

Abstract
A great majority of gastrointestinal stromal tumors (GISTs) are primarily driven by gain-of-function KIT receptor tyrosine kinase mutations that subsequently lead to activation of phosphatidiylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway, a downstream effector of KIT signaling. KIT tyrosine kinase inhibitor, imatinib mesylate, has been successfully used for the treatment of primary, advanced, and disseminated GISTs. Recently, activation of mTOR pathway independent of KIT signaling was demonstrated in imatinib mesylate naïve malignant GISTs and treatment-resistant metastatic tumors. This activation was attributed to oncogenic mutations in PIK3CA encoding PI3K 110α subunit, or to the inactivation of PTEN tumor suppressor, a potent mTOR negative regulator. In this study, mTOR pathway genes were evaluated in 14 imatinib mesylate naïve, KIT-mutant, malignant small intestinal GISTs using next-generation sequencing. Mutations were detected in 3 (21%) of 14 analyzed tumors: (1) c.3200A > T substitution in PIK3CB encoding PI3K 110β subunit, (2) c.1040A > G substitution in tuberous sclerosis complex (TSC2) encoding tuberin, mTOR down-regulator (3) c.6625C > G substitution in mTOR. At the protein level, these changes were predicted to cause, respectively, PIK3CB p.D1067V, TSC2 p.K347R, and mTOR p.L2209V mutations. Previously reported “in vitro” experiments with mouse 3T3 fibroblasts demonstrated oncogenic potential of PIK3CB p.D1067V and mTOR p.L2209V mutants; whereas, PolyPhen-2 software analysis predicted TSC2 p.K347R mutation to likely have a damaging impact on tuberin function. The results of this and previous studies indicate diversity of genetic changes leading to activation of PI3K-AKT-TSC-mTOR pathway in malignant GISTs. Extensive genotyping of the genes involved in mTOR pathway demonstrates common alterations that need to be considered in targeted treatment.
Keywords: GIST, PIK3CB, TSC2, mTOR, mutation, Ion Torrent next-generation sequencing, Sanger sequencing
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract.1 A great majority of GISTs are primarily driven by gain-of-function mutations in KIT receptor tyrosine kinase that subsequently lead to activation of phosphatidiylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway, a downstream effector of KIT signaling.2,3 KIT tyrosine kinase inhibitor, imatinib mesylate, has been successfully used for the treatment of primary, advanced, and disseminated GISTs.4 However, resistance could occur because of the secondary mutations in KIT tyrosine kinase domain.5
PI3K is a family of enzymes, critical modulators of biological processes such as cellular survival, proliferation, and migration.6 PI3Ks represent heterodimers of regulatory and catalytic subunits. Class IA PI3Ks consist of 5 regulatory (p85α, p55α, p50α, p85β, and p55γ) and 3 catalytic (p110α, p110 β, and p110δ) subunits encoded, by PIK3R-1, PIK3R-2, and PIK3R-3 and by PIK3C-A, PIK3C-B, and PIK3C-D, respectively. Class IB PI3Ks consist of 1 regulatory and 1 catalytic (p101 and p110γ) subunit encoded by a single gene each, PIK3R5 and PIK3CG.7 Deregulation of the PI3K signaling pathway triggers activation of downstream kinases such as PDK1 (phosphoinositide-dependent kinase 1) and AKT (V-AKT murine thymoma viral oncogene homolog 1) and subsequently mTOR signaling, promoting protein synthesis, and cell growth.8–11
mTOR encodes an evolutionarily conserved serinethreonine kinase, a member of (phosphoinositide 3-kinase), PI3K-related kinase family that assembles into 2 distinct complexes: mTORC1 and mTORC2. These complexes are essential regulators of a wide range of cell functions such as metabolism, proliferation, survival, regulation of immune response, and actin and intermediate filament organization. Dysfunction of mTORC1 has been implicated in cancer and different metabolic, neurological, and genetic disorders.12
Recently, pathologic activation of PI3K/mTOR signaling pathway has been documented in metastatic KIT-mutant GIST xenografts.13 Parallel studies identified inactivation of PTEN (phosphatase and tensin homolog), a potent negative mTOR regulator and oncogenic mutations in PIK3CA encoding PI3K 110α subunit in imatinib naïve malignant GISTs and treatment-resistant metastatic tumors.10,14–18
Yet, no systematic genotyping of other PI3K/mTOR pathway genes has been carried out. This study examined a panel of mTOR pathway genes for mutations in imatinib naïve malignant GISTs using next-generation sequencing (NGS). The results, finding of mutations in PIK3CB, tuberous sclerosis complex (TSC2), and mTOR, highlight divergent molecular mechanisms underlying pathologic activation of mTOR signaling pathway in malignant GISTs.
MATERIALS AND METHODS
Study Design
Fourteen well-characterized clinically malignant intestinal GISTs were analyzed in this study.19 In all cases, clinicopathologic, immunohistochemical and molecular genetic profile, and complete follow-up data were available. Tumor DNA samples were screened for mutations using NGS technology. Subsequently, targeted polymerase chain reaction (PCR) amplification followed by Sanger sequencing of PCR products was used to confirm the NGS results.
Molecular Studies
Ten 5-μm-thick sections of formalin-fixed paraffin-embedded tissue samples were submitted for DNA extraction. DNA was extracted using formalin-fixed paraffin-embedded DNA kit and an automated nucleic acid purification system, Maxwell Rapid Sample Concentrator (Promega, Madison, WI).
NGS was performed by MacrogenUSA (Rockville, MD) using the Ion Torrent NGS platform and Ion AmpliSeq Comprehensive Cancer Panel (Life Technologies/Thermo Fisher Scientific, Waltham, MA) of 409 genes frequently mutated in cancer including several PI3K/mTOR pathway genes. Bioinformatics of NGS-data was done at the Department of Molecular Diagnostics, Holycross Cancer Center (Kielce, Poland) as previously described.17
The NGS results were confirmed by targeted PCR amplification performed on the same DNA templates following standard 3-temperature protocol with denaturing at 94°C, annealing at 49°C for PIK3CB, 53°C for TSC2, and at 40 to 50°C gradient for mTOR, and extension at 72°C. AmpliTaq Gold DNA polymerase (Applied Biosystems by Life Technologies, Austin, TX) and following pair of primers were used: (1) TSC11.1F 5′-ACAGCAAGCAAGCAGCTCTG-3′ and TSC11.2R 5′-GAGCCGTTCGATGATGTTCA-3′, (2) PIK3CB24.1F 5′-AGGACTCTCTTGCATTAGGG-3′ and PIK3CB24.3R 5′-TCTCTAACAGGGTCATGTTC-3′, (3) TOR47.1F 5′-AAAGGCCATGAAGATCTGCG-3′ and TOR47.2R 5′-CTACACGAGACAAATGTAGG-3′. PCR amplification products were purified using QIAquick Gel Extraction Kit (Qiagen Inc., Valencia, CA) following agarose gel electrophoresis and sequenced directly with forward and reverse primers. Sanger sequencing was completed by MacrogenUSA. PIK3CB (Gene ID: 5291), TSC2 (Gene ID: 7249), and mTOR (Gen ID: 2475) reference sequences were obtained from NCBI database (http://www.ncbi.nlm.nih.gov).
RESULTS
Clinicopathologic Profile of Analyzed GISTs
Fourteen small intestinal GISTs from 9 men (age, 43 to 79 y) and 5 women (age, 37 to 77 y) were analyzed. Tumors predominantly revealed spindle cell morphology with a few cases (n=3) showing both spindle cell and epithelioid features. Immunohistochemical KIT (CD117) expression was documented in all cases. Tumor size varied from 5.5 to 18 (mean, 10.4; median, 9.25) cm. Mitotic count was 1 to 100 (mean, 25; median, 13) per 5 mm2. These GISTs were considered to be moderate [3a (n=1)] to high [3b (n=1), 5 (n=1), and 6a/b (n=11)] risk tumors, on the basis of tumor size/mitotic activity risk stratification.1 KIT mutations were detected in exon 11 (n=13) and in exon 9 (n=1) in all tumors. Complete follow-up, available for the patients, revealed survival from 3 to 54 (mean, 46; median, 16.5) months.
Molecular Studies
NGS detected mTOR pathway gene mutations, TSC2 c.1040A > G, PIK3CB c.3200A > T and mTOR c.6625C > G substitutions, one of each, in 3 (21%) of 14 analyzed tumors. Presence of these mutations was confirmed by targeted PCR amplification and Sanger sequencing of PCR products. Representative NGS images and Sanger sequencing chromatograms are shown in Figure 1.
FIGURE 1.
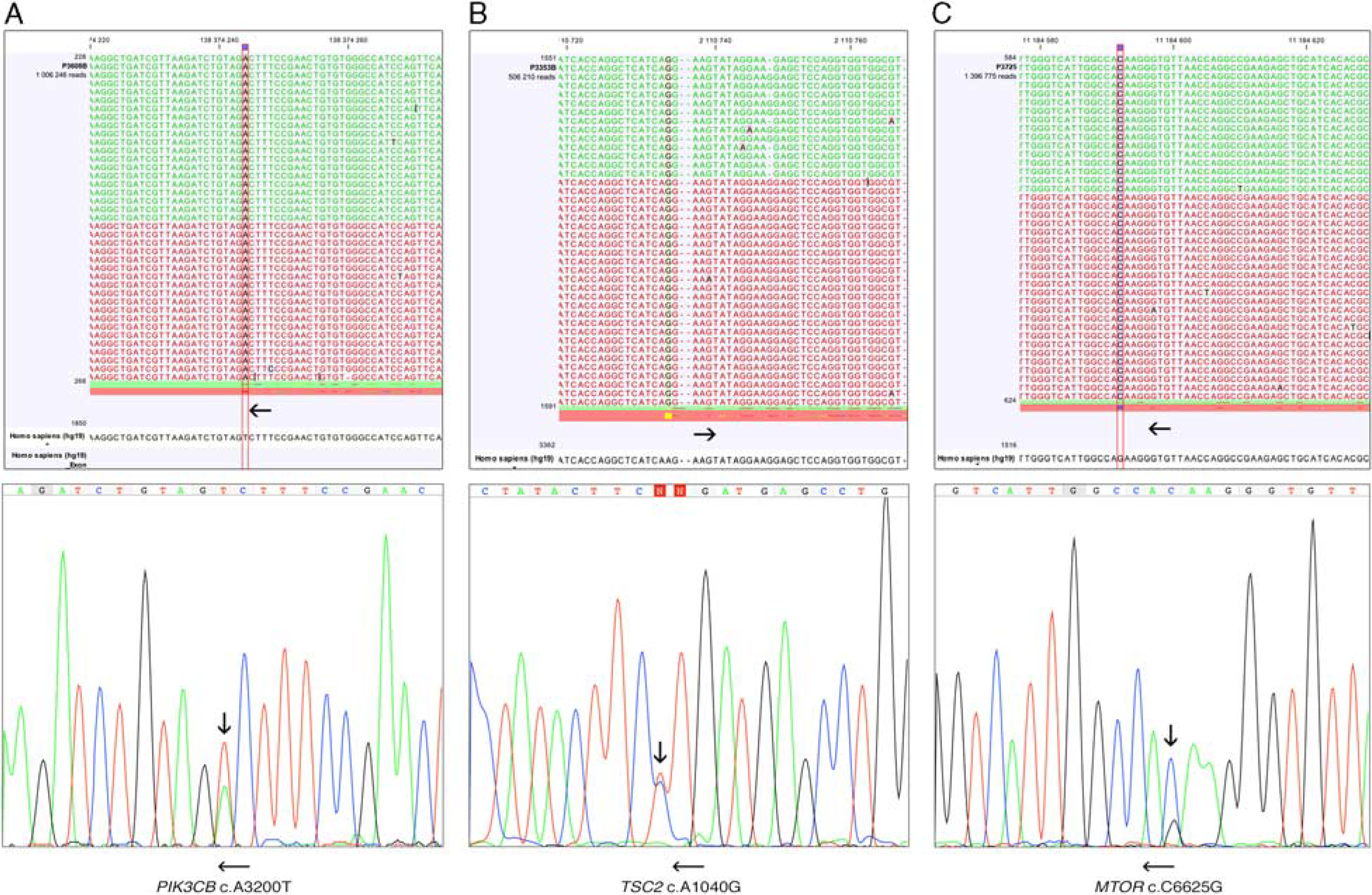
Screen shots of the next-generation sequencing data analysis and Sanger sequencing chromatograms. PIK3CB c.3200A > T mutation (A) TSC2 c.1040A > G mutation (B), mTOR c.6625C > G mutation (C). Vertical arrows indicate substitutions. Horizontal arrows indicate forward and reverse orientations.
Clinicopathologic Profile of PIK3CB, TSC2, and mTOR Mutant Tumors
PIK3CB c.A3200T substitution, predicted to cause p.D1067V mutation, was found in a 17 cm tumor with high (22/5 mm2) mitotic index and spindle/epithelioid cell morphology. KIT expression and typical GIST KIT mutation, p.W557G were previously identified. Immunostainings for CD34, SMA, desmin, and S100 protein were negative. Patient died of the disease 54 months after primary diagnosis.
TCS2 c.1040 A > G substitution predicted to cause TSC2 p.K347R mutation was found in a 5.5 cm tumor with a mitotic rate of 12/5 mm2 and spindle/epithelioid morphology. KIT expression and typical for GISTs KIT mutation, p.W557G were previously identified. Immunostainings for CD34, SMA, desmin, and S100 protein were negative. Patient died of disease 19 months after the primary diagnosis.
mTOR c.C6625G substitution, predicted to cause p.L2209V mutation, was found in a 5 cm GIST with a high (33/5 mm2) mitotic index and spindle cell morphology. KIT expression and typical for GISTs KIT mutation, p.L558_V560del were previously identified. The tumor showed focal CD34 expression and was negative for SMA, desmin, and S100 protein. The patient died of the disease 3 months after the primary diagnosis.
Representative histologic and immunohistochemical images are shown in Figure 2.
FIGURE 2.
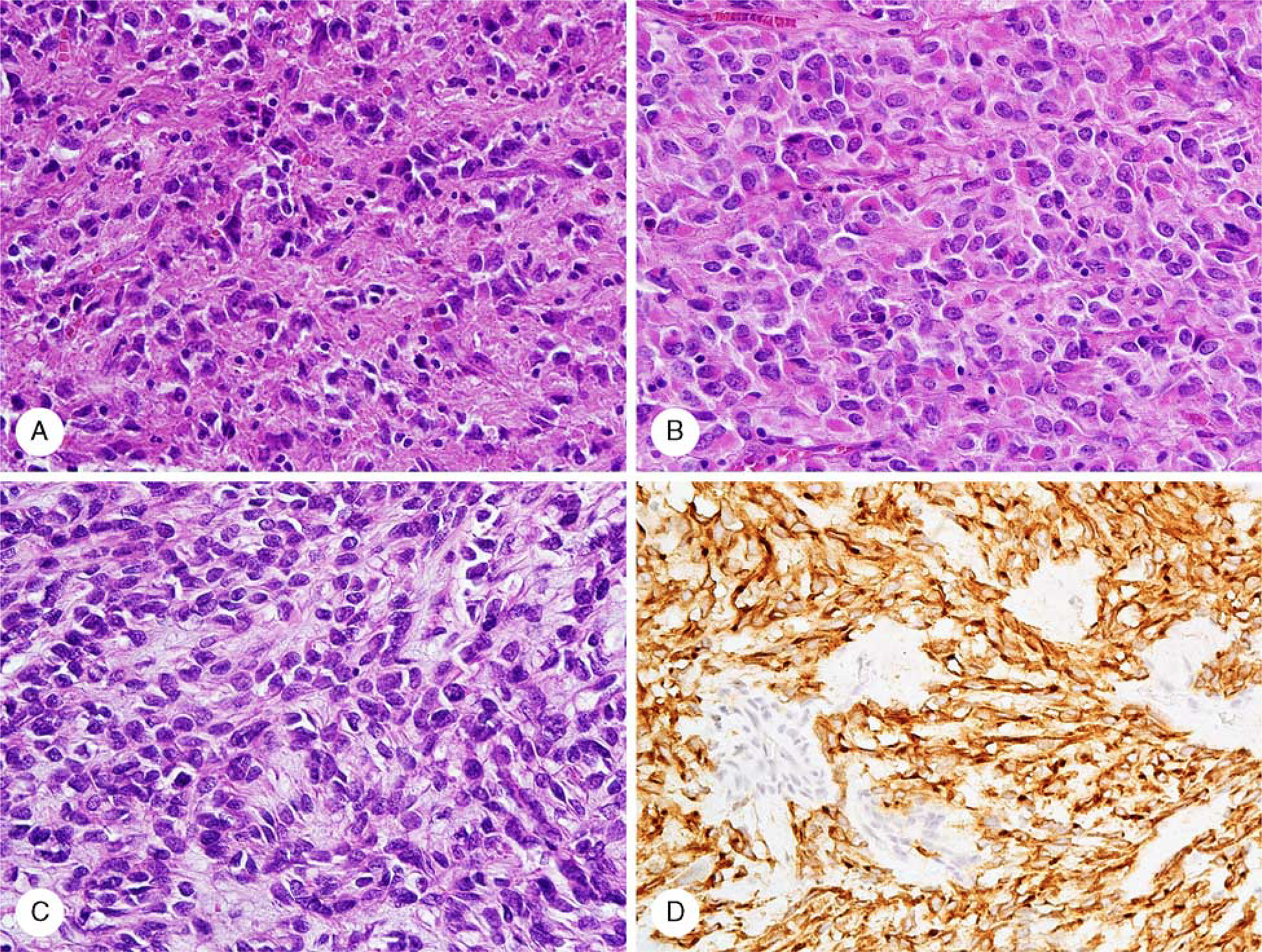
Histologic and immunohistochemical images of PIK3CB (A), TSC (B), and mTOR (C) mutant GISTs. PIK3CB and TSC2 mutants revealed varied spindle and epithelioid (the latter shown in the figure) morphology. mTOR-mutant GIST displayed spindle cell morphology with nuclear pseudopalisading. Immunohistochemically detected KIT expression (D) exemplified by mTOR-mutant GIST was seen in all tumors. GIST Indicates gastrointestinal stromal tumor; mTOR, mammalian target of rapamycln; TSC, tuberous sclerosis complex.
DISCUSSION
In this study, the occurrence of mTOR pathway gene mutations was evaluated using NGS in KIT-mutant malignant GISTs. Genomic mutations in PIK3CB encoding PI3K 110β subunit, TSC2 encoding tuberin, mTOR down-regulator and in mTOR were identified in analyzed tumors. Previous studies documented genetic alteration in PTEN, potent mTOR negative regulator, and in PIK3CA encoding in PI3K 110α subunit in both imatinib naïve GISTs and kinase inhibitor resistant tumors.3,17,18
Mutational activation of PI3K pathway has been detected in different types of cancer, most frequently in breast, colorectal, endometrial, head and neck, lung and prostate carcinomas, and glioblastoma multiforme.20 Oncogenic activation of PI3K pathway signaling is often associated with aggressive tumor phenotype and has been linked to poor survival outcomes in different types of cancer.20,21 Typically, PI3K p110α and p85α subunits are affected, although mutations in 110β subunit were also documented.21,22 PIK3CB p.D1067V mutation discovered in malignant GIST in current study was recently reported in EGFR-mutant lung adenocarcinoma.21 Previously published functional studies revealed that this mutation activates PI3K downstream signaling, enhances NIH-3T3 cells growth “in vitro” and promote tumor growth “in vivo.” Thus, it could function as an oncogenic alteration in cancer.21,23
In the present study, PIK3CB p.D1067V mutant-GIST carried oncogenic KIT mutation. Two of the PIK3CB mutant tumors (melanoma and thyroid carcinoma) listed by The Cancer Genome Atlas (https://cancergenome.nih.gov), harbored BRAF p.V600E mutation.21 Both, KIT p.W557G and BRAF p.V600E, mutants promote oncogenesis through the PI3K pathway activation. Synergistic pathologic activation of KIT and PI3K signaling might give proliferative advantage of the double mutant clone leading to the domination during genetic evolution. Such clones may arise with or without the selection pressure of KIT inhibitors or BRAF inhibitors. Previously, PIK3CA and KIT, and PIK3CA and BRAF double mutants were identified, respectively, in imatinib naïve high-grade GISTs and in metastatic tumor with acquired resistance to BRAF-inhibitor treatment.15,17
Thus, in GIST, at least 2 PI3K subunits, p.110α and p.110β could be affected by additional genetic event that develops naturally during tumor progression and over time leads to clonal selection. A selection pressure caused by the kinase inhibitor treatment can accelerate expansion of the clones carrying additional oncogenic mutations causing resistance to primary KIT-inhibitor treatment.
TSC is a genetic disorder clinically characterized by multiple hamartomas, epilepsy, autism, mental retardation, and hypopigmented macules.24 Constitutive activation of mTORC1 is implicated for this condition and attributed to the loss-of-function mutation in either TSC1 or TSC2 gene, which encode mTORC1 down-regulators hamartin and tuberin, respectively.25 In mouse models, loss of TSC1/TSC2 was linked to tumorigenesis including development of kidney, prostate, lung carcinomas, and mesothelioma. Also, TSC1/TSC2 inactivating mutations were identified in small frequency in different types of human cancer as documented by The Cancer Genome Atlas.26
A TSC2 p.K347R mutation described in current study was previously identified in peripheral blood of a TSC patient (TSC mutation database, Leiden Open Variation Database) and considered to likely have damaging impact on the TSC2 structure and function as predicted by PolyPhen-2 software analysis.27
Present study, for the first time, documented mTOR p.L2209V oncogenic mutations in KIT-mutant, imatinib naïve, malignant GISTs. The Cancer Genome Atlas reported this mutation in an urothelial carcinoma. Recently, a large cell neuroendocrine carcinoma carrying such a mutation was also described.28 “In vitro” studies confirmed transforming ability of the mTOR p.L2209V mutation when expressed in mouse 3T3 fibroblasts and potential to activate mTOR downstream signaling molecules such as p70S6K, S6RP, and 4EBP1 in these cells. However, in the nude mouse tumorigenicity assay, 3T3 mTOR mutant cells did not form tumor in the same observation period as 3T3 v-Ras mutant cells.28
Little is known about frequency of genetic mutations affecting different mTOR pathway genes. Previous study estimated the frequency of PIK3CA mutants among large (> 10 cm) and advanced, imatinib naïve GISTs at 5% to 6%,17 whereas PTEN mono-allelic loss was reported in 9% and 39% of imatinib naïve and imatinib resistant GISTs, respectively.18 In the present study, the frequency of PIK3CB, TSC2, and mTOR mutations was 7% for each mutation. Thus, overall frequency of mutations affecting different genes involved in PI3K/mTOR pathway might be ≥35%.
The identification of mutations activating the PI3K/mTOR pathway has significant implications, as effective treatment may require mutant-specific inhibitors. Recent studies on D1067V-mutant non–small cell lung cancer resistant to erlotinib showed that PIK3CB-selective inhibitor was more effective than a pan-PI3K inhibitor.21 Also, different classes of mTOR inhibitors have been developed and recently used in clinical cancer trials. However, predictive biomarkers remain elusive.26,29,30
In summary, the present study reports for the first time PIK3CB, TSC2, and mTOR gene mutations in KIT-mutant GISTs. These mutations represented additional oncogenic events developing without selection pressure of the kinase inhibitor treatment in imatinib naïve, clinically aggressive tumors. Presence of such mutations could affect mTOR signaling pathway and allow tumor cells to bypass KIT inhibitors creating imatinib resistance. This finding has implications for genetic testing and management of GISTs via targeted therapy with KIT, PI3K, and mTOR inhibitors.
Footnotes
The authors declare no conflict of interest.
REFERENCES
- 1.Miettinen M, Lasota J. Gastrointestinal stromal tumors. Gastroenterol Clin North Am. 2013;42:399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998; 279:577–580. [DOI] [PubMed] [Google Scholar]
- 3.Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999–4006. [DOI] [PubMed] [Google Scholar]
- 4.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. [DOI] [PubMed] [Google Scholar]
- 5.Liegl B, Kepten I, Le C, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. [DOI] [PubMed] [Google Scholar]
- 7.Hawkins PT, Anderson KE, Davidson K, et al. Signaling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006; 34(pt 5):647–662. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Top Microbiol Immunol. 2010;347:21–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13:140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Looy T, Wozniak A, Floris G, et al. Phosphoinositide 3-kinase inhibitors combined with imatinib in patient-derived xenograft models of gastrointestinal stromal tumors: rationale and efficacy. Clin Cancer Res. 2014;20:6071–6082. [DOI] [PubMed] [Google Scholar]
- 14.Daniels M, Lurkin I, Pauli R, et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011;312:43–54. [DOI] [PubMed] [Google Scholar]
- 15.Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serrano C, Wang Y, Mariüo-Enriquez A, et al. KRAS and KIT gatekeeper mutations confer polyclonal primary imatinib resistance in GI stromal tumors: relevance of concomitant phosphatidylinositol 3-kinase/AKT dysregulation. J Clin Oncol. 2015;33:e93–e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasota J, Felisiak-Golabek A, Wasag B, et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: molecular genetic study of 529 cases. Mod Pathol. 2016;29:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quattrone A, Wozniak A, Dewaele B, et al. Frequent mono-allelic loss associated with deficient PTEN expression in imatinib-resistant gastrointestinal stromal tumors. Mod Pathol. 2014;27:1510–1520. [DOI] [PubMed] [Google Scholar]
- 19.Miettinen M, Makhlouf H, Sobin LH, et al. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol. 2006; 30:477–489. [DOI] [PubMed] [Google Scholar]
- 20.LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2013;34:3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pazarentzos E, Giannikopoulos P, Hrustanovic G, et al. Oncogenic activation of the PI3-kinase p110β isoform via the tumor-derived PIK3Cb(D1067V) kinase domain mutation. Oncogene. 2016;35: 1198–1205. [DOI] [PubMed] [Google Scholar]
- 22.Knobbe CB, Reifenberger G. Genetic alterations and aberrant expression of genes related to the phosphatidyl-inositol-3’-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003;13:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakanishi Y, Walter K, Spoerke JM, et al. Activating mutations in PIK3CB confer resistance to PI3K inhibition and define a novel oncogenic role for p110β. Cancer Res. 2016;76:1193–1203. [DOI] [PubMed] [Google Scholar]
- 24.Wataya-Kaneda M Mammalian target of rapamycin and tuberous sclerosis complex. J Dermatol Sci. 2015;79:93–100. [DOI] [PubMed] [Google Scholar]
- 25.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 2005;37:19–24. [DOI] [PubMed] [Google Scholar]
- 26.Kwiatkowski DJ, Choueiri TK, Fay AP, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2016;22:2445–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;chapter 7:unit 7.20:1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaguchi H, Kawazu M, Yasuda T, et al. Transforming somatic mutations of mammalian target of rapamycin kinase in human cancer. Cancer Sci. 2015;106:1687–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zarogoulidis P, Lampaki S, Turner JF, et al. mTOR pathway: a current, up-to-date mini-review. Oncol Lett. 2014;8:2367–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng Y, Jiang Y. mTOR inhibitors at a glance. Mol Cell Pharmacol. 2015;7:15–20. [PMC free article] [PubMed] [Google Scholar]