

Abstract
Antibody-recruiting molecules (ARMs) are one of the most promising tools to redirect the immune response towards cancer cells. In this review, we aim to highlight the recent advances in the field. We will illustrate the advantages of different ARM approaches and emphasize the importance of a multivalent presentation of the binding units.
Antibody-recruiting molecules (ARMs) are one of the most promising tools to redirect the immune response towards cancer cells.
Introduction
According to the World Health Organisation, cancer is the second leading cause of death in the world.1 Cancer treatment has long been based on the direct attack to tumors by surgery or using radiation and chemical agents. Nowadays, cancer immunotherapy is stepping into the game together with these traditional therapies which still suffer from severe limitations and side effects.2 The purpose of immunotherapy is to trigger the patient's immune system against the cancer cells in a selective manner. For this reason, monoclonal antibodies (mAb)3 have been one of the breakthrough discoveries of the last century and they achieved enormous success in the treatment of cancer. Nevertheless, these high molecular weight proteins have serious drawbacks: their production is extremely expensive,4,5 they possess poor pharmacokinetic properties and they often provoke severe side effects for patients.6 Besides mAb, pro-inflammatory cytokines, such as interferon-α and interleukin-2, have been proposed as immunotherapeutics due to their regulative activity of the cancer cell cycle.7 However, because of their modest immunotherapeutic effect, which in addition is most often offset by their cytotoxicity, cytokines are now less used compared to immune checkpoint inhibitors (against TNF-α or TGF-β for example) that proved to be effective in preventing tumorigenesis.8
More recently, synthetic chemists investigated whether they could develop fully synthetic molecules for immunotherapy. These compounds are indeed often easier to prepare than biological species, which require long and costly biotechnological processes. Moreover, unlike mAb and/or proteins, they are easy to administrate and nonimmunogenic9, hence, potentially overcoming obstacles found during the clinical trials,10 such as hypersensitivity reactions.11 With the aim to switch the healthy human serum into a cytotoxic weapon against tumors,12 Antibody Recruiting Molecules (ARMs)13 have been proposed. By virtue of a careful design, ARMs have the ability to exploit endogenous antibodies that are naturally found in the human blood stream and guide them towards cancer cells. ARMs are typically covalently linked bifunctional molecules possessing a Tumor Binding Module (TBM), interacting with cancer-specific biomarkers, and an Antibody Binding Module (ABM) aiming to bind endogenous antibodies (Fig. 1A). These modules first allow the initial formation of a ternary complex between the antibody and cancer cell (Fig. 1B) which can be subsequently recognized by different immune effectors to promote the tumor destruction by various mechanisms. In the complement-mediated cytotoxicity (CDC),14 the C1q complement protein binds to the Fc part of the antibodies and activate the complement cascade, leading to the formation of a membrane attack complex at the cancer cell surface and its lysis. On the other hand, the Fc part of the antibodies can also interact with the Fc gamma receptors (FcγR) of macrophages to elicit phagocytosis and lysosomal degradation of tumor cells (in the antibody-dependent cellular phagocytosis, ADCP).15 Finally, in the antibody-dependent cellular cytotoxicity (ADCC),16 natural killer (NK) cells similarly bind the Fc portion of the antibody to activate the tumor cell lysis. In this review, we aim at describing the structural parameters required for the development of potent ARMs against cancers. During the preparation of the present report, the De Geest group has published an excellent review on multivalent antibody-recruiting molecules17 which is fully complementary to our work.
Fig. 1. (A) ARMs are composed of two domains: TBM (red circle) and ABM (green square). Of note, these two domains are also referred to as the target-binding terminus (TBT) and the antibody-binding terminus (ABT).9,13 (B) Action mode of ARMs: (1) ARM recognition of cancer cells, antibody recruitment and formation of ternary complexes; (2) interactions of the complex with an immune actor (here an effector cell); and (3) destruction of cancer cells by immune-mediated clearance.

The design of ARMs relies on several key parameters. First, all individuals are daily exposed to non-self antigens leading to the presence of a high amount of specific IgA, IgM and/or IgG.18 These antibodies stay in the blood stream during the lifetime and can therefore be exploited in the ARM concept. For example, different studies6,19 have shown that one of the most abundant endogenous antibodies in the human serum is directed against l-rhamnose (l-Rha),20 a carbohydrate antigen found in the bacteria wall.21 In addition, Galili antigens exposed to natural microbiotic fauna22 and mammalian foodstuff23 led to the production of a large amount of anti-αGal antibodies which are notably responsible for hyperacute transplant rejection. Finally, dinitrophenol derivatives (DNP) are another class of non-self antigens responsible of the presence of anti-DNP antibodies in the human serum. One possible reason is the utilization of DNP as wood preservatives and pesticides and the dietary ingestion of proteins containing nitroaromatic amino acids formed in food during cooking.6,24,25 These different antigens therefore represent excellent binding units as ABMs. Secondly, a large variety of specific ligands for overexpressed cancer-related receptors26 has been reported for cell targeting and drug delivery.27,28 These ligands represent the simplest structural element usually chosen as the TBM to target selectively tumor cells and avoid damage to healthy tissues or cells.29 This allows ARMs to bind to the tumor surface that is originally not recognized by the endogenous human antibodies so that they induce tumor destruction by immune effectors. Alternatively, because these receptors are not always specific, it has been demonstrated that either a lipid anchor or tumor metabolism can be used for membrane labelling.30
Another crucial parameter in the design of ARMs is the architecture and geometry of the binding units.17 In particular, a multivalent display is essential to improve the interaction of ABM with antibodies. Multivalency is a well-known concept in biology that can be defined as a multiple and simultaneous noncovalent binding between two systems e.g., a receptor and a ligand.31 Its relevance was demonstrated for biological processes such as cell–cell communication, fertilization, cancer metastasis, host–pathogen recognition and signal transduction.32 Cell surface receptors, for example, tend to aggregate in clusters on the membrane upon binding, like in the case of lectins33 and integrins34 which initiate the corresponding biological effect. Unlike monovalent ligands, which have a restricted possibility of interactions, multivalent conjugates can exploit several mechanisms to interact with their receptors, as extensively addressed by L. Kiessling.35 The so-called chelate effect is found when oligomeric receptors have their binding sites occupied by multiple binding ligands, while monomeric receptors could clusterize to accommodate multivalent partners (clustering effect). Receptors could also possess secondary binding sites exploitable for binding. Finally, the rebinding or statistical association phenomena occur when multivalent ligands display high epitope concentrations leading to an increase of apparent binding affinity. Immunoglobulins are themselves subjected to multiple interactions with clustered ligands. For example, the monomeric form of IgM generally presents equilibrium dissociation constant (KD) values in the μM range for their carbohydrate epitopes,36 mostly due to the high degree of solvent exposure.37 To overcome this issue, IgM possesses the ability to pentamerize and use simultaneously ten binding sites to interact with multivalent compounds.38,39 Therefore, the design of ARMs should consider valency and ligand geometry40 as fundamental key parameters.
Targeting of the cancer cell membrane with specific ligands
The overexpression of proteins at the surface of cancer cells during neoplasia remains essential targets for diverse antitumoral therapies. Rullo et al.41 exploited the mechanism of the serine protease urokinase (uPA) involved in the tumor progression. The uPA binding to its receptor uPAR initiates proteolysis of the extracellular matrix, the spread of tumor cells and, therefore, the metastasis formation.42 After a first generation ARM-U1 compound composed of uPA itself as the TBM,43 the authors synthetized a second generation ARM possessing an analogue of IPR-803, a previously identified uPAR inhibitor. Following initial docking studies, several IPR-803 derivatives were produced and tested by ELISA assays to identify an aryl sulfonate derivative as the more suitable TBM (Table 1A). The importance of the sulfonic acid moiety as a major motif was further confirmed for the interaction with uPAR by co-crystallising the receptor and the inhibitor. The final ARM, composed of this uPAR inhibitor as the TBM and DNP as the ABM, was named ARM-U2 (Fig. 2(1)). In cellulo assays on the uPAR-expressing A172 glioblastoma cell line revealed a high cytotoxicity at 100 nM ARM concentration due to ADCP from IFN-γ-activated U937 effector cells as well an ADCC. In addition, an in vivo test was performed on DNP-immunized mice injected with melanoma B16 and uPAR, showing inhibition of the tumor progression without side effects observed with doxorubicin used as a comparative treatment.44
Examples of ARMs against cancer cells. The ABM module is represented in blue color while the TBM is in red color.

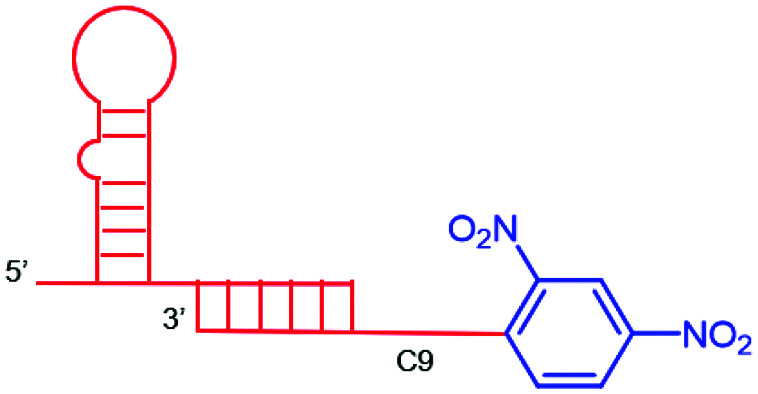


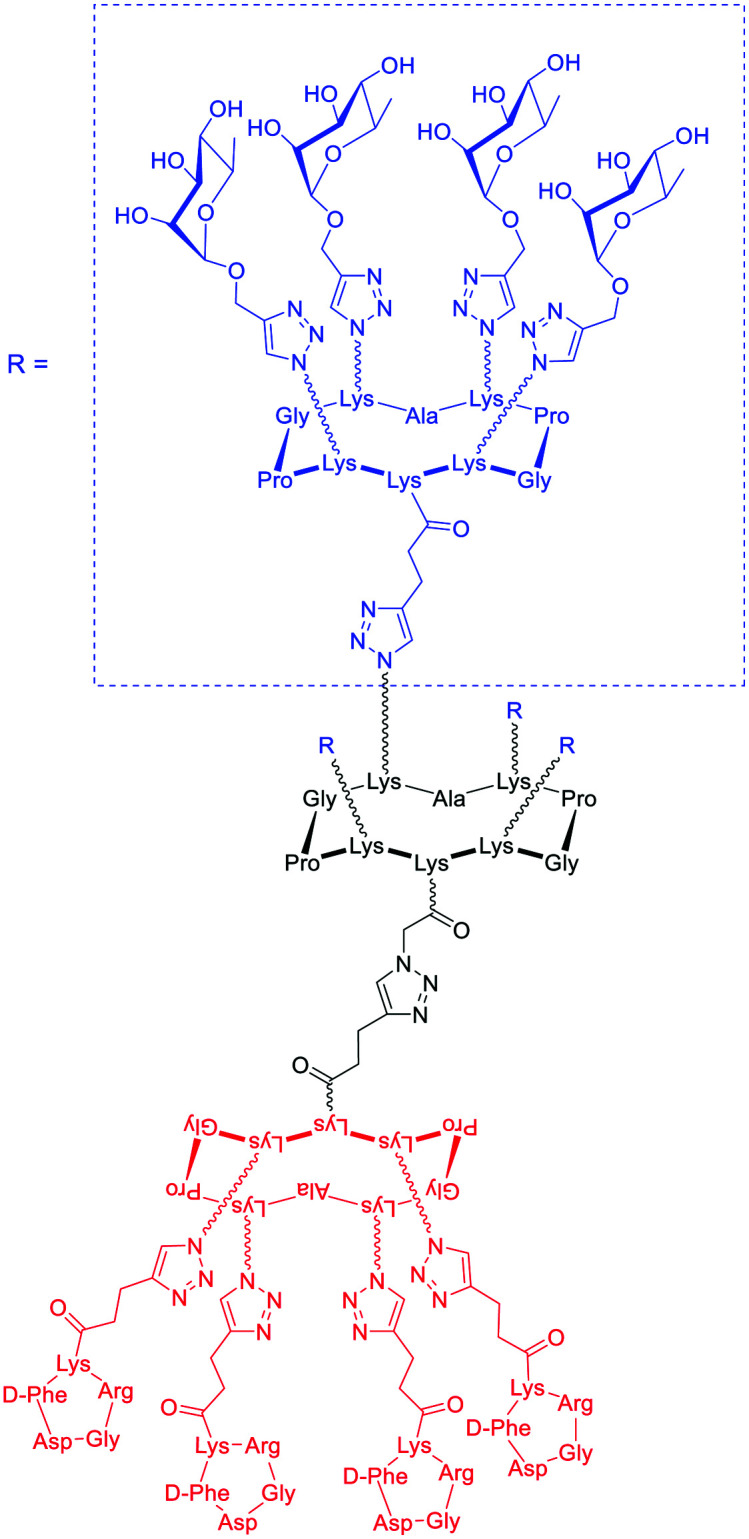
Fig. 2. Targeting of the cancer cell membrane with specific ligands: dinitrophenol (DNP) and the anti-DNP antibody (in black); l-rhamnose (l-Rha) and the anti-Rha antibody (in grey); general endogenous antibody (in red/violet). (1) Serine protease urokinase receptor (uPAR, in green); (2) vascular endothelial growth factor (VEGF) receptor (in orange); (3) folate receptor (in violet); (4) prostate-specific membrane antigen (PSMA, in red); and (5) αvβ3 integrin (in blue).

Another interesting approach involves the use of folded single-stranded nucleic acids, namely aptamers,45 as the TBM. B. Schrand et al.46 have exploited anti-vascular endothelial growth factor (VEGF)47 and anti-osteopontin (OPN) aptamers48 conjugated through hybridization via complementary sequences, with DNP as the ABM (Table 1B). VEGF and OPN are two proteins secreted by tumors which bind, respectively, to proteoglycans49 and integrins/CD44 receptors.50 In this study, immunohistochemical analysis of the H-2d Balb/c-derived 4T1 breast carcinoma-bearing mice and H-2b C57BL6 MC38 colorectal cancer-bearing mice, that were previously immunized against DNP and treated with the VEGF-DNP ARM (Fig. 2(2)), showed the accumulation of endogenous antibody anti-DNP in the proximity of the tumor. Moreover, a significant inhibition of tumor growth has been observed with the treated mice compared to the untreated control (Fig. 3A), without critical toxicity for the animal. Finally, the authors showed that the tumor inhibition was dependent on the CD4+ Th1 pro-inflammatory cells. Altogether, these results showed the interest of using aptamers as tumoral binding ligands in targeted cancer immunotherapy.
Fig. 3. (A) Tumor volume decreases after vascular endothelial growth factor (VEGF)-dinitrophenol DNP (KLH) treatment (reproduced from ref. 46 with permission from Springer Nature, copyright 2018). (B) Tumor volume decrease after Fc-ARM+ intravenous immunoglobulin (IVIG) treatment (reproduced from ref. 51 with permission from Royal Society of Chemistry, copyright 2020). (C) 4–16 ARM (right panel) better antibody recruitment compared to the 4–4 ARM (left panel, reproduced from ref. 56 with permission from John Wiley and Sons, copyright 2019).

A novel strategy was explored by Sasaki et al.51 This group designed the so-called Fc-ARM with the aim to target the largest population and isotypes of endogenous antibodies by using the ligand of the crystallizable fragment (Fc). The Fc-ARM was composed of folic acid (as the TBM) linked via a hexaethylene glycol linker to an Fc-binding cyclic peptide52,53 (as the ABM) (Table 1C and Fig. 2(3)). A preliminary surface plasmon resonance (SPR) assay proved an nM affinity between the Fc-ARM and Trastuzumab, an anti-HER2 IgG1 mAb. Subsequently, flow cytometry and fluorescence microscopy have shown the recruitment of IgG to the human ovarian carcinoma cell line IGROV-1, only in the presence of the Fc-ARM. Next, in order to assess whether Fc-ARM antibody recruitment was able to activate human NK cells and induce ADCC, IGROV-1 cells were cultivated with NK cells in the presence of the Fc-ARM and Ofatumumab, a mAb with certified ADCC ability. The released lactate dehydrogenase (LDH) was measured as the evidence of the effective killing of carcinoma cells. The same assay performed with serum only showed that the endogenous antibodies can also be recruited by the Fc-ARM and induce ADCC. The researchers went one-step further and performed in vivo assay on IGROV-1 tumor-bearing BALB/c nu/nu mice. Intravenous immunoglobulin (IVIG) was used to simulate the IgG concentration of the human blood. After Fc-ARM injections, time-course imaging in vivo showed an improvement of the blood circulation time along with the accumulation of the Fc-ARM in the tumor, without significant weight loss of the animal. Finally, the effect of the strategy on the inhibition of tumoral growth by NK activation was observed by measuring the decrease of the tumor volume (Fig. 3B), proving the therapeutic potential of the Fc-ARM approach.
More recently, the same group54 investigated the applicability of the Fc-ARM approach by targeting prostate cancer and hence obtaining a new family of molecules Fc-ARM-Ps by varying the length between the Fc-III4C peptide and the 2-[3-(1,3-dicarboxypropyl)-ureido]pentanedioic acid (DUPA) ligand for prostate-specific membrane antigen (PSMA) targeting. FACS and confocal microscopy analysis revealed that IgG were successfully recruited onto PSMA positive LNCaP cells in an Fc-ARM-Ps concentration dependent way. Next, to evaluate ADCC cytotoxicity by LDH quantification, researchers exploited Mogamulizumab (anti-CCR4) and Ofatumumab (anti-CD20) mAbs, because of their low binding towards LNCaP, together with NK cells. Surprisingly, the most effective Fc-ARM-P in recruiting antibodies failed to induce ADCC when using Ofatumumab, which contains unmodified N-glycan, while it successfully induced cytotoxicity with the defucosylated N-glycans containing Mogamulizumab. Even though Fc-ARM compounds can clearly recruit any endogenous antibody by exploiting their Fc region, further experiments are ongoing to address the unexpected binding properties.
B. Lake et al. have recently reported a novel strategy based on the covalent labelling of endogenous antibodies directly in human serum with the TBM to optimise the ternary complex formation.55 After docking analysis, a Covalent Immune Recruiter (CIR) composed of three modules has been designed: (i) DNP for recognition with anti-DNP IgG; (ii) a reactive Antibody Labelling Domain (ALD) containing acylimidazole function to label selectively anti-DNP immunoglobulins at their lysine side chain; (iii) biotin (for experiments on streptavidin coated beads) or glutamate urea as the ligand of the Prostate-Specific Membrane Antigen (PSMA), an overexpressed transmembrane protein involved in cancer cell growth/invasion, to bind selectively an antibody–TBM conjugate (CIR-Ab) to tumor cells (Table 1D and Fig. 2(4)). The covalent tagging of endogenous antibodies led to the subsequent removal of DNP after an intramolecular reaction. In most of the experiments, DNP-Gly was added in excess in order to discriminate the covalent conjugate from the non-covalent one. Preliminary biolayer interferometry (BLI) analysis with a CIR derivative and purified polyclonal anti-DNP, followed by non-reducing SDS-PAGE assay with human serum, was employed to assess the labelling selectivity and confirm the formation of the CIR-Ab conjugate. Its ability to mediate ADCP was next evaluated first on streptavidin coated beads and with human monocyte phagocytic cells. By changing the CIR-Ab concentration, a dissociation constant of ≈50 nM was determined by dual colour flow cytometry. Finally, the same experiment was performed on transfected HEK293T cells expressing high levels of PSMA, endorsing the CIR ability to covalently bind the anti-DNP antibody. This new ARM tool may also have interesting application as an immune probe to follow the ARM clearance and quantify human endogenous antibody concentration during immunotherapy.
In a recent study, ARMs displaying multiple copies of l-Rha as the ABM have been developed to evaluate the importance of multivalency for binding with Abs.56 These ABMs have been synthesized from 4- and 16-valent peptide scaffolds using click chemistry. Preliminary ELISA study and competitive assay with human serum highlighted the ability of each ABM to bind to endogenous anti-Rha IgM with a clear improvement of the structure presenting 16 Rha (Table 1E). These ABMs have been combined with tetravalent peptide cRGDfV (cRGD), a well-known binding module for the αvβ3 integrin overexpressed in diverse cancer cell lines.57–59 Two ARMs displaying 4 copies or 16 copies of Rha and a cluster of cRGD (4–4 and 4–16 ARM, respectively, Fig. 2(5)) at their opposite side have been prepared by click chemistry. Each compound has been tested against the αvβ3 integrin expressing M21 tumor cell line and incubated with 50% human serum. FACS and confocal microscopy have finally confirmed the formation of a ternary complex between Abs, ARM and M21 cells, with an increase of antibody recruitment for the 4–16 ARM compared to the 4–4 ARM (Fig. 3C). This study therefore highlights the impact of multivalency for the formation of this key complex for inducing potent antitumoral immune response.
Unspecific targeting of the cancer cell membrane
As previously discussed, most of the ARMs reported so far target receptors overexpressed on the cancer cells with specific ligands. Nevertheless, some disadvantages of this approach have to be accounted. The interaction between ARM compounds and some receptors (e.g. folate receptors60,61 and αvβ3 integrin62) can lead to the endocytosis mechanism upon binding, precluding therefore an efficient recruitment of antibodies and the subsequent clearance by the immune system.63 Moreover, the formation and stability of the ternary complex are, indeed, highly dependent on both concentrations and dissociation constants of each partner, making it difficult to determine experimental conditions for optimal efficiency.55 To circumvent these drawbacks, other studies have investigated the use of lipid anchors or membrane covalent bonds that allow the multivalent display of antibody ligands at the cell surface.
With a lipid anchor
De Coen et al.12 have proposed to anchor ARMs to membrane cells using a lipophilic module. This new ARM family named ARGPs (Antibody Recruiting GlycoPolymers) is composed of a rhamnosylated polymer, which was functionalized with cholesterylamine (CholA) (Table 2A) via amide bond formation for cell surface anchoring (Fig. 4(1)). The glycopolymer was prepared via the Reversible Addition–Fragmentation Chain-transfer (RAFT) polymerisation64 from a monomeric rhamnosyl building block using a pentafluorophenyl (PFP) ester-functionalized chain transfer agent. Insertion of the lipophilic motif into cell membranes of Jurkat T cells was performed by simple incubation, thus allowing l-rhamnose moieties to be exposed at their surface. FACS and confocal microscopy analysis have confirmed the ARM localisation at the membrane at 4 °C and at 37 °C, revealing only poor ARM internalisation. Subsequent incubation with human serum led to the recognition by IgG, with a huge enhancement of the recognition due to the multivalency (Fig. 5). This study highlights once again the importance of a multivalent rhamnose exposition to ensure an efficient antibody recruitment.
Example of ARMs using unspecific targeting of the cancer cell membrane with a lipid anchor. The ABM module is represented in blue color while the TBM is in red color.


Fig. 4. Unspecific targeting of the cancer cell membrane via lipid anchoring for local intra-tumoral administration: (1) cholesterol-based polyRha ARM: l-Rha and the anti-Rha antibody (in grey); cholesterol (in violet). (2) Dialkyl-polyDNP ARM: DNP and the anti-DNP antibody (in black); dialkyl motif (in orange).

Fig. 5. Antibody recruitment with polymers (CholA-p(RhamEAm) versus different concentrations of a single rhamnose moiety linked to CholA (CholA-SQA-Rham), with CholA-p(GalEAm) used as the negative control (reproduced from ref. 12 with permission from American Chemical Society, copyright 2020).

Similarly, A. Uvyn et al.65 have used a lipid anchor as the TBM and have investigated the multivalent presentation of DNP as the ABM. By using the RAFT polymerisation64 with pentafluorophenyl acrylate (PFPA), they were able to control the degree of polymerisation (DP) of the backbone and reach 100 DP, with a maximum of 10 DNP motif substitution per 100 PFPA units (Fig. 4(2)). Firstly, the interaction between the biotinylated polyDNP, coated on a streptavidin surface, and the commercial rabbit anti-DNP polyclonal antibody was quantified by BLI. A 4-log increase of the KD of a decameric PFPA substitution in comparison to the corresponding monomeric was observed. Subsequently, the in cellulo assay on CT26 mouse colon cancer cells using the ARM functionalized with three different lipids proved the effectiveness of the multivalent approach. Cholesterol and monoalkyl- or dialkyl-lipid modules were tested in order to determine the optimal valency of the membrane anchoring and displaying. According to flow cytometry and confocal microscopy analysis, the dialkyl-lipid structure proved to be the more efficient one. The authors next investigated the ability of the different lipid–polymer ARMs to bind polyclonal anti-DNP antibodies to the cell surface. According to the binding of the lipid–polymer module itself, the dialkyl-lipid containing ARM was identified as the most proficient in anti-DNP antibody recruitment (Table 2B). The lower efficiencies observed with the other constructs were attributed to the low binding affinity for the monoalkyl lipid structure, while the cholesterol was probably too quickly endocytosed by the cell membrane. The benefit of multivalent presentation of DNP motifs was also clearly shown since a monovalent DNP-dialkyl lipid used as a control conjugate revealed a very poor anti-DNP recruiting capacity. After proving that the dialkyl-polyDNP ARM was able to bind antibodies even after quite a few days, its ADCP ability was tested against A431 human squamous carcinoma cells, incubated with monoclonal human anti-DNP IgG1 and macrophages derived by CD14+ blood monocytes. The dialkyl-polyDNP ARM was shown to have an ADCP potency comparable to that of the mAb Cetuximab, proving the clinical potential of the receptor-independent approach. However, no experimental data with human serum have been reported so far. Despite obtaining the positive results, summarised in Table 2, it has to be noted that the lipid anchoring strategy is not selective towards cancer cells. For this reason, the only possible utilization of this approach has to be the ARM local intra-tumoral administration.
With covalent linkage
A variety of studies has shown that cell metabolism can be exploited efficiently to expose chemical moieties covalently at the cell surface glycocalyx. This approach was used successfully by Lin et al.66 who have exploited the sialylation pathways67 to incorporate DNP on the membrane of different tumors. It is well-known that tumors take advantage of their surface hypersialylation to hijack the immune system and facilitate the metastasis process.68 By using conjugated sialic acid (DNP-Sia, Table 3A) with hydrophobic group substitution in position C-969,70 for optimal cellular uptake, this group showed the selective incorporation of DNP-Sia into glycoconjugates decorating the cancer cell surface (Fig. 6A). Confocal microscopy, flow cytometry and Western blotting studies with B16F10 melanoma cells have revealed an over-exposition of DNP-Sia at the cell surface. Analysis of the DNP-Sia uptake and membrane presentation with other cancer cell lines, such as Raw 264.7 macrophages, HeLa, L929, SMMC-7721 and U87-MG, confirmed the potency of this approach. Subsequently, C57BL/6 mice pre-immunized with a carrier protein functionalized with DNP (DNP-KLH71) and inoculated with B16F10 cells showed a 60–90% tumor suppression 6–9 days after intravenous injection of DNP-Sia (Fig. 7A). Moreover, when the DNP-KLH-immunized mice were treated with DNP-Sia-displaying B16F10 cells, the metastasis size in the lung was found to be smaller than that in the untreated control. To assess the selectivity of the treatment for cancer cells, DNP-Sia was administrated to C57BL/6 mice bearing subcutaneous B16F10 cells. Fluorescence immunostaining reported a strong fluorescence surrounding cancer cells and the minor fluorescence found in the heart and kidneys decreased 4h after the injection, proving a selective DNP-Sia incorporation by the cancer cells. Finally, no toxicity was observed in mice even at high concentration of DNP-Sia.
Chemical structures for binding to the cancer cell membrane with covalent linkage. The ABM module is represented in blue color while the TBM for cell metabolism is in red color.
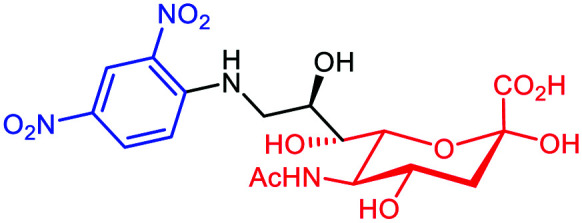
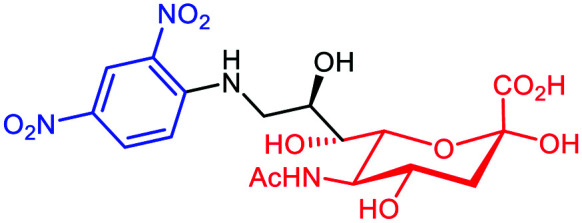
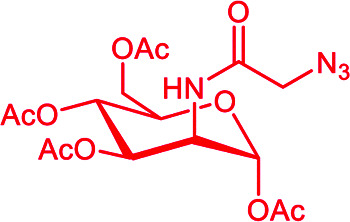
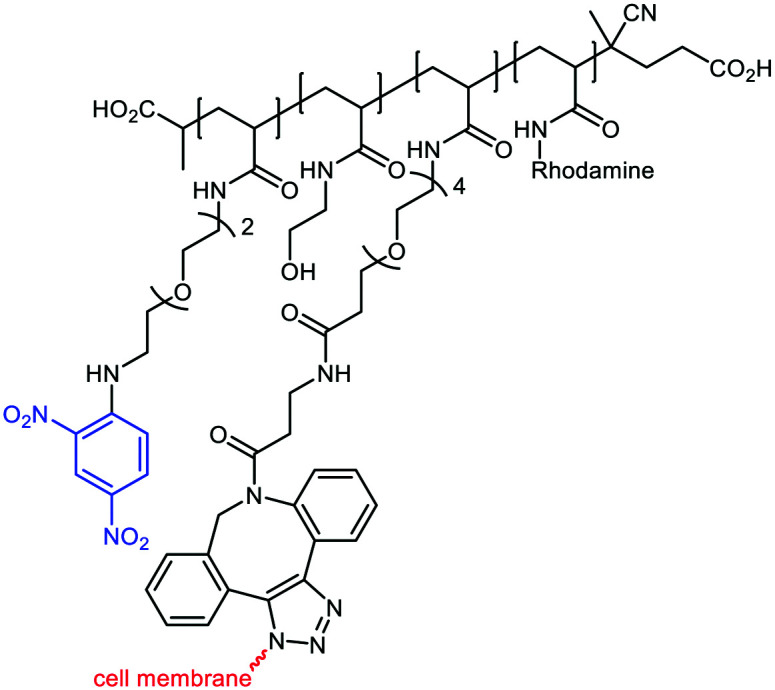
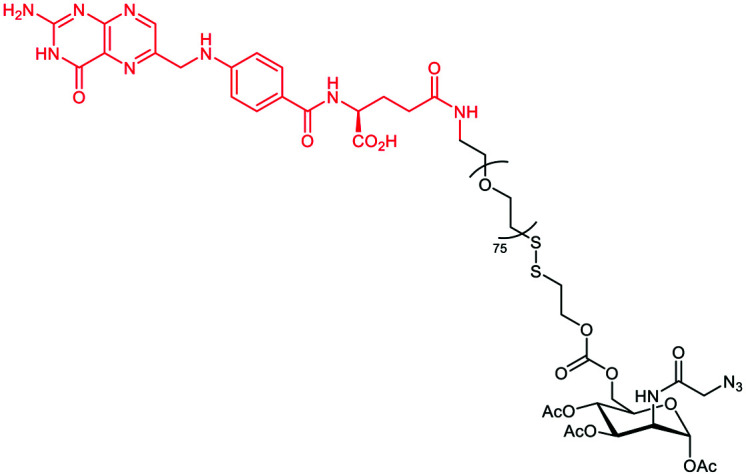

Fig. 6. Binding to the cancer cell membrane via covalent linkage: (A) DNP (black sphere) conjugated to sialic acid (in pink) and the consequent carbohydrate sialylation. (B) N-Azidoacetylmannosamine (Ac3ManN3) is converted intracellularly into sialic acid and exposed at the cell surface for a subsequent SPACC reaction with an alkyne-DNP polymer (in black). (C) Ac4Man (in green) conjugated to folic acid (f-PEG-Mz, in violet) is converted intracellularly into sialic acid and exposed at the cell surface for a subsequent SPACC reaction with an alkyne-l-Rha (in grey).

Fig. 7. (A) Tumor volume comparison with and without ARM (DNP-Sia) treatment (reproduced from ref. 66 with permission from Royal Society of Chemistry, copyright 2016). (B) CDC assay with f-PEG-Mz as the ARM on different cell lines (reproduced from ref. 76 with permission from American Chemical Society, copyright 2018).

Although promising, this approach based on sialic acid is often associated with several synthetic difficulties inherent to this sugar unit.72 For this reason, several groups used the well-known protocol for the metabolic labelling of cells (either ex vivo or in vivo) using N-azidoacetylmannosamine derivatives such as Ac3ManN3 or Ac4ManN3 as the precursor of sialic acid.73 Its incorporation by the glycan biosynthetic machinery in the glycocalyx leads to the expression of the azido group at the cell surface which can be further functionalized with any probe molecule by using the Strain-Promoted Azide–Alkyne Cycloaddition (SPAAC). For example, Uvyn et al.74 investigated the covalent tagging by using Ac4ManN3 followed by the orthogonal with the multivalent DNP-cyclooctyne derivative (Table 3B and Fig. 6B). High multivalency was reached by using 13 SPAAC reactive groups within the multivalent backbone obtained by RAFT polymerisation (DP = 100).65 Dibenzocyclooctyne (DBCO) was chosen as cyclooctyne because of its high selective reactivity with azide groups without requiring a cytotoxic copper catalyst.75 Confocal microscopy and flow cytometry experiments revealed that 25 μM of Ac4ManN3 was the optimal concentration of azide-sugar for an efficient SPAAC conjugation with DBCO-polymers to Jurkat T cells. The ultimate ARM was then composed of a DP 100 polyPFPA backbone functionalised with 5 units of DBCO and 5 units of DNP. Again, confocal microscopy and flow cytometry revealed that the DNP valency was suitable to bind anti-DNP antibodies to the targeted cell surface. Lastly, a 4T1 cancer-3D spherical structure was employed to mimic a solid tumor. The experiment was performed under the same aforementioned conditions and confocal imaging shows an effective recruitment of the anti-DNP antibody at the tumor surface.
Li et al.76 extended the previous strategy to label cancer cells selectively towards normal cells. In their work, the targeting strategy profited from the overexpression of the folate receptor (FR) on the cancer cell surface to deliver Ac3ManN3 using a cleavable folate linker (Table 3C and Fig. 6C). The researchers hence conjugated Ac3ManN3 to a PEG–folic acid via a disulfide bridge that can be reduced in the intracellular environment to release the azido sugar. Prior to this, the optimal concentration of Ac3ManN3 (50 μM) and the subsequent sialic exposition were determined on the KERATIN-forming tumor cell line HeLa (KB cells). After 24 h incubation, the exposed azido-modified sialic acid was then conjugated to DBCO-Rha (50 μM) via SPAAC and flow cytometry revealed an efficient recognition by purified anti-Rha. The CDC response was next assessed by incubating the resulting Rha-labelled cells with 20% human serum supplemented with 1% (v/v) human complement. 50% of the cells previously treated with Ac3ManN3 ended up dead. Finally, Ac3ManN3 conjugated with PEG–folic acid (f-PEG-Mz) was tested. FR up-regulated (+) KB cells were incubated with f-PEG-Mz and the subsequent addition of fluorescent DBCO revealed an effective cleavage and the selective release of the azido sugar inside the cancer cells. FR+ KB cells were finally incubated with f-PEG-Mz for 48 h and cultured with DBCO-Rha and anti-Rha. Flow cytometry revealed an intense binding compared to other cancer cell lines that do not overexpress the folate receptor (KB, MCF7, and HEK-293). CDC assay was performed under the same conditions and gave the same percentage of killed cancer cells (Fig. 7B). In addition, the ADCP mechanism was also investigated and 30% of phagocytosis was observed.
Conclusions and perspectives
The different ARM strategies presented here share the same common objective of directing immune effectors against cancer cells by using synthetic molecules to mediate their selective clearance. ARMs clearly represent promising alternatives in antitumoral immunotherapy where biotechnology still remains the major exploited strategy. However, if all the reported approaches have been revealed to be highly efficient to reach these challenges, they still suffer from limitations. The first ‘non-covalent ARMs’ aim to directly target membrane features of tumors to form a ternary complex leading to the activation of the immune system by several mechanisms. However, the internalisation that may occur during the binding process with non-covalent ARMs and their rapid in vivo clearance could represent major hurdles for therapeutic applications. To circumvent these problems, the non-specific lipid anchoring and the covalent labelling of the cancer cell membrane have been proposed and might certainly represent promising strategies to be explored in the future. In addition, regardless of the approach, the multimeric antigen presentation was shown to be a key structural requirement for effective immune activation by means of multivalent interactions. Due to the diversity in antibody specificity and isotype distributions found in humans, the development of libraries of multivalent ABMs (i.e. with different geometries, linkers, and decorated with various haptens) will be crucial to achieve high affinity for these antibodies and thus cover a large population of patients.
To generalize the utilisation of ARMs in cancer immunotherapy, selectivity issues also appear as an important problem to be addressed. To this aim, the panel of TBMs has to be expanded with new selective ligands to the cancer cell surface. Moreover, the linker between both the ABM and TBM in ARM constructions represents another important parameter to be optimized to increase ARM binding to cell surface receptors and antibodies. Finally, another highly complementary approach derived from the ARM concept and based on constructs possessing both the TBM and effector cell activating modules was shown to elicit selective cancer cell phagocytosis.77 Such an approach could be used as an alternative to the ARM for individuals having low antibody abundance. Surfing on the recent progress in the synthesis of well-defined multitopic supramolecular systems, there is no doubt that new generation of molecules embedded with selective recognition and optimized immunomodulation properties will be reported in near future.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by CNRS, Université Grenoble Alpes, ICMG FR 2607, the French ANR project Glyco@Alps (ANR-15-IDEX-02), Labex ARCANE and CBH-EUR-GS (ANR-17-EURE-0003). O. R. acknowledges the European Research Council Consolidator Grant “LEGO” (647938) for S. A.
References
- WHO, Cancer. Available from:https://www.who.int/news-room/fact-sheets/detail/cancer
- Farkona S. Diamandis E. P. Blasutig I. M. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73. doi: 10.1186/s12916-016-0623-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/27151159
- Diamond B. Scharff M. D. Monoclonal Antibodies. J. Am. Med. Assoc. 1982;248(23):3165–3169. doi: 10.1001/jama.1982.03330230065038. doi: 10.1001/jama.1982.03330230065038. [DOI] [PubMed] [Google Scholar]
- Scott A. M. Allison J. P. Wolchok J. D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/22896759
- Guo C. Manjili M. H. Subjeck J. R. Sarkar D. Fisher P. B. Wang X.-Y. Therapeutic cancer vaccines: Past, Present, and future. Adv. Cancer Res. 2013;119:421–475. doi: 10.1016/B978-0-12-407190-2.00007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/23870514
- Jakobsche C. E. Parker C. G. Tao R. N. Kolesnikova M. D. Douglass Jr EF. Spiegel D. A. Exploring binding and effector functions of natural human antibodies using synthetic immunomodulators. ACS Chem. Biol. 2013;8(11):2404–2411. doi: 10.1021/cb4004942. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/24053626
- Lee S. J. Chinen J. Kavanaugh A. Immunomodulator therapy: Monoclonal antibodies, fusion proteins, cytokines, and immunoglobulins. J. Allergy Clin. Immunol. 2010;125(2):S314–S323. doi: 10.1016/j.jaci.2009.08.018. [DOI] [PubMed] [Google Scholar]
- Berraondo P. Sanmamed M. F. Ochoa M. C. Etxeberria I. Aznar M. A. Pérez-Gracia J. L. et al., Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120(1):6–15. doi: 10.1038/s41416-018-0328-y. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEnaney P. J. Parker C. G. Zhang A. X. Antibody-recruiting small molecules: Synthetic constructs as immunotherapeutics. Annu. Rep. Med. Chem. 2017;50:481–518. [Google Scholar]; . Available from: https://www.sciencedirect.com/science/article/pii/S0065774317300118
- Kudrin A. Overview of cancer vaccines: Considerations for development. Hum. Vaccin. Immunother. 2012;8(9):1335–1353. doi: 10.4161/hv.20518. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/22894970
- Johnson D. E. Biotherapeutics: Challenges and opportunities for predictive toxicology of monoclonal antibodies. Int. J. Mol. Sci. 2018;19(11):3685. doi: 10.3390/ijms19113685. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/30469350
- De Coen R. Nuhn L. Perera C. Arista-Romero M. Risseeuw M. D. P. Freyn A. et al., Synthetic rhamnose glycopolymer cell-surface receptor for endogenous antibody recruitment. Biomacromolecules. 2019;21(2):793–802. doi: 10.1021/acs.biomac.9b01483. [DOI] [PubMed] [Google Scholar]
- McEnaney P. J. Parker C. G. Zhang A. X. Spiegel D. A. Antibody-recruiting molecules: An emerging paradigm for engaging immune function in treating human disease. ACS Chem. Biol. 2012;7(7):1139–1151. doi: 10.1021/cb300119g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev M. Markiewski M. M. Targeting complement-mediated immunoregulation for cancer immunotherapy. Semin. Immunol. 2018;37:85–97. doi: 10.1016/j.smim.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/29454575
- Pollara J. Tay M. Z. Antibody-dependent cellular phagocytosis in antiviral immune responses. Front. Immunol. 2019;10:332. doi: 10.3389/fimmu.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez Román V. R., Murray J. C. and Weiner L. M., in Chapter 1 – antibody-dependent cellular cytotoxicity (ADCC), ed. M. E. Ackerman and F. B. T.-A. F. Nimmerjahn, Academic Press, Boston, 2014, pp. 1–27. Available from: http://www.sciencedirect.com/science/article/pii/B9780123948021000017 [Google Scholar]
- Uvyn A. de Geest B. Multivalent antibody-recruiting macromolecules: Linking increased binding affinities with enhanced innate immune killing. ChemBioChem. 2020;21:3036. doi: 10.1002/cbic.202000261. doi: 10.1002/cbic.202000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. Ding J. L. Natural antibodies bridge innate and adaptive immunity. J. Immunol. 2015;194(1):13–20. doi: 10.4049/jimmunol.1400844. [DOI] [PubMed] [Google Scholar]; . Available from: http://www.jimmunol.org/content/194/1/13.abstract
- Wang X. Chen H. Chiodo F. Tefsen B. Detection of human IgM and IgG antibodies by means of galactofuranose-coated and rhamnose-coated gold nanoparticles. Matters. 2019;5(8):e201908000004. [Google Scholar]
- Hossain M. K. Vartak A. Karmakar P. Sucheck S. J. Wall K. A. Augmenting vaccine immunogenicity through the use of natural human anti-rhamnose antibodies. ACS Chem. Biol. 2018;13(8):2130–2142. doi: 10.1021/acschembio.8b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistou M.-Y. Sutcliffe I. C. van Sorge N. M. Bacterial glycobiology: Rhamnose-containing cell wall polysaccharides in Gram-positive bacteria. FEMS Microbiol. Rev. 2016;40(4):464–479. doi: 10.1093/femsre/fuw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galili U. Mandrell R. E. Hamadeh R. M. Shohet S. B. Griffiss J. M. Interaction between human natural anti-alpha-galactosyl immunoglobulin G and bacteria of the human flora. Infect. Immun. 1988;56(7):1730–1737. doi: 10.1128/iai.56.7.1730-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger C. Fischer J. Wölbing F. Biedermann T. Role and mechanism of galactose-alpha-1, 3-galactose in the elicitation of delayed anaphylactic reactions to red meat. Curr. Allergy Asthma Rep. 2019;19(1):3. doi: 10.1007/s11882-019-0835-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju K.-S. Parales R. E. Nitroaromatic compounds, from synthesis to biodegradation. Microbiol. Mol. Biol. Rev. 2010;74(2):250–272. doi: 10.1128/MMBR.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper C. S. and Grover P. L., Chemical carcinogenesis and mutagenesis II, Springer, 1990 [Google Scholar]
- Santos R. Ursu O. Gaulton A. Bento A. P. Donadi R. S. Bologa C. G. et al., A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery. 2017;16(1):19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/27910877
- Jiang Z. Guan J. Qian J. Zhan C. Peptide ligand-mediated targeted drug delivery of nanomedicines. Biomater. Sci. 2019;7(2):461–471. doi: 10.1039/C8BM01340C. [DOI] [PubMed] [Google Scholar]
- Sis M. J. Webber M. J. Drug delivery with designed peptide assemblies. Trends Pharmacol. Sci. 2019;40(10):747–762. doi: 10.1016/j.tips.2019.08.003. doi: 10.1016/j.tips.2019.08.003. [DOI] [PubMed] [Google Scholar]
- Porkolab V. Pifferi C. Sutkeviciute I. Ordanini S. Taouai M. Thepaut M. et al., Development of c-type lectin oriented surfaces for high avidity glycoconjugates: Towards mimicking multivalent interactions on the cell surface. bioRxiv. 2019:780452. doi: 10.1039/d0ob00781a. [DOI] [PubMed] [Google Scholar]; . Available from: http://biorxiv.org/content/early/2019/09/24/780452.abstract
- Luengo A. Gui D. Y. Vander Heiden M. G. Targeting metabolism for cancer therapy. Cell. Chem. Biol. 2017;24(9):1161–1180. doi: 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/28938091
- Tjandra K. C. Thordarson P. Multivalency in drug delivery–when is it too much of a good thing? Bioconjugate Chem. 2019;30(3):503–514. doi: 10.1021/acs.bioconjchem.8b00804. doi: 10.1021/acs.bioconjchem.8b00804. [DOI] [PubMed] [Google Scholar]
- Mammen M. Choi S.-K. Whitesides G. M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem., Int. Ed. 1998;37(20):2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO. [DOI] [PubMed] [Google Scholar]
- Monsigny M. Mayer R. Roche A.-C. Sugar-lectin interactions: Sugar clusters, lectin multivalency and avidity. Carbohydr. Lett. 2000;4:35–52. [PubMed] [Google Scholar]
- Lepzelter D. Bates O. Zaman M. Integrin clustering in two and three dimensions. Langmuir. 2012;28(12):5379–5386. doi: 10.1021/la203725a. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/22204631
- Kiessling L. L. Gestwicki J. E. Strong L. E. Synthetic multivalent ligands as probes of signal transduction. Angew. Chem., Int. Ed. 2006;45(15):2348–2368. doi: 10.1002/anie.200502794. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/16557636
- Haji-Ghassemi O. Blackler R. J. Martin Young N. Evans S. V. Antibody recognition of carbohydrate epitopes. Glycobiology. 2015;25(9):920–952. doi: 10.1093/glycob/cwv037. doi: 10.1093/glycob/cwv037. [DOI] [PubMed] [Google Scholar]
- Tommasone S. Allabush F. Tagger Y. K. Norman J. Köpf M. Tucker J. H. R. et al., The challenges of glycan recognition with natural and artificial receptors. Chem. Soc. Rev. 2019;48(22):5488–5505. doi: 10.1039/C8CS00768C. [DOI] [PubMed] [Google Scholar]
- Janeway C. A., Capra J. D., Travers P. and Walport M., Immunobiology: the immune system in health and disease, 1999 [Google Scholar]
- Hiramoto E. Tsutsumi A. Suzuki R. Matsuoka S. Arai S. Kikkawa M. et al., The IgM pentamer is an asymmetric pentagon with an open groove that binds the AIM protein. Sci. Adv. 2018;4(10):eaau1199. doi: 10.1126/sciadv.aau1199. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: http://advances.sciencemag.org/content/4/10/eaau1199.abstract
- Johannssen T. Lepenies B. Glycan-based cell targeting to modulate immune responses. Trends Biotechnol. 2018;35(4):334–346. doi: 10.1016/j.tibtech.2016.10.002. doi: 10.1016/j.tibtech.2016.10.002. [DOI] [PubMed] [Google Scholar]
- Rullo A. F. Fitzgerald K. J. Muthusamy V. Liu M. Yuan C. Huang M. et al., Re-engineering the immune response to metastatic cancer: Antibody-recruiting small molecules targeting the urokinase receptor. Angew. Chem., Int. Ed. 2016;55(11):3642–3646. doi: 10.1002/anie.201510866. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/26879524
- Mahmood N. Mihalcioiu C. Rabbani S. A. Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front. Oncol. 2018;8:24. doi: 10.3389/fonc.2018.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/29484286
- Jakobsche C. E. McEnaney P. J. Zhang A. X. Spiegel D. A. Reprogramming urokinase into an antibody-recruiting anticancer agent. ACS Chem. Biol. 2012;7(2):316–321. doi: 10.1021/cb200374e. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/22098560
- Shafei A. El-Bakly W. Sobhy A. Wagdy O. Reda A. Aboelenin O. et al., A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017;95:1209–1218. doi: 10.1016/j.biopha.2017.09.059. [DOI] [PubMed] [Google Scholar]; . Available from: http://www.sciencedirect.com/science/article/pii/S0753332217330998
- Ma H. Liu J. Ali M. M. Mahmood M. A. I. Labanieh L. Lu M. et al., Nucleic acid aptamers in cancer research, diagnosis and therapy. Chem. Soc. Rev. 2015;44(5):1240–1256. doi: 10.1039/C4CS00357H. [DOI] [PubMed] [Google Scholar]
- Schrand B. Clark E. Levay A. Capote A. R. Martinez O. Brenneman R. et al., Hapten-mediated recruitment of polyclonal antibodies to tumors engenders antitumor immunity. Nat. Commun. 2018;9(1):3348. doi: 10.1038/s41467-018-05566-x. doi: 10.1038/s41467-018-05566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo C. A. Nery A. A. Alves J. M. Martins A. H. Ulrich H. Development of the anti-VEGF aptamer to a therapeutic agent for clinical ophthalmology. Clin. Ophthalmol. 2007;1(4):393–402. [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/19668516
- Mi Z. Guo H. Russell M. B. Liu Y. Sullenger B. A. Kuo P. C. RNA aptamer blockade of osteopontin inhibits growth and metastasis of MDA-MB231 breast cancer cells. Mol. Ther. 2009;17(1):153–161. doi: 10.1038/mt.2008.235. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/18985031
- Johns S. C. Yin X. Jeltsch M. Bishop J. R. Schuksz M. El Ghazal R. et al., Functional Importance of a Proteoglycan Coreceptor in Pathologic Lymphangiogenesis. Circ. Res. 2016;119(2):210–221. doi: 10.1161/CIRCRESAHA.116.308504. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/27225479
- Wei R. Wong J. P. C. Kwok H. F. Osteopontin – a promising biomarker for cancer therapy. J. Cancer. 2017;8(12):2173–2183. doi: 10.7150/jca.20480. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/28819419
- Sasaki K. Harada M. Miyashita Y. Tagawa H. Kishimura A. Mori T. et al., Fc-binding antibody-recruiting molecules exploit endogenous antibodies for anti-tumor immune responses. Chem. Sci. 2020;11(12):3208–3214. doi: 10.1039/D0SC00017E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K. Miyashita Y. Asai D. Funamoto D. Sato K. Yamaguchi Y. et al., A peptide inhibitor of antibody-dependent cell-mediated cytotoxicity against EGFR/folate receptor-α double positive cells. MedChemCommun. 2018;9(5):783–788. doi: 10.1039/C8MD00010G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. Ultsch M. H. Wells J. A. Convergent solutions to binding at a protein-protein interface. Science. 2000;287(5456):1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Sasaki K. Harada M. Yoshikawa T. Tagawa H. Harada Y. Yonemitsu Y. et al., Fc-binding antibody-recruiting molecules targeting prostate-specific membrane antigen: defucosylation of antibody for efficacy improvement. ChemBioChem. 2021;22:496. doi: 10.1002/cbic.202000577. doi: 10.1002/cbic.202000577. [DOI] [PubMed] [Google Scholar]
- Lake B. Serniuck N. Kapcan E. Wang A. Rullo F. A covalent immune recruiters: Tools to gain chemical control over immune recognition. ACS Chem. Biol. 2020;15(4):1089–1095. doi: 10.1021/acschembio.0c00112. doi: 10.1021/acschembio.0c00112. [DOI] [PubMed] [Google Scholar]
- Liet B. Laigre E. Goyard D. Todaro B. Tiertant C. Boturyn D. et al., Multifunctional glycoconjugates for recruiting natural antibodies against cancer cells. Chem. – Eur. J. 2019;25(68):15508–15515. doi: 10.1002/chem.201903327. doi: 10.1002/chem.201903327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierschbacher M. D. Ruoslahti E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature. 1984;309(5963):30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- Nieberler M. Reuning U. Reichart F. Notni J. Wester H.-J. Schwaiger M. et al., Exploring the role of RGD-recognizing integrins in cancer. Cancer. 2017;9(9):116. doi: 10.3390/cancers9090116. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/28869579
- Boturyn D. Coll J.-L. Garanger E. Favrot M.-C. Dumy P. Template assembled cyclopeptides as multimeric system for integrin targeting and endocytosis. J. Am. Chem. Soc. 2004;126(18):5730–5739. doi: 10.1021/ja049926n. [DOI] [PubMed] [Google Scholar]
- Sabharanjak S. Mayor S. Folate receptor endocytosis and trafficking. Adv. Drug Delivery Rev. 2004;56(8):1099–1109. doi: 10.1016/j.addr.2004.01.010. [DOI] [PubMed] [Google Scholar]; . Available from: http://www.sciencedirect.com/science/article/pii/S0169409X04000158
- Bandara N. A. Hansen M. J. Low P. S. Effect of receptor occupancy on folate receptor internalization. Mol. Pharmaceutics. 2014;11(3):1007–1013. doi: 10.1021/mp400659t. doi: 10.1021/mp400659t. [DOI] [PubMed] [Google Scholar]
- Paul N. R. Jacquemet G. Caswell P. T. Endocytic trafficking of integrins in cell migration. Curr. Biol. 2015;25(22):R1092–R1105. doi: 10.1016/j.cub.2015.09.049. doi: 10.1016/j.cub.2015.09.049. [DOI] [PubMed] [Google Scholar]
- Chew H. Y. De Lima P. O. Gonzalez Cruz J. L. Banushi B. Echejoh G. Hu L. et al., Endocytosis inhibition in humans to improve responses to ADCC-mediating antibodies. Cell. 2020;180(5):895–914. doi: 10.1016/j.cell.2020.02.019. doi: 10.1016/j.cell.2020.02.019. [DOI] [PubMed] [Google Scholar]
- Lowe A. B. McCormick C. L. Reversible addition–fragmentation chain transfer (RAFT) radical polymerization and the synthesis of water-soluble (co)polymers under homogeneous conditions in organic and aqueous media. Prog. Polym. Sci. 2007;32(3):283–351. doi: 10.1016/j.progpolymsci.2006.11.003. [DOI] [Google Scholar]; . Available from: http://www.sciencedirect.com/science/article/pii/S0079670006001353
- Uvyn A. De Coen R. Gruijs M. Tuk C. W. De Vrieze J. van Egmond M. et al., Efficient innate immune killing of cancer cells triggered by cell-surface anchoring of multivalent antibody-recruiting polymers. Angew. Chem., Int. Ed. 2019;58(37):12988–12993. doi: 10.1002/anie.201905093. doi: 10.1002/anie.201905093. [DOI] [PubMed] [Google Scholar]
- Lin B. Wu X. Zhao H. Tian Y. Han J. Liu J. et al., Redirecting immunity via covalently incorporated immunogenic sialic acid on the tumor cell surface. Chem. Sci. 2016;7(6):3737–3741. doi: 10.1039/C5SC04133C. [DOI] [PMC free article] [PubMed] [Google Scholar]; . Available from: https://pubmed.ncbi.nlm.nih.gov/29997860
- Vajaria B. N. Patel K. R. Begum R. Patel P. S. Sialylation: An avenue to target cancer cells. Pathol. Oncol. Res. 2016;22(3):443–447. doi: 10.1007/s12253-015-0033-6. [DOI] [PubMed] [Google Scholar]
- Rodrigues E. Macauley M. S. Hypersialylation in cancer: Modulation of inflammation and therapeutic opportunities. Cancer. 2018;10(6):207. doi: 10.3390/cancers10060207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X. Tian Y. Yu M. Lin B. Han J. Han S. A fluorescently labelled sialic acid for high performance intraoperative tumor detection. Biomater. Sci. 2014;2(8):1120–1127. doi: 10.1039/C4BM00028E. doi: 10.1039/C4BM00028E. [DOI] [PubMed] [Google Scholar]
- Wu X. Yu M. Lin B. Xing H. Han J. Han S. A sialic acid-targeted near-infrared theranostic for signal activation based intraoperative tumor ablation. Chem. Sci. 2015;6(1):798–803. doi: 10.1039/C4SC02248C. doi: 10.1039/C4SC02248C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmers F. de Haas N. Scholzen A. Schreibelt G. Simonetti E. Eleveld M. J. et al., Monitoring of dynamic changes in Keyhole Limpet Hemocyanin (KLH)-specific B cells in KLH-vaccinated cancer patients. Sci. Rep. 2017;7(1):43486. doi: 10.1038/srep43486. doi: 10.1038/srep43486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H. Synthetic approach toward complexity of sialic acid-containing glycans. Biosci. Biotechnol. Biochem. 2015;79(3):343–346. doi: 10.1080/09168451.2014.990228. [DOI] [PubMed] [Google Scholar]
- Collins B. E. Fralich T. J. Itonori S. Ichikawa Y. Schnaar R. L. Conversion of cellular sialic acid expression from N-acetyl-to-N-glycolylneuraminic acid using a synthetic precursor, N-glycolylmannosamine pentaacetate: inhibition of myelin-associated glycoprotein binding to neural cells. Glycobiology. 2000;10(1):11–20. doi: 10.1093/glycob/10.1.11. [DOI] [PubMed] [Google Scholar]
- Uvyn A. De Coen R. De Wever O. Deswarte K. Lambrecht B. N. De Geest B. G. Cell surface clicking of antibody-recruiting polymers to metabolically azide-labeled cancer cells. Chem. Commun. 2019;55(73):10952–10955. doi: 10.1039/C9CC03379C. [DOI] [PubMed] [Google Scholar]
- Jewett J. C. Sletten E. M. Bertozzi C. R. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 2010;132(11):3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Yu B. Wang J. Zheng Y. Zhang H. Walker J. et al., Biomarker-based metabolic labeling for redirected and enhanced immune response. ACS Chem. Biol. 2018;13(6):1686–1694. doi: 10.1021/acschembio.8b00350. [DOI] [PubMed] [Google Scholar]
- McEnaney P. J. Fitzgerald K. J. Zhang A. X. Douglass E. F. Shan W. Balog A. Kolesnikova M. D. Spiegel D. A. Chemically synthesized molecules with the targeting and effector functions of antibodies. J. Am. Chem. Soc. 2014;136(52):18034–18043. doi: 10.1021/ja509513c. [DOI] [PMC free article] [PubMed] [Google Scholar]