.5 mM.19 For adjustments, the dissociation constants (KD) for dNTP and Mg2+ were assumed to be the same as that of ATP (KD= 89.1 × 10–6 M).18 A third loop contained 20 μL of a solution containing 0.5 M EDTA (pH 8). Reactions were initiated by rapidly mixing the solutions in loops 1 and 2 and then terminated by mixing with loop 3 at time points ranging from 5 ms to 2 s. Specific time points depended on the incorporation rates of the individual analogues. The products were mixed with an equal volume of 2× loading buffer (90% formamide, 0.025% each bromophenol blue, and xylene cyanol) and resolved on 20% polyacrylamide-7 M urea denaturing PAGE gels as described.40 Gels were dried and then exposed to phosphorimager screens. Quantification was performed using a Fujifilm FLA7000 phosphorimager and Fuji ImageQuant software. First-order-rate constants for incorporation (kobs) were determined using SigmaPlot by plotting the concentration of the extended starting material versus time and fitting the data to a single exponential equation41
where a is the amplitude, k (kobs in this case) is the rate, and C is the end point. In some cases, especially for analogues with slower incorporation rates, the data fit better to a simpler equation
For some analogues, rate constants at different analogue concentrations were used to determine the equilibrium dissociation constant and maximum rate of nucleotide addition (KD and kpol, respectively) by plotting kobs versus the concentration of the nucleotide and fitting to a hyperbolic equation for ligand binding with single site saturation
where Bmax corresponds to kpol. Standard error values for KD and kpol were generated by the program.
Inhibitor Analysis during Primer Extension on a Long Template
Primer extension reactions were performed to study the inhibition of extension by thymidine analogues as described previously.19 Briefly, the 15 nM 5′-32P-labeled primer was hybridized with the 22.5 nM template at a molar ratio of 1:1.5 as described above. Hybrids were preincubated for 3 min at 37 °C in 8.5 μL of buffer 1, all 4 dNTPs, and 1 of the analogues (5 μM each and 3 μM, respectively (final concentrations in reactions)). Extension was initiated by adding 4 μL of HIV RT (final concentration 100 nM) in buffer 1. After extension for 30 min, the reactions were terminated by adding 12.5 μL of 2× gel loading buffer and samples were resolved on an 8% denaturing urea gel and processed as described above.
Acknowledgments
This work was supported by the National Institutes of Health [grant numbers R01AI150480]. The sponsor was not involved in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication. We thank Dr. Stephen Hughes (National Institutes of Health) for 4′-C-methyl, 4′-C-ethyl, and d-carba dTTP.
Author Present Address
§ Vigene Biosciences, Rockville, Maryland 20850, United States (C.R.D.).
The authors declare no competing financial interest.
References
- Sarafianos S. G.; Marchand B.; Das K.; Himmel D. M.; Parniak M. A.; Hughes S. H.; Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009, 385, 693–713. 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.; Marchand B.; Kirby K. A.; Michailidis E.; Sarafianos S. G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606–638. 10.3390/v2020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holec A. D.; Mandal S.; Prathipati P. K.; Destache C. J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2017, 15, 411–421. 10.2174/1570162X15666171120110145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama E. I.; Kohgo S.; Kitano K.; Machida H.; Gatanaga H.; Shigeta S.; Matsuoka M.; Ohrui H.; Mitsuya H. 4′-Ethynyl nucleoside analogs: potent inhibitors of multidrug-resistant human immunodeficiency virus variants in vitro. Antimicrob. Agents Chemother. 2001, 45, 1539–1546. 10.1128/AAC.45.5.1539-1546.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrui H.; Kohgo S.; Hayakawa H.; Kodama E.; Matsuoka M.; Nakata T.; Mitsuya H. 2’-Deoxy-4’-C-ethynyl-2-fluoroadenosine: a nucleoside reverse transcriptase inhibitor with highly potent activity against all HIV-1 strains, favorable toxic profiles and stability in plasma. Nucleosides, Nucleotides Nucleic Acids 2007, 26, 1543–1546. 10.1080/15257770701545218. [DOI] [PubMed] [Google Scholar]
- Kawamoto A.; Kodama E.; Sarafianos S. G.; Sakagami Y.; Kohgo S.; Kitano K.; Ashida N.; Iwai Y.; Hayakawa H.; Nakata H.; Mitsuya H.; Arnold E.; Matsuoka M. 2′-deoxy-4’-C-ethynyl-2-halo-adenosines active against drug-resistant human immunodeficiency virus type 1 variants. Int. J. Biochem. Cell Biol. 2008, 40, 2410–2420. 10.1016/j.biocel.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Michailidis E.; Marchand B.; Kodama E. N.; Singh K.; Matsuoka M.; Kirby K. A.; Ryan E. M.; Sawani A. M.; Nagy E.; Ashida N.; Mitsuya H.; Parniak M. A.; Sarafianos S. G. Mechanism of inhibition of HIV-1 reverse transcriptase by 4′-Ethynyl-2-fluoro-2′-deoxyadenosine triphosphate, a translocation-defective reverse transcriptase inhibitor. J. Biol. Chem. 2009, 284, 35681–35691. 10.1074/jbc.M109.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui M. A.; Hughes S. H.; Boyer P. L.; Mitsuya H.; Van Q. N.; George C.; Sarafinanos S. G.; Marquez V. E. A 4’-C-ethynyl-2′,3′-dideoxynucleoside analogue highlights the role of the 3’-OH in anti-HIV active 4’-C-ethynyl-2′-deoxy nucleosides. J. Med. Chem. 2004, 47, 5041–5048. 10.1021/jm049550o. [DOI] [PubMed] [Google Scholar]
- Cheng Y. C.; Dutschman G. E.; Bastow K. F.; Sarngadharan M. G.; Ting R. Y. Human immunodeficiency virus reverse transcriptase. General properties and its interactions with nucleoside triphosphate analogs. J. Biol. Chem. 1987, 262, 2187–2189. 10.1016/S0021-9258(18)61637-0. [DOI] [PubMed] [Google Scholar]
- Herdewijn P.; Pauwels R.; Baba M.; Balzarini J.; De Clercq E. Synthesis and anti-HIV activity of various 2′- and 3′-substituted 2′,3′-dideoxyadenosines: a structure-activity analysis. J. Med. Chem. 1987, 30, 2131–2137. 10.1021/jm00394a034. [DOI] [PubMed] [Google Scholar]
- Okano H.; Baba M.; Hidese R.; Iida K.; Li T.; Kojima K.; Takita T.; Yanagihara I.; Fujiwara S.; Yasukawa K. Accurate fidelity analysis of the reverse transcriptase by a modified next-generation sequencing. Enzyme Microb. Technol. 2018, 115, 81–85. 10.1016/j.enzmictec.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Achuthan V.; Keith B. J.; Connolly B. A.; DeStefano J. J. Human immunodeficiency virus reverse transcriptase displays dramatically higher fidelity under physiological magnesium conditions in vitro. J. Virol. 2014, 88, 8514–8527. 10.1128/JVI.00752-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout E.; Rebeille F.; Douce R.; Bligny R. Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E4560–E4567. 10.1073/pnas.1406251111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E.; Freudenrich C. C.; Levy L. A.; London R. E.; Lieberman M. Monitoring cytosolic free magnesium in cultured chicken heart cells by use of the fluorescent indicator Furaptra. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 2981–2984. 10.1073/pnas.86.8.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delva P.; Pastori C.; Degan M.; Montesi G.; Lechi A. Intralymphocyte free magnesium and plasma triglycerides. Life Sci. 1998, 62, 2231–2240. 10.1016/S0024-3205(98)00201-X. [DOI] [PubMed] [Google Scholar]
- Delva P.; Pastori C.; Degan M.; Montesi G.; Lechi A. Catecholamine-induced regulation in vitro and ex vivo of intralymphocyte ionized magnesium. J. Membr. Biol. 2004, 199, 163–171. 10.1007/s00232-004-0686-7. [DOI] [PubMed] [Google Scholar]
- Gupta R. K.; Benovic J. L.; Rose Z. B. The determination of the free magnesium level in the human red blood cell by 31P NMR. J. Biol. Chem. 1978, 253, 6172–6176. 10.1016/S0021-9258(17)34595-7. [DOI] [PubMed] [Google Scholar]
- Goldschmidt V.; Didierjean J.; Ehresmann B.; Ehresmann C.; Isel C.; Marquet R. Mg2+ dependency of HIV-1 reverse transcription, inhibition by nucleoside analogues and resistance. Nucleic Acids Res. 2006, 34, 42–52. 10.1093/nar/gkj411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achuthan V.; Singh K.; DeStefano J. J. Physiological Mg(2+) Conditions Significantly Alter the Inhibition of HIV-1 and HIV-2 Reverse Transcriptases by Nucleoside and Non-Nucleoside Inhibitors in Vitro. Biochemistry 2017, 56, 33–46. 10.1021/acs.biochem.6b00943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond T. L.; Roshal M.; Jamburuthugoda V. K.; Reynolds H. M.; Merriam A. R.; Lee K. Y.; Balakrishnan M.; Bambara R. A.; Planelles V.; Dewhurst S.; Kim B. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 2004, 279, 51545–51553. 10.1074/jbc.M408573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy E. M.; Gavegnano C.; Nguyen L.; Slater R.; Lucas A.; Fromentin E.; Schinazi R. F.; Kim B. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J. Biol. Chem. 2010, 285, 39380–39391. 10.1074/jbc.M110.178582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amie S. M.; Noble E.; Kim B. Intracellular nucleotide levels and the control of retroviral infections. Virology 2013, 436, 247–254. 10.1016/j.virol.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P. L.; Julias J. G.; Ambrose Z.; Siddiqui M. A.; Marquez V. E.; Hughes S. H. The nucleoside analogs 4’C-methyl thymidine and 4’C-ethyl thymidine block DNA synthesis by wild-type HIV-1 RT and excision proficient NRTI resistant RT variants. J. Mol. Biol. 2007, 371, 873–882. 10.1016/j.jmb.2007.05.043. [DOI] [PubMed] [Google Scholar]
- Jamburuthugoda V. K.; Guo D.; Wedekind J. E.; Kim B. Kinetic evidence for interaction of human immunodeficiency virus type 1 reverse transcriptase with the 3’-OH of the incoming dTTP substrate. Biochemistry 2005, 44, 10635–10643. 10.1021/bi050611+. [DOI] [PubMed] [Google Scholar]
- Kerr S. G.; Anderson K. S. Pre-steady-state kinetic characterization of wild type and 3′-azido-3′-deoxythymidine (AZT) resistant human immunodeficiency virus type 1 reverse transcriptase: implication of RNA directed DNA polymerization in the mechanism of AZT resistance. Biochemistry 1997, 36, 14064–14070. 10.1021/bi9713862. [DOI] [PubMed] [Google Scholar]
- Kellinger M. W.; Johnson K. A. Role of induced fit in limiting discrimination against AZT by HIV reverse transcriptase. Biochemistry 2011, 50, 5008–5015. 10.1021/bi200204m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.; Wang J.; Cheng Y.; Dutschman G. E.; Tanaka H.; Baba M.; Cheng Y.-C. Mechanism of inhibition of human immunodeficiency virus type 1 reverse transcriptase by a stavudine analogue, 4′-ethynyl stavudine triphosphate. Antimicrob. Agents Chemother. 2008, 52, 2035–2042. 10.1128/AAC.00083-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs R.; Immendorfer U.; Thrall S. H.; Wohrl B. M.; Goody R. S. Single-step kinetics of HIV-1 reverse transcriptase mutants responsible for virus resistance to nucleoside inhibitors zidovudine and 3-TC. Biochemistry 1997, 36, 10292–10300. 10.1021/bi970512z. [DOI] [PubMed] [Google Scholar]
- Salie Z. L.; Kirby K. A.; Michailidis E.; Marchand B.; Singh K.; Rohan L. C.; Kodama E. N.; Mitsuya H.; Parniak M. A.; Sarafianos S. G. Structural basis of HIV inhibition by translocation-defective RT inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA). Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9274–9279. 10.1073/pnas.1605223113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu Y.; Sohl C. D.; Mislak A. C.; Mitsuya H.; Sarafianos S. G.; Anderson K. S. Probing the molecular mechanism of action of the HIV-1 reverse transcriptase inhibitor 4′-ethynyl-2-fluoro-2′-deoxyadenosine (EFdA) using pre-steady-state kinetics. Antiviral Res. 2014, 106, 1–4. 10.1016/j.antiviral.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer P. L.; Vu B. C.; Ambrose Z.; Julias J. G.; Warnecke S.; Liao C.; Meier C.; Marquez V. E.; Hughes S. H. The nucleoside analogue D-carba T blocks HIV-1 reverse transcription. J. Med. Chem. 2009, 52, 5356–5364. 10.1021/jm801176e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astatke M.; Grindley N. D.; Joyce C. M. How E. coli DNA polymerase I (Klenow fragment) distinguishes between deoxy- and dideoxynucleotides. J. Mol. Biol. 1998, 278, 147–165. 10.1006/jmbi.1998.1672. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Yang W. Capture of a third Mg+ is essential for catalyzing DNA synthesis. Science 2016, 352, 1334–1337. 10.1126/science.aad9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Shock D. D.; Wilson S. H. Observing a DNA polymerase choose right from wrong. Cell 2013, 154, 157–168. 10.1016/j.cell.2013.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Zhao Y.; Yamagata Y.; Hua Y. J.; Yang W. Watching DNA polymerase eta make a phosphodiester bond. Nature 2012, 487, 196–201. 10.1038/nature11181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas R.; Reed A. J.; Tokarsky E. J.; Suo Z. Viewing Human DNA Polymerase beta Faithfully and Unfaithfully Bypass an Oxidative Lesion by Time-Dependent Crystallography. J. Am. Chem. Soc. 2015, 137, 5225–5230. 10.1021/jacs.5b02109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B. D.; Beard W. A.; Perera L.; Shock D. D.; Kim T.; Schlick T.; Wilson S. H. Uncovering the polymerase-induced cytotoxicity of an oxidized nucleotide. Nature 2015, 517, 635–639. 10.1038/nature13886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher R. S.; Holleschak G.; Nagy E.; Arion D.; Borkow G.; Gu Z.; Wainberg M. A.; Parniak M. A. Single-step purification of recombinant wild-type and mutant HIV-1 reverse transcriptase. Protein Expression Purif. 1996, 7, 27–32. 10.1006/prep.1996.0004. [DOI] [PubMed] [Google Scholar]
- Hou E. W.; Prasad R.; Beard W. A.; Wilson S. H. High-level expression and purification of untagged and histidine-tagged HIV-1 reverse transcriptase. Protein Expression Purif. 2004, 34, 75–86. 10.1016/j.pep.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Sambrook J.; Russell D. W.. Molecular Cloning: A Laboratory Manual. 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, 2001. [Google Scholar]
- Johnson K. A. Rapid quench kinetic analysis of polymerases, adenosinetriphosphatases, and enzyme intermediates. Methods Enzymol. 1995, 249, 38–61. 10.1016/0076-6879(95)49030-2. [DOI] [PubMed] [Google Scholar]