

Abstract
The innate lymphoid cell (ILC) is a group of effector cells with diverse important cellular functions in both health and disease states. In comparison with healthy controls, there were increases in circulating ILC in SLE patients. The proportion of ILC1 significantly increased with significant decreases of ILC2 in SLE patients and ILC3 in SLE patients with moderate to severe activity. IL-12, IL-18, IL-25, IL-33, IL-23, IL-1β and IFN-γ were significantly increased in SLE patients. Moreover, IL-12, IL-18 and IL-1β but not IFN-γ correlated significantly with SLEDAI. Successful treatments rapidly reduced them and with certain normalization of the ILC subsets. In addition to increases in ILC1 numbers, ~ 80% of the ILC1 in SLE patients were positive for synthesis of IFN-γ. Plasma from SLE patients were shown to be potent in inducing ILC1. Thus, increased circulating ILC1 might contribute to the pathogenesis of SLE through mounting type 1 immune response.
Keywords: Innate lymphoid cell, Systemic lupus erythematosus, IFN-γ, Type 1 immune response
1. Introduction
The innate lymphoid cell (ILC) family is a novel group of innate effector cells which provide early and prompt defensive immune response to protect epithelial integrity and tissue immunity throughout the body [1]. They are defined by their lymphoid morphology and the lack of antigen specificity for T and B cells or myeloid and dendritic cell phenotypical markers [2]. ILCs are composed of “cytotoxic” ILCs (natural killer cells) and “helper” ILCs [3]. The latter are characterized by CD127+ phenotype and are divided into three groups including type 1, 2 and 3 ILC subsets according to the expression of cytokines and transcription factors [3].
ILCs have been proposed to be classified as the counterparts of T helper (Th) cells [2]. In this classification scheme, type 1 ILCs (ILC1) express T-bet and produce T helper 1-associated cytokine IFN-γ in response to IL-12 and IL-18 [2]. Type 2 ILCs (ILC2) express high levels of GATA3 and are able to produce Th2-associated cytokines in response to thymic stromal lymphopoietin (TSLP), IL-25 and IL-33. Type 3 ILCs (ILC3) express the transcription factor RORγt and make IL-17 [2]. Some of the ILC3 cells in the skin and in the gut also make IFN-γ and IL-22 [2].
Since their discovery, ILCs have been studied extensively [1,4,5]. In addition to contributing to multiple immune pathways, they are important in tissue homeostasis, morphogenesis, metabolism, repair and regeneration. Therefore it is not surprising that pathogenic roles of ILCs in autoimmune diseases have been explored. Because of their roles in maintaining mucosal barriers and the availability of tissue from affected patients, significant information on their roles in the pathogenesis of Crohn’s disease has been generated. In inflamed intestines of these patients, frequency of IFN-γ-producing ILC1 was much higher than that of healthy intestines and the frequency of IL-22-producing ILC3 decreased, indicating a role for ILC1 in the pathogenesis of mucosal inflammation [6]. This conclusion is congruent with the fact that anti-IL-17 therapy is not effective in the treatment of Crohn’s disease [7]. Although psoriasis has been thought to be a Th17 prototypic disease, proportions of NKp44+ ILC3 have been shown to be significantly increased in the skin lesions and peripheral blood suggesting that ILC3 cells participate in psoriasis through producing IL-22 and IL-17 [8]. For ANCA associated vasculitis, circulating ILC1 were increased with decreases in ILC2 and ILC3 and the imbalance of ILCs returned to normal when the disease was in remission [9].
In systemic lupus erythematosus (SLE), limited information is in the literature regarding the role of ILCs in its pathogenesis. In the present study, circulating ILCs subsets as defined by the current nomenclature in patients with active disease activity were interrogated and correlated with circulating cytokine levels. The results suggest that the marked increases in ILC1 in the ILCs population may contribute to the pathogenesis of SLE through the mounting of a Th1 immune response.
2. Methods
2.1. Study subjects
Forty-nine SLE patients were enrolled from the First Affiliated Hospital, Sun Yat-sen University. All SLE patients fulfilled the American College of Rheumatology criteria for the classification of SLE. SLE Disease Activity Index (SLEDAI) was calculated to classify the SLE patients into patients with inactive disease (SLEDAI: 0–4, n = 4), with mild disease activity (SLEDAI: 5–9, n = 10), with moderate disease activity (SLEDAI: 10–14, n = 16) and with severe disease activity (SLEDAI > 15, n = 19). Enrolled SLE patients included newly diagnosed patients and those experiencing relapse because of discontinuation of steroid and immunosuppressive agents for at least three months. Patients with complications such as infection or tumor were excluded. Twenty age and sex matched healthy controls were recruited. This study was approved by the Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University. Informed consents were obtained from all patients and healthy controls. The demographic and clinical features of the SLE patients and healthy controls were summarized in Supplementary Table 1.
2.2. PBMC isolation and stimulation
Blood from SLE patients and healthy controls were drawn into tubes coated with heparin. PBMCs were isolated using density-gradient centrifugation on Ficoll-Paque™ PLUS (GE Healthcare, Chicago, IL, USA). Single cell suspensions were prepared and subjected to flow cytometry or in vitro stimulation. PBMCs from 5 healthy controls were seeded in 12-well plates with a concentration of 1 × 106 cells/ml and cultured with plasma from healthy controls or SLE patients for 3 days. In separate experiments, PBMCs from 11 healthy controls and 11 SLE patients were seeded in 12-well plates with a concentration of 1 × 106 cells/ml and cultured with RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA), 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% heat-inactivated FBS (Thermo Fisher Scientific, Waltham, MA, USA). Although IL-2 itself does not activate ILC1, it enhances the activation of ILC1 by IL-12/IL-18 [6]. Fifty ng/ml recombinant human IL-2 (PeproTech, Rocky Hill, NJ, USA) was added to the cell cultures together with 50 ng/ml recombinant human IL-12 (PeproTech, Rocky Hill, NJ, USA) and 50 ng/ml recombinant human IL-18 (Invivogen, San Diego, CA, USA) for 3 days [6]. After stimulation, PBMCs were collected and stained for flow cytometric analysis. The choice of 3 days’ incubation was carried out according to the protocol of Bernink et al. [6].
2.3. Flow cytometry for ILCs
For ILCs identification, freshly isolated PBMCs were blocked with Human TruStain FcX™ (Fc Receptor Blocking Solution) (Biolegend, San Diego, CA, USA) for 15 min at room temperature. Following blocking, PBMCs were stained with Brilliant Violet 510™ anti-human CD45 antibody (Biolegend, San Diego, CA, USA), Pacific Blue™ anti-human Lineage Cocktail (CD3, CD14, CD16, CD19, CD20 and CD56) (Lin, Biolegend, San Diego, CA, USA), Alexa Fluor® 488 anti-human CD127 (IL-7Rα) antibody (Biolegend, San Diego, CA, USA), PE anti-human CD294 (CRTH2) antibody (Biolegend, San Diego, CA, USA), APC anti-human CD117 (c-kit) antibody (Biolegend, San Diego, CA, USA) and PerCP/Cy5.5 anti-human CD336 (NKp44) antibody (Biolegend, San Diego, CA, USA). For intracellular cytokine staining, in order to block cytokine secretion, Brelfeldin A solution (1000×) (Biolegend, San Diego, CA, USA) was added to freshly isolated PBMCs for 6 h before flow cytometry or stimulated PBMCs for the last 6 h. PBMCs were then collected and stained with the above antibodies and APC/Cy7 anti-human IFN-γ antibody (Biolegend, San Diego, CA, USA).
2.4. Elisa
Plasma was collected from SLE patients and healthy controls. IL-12, IL-1β, IL-25, IL-33, IL-23 and IL-18 were measured using ELISA kits (Cusabio, Wuhan, China) according to the manufacturer’s instructions. IFN-γ (Multi Sciences, Hangzhou, China) was also measured.
2.5. Statistical analysis
Statistical analysis was performed using SPSS 20.0 software. Normally distributed data were presented as mean ± SD and non-normally distributed data were presented as median ± interquartile range. Differences were determined with a two-tailed unpaired t-test, Mann-Whitney U test, paired t-test or Chi-squared test. Correlations were analyzed using Spearman correlation coefficient with two-tailed p value. P values < .05 were considered statistically significant.
3. Results
3.1. Frequency of ILCs increased in the peripheral blood of SLE patients
In the present study, 49 SLE patients and 20 healthy controls were included. No significant differences in age and gender were observed between these two groups (Supplementary Table 1). Selected laboratory test results and organ involvement in SLE patients are presented in Supplementary Table 1.
Isolated PBMCs from patients and healthy controls were stained and subjected to flow cytometry for analysis of ILC subsets according to the gating strategy as outlined by Munneke J et al. [10]. The lineage makers (CD3, CD14, CD16, CD19, CD20 and CD56) are those for B, T, monocytes, dendritic and NK cells. Lin−CD127+ cells gated on CD45+ lymphocytes were considered to be ILCs (Fig. 1A–C). For further classification, ILCs expressing CRTH2 were defined as ILC2 (Fig. 1B and C). CRTH2−CD117− ILCs were designated as ILC1 and CRTH2−CD117+ ILCs can also be subdivided into NKp44+ ILC3 and NKp44− ILC3 according to the expression of NKp44 (Fig. 1B and C). As shown in Fig. 1B and C from a typical healthy individual and a SLE patient, ILC1, ILC2 and NKp44− ILC3 were the major components of ILCs in the peripheral blood of both healthy controls and SLE patients, while NKp44+ ILC3 was rarely seen in both groups. The data show that the ratios of these ILCs are altered in SLE patients. This later point will be discussed later. It suffices to state that the percentages of ILC/total PBMCs (Total cells) (Fig. 1D) and ILC/lymphocytes (Fig. 1E) were significantly increased in SLE patients compared to controls from a ratio of 0.45% to 0.89% and from 0.84% to 1.77% respectively.
Fig. 1.

Increased frequency of ILCs in peripheral blood of SLE patients. PBMCs from healthy controls and SLE patients were stained for flow cytometry. (A) Single CD45+ lymphocytes were selected for ILCs analysis. ILCs were defined as Lin−CD127+ cells. Type 1, 2 and 3 ILC are identified by the expression of CRTH2, CD117 and NKp44. Representative gating strategy of ILCs in a healthy control (B) and a SLE patient (C). Percentages of ILC/Total cells (PBMC) (D) and ILC/Lymphocytes (E) in SLE patients (N = 49) and healthy controls (N = 20).
3.2. ILC subsets were altered in SLE patients
In addition to an increase in ILCs in the peripheral blood of SLE patients, their ILC subsets were markedly altered. In healthy controls, ILC2 accounted for about 40% ILCs as the major ILC subset in the peripheral blood (Fig. 2B). In SLE, ILC1 were significantly increased and became the dominant ILC subset (Fig. 2A) while ILC2 (Fig. 2B) and ILC3 (Fig. 2C) subsets decreased significantly.
Fig. 2.

Alteration of ILC subset in SLE patients. PBMCs were collected from SLE patients and healthy controls. 49 SLE patients were scored and classified into inactive disease (SLEDAI: 0–4, n = 4) (data not shown), mild disease activity (SLEDAI: 5–9, n = 10), moderate disease activity (SLEDAI: 10–14, n = 16) and severe disease activity (SLEDAI > 15, n = 19), Percentages of ILC1/ILCs (A, D), ILC2/ILCs (B, E), ILC3/ILCs (C, F) in both groups are shown. Correlations between ILC1/ILCs (G), ILC2/ILCs (H), ILC3/ILCs (I) and SLEDAI are presented.
SLE patients with moderate and severe disease activities had increases in ILC1/ILC and decreases in ILC2/ILC and ILC3/ILC (Fig. 2D–F). In the case of patients with mild disease activity, there were significant increases in ILC1/ILC and decreases in ILC2/ILC but the proportion of ILC3/ILCs in patients with mild active SLE was not significantly different from that of the control group (Fig. 2F). The subset alterations correlated with disease activities as measured by SLEDAI except ILC3/ILCs (Fig. 2G–I). As a group, SLE patients’ SLEDAI scores correlates positively with the increases in ILC1 and negatively with decrease in ILC2 (Fig. 2G and H). SLEDAI scores were not significantly correlated with changes in the ILC3 subset (Fig. 2I).
The ratio of ILC/total PBMCs and ILC/lymphocytes were not statistically different between patients with or without renal lesions (Supplementary Fig. 1A and B). However, the ratio of ILC1/ILC was significantly higher and the ratio of ILC2/ILC was significantly lower in patients with renal lesions compared to those SLE patients without renal lesions. ILC3/ILC was not significantly different between patients with or without renal lesions (Supplementary Fig. 1C–E).
3.3. Levels of IL-12 and IL-18 increased and positively correlated with ILC1/ILCs
IL-12, IL-18, IL-25, IL-33, IL-23 and IL-1β are the cytokines that are closely associated with ILC subset induction [4]. These cytokines were significantly increased in SLE patients (Fig. 3 and Supplementary Fig. 2). IL-12, IL-18 and IL-1β were positively correlated with SLEDAI (Fig. 3E–G). Similar positive correlations were also observed between IL-25 and SLEDAI (Supplementary Fig. 2A). However, there were no significant correlations between IFN-γ, IL-23 or IL-33 and SLEDAI (Fig. 3H, Supplementary Fig. 2B and C).
Fig. 3.

IL-12 and IL-18 positively correlated with SLEDAI and ILC1/ILCs. Plasma from healthy controls and SLE patients were collected. Levels of IL-12 (A), IL-18 (B), IL-1β (C) and IFN-γ (D) in both groups and respective correlation with SLEDAI (E-H) and ILC subsets (I-L) are shown. Each point represents an individual subject.
Regarding cytokines that are relevant to the induction of ILC subsets [4], levels of IL-12 and IL-18 were positively correlated with ILC1 (Fig. 3I and J). However, IL-25 and IL-33 did not correlate with ILC2 (Supplementary Fig. 2A and B). IL-1β negatively correlated with ILC3 (Fig. 3K) while there was no significant correlation between IL-23 and ILC3 (Supplementary Fig. 2C). In addition, the level of IFN-γ did not correlate with ILC1 (Fig. 3L).
3.4. Treatment restored ILC1/ILCs and ILC2/ILCs in SLE patients
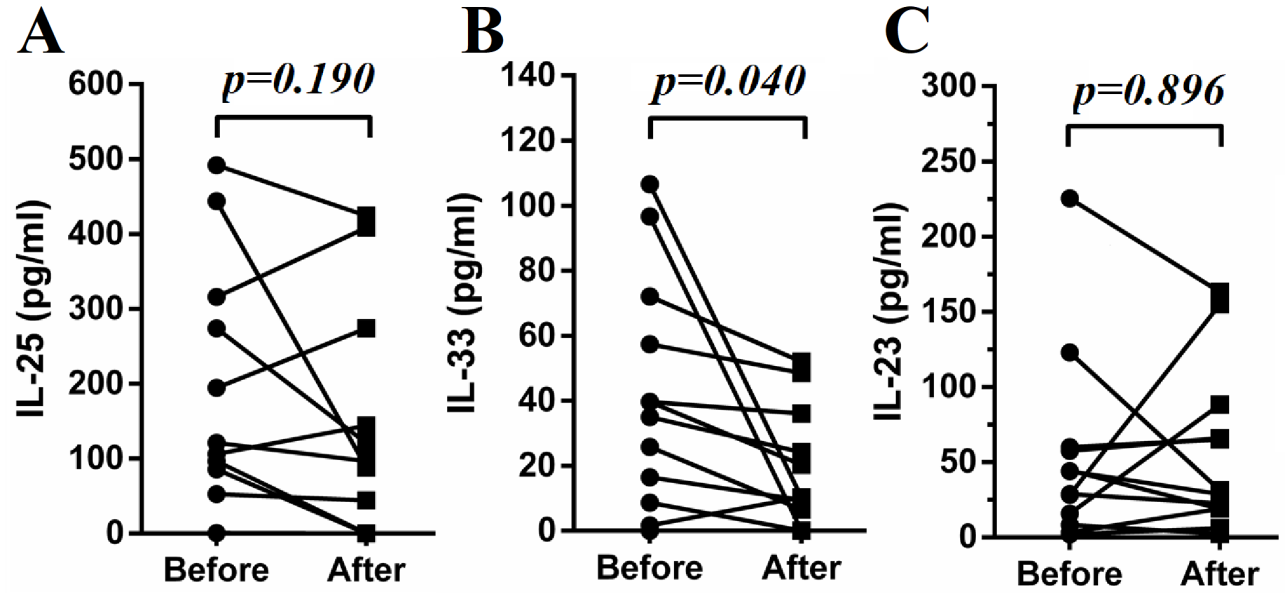
Twelve SLE patients with lupus nephritis were followed up for 10–12 days after the initiation of treatments with steroid and cyclophosphamide. The plasma samples of these patients before and after the initiation of therapy were analyzed for cytokines of interest. Some of these patients’ PBMC were analyzed regarding their changes in ILC subsets. As shown in Fig. 4A–C, ILC1 were significantly decreased with concomitant increases in ILC2 while no significant change was found in ILC3. In addition, ILC1 inducing cytokines IL-12 and IL-18 were significantly decreased (Fig. 4D). IL-33 and IL-1β were also decreased following treatment, but IL-25 and IL-23 did not change significantly (Fig. 4D and Supplementary Fig. 3). Levels of IFN-γ were also decreased (Fig. 4D). In addition, lower SLEDAI scores were achieved following the treatments (Fig. 4C).
Fig. 4.

Treatment restored proportions of ILCs subsets. Fresh blood samples from 12 SLE patients with lupus nephritis were collected before and 10–12 days after treatment. PBMCs of some of these patients were subjected to flow cytometry. Representative flow cytometry results of SLE patients before treatment (A) and after treatment (B). (C) Proportions of ILC1/ILCs, ILC2/ILCs and ILC3/ILCs and SLEDAI before and after treatment are compared. (D) Plasma was subjected to ELISA analysis. Levels of IL-12, IL-18, IL-1β and IFN-γ before and after treatment are presented. Each point represents an individual subject.
3.5. ILC1 presented potent ability to secrete IFN-γ in SLE
In order to ascertain the function of ILC1 in the peripheral blood of SLE patients, PBMCs from 11 healthy controls and 11 SLE patients were prepared for intracellular cytokine staining. As shown in Fig. 5A and B, ILC1 from healthy controls and SLE patients were able to secret IFN-γ. ILC1 from SLE patients secreted larger amount of IFN-γ as presented by the elevated MFI of IFN-γ (Fig. 5C and D). In healthy controls, only about 20% of ILC1 were positive for IFN-γ (Fig. 5B and E). In contrast, over 80% of ILC1 were positive for IFN-γ in SLE patients (Fig. 5B and E).
Fig. 5.

ILC1 presented potent ability to secrete IFN-γ in SLE. PBMCs from 11 healthy controls and 11 SLE patients were isolated and cultured in RPMI1640 supplemented with 10%FBS in the presence of Brelfeldin A for 6 h. Intracellular IFN-γ staining was performed. (A). Gating strategy is presented. (B). The production of IFN-γ is measured by flow cytometry in cells gated on the ILC1 population. (C, D) Comparison of intensity of IFN-γ between control and SLE groups are shown. (E) Frequencies of IFN-γ positive ILC1 in control and SLE patients groups are compared.
3.6. Plasma from SLE patients promoted the production of IFN-γ in ILC1
PBMCs from healthy controls or SLE patients were treated with different stimuli to explore the responsible factors for the activation of ILC1 in SLE patients. Gating strategy is shown in Fig. 6A. PBMCs from 5 healthy controls were stimulated with plasma from healthy controls or with plasma from SLE patients for 3 days. In these groups, stimulations of SLE plasma or IL-2/IL-12/IL-18 significantly increased the amounts of IFN-γ secreted by ILC1 presented as increased MFI of IFN-γ (Fig. 6B–D). 10–40% of the ILC1 cells in control PBMCs incubated with control plasma were positive for IFN-γ (Fig. 6E). After incubation with SLE plasma, percentages of IFN-γ positive ILC1 increased to 50–90% (Fig. 6E). Similarly, IL-2/IL-12/IL-18 significantly increased the percentages of IFN-γ positive ILC1 in PBMCs from both healthy volunteers and SLE patients (Fig. 6F and G).
Fig. 6.

IL-2/IL-12/IL-18 promoted the production of IFN-γ in ILC1. PBMCs from healthy controls or SLE patients were treated with different stimuli for 3 days. Brelfeldin A was added in the last 6 h of the incubation period. Cells were collected and stained for flow cytometry. (A) Gating strategy is shown. MFI of IFN-γ gated on ILC1 and frequencies of IFN-γ positive ILC1 are presented: PBMCs from 5 healthy controls were stimulated with plasma from healthy controls or SLE patients (B, E); PBMCs from 11 healthy controls were stimulated with PBS or IL-2/IL-12/IL-18 (C, F); PBMCs from 11 SLE patients were stimulated with PBS or IL-2/IL-12/IL-18 (D, G). Each PBMC was divided into two parts and received different stimuli.
4. Discussion
ILCs are functionally diverse and developmentally related innate lymphocytes with phenotypes that resemble those of polarized T cell subsets [11]. They are enriched at barrier surfaces in tissues such as skin, lung and intestine. They respond to cytokines and microbial signals promptly [12]. Mature ILCs secret a series of type 1, type 2 and type 17 immune response cytokines to mount pro-inflammatory responses, regulate tissue homeostasis and shape the adaptive immune responses [1]. In the present study, we found that ILC subsets were significantly altered with an increase of ILC1 and decrease of ILC2 and ILC3 in SLE. In addition, functional ILC1 increased and presented with robust IFN-γ secretion.
Our results showed that about 0.1% lymphocytes in the peripheral blood were ILCs and ILC2 was the major subset of ILCs in healthy controls. This percentage of ILCs was consistent with that reported in the literature [13]. In SLE, the frequencies of ILCs increased to about 0.2% and ILC1 increased from 30% to near 70% accompanied with a decrease in ILC2. Correlation analysis showed a positive correlation between ILC1 and SLEDAI and a negative correlation between ILC2 and SLEDAI. This added the credence that type 1 immune response was the prominent immune response in SLE. Such a change in the proportion of ILC subsets was reversed after immunosuppressive treatment of SLE. These findings indicate that ILC1 may be a useful marker to predict disease activity and to monitor disease activity in SLE.
In this study, the cytokines that are related to the induction of ILC subsets such as IL-12 and IL-18 for ILC1, IL-25 and Il-33 for ILC2 and IL-23 and IL-1β for ILC3 are elevated in SLE patients. Except for IL-12 and IL-18 for ILC1, the elevation of circulating cytokines does not correlate with the elevation of related ILC subsets. This observation is concurrent with the observation that induction of ILC2 and ILC3 are under the control of additional cytokines [4]. In addition, it suggests that there are local factors determining the induction of these cells. This complexity is illustrated by the subset of NKp44−ILC3. NKp44− ILC3 in the peripheral blood differ dramatically from NKp44− ILC3 in the gut. The NKp44−ILC3 in the gut do not express RORγt and yet they are able to differentiate to functional ILC1, ILC2 or ILC3 in the presence of cytokines that drive ILC subset development, indicating that NKp44− ILC3 was a circulating pool of partially committed ILC progenitors [3]. Even in the gut, CD117+NKp44− ILC3 are able to differentiate to NKp44+ ILC3 in the presence of IL-23 and IL-1β [7]. These findings demonstrated that ILC have plasticity and may present certain types of immune responses in specific microenvironments. This may explain why ILC3 did not correlate well with relevant cytokines in SLE patients.
The rapid rate of changes in circulating cytokines after the initiation of treatment in SLE was unexpected. The rates of the changes vary greatly from one patient to another. It is tempting to speculate that the changes in cytokine profiles may influence the changes in ILC subsets. In addition, whether the rates of changes in cytokine concentrations have any prognostic significance should be investigated with serials studies of circulating cytokines in SLE patients during the course of their treatments.
IFN-γ is the hallmark cytokine of the Th1 immune response. IFN-γ has been implicated in the pathogenesis of SLE. IFN-γ genetic polymorphism is associated with susceptibility to SLE [14]. IFN-γ receptor deletion prevents autoantibody production and protects NZB/W F1 mice from the development of lupus nephritis [15]. Recent studies showed that IFN-γ activity preceded autoantibody accrual and elevated IFN-α activity and SLE clinical manifestation [16]. In the present study, we found that plasma IFN-γ was elevated in SLE patients and decreased following treatment. However, the level of IFN-γ did not correlate with disease activity or ILC1/ILCs. This might be explained by the fact that IFN-γ are produced by a variety of cells including activated Th1 CD4+ lymphocytes, CD8+ lymphocytes NK cells, NKT cells, and professional antigen-presenting cells as well as B cells [17]. On the other hand, ~ 80% of circulating ILC1 expressed IFN-γ in SLE patients. In contrast, only ~20% of circulating ILC1 cells were positive for this cytokine. We also showed that plasma from SLE patients were potent in the induction of IFN-γ positive ILC1 cells and IFN-γ production by the circulating ILC1 isolated from normal controls. This activity may reflect the increase in IL-12 and IL-18 in SLE patients’ plasma. While we were not able to determine the relative contribution of ILC1 to the elevation of circulating IFN-γ in SLE, it is reasonable to conclude that increased circulating ILC1 might contribute to the pathogenesis of SLE through mounting type 1 immune response.
IL-12 is mainly produced by monocytes and macrophages, and to a less extent by dendritic cells [18,19]. However, its role in the pathogenesis of SLE is conflicting. Many studies reported that the blood levels of IL-12 were higher in SLE patients than those of healthy subjects [20,21]. Tucci et al. showed that levels of both IL-12 in sera and urine were elevated and reflect glomerular production and parallel Th1 polarization and high IFN-γ production in SLE [22]. In contrast, Liu et al. reported impaired production of IL-12 in SLE [23]. IL-1β was also significantly increased in the present study and positively correlated with disease activity. Maturation and secretion of IL-1β and IL-18 was mediated by NLRP3 inflammasome [24]. Blocking the NLRP3 inflammasome by antagonists against upstream molecules was shown to be effective in controlling progression of LN [25–27], indicating that the NLRP3 inflammasome might be a potential target to treat lupus. Recently, blood levels of IL-18 were shown to be increased in SLE patients and were potentially associated with active renal disease while IL-β was not significantly elevated [28]. More studies will be needed to clarify the relative role of these two cytokines in the pathogenesis of SLE.
Circulating ILCs may accumulate in certain organs and their accumulation may be a predictor of the ongoing processes. NKp44+ ILC3 accumulated in skin lesions in psoriasis patients and expanded in peripheral blood, driving epidermal thickening through secreting IL-22 [8,29]. Wohlfahrt et al. documented that the frequency of ILC2 was significantly increased in skin sections and in circulation of patients with systemic sclerosis compared with healthy controls, suggesting that ILC2 might be the pathogenic factor triggering fibroblast activation and tissue fibrosis [30]. In the present studies, we found that the proportion of ILC1 was higher while ILC2 was lower in PBMC of SLE with renal involvement compared to those without renal involvement, suggesting that ILC1 might be a pathogenic player in renal damage. This was consistent with previous reports showing that type 1 immune response plays a critical role in the pathogenesis of lupus nephritis. Predominance of Th1 response in both peripheral and renal tissues was observed in diffuse proliferative lupus nephritis [31,32]. The numbers of renal IFN-γ producing CD4+ Th1 cells gradually increased in the kidneys of MRL/lpr and NZB/W F1 mice over time [33]. Further studies of renal ILC subsets may provide novel insights in the pathogenesis of human and mouse lupus.
It will be remiss not to discuss the role of NK cells in the pathogenesis of SLE. There are multiple publications on NK cells in SLE [34]. Phenotypic analyses of NK cells in SLE have not yielded consistent results. For example, Schepis et al., reported the increase in CD56 bright NK cells in SLE patients irrespective of their disease activities [35]. However, Hervier et al. showed that the absolute NK cell count was decreased in patients with active SLE but the relative frequency of CD56 bright/dim NK cells were unaffected [36]. The divergent results on the NK cell populations may be due to the heterogeneity and the treatments of the SLE patient populations as well as the methods of gating the cell populations. Phenotypic analyses further showed there was reduction of expression of inhibitory receptors on the NK cell population in SLE with variable expression of activating receptors. These results do not support the thesis that the NK cells are more active in SLE. At any rate, most if not all the studies on NK cells were carried out before the classification of ILC. Their relevance to the present study is not apparent.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
All authors were involved in drafting and revising the article, and all authors approved the final version to be published. Dr. Yang had full access to all of the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by the National Natural Science Foundation of China [grant numbers 81671593, 81471598, 81701595, 81801614]; the Guangdong Natural Science Foundation [grant numbers 2014A030313096, 2016A030310172]; the Guangzhou Science and Technology Planning Program [grant number 201707010093]; the National Institutes of Health [grant number R01 AR-047988]; and the Lupus Research Alliance [grant numbers TIL332635 and 2016 Dr. William E. Paul Distinguished Innovator Award # 514370].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.clim.2019.03.008.
Study conception and design
C. Guo, M. Zhou, S. Zhao, N. Yang and S. Fu.
Acquisition of data
C. Guo, M. Zhou, S. Zhao, Y. Huang, S. Wang, R. Fu, M. Li, T. Zhang, F. Gaskin and N. Yang.
Analysis and interpretation of data
C. Guo, M. Zhou, S. Zhao, Y. Huang, S. Wang, R. Fu, M. Li, T. Zhang, F. Gaskin, N Yang and S. Fu.
Declarations of interest
None.
References
- [1].Klose CS, Artis D, Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis, Nat. Immunol 17 (2016) 765–774, 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- [2].Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E, Innate lymphoid cells–a proposal for uniform nomenclature, Nat. Rev. Immunol 13 (2013) 145–149, 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- [3].Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, Masse-Ranson G, David E, Strick-Marchand H, Le Bourhis L, Cocchi R, Topazio D, Graziano P, Muscarella LA, Rogge L, Norel X, Sallenave JM, Allez M, Graf T, Hendriks RW, Casanova JL, Amit I, Yssel H, Di Santo JP, Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation, Cell 168 (2017) 1086–1100 e1010 10.1016/j.cell.2017.02.021. [DOI] [PubMed] [Google Scholar]
- [4].Shikhagaie MM, Germar K, Bal SM, Ros XR, Spits H, Innate lymphoid cells in autoimmunity: emerging regulators in rheumatic diseases, Nat. Rev. Rheumatol 13 (2017) 164–173, 10.1038/nrrheum.2016.218. [DOI] [PubMed] [Google Scholar]
- [5].Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H, Lnnate lymphoid cells: 10 years on, Cell 174 (2018) 1054–1066, 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- [6].Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H, Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues, Nat. Immunol 14 (2013) 221–229, 10.1038/ni.2534. [DOI] [PubMed] [Google Scholar]
- [7].Fragoulis GE, Siebert S, McInnes IB, Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases, Annu. Rev. Med 67 (2016) 337–353, 10.1146/annurev-med-051914-021944. [DOI] [PubMed] [Google Scholar]
- [8].Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, Cheuk S, Brouwer MWD, Menting SP, Eidsmo L, Spits H, Hazenberg MD, Mjosberg J, Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients, J. Investig. Dermatol 134 (2014) 2351–2360, 10.1038/jid.2014.146. [DOI] [PubMed] [Google Scholar]
- [9].Braudeau C, Amouriaux K, Neel A, Herbreteau G, Salabert N, Rimbert M, Martin JC, Hemont C, Hamidou M, Josien R, Persistent deficiency of circulating mucosal-associated invariant T (MAIT) cells in ANCA-associated vasculitis, J. Autoimmun 70 (2016) 73–79, 10.1016/j.jaut.2016.03.015. [DOI] [PubMed] [Google Scholar]
- [10].Munneke JM, Bjorklund AT, Mjosberg JM, Garming-Legert K, Bernink JH, Blom B, Huisman C, van Oers MH, Spits H, Malmberg KJ, Hazenberg MD, Activated innate lymphoid cells are associated with a reduced susceptibility to graft-versus-host disease, Blood 124 (2014) 812–821, 10.1182/blood-2013-11-536888. [DOI] [PubMed] [Google Scholar]
- [11].Bando JK, Colonna M, Innate lymphoid cell function in the context of adaptive immunity, Nat. Immunol 17 (2016) 783–789, 10.1038/ni.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sonnenberg GF, Artis D, Innate lymphoid cells in the initiation, regulation and resolution of inflammation, Nat. Med 21 (2015) 698–708, 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hazenberg MD, Spits H, Human innate lymphoid cells, Blood 124 (2014) 700–709, 10.1182/blood-2013-11-427781. [DOI] [PubMed] [Google Scholar]
- [14].Kim K, Cho SK, Sestak A, Namjou B, Kang C, Bae SC, Interferon-gamma gene polymorphisms associated with susceptibility to systemic lupus erythematosus, Ann. Rheum. Dis 69 (2010) 1247–1250, 10.1136/ard.2009.117572. [DOI] [PubMed] [Google Scholar]
- [15].Haas C, Ryffel B, Le Hir M, IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice, J. Immunol 160 (1998) 3713–3718. [PubMed] [Google Scholar]
- [16].Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, Niewold TB, Tsokos GC, Keith MP, Harley JB, James JA, Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification, Ann. Rheum. Dis 75 (2016) 2014–2021, 10.1136/annrheumdis-2015-208140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schroder K, Hertzog PJ, Ravasi T, Hume DA, Interferon-gamma: an overview of signals, mechanisms and functions, J. Leukoc. Biol 75 (2004) 163–189, 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- [18].Dorman SE, Holland SM, Interferon-gamma and interleukin-12 pathway defects and human disease, Cytokine Growth Factor Rev. 11 (2000) 321–333. [DOI] [PubMed] [Google Scholar]
- [19].Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G, Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells, Eur. J. Immunol 26 (1996) 659–668, 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- [20].Lauwerys BR, Van Snick J, Houssiau FA, Serum IL-12 in systemic lupus erythematosus: absence of p70 heterodimers but presence of p40 monomers correlating with disease activity, Lupus 11 (2002) 384–387, 10.1191/0961203302lu213oa. [DOI] [PubMed] [Google Scholar]
- [21].Tokano Y, Morimoto S, Kaneko H, Amano H, Nozawa K, Takasaki Y, Hashimoto H, Levels of IL-12 in the sera of patients with systemic lupus erythematosus (SLE)–relation to Th1- and Th2-derived cytokines, Clin. Exp. Immunol 116 (1999) 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tucci M, Lombardi L, Richards HB, Dammacco F, Silvestris F, Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis, Clin. Exp. Immunol 154 (2008) 247–254, 10.1111/j.1365-2249.2008.03758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu TF, Jones BM, Wong RW, Srivastava G, Impaired production of IL-12 in systemic lupus erythematosus. III: deficient IL-12 p40 gene expression and cross-regulation of IL-12, IL-10 and IFN-gamma gene expression, Cytokine 11 (1999) 805–811, 10.1006/cyto.1999.0512. [DOI] [PubMed] [Google Scholar]
- [24].Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E, Targeting the NLRP3 inflammasome in inflammatory diseases, Nat. Rev. Drug Discov 17 (2018) 588–606, 10.1038/nrd.2018.97. [DOI] [PubMed] [Google Scholar]
- [25].Zhao J, Wang H, Huang Y, Zhang H, Wang S, Gaskin F, Yang N, Fu SM, Lupus nephritis: glycogen synthase kinase 3beta promotion of renal damage through activation of the NLRP3 inflammasome in lupus-prone mice, Arthritis Rheum. 67 (2015) 1036–1044, 10.1002/art.38993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhao J, Wang H, Dai C, Zhang H, Huang Y, Wang S, Gaskin F, Yang N, Fu SM, P2X7 blockade attenuates murine lupus nephritis by inhibiting activation of the NLRP3/ASC/caspase 1 pathway, Arthritis Rheum. 65 (2013) 3176–3185, 10.1002/art.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fu R, Guo C, Wang S, Huang Y, Jin O, Hu H, Chen J, Xu B, Zhou M, Zhao J, Sung SJ, Wang H, Gaskin F, Yang N, Fu SM, Podocyte activation of NLRP3 Inflammasomes contributes to the development of proteinuria in lupus nephritis, Arthritis Rheum. 69 (2017) 1636–1646, 10.1002/art.40155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mende R, Vincent FB, Kandane-Rathnayake R, Koelmeyer R, Lin E, Chang J, Hoi AY, Morand EF, Harris J, Lang T, Analysis of serum interleukin (IL)-1beta and IL-18 in systemic lupus Erythematosus, Front. Immunol 9 (2018) 1250, 10.3389/fimmu.2018.01250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P, Nestle FO, Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis, J. Investig. Dermatol 134 (2014) 984–991, 10.1038/jid.2013.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wohlfahrt T, Usherenko S, Englbrecht M, Dees C, Weber S, Beyer C, Gelse K, Distler O, Schett G, Distler JH, Ramming A, Type 2 innate lymphoid cell counts are increased in patients with systemic sclerosis and correlate with the extent of fibrosis, Ann. Rheum. Dis 75 (2016) 623–626, 10.1136/annrheumdis-2015-207388. [DOI] [PubMed] [Google Scholar]
- [31].Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, Arinobu Y, Yamaoka K, Niiro H, Shinozaki M, Hirakata H, Horiuchi T, Otsuka T, Niho Y, Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus, Arthritis Rheum. 42 (1999) 1644–1648, . [DOI] [PubMed] [Google Scholar]
- [32].Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, Fukuda H. Kanai, H. Nakashima, T. Otsuka, H. Hirakata, Predominance of Th1 immune response in diffuse proliferative lupus nephritis, Arthritis Rheum. 44 (2001) 2097–2106, . [DOI] [PubMed] [Google Scholar]
- [33].Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, Koyro T, Peters A, Velden J, Hunemorder S, Haag F, Steinmetz OM, Mittrucker HW, Stahl RA, Panzer U, Function of the Th17/interleukin-17A immune response in murine lupus nephritis, Arthritis Rheum. 67 (2015) 475–487, 10.1002/art.38955. [DOI] [PubMed] [Google Scholar]
- [34].Spada R, Rojas JM, Barber DF, Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus, J. Leukoc. Biol 98 (2015) 479–487, 10.1189/jlb.4RU0315-081RR. [DOI] [PubMed] [Google Scholar]
- [35].Schepis D, Gunnarsson I, Eloranta ML, Lampa J, Jacobson SH, Karre K, Berg L, Increased proportion of CD56bright natural killer cells in active and inactive systemic lupus erythematosus, Immunology 126 (2009) 140–146, 10.1111/j.1365-2567.2008.02887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, Musset L, Debré P, Amoura Z, Vieillard V, Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-γ production in patients with active disease, Arthritis Rheum. 63 (2011) 1698–1706, 10.1002/art.30313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.