

Summary
Single-cell transcriptomics (scRNA-seq) has revolutionized the understanding of the spatial architecture of tissue structure and function. Advancing the “transcript-centric” view of scRNA-seq analyses is presently restricted by the limited resolution of proteomics and genome-wide techniques to analyze post-translational modifications. Here, by combining spatial cell sorting with transcriptomics and quantitative proteomics/phosphoproteomics, we established the spatially resolved proteome landscape of the liver endothelium, yielding deep mechanistic insight into zonated vascular signaling mechanisms. Phosphorylation of receptor tyrosine kinases was detected preferentially in the central vein area, resulting in an atypical enrichment of tyrosine phosphorylation. Prototypic biological validation identified Tie receptor signaling as a selective and specific regulator of vascular Wnt activity orchestrating angiocrine signaling, thereby controlling hepatocyte function during liver regeneration. Taken together, the study has yielded fundamental insight into the spatial organization of liver endothelial cell signaling. Spatial sorting may be employed as a universally adaptable strategy for multiomic analyses of scRNA-seq-defined cellular (sub)-populations.
Keywords: transcriptomics, proteomics, phosphoproteomics, liver endothelial cell (L-EC), vascular zonation, angiocrine factors, Tie1, Tie2, Wnt
Graphical abstract
Highlights
-
•
ScRNA-seq-guided spatial sort enables multiomic dissection of the liver vasculature
-
•
Liver sinusoidal endothelial cells have a hybrid vascular-lymphatic phenotype
-
•
Tyrosine phosphorylation of endothelial cell molecules is enriched on central vein
-
•
Endothelial Tie1 shapes hepatic Wnt signal zonation and promotes liver regeneration
Inverso, Shi et al. generate a multiomic encyclopedia of liver endothelial cells (L-ECs) with spatial resolution of transcriptome, proteome, and phosphoproteome. The study provides insight into liver vascular zonation and a template for scRNA-seq-data-guided spatial proteome and phosphoproteome analyses.
Introduction
The liver is endowed with a unique dual blood supply with oxygenated blood entering through the hepatic artery and hypoxic blood arriving from the intestine via the hepatic portal vein. Blood from both vessels merges in the liver capillaries, called sinusoids, to generate a low-oxygen and low-pressure system that eventually exits the liver via the centro-lobular vein (Gebhardt, 1992). Recent developments in single-cell biology have enabled the dissection of the complex transcriptomic heterogeneity of hepatocytes and liver endothelial cells (L-ECs) along the axis of the liver lobule from the portal to the central vein, referred to as “hepatic lobular zonation” (Halpern et al., 2018; Halpern et al., 2017). Beyond their rheological functions, L-ECs are now well established to exert instructive functions on the spatial organization of the liver parenchyma and in maintaining liver homeostasis: L-ECs do not just act as a filter regulating metabolite trafficking from the gut to hepatocytes, they also control the immune response to viral infections through their fenestrations independently of leukocyte extravasation (Guidotti et al., 2015) and, as shown recently, their onco-fetal reprogramming during tumor growth promotes an immune suppressive environment (Sharma et al., 2020).
Although much has been learned about instructive L-EC functions in recent years, the high-resolution molecular analysis of spatial signaling mechanisms is mostly limited to a transcript-centric view of gene regulation based on single-cell transcriptomic (scRNA-seq) analyses. Yet, while gene expression correlates for most molecules strongly with protein abundance, protein function and eventual biological outcome are regulated in multi-layered processes of post-translational modifications (PTM), which are not reflected in the current scRNA-seq-defined spatial organization and biochemical division of labor in the liver. This lack of information hampers our understanding of fundamental biological features of the liver. For example, although the strongly localized expression of short-range acting L-EC-derived Wnt ligands in the central vein area is well known (Rocha et al., 2015; Wang et al., 2015), the molecular determinants of this process are yet to be elucidated.
As proteomics and even more so phosphoproteomics are still beyond the boundaries of single-cell resolution (Marx, 2019), this study was aimed at overcoming these limitations by employing a spatial cell sorting strategy to enrich L-ECs from distinct zones of the hepatic lobule in combination with transcriptomic, proteomic, and phosphoproteomic analyses, thereby providing a draft of the anatomical organization of protein regulation in the liver vasculature. In doing so, we identified an unexpected zonation of protein activation and discovered the receptor tyrosine kinase Tie1 as a key regulator of hepatic vascular Wnt gradient and in sustaining efficient liver regeneration.
Results
Spatial multiomics of the liver endothelium
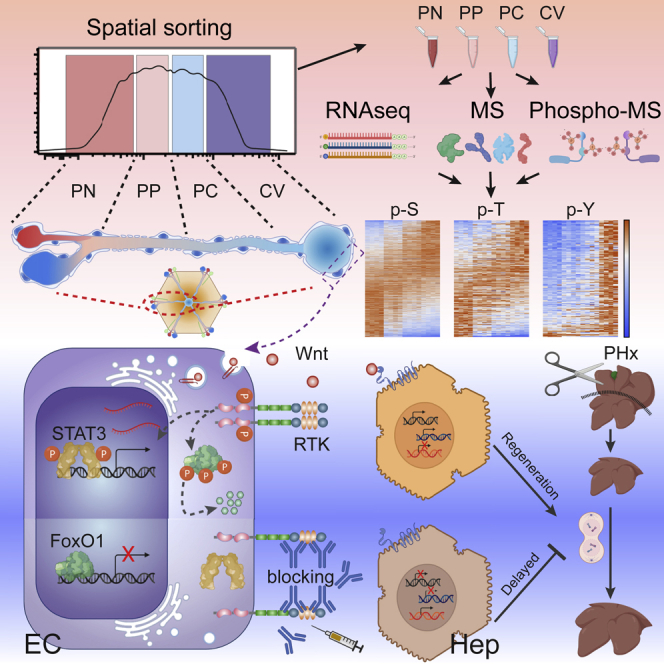
Pre-amplification of genetic material facilitates unparalleled sensitivity all the way to the single-cell level in high-throughput sequencing analyses (van Dijk et al., 2014). In turn, proteomic analyses by mass spectrometry (MS) inherently require much higher input material than transcriptomic studies (Marx, 2019). Phosphoproteomic analysis is even more demanding, as it requires a pre-enrichment of the phosphorylated peptides that otherwise represent only a minimal fraction of the total peptides subject to MS measurement (Fíla and Honys, 2012). To overcome these limitations and at the same time preserve tissue spatial resolution, we datamined the published L-EC scRNA-seq dataset for surface molecules that may enable the isolation of different L-EC subpopulations from consecutive sections of a liver vessel (Halpern et al., 2018). Based on the scRNA-seq-defined gradual increase of cKit expression along the portal-central liver lobule axis, we performed FACS analyses of CD146 magnetic bead-pre-purified L-ECs (purity approximately 95%) with a cKit antibody to sort 4 consecutive pools of L-ECs based on cKit expression intensity (portal node [PN], peri-portal [PP], peri-central [PC], and central vein [CV]) (Figures 1A, S1A, and S1B). Moreover, to standardize the gating strategy, we used another CV landmark gene, thrombomodulin (CD141), whose fluorescence intensity was proportionally increasing across the consecutive cKit gates (Figure S1B). RT-qPCR and western blot analysis of the four sorted subpopulations showed an almost linear gradient of both, cKit mRNA and protein (Figures 1B–1D) across the sorting gates, demonstrating the ability of the employed sorting strategy to maintain the spatial coordinates of the sorted L-ECs. Furthermore, immunostaining co-localization of cKit and glutamine synthetase (GS, to mark the CV area) further confirmed the cKit gradient from portal to central area in situ (Figure 1E).
Figure 1.

Spatial multiomics of the liver endothelium
(A) Scheme of spatial sorting strategy. L-ECs from the portal node to CV were sorted into four consecutive subpopulations depending on their cKit staining gradient. PN, portal node; PP, peri-portal; PC, peri-central; CV, central vein.
(B) Normalized expression of cKit mRNA from spatial sorted L-ECs determined by qPCR normalized to actin and further normalized to group mean. Each point represents an individual animal, dot colors indicate the samples from the same animal. Data are means ± SD.
(C) Expression of cKit protein from the same mice indicated in (B) determined by western blot (WB), normalized to α/β-tubulin and further normalized to group mean. Data are means ± SD.
(D) Representative WB images for (C).
(E) Representative immunofluorescent staining of cKit (red) and glutamine synthetase (GS, gray) on liver section. Central vein areas are indicated by GS staining, and blue circles indicate portal areas. Left: single fluorescence channel of cKit and GS; right: overlay image. Scale bar, 200 μm.
(F) Correlation of spatial sorting RNA-seq to scRNA-seq. RNA expression center-of-mass from single cell (x axis) and spatial sorting (y axis) RNA-seq of 48 genes significantly zonated in both datasets.
(G) Total number of detected genes (gray), proteins (blue), and phospho-sites (red). The left graphs show detected genes, proteins, and phospho-sites in each indicated sorting gate (n = 4). The circles on the right depict cumulative data from all gates.
See also Figure S1.
For comparative transcriptomic, proteomic, and phosphoproteomic analyses, L-ECs from 30 mice were pooled toward as one biological replicate for parallel processing (Tables S1, S2, and S3) with negligible contamination of non-EC populations (Figure S1C). By using the expression center-of-mass (CoM) as an overall zonation score to correlate the spatial information obtained by this approach to 48 zonated genes previously reported by an independent scRNA-seq study (Halpern et al., 2018), we found a nearly linear correlation (r = 0.87) (Figure 1F). Moreover, representative expression profiles of landmark genes showed mostly overlapping patterns (Figure S1D), demonstrating the robustness of the approach to overcome the sensitivity limitations of scRNA-seq, while fully preserving high spatial resolution.
The analytical procedure allowed an almost complete coverage of the L-EC transcriptome (28,727 genes) together with 5,015 proteins and 19,607 phosphosites (p-site) mapped to 3,447 proteins (Figure 1G). Phosphorylation occurred almost exclusively on serine (S), threonine (T), and tyrosine (Y) residues (Sharma et al., 2014), which represented 77%, 20%, and 3% of the identified p-sites, respectively (Figure 1G). Moreover, the comparable coverage across the sorting gates further substantiated the reproducibility of the spatial sorting strategy (Figure 1G). By comparing the differential expression across the four sorted populations, we defined the zonation patterns of all L-EC transcripts, thousands of proteins and the corresponding phosphosites, establishing a comprehensive spatially resolved multiomic map of the protein expression and phosphorylation in a vasculature bed, which is available as an interactive web-tool at https://pproteomedb.dkfz.de for the research community to visualize and further explore the data.
Transcriptome zonation defines distinct L-EC signatures
While previous scRNA-seq-based spatial analysis had identified ∼1,300-L-EC-specific genes, of which 475 were zonated (Halpern et al., 2018), RNA-seq of sorted bulk populations identified more than 28,000 genes. Of the quantitatively analyzed 13,737 genes, 4,943 were significantly zonated in their expression pattern (Figure 2A). The large number of zonated genes thereby allowed to define additional expression patterns not only limited to genes polarized either on portal or central side but also genes enriched in the mid-layers (sinusoids) or in large vessels (Figures 2B–2D). Notably, we identified 365 zonated transcription factors (TFs) (Figures S2A and S2B), including those specifying arteries and veins, Hey1 and Sox7 (Niklason and Dai, 2018), with portal or central expression pattern, respectively (Figure 2E). Intriguingly, the portal zonation of the lymphatic fate determining TF Sox18 (Figure 2E), as well as the lymphatic identity markers VEGFR3 and Lyve1 were enriched in the sinusoidal area (Figures 2C and 2D), delineating a unique hybrid phenotype of liver sinusoidal EC (LSEC) between lymphatic and vascular EC (Niklason and Dai, 2018; Tanaka and Iwakiri, 2016). Furthermore, 74% (381/515) of known kinases (Figures S2C and S2D) and the majority of phosphatases (98/127; Figures S2E and S2F) were expressed, indicating hitherto unappreciated signaling activities in L-ECs (Figure S3). Collectively, the bulk analysis of spatially sorted L-EC populations was capable of increasing the spatial resolution of previous scRNA-seq-defined zonation by an order of magnitude, revealing that approximately one-third of quantifiable L-EC transcripts were, in fact, expressed in a zonated manner.
Figure 2.

Transcriptome zonation defines distinct L-EC signatures
(A) Heatmap representation of the expression profiles of 4,943 genes with significantly zonated expression. Genes are normalized to their maximum expression and sorted by their center-of-mass. Representative central and portal zonated genes are indicated in blue and red, respectively (n = 4).
(B) Expression profiles of 890 genes significantly zonated on large vessels or sinusoids. Genes are normalized to their maximum expression and sorted by their log2 fold change of vessel to sinusoid. Representative vessel and sinusoidal zonated genes are indicated in orange and green, respectively (n = 4).
(C) Representative gene expression profiles for each zonation pattern (as indicated by color) with the corresponding qRT-PCR validation (black). Gene expression is represented by percentage of maximum; patches represent SD (n = 4).
(D) Representative liver immunofluorescence staining for large vessel and sinusoidal zonation patterns. CD31 in gray represents large vessel zonation and Lyve-1 in red represents sinusoidal zonation. Scale bars, 100 μm (left), 50 μm (right).
(E) Expression profiles of representative transcription factors zonated in the L-ECs. Gene expression is represented by percentage of maximum; patches represent SD (n = 4); color scale indicates relative expression of individual samples as in (A).
See also Figures S2 and S3.
Post-transcriptional regulation of protein abundance
Each gene needs to be expressed with an appropriate protein copy number to exert its function, which is regulated by synthesis (transcription and translation) and decay (dilution and degradation) (Hausser et al., 2019). In general, it is thought that the transcriptome reflects the proteome. To check if this assumption is valid for L-ECs, proteome and transcriptome were correlated, yielding a total of 4,169 protein-mRNA pairs. Despite a general positive correlation of RNA and protein abundance, similar transcript abundance could lead to proteins with different abundance in a range of 1,000-fold, reflecting a relevant difference of synthesis and/or decay for different proteins (Figure 3A).
Figure 3.

Post-transcriptional regulation of the protein abundance
(A) Scatterplot of protein and RNA abundance (mean expression across all zones). Red and blue mark the genes with a high and low PTR as defined in (B and C).
(B) Protein-to-transcript ratio (PTR) definition. Regulation on synthesis/decay, which contributes toward high or low PTR are indicated with arrows marked with blue and red, respectively.
(C) Distribution of PTR values of protein-RNA pairs. The red line indicates the median and the red and blue overlay display ±1 SD from the median, defined as high or low PTR range.
(D) Dot plot of the KEGG pathways significantly enriched in the gene sets corresponding to low or high PTR. Pathways (y axis) are ordered from low to high PTR by increasing median PTR value (x axis) of the proteins enriched in the pathway. Dot size and color indicate gene count and −log10 FDR for each pathway, respectively.
(E and F) Interaction network of low (E) and high (F) PTR proteins. Interaction was based on STRING and visualized by Gephi. Node size is proportional to the protein abundance (LFQ) and the edge weight is proportional to the combined interaction score. Proteins (node) and the related interaction (edge) belonging to selected pathways were highlighted as indicated in the figures.
The differential expression of protein and RNA is reflected in different protein-to-transcript ratios (PTR) (Mergner et al., 2020) (Figure 3B), which followed a Gaussian distribution with a shift toward high PTR (Figure 3C). We defined the range for the high and low PTR as one standard deviation distance to the median value and observed 3-fold more high PTR proteins than low PTR proteins (Figure 3C). This observation was consistent with the notion that low PTR genes are evolutionary disadvantaged, as this dynamic requires an increased RNA transcription and, therefore, a higher energy consumption for the protein synthesis (Hausser et al., 2019). Genes with low or high PTR were enriched for different pathways and segregated into different protein networks (Figure 3D; Table S4). Notably, ribosome component proteins formed a prominent cluster among the low PTR proteins (Figures 3D and 3E), indicative of a large RNA reserve for ribosomal proteins (Schwanhäusser et al., 2011). Conversely, high PTR values were detected for proteins involved in metabolism and biosynthesis, implying optimized translational rates and/or protein stability for fundamental cellular activities (Figures 3D and 3F).
Spatial proteomics reveals a differential phosphorylation along the liver vasculature
Similar to the bulk RNA-seq experiments, we next analyzed the spatial distribution of protein expression along the liver sinusoids. In line with the RNA zonation, 25% of the quantified L-EC proteome was found to be expressed zonated along the axis of the liver lobule (Figure 4A). Interestingly, cytochrome P450 pathway molecules were enriched on the portal side (Figures 4B, S4A, and S4B; Table S4), which was in marked contrast to the previously reported zonation pattern of hepatocytes (Ben-Moshe et al., 2019), highlighting a heterogeneous metabolic behavior of different liver cell populations despite their anatomical proximity. Of note, abundant expression of prototypic EC pathway genes (e.g., cell adhesion molecules, tight junction, and trans-endothelial migration [Figures 4C, S4C, and S4D; Table S4]) was enriched in large vessel EC compared with sinusoidal EC, further supporting the notion that liver sinusoids may be a highly specialized EC population with a unique hybrid phenotype. Indeed, this expression pattern may appear contradictory to the fact that many cells are preferentially recruited to the liver sinusoids (Inverso and Iannacone, 2016). However, it was shown that sinusoidal recruitment of neutrophils and CD8 T cells often occurs independently of major homing molecules (Guidotti et al., 2015; McDonald et al., 2008), which is consistent with the observed low expression of these pathways in the sinusoidal area.
Figure 4.

Spatial proteomics reveals a differential phosphorylation along the liver vasculature
(A–C) Heatmap representation of the expression profiles of 1,042 proteins with significantly zonated expression (A), “cytochrome P450” (KEGG pathway mmu00982) (B) and “cell adhesion molecules” (KEGG pathway mmu04514) (C). Proteins are normalized to their maximum expression and sorted by their center-of-mass (A and B) or their vessel to sinusoid log2 fold change (C) (n = 4).
(D) Zonation shift of the protein-RNA pairs. ΔCoM is the difference between the overall protein CoM and RNA CoM, indicative for the zonation shift. Unpaired Student’s t test was used to determine the difference between the protein CoM values from the four biological replicates and the four RNA CoM values. Red dots in the scatter dot plot mark significantly shifted genes, and their percentages were indicated above.
(E) Expression profiles of the indicated proteins-RNA pairs. Expression is represented by percentage of maximum; patches represent SD (n = 4).
(F) Heatmap representation (as described in A) of the expression profiles of 2,828 p-peptides with significantly zonated expression (n = 4).
(G) Scatter dot plot of the ΔCoM and the log10 p value of 7,520 p-peptide-protein pairs, showing zonation shift of p-peptide to protein. Red dots mark significantly shifted proteins.
(H) Expression profiles of the indicated matches of p-peptides (red), proteins (blue), and RNA (black). Expression is represented by percentage of maximum; patches represent SD (n = 4).
(I) Bar graph of the SMART protein domains significantly enriched for portal (red) or for central (blue) zonated phosphorylated proteins. Domains (x axis) are ordered from portal to central by increasing median center-of-mass (y axis) of the proteins enriched for the domain. Bar color indicates the FDR range for the enrichment score.
See also Figure S4.
The variance in PTR values suggested that a different efficiency in RNA translation or protein stability could act as a major regulatory mechanism of protein zonation along the liver sinusoids. In order to define the determinants for protein zonation, we statistically analyzed the zonation shift for each RNA-protein pair by comparing protein and RNA CoM (ΔCoM) in the four biological samples to evaluate if there was a significant zonation shift between the protein and the relative RNA (Figure 4D; Table S5). Indeed, while the vast majority of analyzed proteins exhibited zonation patterns corresponding to their relative transcript zonation, 8% of L-EC proteins were, in their zonation pattern, not concordantly regulated on the mRNA and protein level (Figures 4D and 4E), indicative of post-transcriptional mechanisms regulating protein levels.
The function of many proteins, most notably of molecules involved in signaling, is not primarily regulated by their abundance but rather by their activation state, which is determined by PTM, such as protein phosphorylation (Huttlin et al., 2010). Due to technical limitations and the reversible and sub-stoichiometric nature of signaling events, deep phosphoproteomic analysis remains challenging, particularly when tissue dissociation is a pre-requisite for analysis. Applying the dedicated spatial L-EC isolation workflow, we next performed phosphoproteomic analyses on the same samples that had been employed to establish spatial transcriptomic and proteomic zonation maps. Indeed, this approach enabled the establishment of a comprehensive in vivo phosphoproteomic map of L-EC signaling zonation (Figure 4F; Table S3). Phosphorylation motif analysis (Hornbeck et al., 2015) identified 116 conserved motifs belonging to four categories (proline-directed, acidic, basic, and other) (Villén et al., 2007) (Figure S4E; Table S6). Both categorical classification (Figure S4E) and consensus sequences (Figure S4F) showed high similarities between phospho-serine (p-S) and phospho-threonine (p-T) and marked discrepancies to phospho-tyrosine (p-Y), likely reflecting substrate differences between serine/threonine- and tyrosine kinases. Notably, the consensus sequence from “other” motifs of p-Y was reminiscent of acidic motifs, indicating a possible acidic prone tropism of the L-EC tyrosine-kinome.
Analysis of the spatial distribution of class-I p-peptides revealed significant zonation for 25% of the identified peptides (Figure 4F), which was in line with overall protein zonation. Interestingly, comparing CoM of 7,520 p-peptide-protein pairs, we found a significant zonation shift for 16% of the pairs (Figures 4G and 4H; Table S5), compared with 8% observed for protein-RNA pairs, identifying differential phosphorylation along the sinusoid as a major regulator of protein function. Lastly, SMART protein domain analysis (Letunic and Bork, 2018) revealed a strong enrichment of the tyrosine-kinase catalytic domain restricted to the central area, whereas the serine/threonine catalytic domain was equally represented across the zones (Figure 4I; Table S4). Collectively, these data show that most of the L-EC proteome zonation reflected the spatial distribution of the relative RNA. Still there was about 8% of the proteins with a significant post-transcriptional regulation that led to a different expression pattern between protein and RNA. Moreover, the finding that ∼16% of the detected phosphoprotein had a significant zonation shift compared with the associated protein indicated that a spatial gradient of the phosphorylation status was a major determinant of the L-EC functional signature along the liver sinusoids.
Peri-central compartmentalization of tyrosine phosphorylation
To further characterize the spatial arrangement of protein phosphorylation, we analyzed the distribution of the 3 major phospho-sites. In line with our previous findings, the zonation of p-S and p-T occurrence were equally distributed across the zones, whereas zonated p-Y was restricted at the CV (Figure 5A). This strong enrichment of tyrosine phosphorylation could be a consequence of increased phosphorylation rate or could reflect a similar zonation of the corresponding protein. Comparing the expression profile of the 171 zonated p-Y sites (Figures S5 and S6A) and the relative CoM with the respective protein, 133 out of 171 p-peptides displayed a shifted expression pattern (Figures 5B and 5C), indicating that tyrosine phosphorylation was zonated largely irrespective of the amount of protein.
Figure 5.

Peri-central compartmentalization of tyrosine phosphorylation
(A) Heatmap representations of significantly zonated phospho-serine (P-S), phospho-threonine (P-T), and phospho-tyrosine (P-Y). P-peptides are normalized to their maximum expression and sorted by their center-of-mass (n = 4).
(B and C) Variation of the zonation score of p-Y peptides and corresponding proteins.
(B) Aligned dot plot of the CoM relative to p-Y peptides and corresponding proteins. Before-after connecting lines indicate a shift to central (red) or to portal (blue).
(C) Scatter dot plot of the same groups represented in (B). Data are represented as mean ± SD.
(D and E) Distribution of all class-I p-S, p-T, and p-Y for their strongest expressing zone. Each p-site was assigned to one zone according to the maximum expression, by average expression of four biological replicates (D) or for each replicate (E). Afterward, distribution was calculated for each zone and shown as pie charts (D) or connecting lines (E) with patches indicating SD of the four replicates.
(F) Kinome phylogenetic tree of phosphorylated kinases. Each kinase is represented by a circle and grouped by kinases family. The circle size is proportional to the corresponding TPM. The color represents phosphorylation zonation from portal (red) to central (blue).
(G) Expression profiles of matches of p-peptides (red or orange), proteins (blue), and RNA (black) for the indicated receptor tyrosine kinases. Expression is represented by percentage of maximum; patches represent SD (n = 4).
See also Figures S5 and S6.
The enrichment of p-Y in CV L-ECs could also be observed at the level of relative overall abundance of the three phospho-sites across the zones. Although the area spanning from PN to PC displayed a p-Y percentage (2%–5%) that was in line with previous reports (Huttlin et al., 2010; Van Hoof et al., 2009), the CV fraction was found strongly enriched in p-Y phosphorylation (10%) (Figures 5D and 5E). These data support the idea of an increased tyrosine phosphorylation on the L-ECs surrounding the CV. Consistently, phylogenetic tree analysis of the L-EC kinome (Metz et al., 2018) revealed that receptor tyrosine kinases (RTKs) were the most abundantly expressed kinase family in L-ECs and that phosphorylation of RTK predominantly occurred at the central region, which was in contrast to the portal pattern of serine-threonine kinases (Figures 5F, 5G, and S6B). Collectively, the data showed a sharp compartmentalization of the phosphorylation activity within the liver lobule, with the CV being characterized by a pronounced RTK activity.
CV phosphorylation of the tyrosine kinase Tie1 shapes L-EC zonation and establishes a Wnt9b gradient
When zooming in on individual vascular RTKs, the angiopoietin receptors Tie1 and Tie2/TEK were identified among the top-zonated p-proteins, despite their homogeneous mRNA and protein levels along the axis of the liver lobule (Figure 5G), suggesting a highly localized activation of this pathway and possibly a regulatory role in liver zonation. Therefore, we performed pathway-blocking experiments using a Tie1-function-blocking antibody (Tie1-39) (Singhal et al., 2020). Indeed, systemic treatment of mice with Tie1-39 led within 2 h to detectable transcriptomic changes in L-ECs, which were more pronounced in CV compared with PN L-ECs (Figures 6A and 6B; Table S7). Most notably, the expression of the CV landmark gene Wnt9b was almost completely shut-off (Figures 6C, 6D, and S7A), identifying Tie receptor signaling as a critical regulator of vascular Wnt expression.
Figure 6.

CV phosphorylation of the tyrosine kinase Tie1 shapes L-EC zonation and maintains Wnt9b gradient
(A) Differential gene expression induced by Tie1 blockade. Volcano plots of gene regulations 2 h after Tie1 blockade in spatially sorted L-ECs from portal node (left) and central vein (right), respectively. Red dots mark the significantly regulated genes, indicated by the number in each square.
(B) Histogram of the −log10 q value distribution of regulated genes in portal node and/or central vein 2 h after Tie1 blockade. The effect of Tie1 blockade on PN and CV was compared by Wilcoxon matched-pairs signed rank test of the −log10 q values.
(C) RNA fluorescence in situ hybridization (FISH) analysis of Wnt9b RNA (red) 2 h after treatment of anti-Tie1 antibody, compared with IgG control. Endothelial cells were visualized by Pecam1 RNA (green) FISH staining, CV areas by glutamine synthetase (GS, gray) immunostaining, and cell nuclei (blue) counterstained with DAPI. (i) Overlay image of central vein area; (ii–iv) Zoomed overlay image (ii), Pecam1 RNA (iii), and Wnt9b RNA (iv) of the area indicated in (i). Arrow heads indicate Wnt9b RNA staining. Scale bars, 20 μm (i) and 5 μm (ii–iv).
(D) RNA expression of Wnt9b in the whole liver tissues from anti-Tie1 Ab-treated mice at the indicated time points, normalized to the relative IgG-treated mice (dashed line), significantly regulated time points highlighted in red.
(E) Signaling scheme of FoxO1 and STAT3 activation and nuclear translocation with inactive (top) or active (bottom) RTK signaling.
(F and G) RNA expression of Wnt9b in Stat3iECKO (F) and Foxo1iECKO (G) mice (red bar) normalized to the relative control mice (Cre- littermates, gray bar) from isolated L-ECs.
(C, F and G) RNA expression was determined by qRT-PCR and normalized to Actb. Data are expressed as percentage normalized to the corresponding controls. Each data point represents one animal. Data are means ± SD. Unpaired Student’s t test was used to determine the difference between experimental groups. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
See also Figure S7.
CV-derived Wnt ligands play a key role in the angiocrine regulation of liver zonation (Wang et al., 2015). Indeed, the EC-specific genetic inactivation of the Wnt signaling enhancer Rspo3 abrogates hepatocyte zonation (Rocha et al., 2015). The rapid regulation of L-EC Wnt9b expression consequently prompted us to hypothesize that vascular Tie receptor signaling could act as a key regulator of maintaining liver homeostasis in a Wnt signaling-dependent manner. Temporal analysis of the effect of systemic Tie1 blocking antibody application identified the rapid and transient downregulation of Wnt9b in L-ECs (Figure 6D). Moreover, the regulation of L-EC Wnt9b expression was highly specific for Tie receptor signaling, since neither antibody blockade of VEGFR-2, VEGFR-3, Dll4, integrins-αV, integrin-α5, or PECAM1 in vivo had a similar effect on L-EC Wnt9b expression (Figures S7B–S7E).
The rapid response upon Tie1 blockade suggested direct transcriptional regulation of Wnt9b mediated by Tie1 signaling. In silico analysis of the Wnt9b promoter region for the typical Tie receptor signaling effector molecules revealed putative binding sites for FoxO1 and STAT3 (Figure S7F). However, phosphorylation of FoxO1 and STAT3 has distinctly opposing functional consequences: whereas STAT3 activation promotes nuclear translocation and STAT3-dependent transcription, FoxO1 phosphorylation leads to nuclear exclusion and inactivation of FoxO1-dependent transcription (Figure 6E) (Farhan et al., 2017; Huynh et al., 2019). The potential involvement of both, STAT3- and FoxO1, consequently suggested a fine-tuned balance of the TFs in Tie receptor signaling regulation of Wnt9b expression. Indeed, the in vivo EC-specific conditional genetic inactivation of Stat3 or Foxo1 (Figures S7G and S7H) had opposing effects on L-EC Wnt9b expression (Figures 6F and 6G), validating that STAT3 promoted Wnt9b transcription, whereas FoxO1 acted as a Wnt9b transcriptional repressor. These findings identified distinct intracellular signaling circuits that control the localized vascular production of Wnt ligands within the liver.
Tie1-induced Wnt is required for liver regeneration
During homeostatic cell renewal, new hepatocytes derive from differentiated pre-existing cells (hepatocyte or cholangiocytes) instead of a well-defined stem cell compartment. This feature contributes to support the liver regenerative potential as differentiated cells proliferate in response to tissue loss until the original liver mass is restored (Fausto et al., 2006). Nevertheless, the peri-central Axin2+ Tbx3+ hepatocytes, maintained by locally produced EC-derived Wnt ligands, were shown to exhibit stem-cell-like functions to maintain the liver parenchyma (Wang et al., 2015). Similarly, Lgr5-positive cholangiocytes expand and differentiate during acute liver damage into mature hepatocytes starting from the peri-central area (Huch et al., 2013). As Tie1 specifically regulated L-EC Wnt9b expression, we focused on the contribution of this signaling axis toward liver regeneration. We conditionally inactivated Tie1 in EC (Tie1iECKO) and traced liver regeneration after two-thirds partial hepatectomy (PHx) (Figures 7A and 7B). Two days after PHx, expression of Wnt9b and Wnt2 was significantly reduced in liver lysates of PHx mice compared with control mice. Wnt ligands downregulation was specific for these L-EC-expressed Wnt ligands and not observed for non-endothelial Wnt ligands, including Wnt2b, Wnt4, Wnt 5a, Wnt5b, Wnt7b, Wnt 9a, and Wnt 11 (Figure 7C), furthermore substantiating the specificity of the angiocrine Tie-Wnt crosstalk axis. Consistently, the Wnt-responsive genes Axin2 and Tbx3 were downregulated along with the reduced Wnt expression in Tie1iECKO mice (Figures 7D and 7E). Similar downregulation occurred for Sox9 and Lgr5, defining other liver progenitor cells with high proliferative capacity (Han et al., 2019; Huch et al., 2013) (Figures 7F and 7G). As a consequence, liver regeneration was significantly impaired with a reduced liver-to-body ratio 2 days after surgery (Figure 7H). Lastly, the specificity of these findings was substantiated by Tie1-blocking antibody experiments during PHx, which phenocopied the genetic Tie1 endothelial inactivation experiment (Figures S7I and S7J). Together, these data showed that Tie1-signaling-dependent Wnt production is a critical determinant to sustain the liver regenerative niche and to restore liver mass after injury.
Figure 7.

Tie1-induced Wnt is required for liver regeneration
(A) Experimental schedule for inducible EC-specific knockout of Tie1 (Tie1iECKO) followed by two-thirds partial hepatectomy (PHx).
(B) mRNA expression of Tie1 from whole liver tissues 2 days after two-thirds PHx in Tie1iECKO mice and corresponding controls (Cre- littermates, gray bar).
(C) mRNA expression of Wnt ligands from whole liver tissues 2 days after two-thirds PHx in Tie1iECKO mice and the relative control mice (Cre- littermates, dashed line). Significantly regulated genes were highlighted in red.
(D–G) mRNA expression of Wnt target genes, Axin2 (D), Tbx3 (E), Sox9 (F), and Lgr5 (G) from whole liver tissue 2 days after two-thirds PHx in Tie1iECKO mice (red bar) and corresponding controls (Cre- littermates, gray bar).
(H) Liver-to-body ratio of Tie1iECKO (red bar) and relative controls (Cre- littermates, gray bar) at the indicated time points after two-thirds PHx.
(B–G) mRNA expression was determined by qRT-PCR and normalized to Actb. Data are expressed as percentage normalized to the corresponding controls. Each data point represents one animal. Data are means ± SD. Unpaired Student’s t test was used to determine the difference between experimental groups. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
See also Figure S7.
Discussion
The liver endothelium displays spatial and molecular heterogeneity along the axis of the liver lobule, facilitating its specialized angiocrine functions through which it controls adjacent hepatocytes. The endothelium thereby exerts gatekeeper roles in maintaining liver metabolic zonation (Rocha et al., 2015) and hepatic responses to pathologic challenge including liver regeneration, fibrosis, and cancer (Cao et al., 2017; Hu et al., 2014; Morse et al., 2019). We have, in this study, established comprehensive genome-wide transcriptomic, proteomic, and phosphoproteomic maps of liver endothelium, providing a spatially resolved analysis of protein signaling controlling the activity of individual L-EC molecules and pathways. Specifically, we show that (1) scRNA-seq-data-guided spatial sorting of functionally relevant cellular (sub)-populations can be employed as a powerful and versatile technique for high-resolution bulk multiomic analyses, including phosphoproteomics, (2) comparative RNA, and proteome analyses revealed a high degree of concordance, but up to 10% of transcribed genes were identified as significantly regulated on the post-transcriptional level, (3) phosphoproteomic analysis of spatially sorted cells yields high-resolution and most immediate insight into cellular phenotype and function on the systems and individual molecule level, and (4) prototypic functional exploitation of the data identified a selective and specific Tie-Wnt signaling axis as a critical regulator of vascular Wnt ligand production and angiocrine control of hepatocyte function.
The spatial sorting protocol of L-ECs in 4 different populations along the axis of the liver lobule, on which subsequent bulk transcriptome, proteome, and phosphoproteome analyses were based, was enabled only by retrieving information from previously established L-EC scRNA-seq data (Halpern et al., 2018). Bulk RNA-seq of sorted L-EC populations fully reproduced the spatial organization map as revealed by scRNA-seq. Yet, given the much higher sequencing depth enabled by the bulk approach, this study has yielded more than an order of magnitude higher resolution of L-EC zonation demonstrating that in fact one-third of the L-EC transcriptome is zonated.
ScRNA-seq is most insightful to identify the transcriptome of individual cells for applications aimed at studying the biology of individual cells, for example, in the field of stem cell biology. Yet, many, probably most biological studies in the scRNA-seq field are hitherto aimed at identifying (sub)-populations of molecularly and functionally similar cells. Building on this information, this study can probably serve as a prototypic template on how to overcome analytical limitations of scRNA-seq approaches: by datamining scRNA-seq data for FACS suitable surface molecules with biologically relevant spatial expression pattern, spatial sorting protocols of pre-purified cell populations can, in principle, be deduced from any tSNE or UMAP along any spatial anatomical or biochemical axis of interest. ScRNA-seq and spatial sorting bulk analyses thereby complement each other to apply the power of single-cell and bulk spatial resolution to enable proteomic and, as shown as proof of concept in this study, even phosphoproteomic analyses.
The full coverage of the L-EC transcriptome coupled with the spatial information and the proteome validation allowed us to unambiguously define expression pattern enriched on large vessels or in sinusoidal EC. Of note, sinusoidal EC are positioned between a vascular lumen (sinusoid) and a lymphatic-like space (space of Disse), which is in line with the atypical expression of lymphatic EC identity markers Vegfr3 and Lyve1 in the sinusoidal area but not on the neighboring large vessels. In turn, proteins characterizing typical vascular pathways (i.e., shear stress and cell adhesion molecule) were found polarized on portal and central EC but not in sinusoidal EC. Together, these findings delineate sinusoidal EC as a highly specialized cell population with a unique hybrid phenotype between lymphatic and vascular EC. The biological outcome of this mixed phenotype has important consequences such as a peculiar liver leukocyte trafficking paradigm. Indeed, hyaluronan accumulation in the sinusoidal area, together with the reduced expression of adhesion molecules, generates a docking site dependent on CD44-hyaluronan interaction reported for different cell subsets including neutrophils and effector T cells. Yet, different cell types, such as naive T cells (McNamara et al., 2017) and circulating NKT (Geissmann et al., 2005), require specific trafficking molecules such as LFA1 and CXCR6 to home the sinusoids. Of note, the recent finding that Kupffer cell and resident NKT are enriched on the portal area via CXCR3 (Gola et al., 2021) is confirmed by our data showing that CXCL9 (CXCR3 ligand) is significantly zonated on the portal L-ECs.
The comparative analysis of protein and RNA abundance revealed a large variance of PTR for different genes, indicative of a diverse regulation of protein biosynthesis and decay. PTR in general follows a Gaussian distribution, with ∼80% in a relative restricted range. However, several hundreds of genes strongly deviated from this range, defined as high or low PTR genes. From an evolutionary point of view, a high RNA pool for translation (i.e., lower PTR) reflects the fast adaptation to the cellular need to achieve an appropriate protein copy number. In turn, a high RNA reserve is energy demanding, evidenced by fewer low PTR proteins. Overall, the cell acquires an equilibrium between precise regulation and cost efficiency (Hausser et al., 2019). In line with this, we identified ribosome component proteins enriched among low PTR proteins, ensuring a large RNA reserve ready to adapt to intrinsic and extrinsic challenges, whereas metabolism-related processes are likely to have better protein stability to most cost efficiently maintain the basic cellular activities. Together, L-ECs exhibit a highly diverse and tightly coordinated regulation on post-transcriptional and post-translational levels to balance their energy consumption and adaptability. The determined phosphoproteomic map of L-EC spatial zonation allows to correlate typical expression data as mRNA and protein amount with a functional readout as the phosphorylation status and will serve as a unique resource for the vascular biology and hepatology communities (https://pproteomedb.dkfz.de).
Biologically, the probably most remarkable discovery was the strong enrichment of receptor tyrosine kinase signaling in the CV area of the endothelium. We identified several prominent vascular RTKs whose expression was transcriptionally and translationally not zonated but zonated only on the level of phosphorylation. Notably, the cooperating vascular RTKs Tie1 and Tie2 were discovered as activation-dependently regulated molecules. We retrieved this information from the phosphoproteomic map and prototypically validated it functionally. Indeed, in vivo application of a Tie1-blocking antibody followed by spatial transcriptomic analysis of peri-portal and peri-central L-ECs identified a more prominent gene regulation on CV L-ECs (157 regulated genes) compared with portal L-ECs (88 regulated genes), further validating the peri-central polarization of Tie1 signaling. Notably, Wnt9b and Lhx6, scoring among the top centrally zonated genes, were both identified as the most prominently regulated transcripts indicating that Tie1 signaling acts as a CV specifier. Further, in vivo blocking experiments revealed a high specificity of Tie signaling pathway regulation on L-EC Wnt ligand expression. Moreover, the finding that STAT3 and FoxO1, both known as typical RTK effector molecules, differentially regulated Wnt9b expression, defined an intricate intracellular signaling cascade regulating local Wnt production within the liver lobule. Indeed, phosphorylation of Stat3 and FoxO1 through Tie2 signaling (Li et al., 2019; Kim et al., 2016; Salih and Brunet, 2008) goes functionally in the same direction: Stat3 phosphorylation induces nuclear translocation and subsequent transcriptional activation. Conversely, Foxo1 phosphorylation leads to cytoplasmic translocation and inhibition of FoxO1-driven transcriptional activity. As such, the identified reciprocal regulation of Wnt9b expression by Stat3 and FoxO1 reflects concordant net effects of Tie2 activation on Wnt9b expression.
Validating the identified L-EC Tie-Wnt signaling axis, partial hepatectomy experiments in genetically engineered and antibody-challenged mice identified an essential angiocrine signaling role of the Tie-Wnt axis in liver regeneration. Given the prominent roles of vascular Wnt ligands in the regulation of angiocrine signaling, particularly in the liver along with the recent discovery of LECT2 as Tie1 ligand (Xu et al., 2019), the identification of a Tie-Wnt signaling axis probably hints at a more fundamental role of Tie receptor signaling in determining an organ-specific vascular Wnt code and thereby organ-specific angiocrine functions.
Taken together, the data presented here unambiguously identified tyrosine phosphorylation as a prominently zonated process, and that Tie1 phosphorylation acted as a specifier of the CV area L-EC signature and function. The upstream regulator(s) and the detailed biological consequences of this phosphorylation gradient await to be unraveled in future studies. Different micro-environmental factors may be responsible for the observed differential phosphorylation including differences in metabolite distribution, shear stress, and oxygen gradient along the axis of the sinusoids. Indeed, the liver is characterized by a dual blood supply with blood entering the liver via both hepatic artery (high pressure) and portal vein (low pressure) that generate a sudden drop of blood pressure at the merging point between the two vessels toward the sinusoids. This unique anatomical structure generates heterogeneous hemodynamic conditions along the sinusoids, where the EC probably translate different shear stress conditions into different activation and signaling pathways (Lorenz et al., 2018). Another consequence of the portal circulatory system is that the mixture of arterial and venous blood produces low oxygen levels in the sinusoids and a physiologic hypoxic area around the central lobular area. Lastly, the portal area is exposed to bacterial and metabolite products coming from the gut that contribute to MyD88 activation in the portal area, responsible for a specific homing molecule signature (Gola et al., 2021). Given the complexity of zonated signaling pathways controlling cellular crosstalk between parenchymal and non-parenchymal cells of the liver, the possibility to spatially relate gene expression data to functional biochemical readouts such as protein phosphorylation will be an important step toward a deep mechanistic understanding of the physiologically relevant factors and serve as a solid foundation to further explore pathological conditions.
Limitations of the study
While this study presented the first spatially resolved phosphoproteomic map of a vascular bed, the proteomic analysis depth is still not comparable with RNA-seq. Mass spectrometers usually cover a dynamic range of only 2–3 orders, whereas protein expression varies in a range of 5 to 12 orders of magnitude. Consequently, many low-abundance proteins were not detected, and their role is underappreciated. Future work could consider fractionation of proteins (e.g., SDS-PAGE or 2D gel electrophoresis), provided that sufficient amounts of material is available to begin with.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-α/β-Tubulin Antibody | Cell Signaling Technology | Cat# 2148S; RRID: AB_2288042 |
| Human monoclonal anti-CD141 (Thrombomodulin) PE | Miltenyi Biotec | Cat# 130-116-094; Clone REA964; RRID: AB_2727343 |
| Goat polyclonal anti-CD31 (PECAM1) | R&D Systems | Cat# AF3628; RRID: AB_2161028 |
| Rat monoclonal anti-CD31 (PECAM1) LEAF | BioLegend | Cat# 102412; Clone 390; RRID: AB_312907 |
| Rat monoclonal anti-CD31 (PECAM1) Ultra-LEAF | BioLegend | Cat# 102530; Clone MEC13.3; RRID: AB_2832293 |
| Rat monoclonal anti-CD31 (PECAM1) PE/Cyanine7 | BioLegend | Cat# 102524; Clone MEC13.3; RRID: AB_2572182 |
| Mouse monoclonal anti-CD45.2 Alexa Fluor 488 | BioLegend | Cat# 109816; Clone 104; RRID: AB_492868 |
| Rabbit monoclonal anti-cKit (CD117) | Abcam | Cat# ab256345; Clone EPR22566-344; RRID: AB_2891166 |
| Rat monoclonal anti-cKit (CD117) APC | BioLegend | Cat# 105812; Clone 2B8; RRID: AB_313221 |
| Hamster monoclonal anti-Delta-like protein 4 (DLL4) | Bio X Cell | Cat# BE0127;Clone HMD4-2; RRID: AB_10950366 |
| Rabbit polyclonal anti-Glutamine Synthetase (GS) | Abcam | Cat# ab49873; RRID: AB_880241 |
| Rat monoclonal anti-Integrin-αV (CD51) Ultra-LEAF | BioLegend | Cat# 104110; Clone RMV-7; RRID: AB_2819798 |
| Rat monoclonal anti-Integrin-α5 (CD49e) Ultra-LEAF | BioLegend | Cat# 103910; Clone HMα5-1; RRID: AB_2832321 |
| Rabbit Polyclonal anti-LYVE-1 | Novus Biologicals | Cat# NB600-1008; RRID: AB_10000497 |
| Humanized monoclonal anti-Tie1 | Eli Lilly and Company | Clone Tie1-39 |
| Rat monoclonal anti-VEGFR2 | Bio X Cell | Cat# BP0060; Clone DC101; RRID: AB_1107766 |
| Hamster monoclonal anti-VEGFR3 | BioLegend | Cat# 140902; Clone AFL4; RRID: AB_10680790 |
| Donkey anti-rabbit IgG (H+L) Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Donkey anti-goat IgG (H+L) Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11057; RRID: AB_2534104 |
| Goat polyclonal anti-Rabbit IgG HRP | Dako | Cat# P0448; RRID: AB_2617138 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Benzonase Nuclease | Merck Millipore | Cat# 70746; CAS# 9025-65-4 |
| Blue Loading Buffer Pack | Cell Signaling Technology | Cat# 7722 |
| Fluorescence Mounting Medium | Agilent Dako | Cat# S302380-2 |
| FxCycle Violet Stain | Thermo Fisher Scientific | Cat# F10347 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| Paraformaldehyde (PFA) | Sigma-Aldrich | Cat# P6148; CAS# 30525-89-4 |
| Pierce Lys-C Protease, MS Grade | Thermo Fisher Scientific | Cat# 90307 |
| Pierce Phosphatase Inhibitor Mini Tablets | Thermo Fisher Scientific | Cat# A32957 |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | Cat# 90058 |
| Protease-Inhibitor Mix HP | SERVA | Cat# 39106.03 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat# 89900 |
| Sucrose | Sigma-Aldrich | Cat# 84100; CAS# 57-50-1 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648; CAS# 10540-29-1 |
| Target Retrieval Solution, pH 6 | Agilent Dako | Cat# S1699 |
| Critical Commercial Assays | ||
| Arcturus PicoPure RNA Isolation Kit | Thermo Fisher Scientific | Cat# KIT0214 |
| DirectPCR DNA Extraction System | VWR | Cat# 732-3256 |
| DNA 1000 Kit | Agilent | Cat# 5067-1504 |
| GenElute Mammalian Total RNA Purification Kit | Sigma-Aldrich | Cat# RTN350 |
| Micro BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23235 |
| Mouse cKit (CD117) ELISA kit | Gentaur | Cat# EKC37351 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat# 4368708 |
| Proteinase K | Gerbu | Cat# 1344 |
| Proteome Profiler Mouse Phospho-RTK Array Kit | R&D Systems | Cat# ARY014 |
| QuantiTect Rev. Transcription Kit | Qiagen | Cat# 205313 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32854 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32852 |
| RedTaq ReadyMix PCR Reaction Mix | Sigma-Aldrich | Cat# R2523-100RXN |
| RNase-Free DNase Set | Qiagen | Cat# 79256 |
| RNA 6000 Pico Kit | Agilent | Cat# 5067-1513 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| TaqMan Fast Advanced PCR Master Mix | Thermo Fisher Scientific | Cat# 4444965 |
| TruSeq Stranded mRNA Library Prep | Illumina | Cat# 20020594 |
| ViewRNA Tissue Assay Core Kit | Thermo Fisher Scientific | Cat# 19931 |
| ViewRNA Tissue Probe Set - Wnt9b | Thermo Fisher Scientific | Cat# VX-06; Assay ID: VB1-15880-VT |
| ViewRNA Tissue Probe Set – CD31 (Pecam1) | Thermo Fisher Scientific | Cat# VX-06; Assay ID: VB6-12921-VT |
| Zenon Rabbit IgG Labeling Kit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# Z25302 |
| Deposited Data | ||
| Hepatocyte transcriptomic data | Halpern et al., 2017 | GEO: GSE84498 |
| Genome Reference Consortium Mouse Build 38, GRCm38 | NCBI Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Mouse liver single cell RNA-seq | Xiong et al., 2019 | GEO: GSE129516 |
| Mouse mesenchymal cell data | Dobie et al., 2019 | GEO: GSE137720 |
| Paired-cell sequencing data | Halpern et al., 2018 | GEO: GSE108561 |
| Reference Proteomes - Mus musculus (Mouse) | Uniprot | Uniprot: UP000000589 |
| Spatial sorting L-EC transcriptomic data | This paper | GEO: GSE155797 |
| Spatial sorting L-EC label free proteomic data | This paper | PRIDE: PXD020760 |
| Spatial sorting L-EC phosphoproteomic data | This paper | PRIDE: PXD020805 |
| Experimental Models: Organisms/Strains | ||
| Mouse strain Foxo1tm1Rdp (Foxo1 floxed) | Paik et al., 2007 | MGI:3698867 |
| Mouse strain Stat3tm2Aki (Stat3 floxed) | Takeda et al., 1998 | MGI:1926816 |
| Mouse strain Tie1tm1.1Scba (Tie1 floxed) | Qu et al., 2010 | MGI:4441288 |
| Mouse strain Tg(Cdh5-cre/ERT2)1Rha | Wang et al., 2010 | MGI:3848982 |
| Mouse strain wild type C57BL/6 | Janvier Labs | N/A |
| Oligonucleotides | ||
| Actin-forward primer (Cdh5-cre/ERT2 genotyping PCR control): CAATGGTAGGCTCACTCTGGGA GATGATA |
eurofins | N/A |
| Actin-reverse primer (Cdh5-cre/ERT2 genotyping PCR control): AACACACACTGGCAGGACTG GCTAGG |
eurofins | N/A |
| Cre-forward primer (Cdh5-cre/ERT2 genotyping): GCCTGCATTACCGGTCGATGCAACGA | eurofins | N/A |
| Cre-reverse primer (Cdh5-cre/ERT2 genotyping): GTGGCAGATGGCGCGGCAACACCATT | eurofins | N/A |
| Foxo1ckA primer (Foxo1 floxed genotyping): GCTTAGAGCAGAGATGTTCTCACATT |
eurofins | N/A |
| Foxo1ckB primer (Foxo1 floxed genotyping): CCAGAGTCTTTGTATCAGGCAAATAA | eurofins | N/A |
| Foxo1ckD primer (Foxo1 floxed genotyping): CAAGTCCATTAATTCAGCACATTGA | eurofins | N/A |
| Stat3-flox-forward primer (Stat3 floxed genotyping): CCTGAAGACCAAGTTCATCTGTGTGAC | eurofins | N/A |
| Stat3-flox-reverse primer (Stat3floxed genotyping): CACACAAGCCATCAAACTCTGGTCTCC | eurofins | N/A |
| Tie1-flox-forward primer (Tie1 floxed genotyping): ATGCCTGTTCTATTTATTTTTCCAG | eurofins | N/A |
| Tie1-flox-reverse primer (Tie1 floxed genotyping): TCGGGCGCGTTCAGAGTGGTAT | eurofins | N/A |
| Cxcl9-forward (SybrGreen qPCR primer): CTTCGAGGAACCCTAGTGATAAGG | eurofins | N/A |
| Cxcl9-reverse (SybrGreen qPCR primer): CCTCGGCTGGTGCTGATG |
eurofins | N/A |
| Ace2 (Mm01159006_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Actb (Mm00607939_S1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Angpt2 (Mm00545822_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Axin2 (Mm00443610_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| CD9 (Mm00514275_g1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Cdk1 (Mm00772472_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Esm1 (Mm00469953_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Fgfr2 (Mm01269930_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Foxo1 (Mm00490671_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| IL33 (Mm00505403_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Lcp2 (Mm01187570_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Lgr5 (Mm00438890_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Lhx6 (Mm01333348_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Lyve1 (Mm00475056_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| PDGFb (Mm00440677_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Peg10 (Mm01167724_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| SDC1 (Mm00448918_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Sox9 (Mm00448840_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| STAT3 (Mm01219775_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Tbx3 (Mm01195726_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Tie1 (Mm00441786_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| TIMP3 (Mm00441826_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| VEGFR3 (Flt4) (Mm01292604_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt11 (Mm00437328_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt2 (Mm00470018_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt2b (Mm00437330_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt4 (Mm01194003_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt5a (Mm00437347_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt5b (Mm01183986_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt7b (Mm01301717_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt9a (Mm00460518_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Wnt9b (Mm00457102_m1) TaqMan probe | Thermo Fisher Scientific | Cat#4331182 |
| Software and Algorithms | ||
| Biorender | Biorender.com | https://biorender.com/ |
| FACSDiva | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| Galaxy | Afgan et al., 2018 | (DKFZ internal Galaxy instance) http://dkfzgalaxy/ |
| Gephi | Bastian et al., 2009 | https://gephi.org/ |
| Gitools | Perez-Llamas and Lopez-Bigas, 2011 | http://www.gitools.org/ |
| Graph Pad Prism (v8.0) | Graph Pad | https://www.graphpad.com/scientific-software/prism/ |
| Illustrator | Adobe | https://www.adobe.com/de/products/illustrator.html |
| Imaris | Bitplane | https://imaris.oxinst.com/ |
| Leica Application Suite X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Light Cycler 480 software | Roche | https://lifescience.roche.com/en_de/products/lightcycler14301-480-software-version-15.html |
| Rstudio | RStudio-Team, 2020 | http://www.rstudio.com/ |
| Other | ||
| Resource website for the spatial multiomic data | This paper | http://pproteomedb.dkfz.de/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hellmut G. Augustin (augustin@angioscience.de)
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNAseq FASTAQ files generated during this study are available at the Gene Expression Omnibus (GEO) repository (accession number GEO: GSE155797). The proteomic and phosphoproteomic thermo.raw files are available at the PRIDE database (accession number PRIDE: PXD020760 and PRIDE: PXD020805). All data are also available from the corresponding authors on reasonable request.
Experimental model and subject details
Mice
Tie1tm1.1Scba (MGI:4441288)(Qu et al., 2010), Stat3tm2Aki (MGI:1926816) (Takeda et al., 1998), or Foxo1tm1Rdp (MGI:3698867) (Paik et al., 2007) transgenic mice, carrying floxed alleles of Tie1, Stat3 or Foxo1, respectively, were crossed with Tg(Cdh5-cre/ERT2)1Rha mice (MGI:3848982) (Wang et al., 2010) expressing tamoxifen inducible Cre recombinase under the Cdh5 promoter, to obtain inducible endothelial specific knock out (iECKO) mice for Tie1, Stat3 and Foxo1. All strains were backcrossed to C57BL/6 background. Wild type C57BL/6 mice were purchased from Janvier Labs. Mice were housed at the Laboratory Animal Facility in the German Cancer Research Center (DKFZ) under specific pathogen-free conditions. All animal experiments were approved by the institutional and governmental Animal Care and Use Committees from Regierungspräsidium Karlsruhe, Germany. All experiments were performed in accordance with the institutional guidance for the care and use of laboratory animals. All genotyping was done by PCR. Age-matched male mice were used for all experiments.
Method details
In vivo mouse experiments
Tamoxifen treatment
To induce EC-specific gene deletions, mice were intraperitoneally injected with 2 mg/mouse of tamoxifen (Merck) dissolved in 100 μl of peanut oil (Merck) for 5 consecutive days at 4-6 weeks. Treated mice were used for experiments after adulthood (8-10 weeks). Littermates of Cre- genotypes were used as control for Cre+ experimental group.
Partial hepatectomy
Two-third partial hepatectomy (PHx) was performed according to the methods described by Mitchell and Willenbring to induce liver regeneration (Mitchell and Willenbring, 2008). In brief, mice were anaesthetized with a mixture of ketamin (100 mg/kg body weight) and xylazine (10 mg/kg body weight) by intraperitoneal injection. After resection of falciform and triangle ligaments, the left lateral lobe was ligated with 4-0 silk sutures (Ethicon) and resected. Subsequently, the median lobe was ligated with suture between the gall bladder and suprahepatic vena cava and then resected. During and after surgery, the mice were maintained on a heating pad until waking-up. Metamizole was used as post-surgical analgesic treatment for the first 48 h post-surgery. Mice were euthanized at indicated time points to monitor liver regeneration by determining the liver to body weight ratio.
In vivo blocking experiments
The following blocking antibodies or corresponding IgG controls were diluted in saline solution and injected intravenously at the indicated dosage: anti-Tie1 (clone Tie1-39, Eli Lilly) was provided by Eli Lilly and used at 8 mg/kg. Anti-VEGFR2 (clone DC101, Bio X Cell) 4 mg/kg; anti-VEGFR3 (clone AFL4, Biolegend) 4 mg/kg; anti-Dll4 (clone HMD4-2, Bio X Cell) 4 mg/kg; anti-CD31 antibody (clone MEC13.3 and clone 390, Biolegend) 4 mg/kg; anti-Integrin-αV (clone RMV-7, Biolegend) 4 mg/kg; anti-Integrin-α5 (clone HMα5-1, Biolegend) 4 mg/kg. Injected mice were sacrificed at the indicated time points.
Liver perfusion and isolation of liver non parenchymal cells (NPC)
Liver cell isolation was adapted to minimize flow shear stress and preserve protein phosphorylation (Mederacke et al., 2015). In brief, a 27 G Surflo infusion catheter (Terumo) connected to the tubing system of an IPC pump (Ismatec) was fixed into the vena cava. The liver was perfused with 37°C pre-warmed liver perfusion medium (Gibco) at 4 ml/min for 1 min, followed by 37°C pre-warmed liver digestion medium (Gibco) supplemented with 40 μg/ml LiberaseTM TM (Roche) at 2.7 ml/min for 8 min. The portal vein was cut shortly after the beginning of perfusion to allow blood drainage. After perfusion, livers were explanted into a Petri dish with pre-warmed RPMI medium (Gibco) with 1 mM sodium orthovanadate (Sigma). After removing the liver capsule membrane, tissue was dissociated by gently shaking in a final volume of 40 ml of RPMI. Dissociated liver cells were collected and filtered through a 100 μm cell strainer, centrifuged twice at 50g for 3 min at 4°C and the supernatant containing the NPC was collected. The NPC solution was centrifuged at 300g for 10 min at 4°C. The pellet was washed once in ACK buffer and finally spun at 400g for 5 min to obtain the final NPC pellet. All steps following perfusion were performed on ice in buffers supplemented with 1 mM sodium orthovanadate.
Positive selection of liver endothelial cell (L-EC)
NPC from 30 mice were pooled as a single biological replicate and further processed for L-EC enrichment. Approximately 3x108 NPC were resuspended in 4.5 ml of MACS buffer containing 2mM EDTA, 0.5% BSA, phosphatase inhibitor (Thermo fisher, A32957) and stained with 500 μl of mouse CD146 MicroBeads (Miltenyi Biotec, 130-092-007), for 15 min on ice. NPC were washed twice in cold MACS buffer, resuspended in 5 ml and loaded on a LS column (Miltenyi Biotec, 130-042-401). The column was washed twice and then eluted with 3 ml MACS buffer. Typically, 30 pooled mice yielded approximately 1.2X108 L-EC with purity above 95% and a viability above 90% (Figure S1A).
Flow cytometry and cell sorting
The single cell suspension was stained on ice for 20 min with CD31 PE-Cy7 (BioLegend), CD45 FITC (BioLegend), CD117(c-Kit) APC (BioLegend) and CD141 (Thrombomodulin) PE (Miltenyi Biotec), washed twice and resuspended in 5 ml of FACS buffer. Stained cells were sorted by FACSAria sorter (BD Biosciences) using a 70 μm nozzle.
To obtain L-EC of different zones along the portal-central axis, FACS events were screened through the following nested gates (Figures S1A and S1B): (1) plotting forward side scatter area (FSC-A) against side scatter area (SSC-A) to exclude large clusters and small debris; (2) singlets-set by excluding the margins of FSC-A and FSC-H width plot; (3) live cell gates according to the FxCycle Violet stain; (4) EC, by gating CD31-positive and CD45-negative population, and (5) portal, peri-portal, peri-central and central ECs, inferred from the CD117 histogram. To cross-check the gating of CD117, we used an additional central vein landmark gene CD141 to check that its fluorescence intensity was proportional to CD117.
RNA-sequencing and analysis pipeline
RNA extraction from sorted cells was performed with PicoPure RNA Isolation Kit (Thermo Fisher Scientific) and DNA was removed by on-column treatment with DNase I (RNase-Free DNase Set, Qiagen) according to the manufacturer’s instructions. RNA integrity was measured by RNA 6000 Pico Kit (Agilent) on Bioanalyzer 2100 (Agilent) and the concentration was determined by Qubit RNA HS Assay Kit (Thermo Fisher Scientific). Only samples with RIN above 8 were used for library preparation. RNA from each sample (350 ng each) was used for library generation using the TruSeq Stranded mRNA Library Prep kit (Illumina, 20020594). Quality control of the resulting libraries was performed with DNA 1000 Kit (Agilent) on Agilent Technologies 2100 Bioanalyzer and the concentration was determined by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific). 10 nM of 8 libraries was pooled using unique dual (UD) i7 index adapters (IDT for Illumina–Nextera DNA UD Indexes) and sequenced with NextSeq 550 Single-Read 75bp High-Output.
Illumina output files were demultiplexed with bcl2fastq2 Conversion Software v2.20 (Illumina). The resulting FASTQ files were analysed on the pipeline built on the DKFZ internal Galaxy (Afgan et al., 2018) instance. Adapter sequences were removed by Cutadapt (Martin, 2011) v1.16.6. The trimmed output sequences were aligned to the transcriptome index of the GRCm38.92 (Ensembl) using the RNA STAR (Dobin et al., 2013) v2.7.2b and gene expression was measured by featureCounts (Liao et al., 2014) v1.6.3. Gene count outputs were normalized to the relative gene length and transcripts per million (TPM) were calculated for each sample for further analysis.
Protein extraction
For label free mass spectrometry, cell pellets were lysed in RIPA lysis and extraction buffer (Thermo Fisher Scientific) supplemented with phosphatase inhibitor and protease inhibitor mix (Serva Electrophoresis) according to manufacturer’s instructions. For phospho-enrichment, cell pellets were lysed as previously described (Potel et al., 2018). In brief, the pellet was lysed with 7M urea lysis buffer containing: 100 mM Tris-HCl pH 8.5, 7 M Urea, 1% Triton, 10 U/ml DNase I, 1 mM magnesium chloride, 1% Benzonase (Sigma), 1 mM sodium orthovanadate, phosphatase inhibitors and protease Inhibitor. Dissolved pellets were sonicated at 10% output with 1s-on-1s-off for 1 min on ice (Sonifier W-250 D, G. Heinemann). The residual cell debris was removed by centrifugation at 18,000g for 1h at 4°C. The sample was then incubated for 2 h at room temperature for Benzonase digestion. The protein concentration was determined by the micro BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Protein digestion and peptide labelling
For label-free mass spectrometry, 10 μg protein extracts were processed via tryptic in-gel digestion. Briefly, proteins were loaded on an SDS-PAGE-gel and ran 0.5 cm in the gel. After Commassie staining, the sample underwent tryptic digestion as previously described (Shevchenko et al., 2006), adapted to a DigestPro MSi robotic system (INTAVIS Bioanalytical Instruments AG).
For phospho-enrichment, protein reduction and alkylation were done with 10 mM TCEP and 40 mM CAA for 30 min at RT. For each sample, 200 μg protein was digested first by Lys-C at 1:100 enzyme:protein ratio for 4 h at 37°C at pH 8.5. The solution was then diluted 1:5 with 50 mM TEAB (pH 8.5) and further digested with trypsin at 1:50 enzyme:protein ratio over night at 37°C. The digestion was stopped by adding TFA to 0.4% (vol/vol), then centrifuged at 2,500g for 10 min. The supernatants were collected and pH-verified to be below 2 and then processed with SepPak tC18 100 mg 1cc (Waters) for desalting and dimethyl labelling. Briefly, the cartridge was conditioned with 3 ml ACN followed by 1 ml 50% ACN 0.5% HAcO, then equilibrated with 3 ml 0.1% TFA before loading the acidified peptide samples. The samples were washed with 3 ml 0.1% TFA and then 500 μl light labelling reagent (50 mM sodium phosphate buffer pH 7.5, 4% Formaldehyde (CH2O), 0.6 M Cyanoborohydride (NaBH3CN)). Thereafter, the cartridges were again washed with 3 ml 0.1% TFA and the labelled peptides were eluted with 750 μl 50% ACN 0.5% HAcO. In parallel, an internal control (L-EC pooled from all sorting gates) was processed following the same protocol except labelled with medium labelling reagent (50 mM sodium phosphate buffer pH 7.5, 4% deuterated Formaldehyde (CD2O), 0.6 M Cyanoborohydride (NaBH3CN)). Equal amount of internal control was spiked into each sample and then the sample/spike mix was vacuum dried before processing for phosphopeptide enrichment.
Phosphopeptide enrichment
An automated phosphopeptide enrichment protocol based on Immobilized metal affinity chromatography (IMAC) using a nitrilotriacetic acid (NTA) chelating ligand functionalized with Fe(III) was applied to the mixture of labelled peptides and internal controls, with AssayMap Bravo platform (Agilent Technologies). Each sample was reconstituted with 110 μL 80% ACN, 0.1% TFA buffer. They were gently sonicated in water bath until complete dissolution and then they were transferred manually to the Greiner 96-well full skirt PolyPro PCR plate. The Agilent AssayMAP Phosphopeptide Enrichment v2.0 App, included with the Agilent AssayMAP Bravo Protein Sample Prep Workbench v2.0 software suite, was run using AssayMAP Fe(III)-NTA cartridges (Agilent Technologies). Briefly, the cartridges were firstly primed with 100 μL 50% ACN, 0.1% TFA, then equilibrated with 50 μL of 80% ACN, 0.1% TFA. Each sample was loaded onto the Fe(III)-NTA cartridges and then they were washed with 50 μL 80% ACN, 0.1% TFA buffer. Finally, phosphopeptides were eluted with 20 μL of 1% NH4OH buffer (pH ∼11), acidified with 2 μl of formic acid (pH 3) and dried down for MS analysis.
LC-MS/NS analysis
Nanoflow LC-MS/MS was performed by coupling a Dionex 3000 (Thermo Fisher Scientific) to a QExactive Orbitrap HF-X (Thermo Fisher Scientific). Samples for the proteome or phosphoproteome analysis were re-suspended in loading buffer containing 2.5% 1,1,1,3,3,3-Hexafluoro-2-propanol, 0.1% TFA in water or 50 mM citrate and 0.1% TFA, respectively. Peptide loading and washing were done on a trapping cartridge (Acclaim PepMap300 C18, 5μm, 300Å wide pore, Thermo Fisher Scientific) and washed for 3 min with 0.1% TFA in water at a flow rate of 30 μl/min. Peptide separation was performed on an analytical column (nanoEase, 300Å, 1.7 μm, 75 μm x 200 mm, Waters) at a flow rate of 300 nl/min using a three step 210 min gradient consisting of the following steps: 2-8% solvent B (80% acetonitril, 20% water with 0.1% formic acid) in 15 min, 8-25% in 135 min and 25-40% in 30 min followed by a washing and an equilibration step with solvent A being 0.1% formic acid in water. In order to accompany for the more hydrophilic nature of phospho-peptides, the 210 min method was adjusted as follows: 2-25% solvent B in 150 min, 25-40% in 30 min followed by washing and an equilibration step. Peptides were ionized using a spray voltage of 2.2 kV and a capillary temperature of 275°C. The instrument was operated in data-dependent mode. For the full proteome samples, full scan MS spectra (m/z 375–1,500) were acquired with a maximum injection time of 54 ms at 120,000 resolution and an automatic gain control (AGC) target value of three million charges. MS/MS scans were triggered for the top 35 precursor ions, high-resolution MSMS spectra were acquired in the orbitrap with a maximum injection time of 22 ms at 15,000 resolution (isolation window 1.6 m/z), an AGC target value of 100 000 ions and normalized collision energy of 27. Dynamic exclusion was set to 60 s (16 s phospho samples). Undetermined charge states and single charged species were excluded from fragmentation.
Peptide and protein identification and quantification
Data analysis was carried out by MaxQuant (Tyanova et al., 2016a) v1.6.3.3. Carbamidomethylation of cysteines was set as fixed modification. Phosphorylation of serine, threonine or tyrosine as well as oxidation of methionines and N-terminal acetylation were set as variable modifications. Identification FDR cutoffs were 0.01 on peptide level and 0.01 on protein level.
The LFQ based full proteome analysis was carried out with organism specific UniProt database UP000000589 (Mus musculus; Dec, 2017; 60715 sequences) and enabled ‘match-between-runs’ function. The LFQ option was enabled and left at default parameters.
The phosphoproteome analysis samples was carried out with organism specific UniProt database UP000000589 (Mus musculus; Feb, 2020; 55421 sequences). The multiplicity was set to ‘2’ with ‘DimethLys0’ and ‘DimethNter0’ as light labels and ‘DimethLys4’ and ‘DimethNter4’ as heavy labels. The ‘match-between-runs’ function was enabled and fractions were assigned so that the function applied separately within the phospho fractions (fraction 1) and the full proteome fraction (fraction 11). The ‘Re-quantify’ option was enabled.
Phospho-receptor tyrosine kinase (RTK) array
Mouse phospho-RTK array kits were purchased from R&D systems. Spatially sorted L-EC were lysed with the provided lysis buffer supplemented with phosphatase inhibitor and protease inhibitor mix. Protein concentration was quantified with Micro BCA Protein Assay Kit according to the manufacturer’s instructions. Protein lysates (30 μg) were loaded for each membrane. The array was performed according to the manufacturer’s instructions, except that the Chemi Reagent Mix was replaced with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific). The images were acquired with Amersham Imager 600 (GE Healthcare).
RNA extraction, cDNA synthesis and qRT-PCR
RNA extraction from sorted cells was performed with PicoPure RNA Isolation Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Whole liver samples were homogenized by TissueLyzer (Retsch) in lysis solution and RNA extraction was performed with GenElute Mammalian Total RNA Purification Kit (Sigma) according to the manufacturer’s instructions. Total RNA was reverse transcribed into cDNA using the QuantiTect Reverse transcription kit (Qiagen) according to the manufacturer’s instructions. qRT-PCR reaction was performed with TaqMan Fast Advanced PCR Master Mix (Thermo Fisher Scientific) or Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) and read by LightCycler 480 (Roche). Gene expression levels were calculated based on the ΔΔCt relative quantification method, normalized to Actb expression.
Immunofluorescence and confocal microscopy
Paraffin sections (30 μm) were cut on a HM355S microtome (Thermo Fisher Scientific) and allowed to adhere to Superfrost Plus slides (Thermo Fisher Scientific). Sections were permeabilized and blocked in PBS containing 0.3% Triton X-100 (Sigma-Aldrich) and 10% FBS followed by staining in the same blocking buffer. The following primary antibodies were used for staining: goat anti-mouse CD31 (1:100, R&D Systems); rabbit anti-mouse LYVE1 (1:200, Novus Biologicals); rabbit anti-cKit (CD117) (1:100, Abcam). The following secondary antibodies were used for staining: Alexa Fluor 647 donkey anti-rabbit IgG (H+L), Alexa Fluor 568 donkey anti-goat IgG (H+L) (Thermo Fisher Scientific). Rabbit anti-mouse Glutamine Synthetase (AbCam) was directly conjugated with Zenon Alexa Fluor 488 Rabbit-IgG.
Stained slides were mounted with Fluorescence Mounting Medium (Agilent Dako) and images were acquired on an inverted Leica microscope (TCS STED CW SP8, Leica Microsystems) with a motorized stage for tiled imaging. To minimize fluorophore spectral spillover, we used the Leica sequential laser excitation and detection modality. The bleed-through among sequential fluorophore emission was removed applying simple compensation correction algorithms to the acquired images. Lif files were imported into Imaris (Bitplane) for background adjustment and exported as tiff images.
Fluorescence In situ hybridization (FISH)
FISH of Wnt9b was performed using the ViewRNA ISH Tissue Assay Core Kit (Thermo Fisher Scientific). Cryosections (7 μm) were fixed with 4% paraformaldehyde overnight in the dark at 4°C. The sections were then washed with PBS, dehydrated in ethanol, baked for 1h at 60°C, boiled for 15 min in pre-treatment solution from the kit, and digested for 15 min in protease solution provided by the manufacturer (Thermo Fisher Scientific). Following protease treatment, the sections were hybridized for 2 h at 40°C with the Wnt9b (VB1-15880-VT) and Pecam1 (VB6-12921-VT) probes (Thermo Fisher Scientific). The hybridized sections were pre-amplified and amplified according to the manufacturer’s manual. For visualization of the FISH probe, the labelled probe conjugated to alkaline phosphatase type 1 and fast red substrate was treated to detect Wnt9b expression (Thermo Fisher Scientific). Immunofluorescent (IF) co-staining of glutamine synthetase was immediately performed after the initial FISH steps. Briefly, the sections were blocked, stained with rabbit anti-mouse Glutamine Synthetase antibody (Abcam) followed by AF647 goat anti-rabbit (Thermo Fisher Scientific) for secondary detection. Images were acquired as described before for the confocal microscopy.
Western blotting (WB)
Protein lysates were adjusted to the same concentration, mixed with 3× Blue Loading Buffer (Cell Signaling Technology), heated at 95°C for 5 minutes, and subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to PVDF membranes by a wet tank transfer system. Membranes were blocked with 3% BSA in TBST buffer and probed with primary antibodies as indicated in Figure 1D. Afterwards, membrane were incubated with HRP-conjugated secondary antibodies, developed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific), and images acquired by Amersham Imager 600 (GE Healthcare).
Data analysis
Analysis of non-EC contaminants in RNAseq and mass spectrometry
In order to assure that the selected FACS gates were greatly enriched for EC and, moreover, that there were no zonation confounding effects due to non-EC contamination, we estimated the fraction of non-EC markers in the RNA and protein samples. To this end, we compiled a list of previously published transcriptomes of different liver cell types. Liver immune cell types and EC expression were taken from (Halpern et al., 2018). The hepatocyte transcriptome was retrieved from (Halpern et al., 2017). Cholangiocyte data were extracted from averaging expression data of single cholangiocytes resulting from single cell sequencing of mouse healthy liver as reported by (Xiong et al., 2019). Hepatic stellate cells and fibroblast expression data were taken from (Dobie et al., 2019). Expression data were normalized to the sum of each cell type, resulting in the expression fraction of each gene in each of the cell types’ transcriptomes.
The 5,000 most highly expressed genes from each cell types were pooled together into 11,617 unique genes. These genes were further filtered and were considered as non-EC markers if the fraction of gene in EC was less than 1x10-5 and if the ratio between the expression in at least one non-EC type and EC was 10-fold or higher. A special case was hepatocyte, which are substantially larger than EC and higher in RNA content. Hepatocyte markers therefore passed our filter if the expression ratio between hepatocyte and EC was 2-fold or higher.
In total, 2,626 genes passed this filter. We then calculated the fraction of these non-EC markers out of the total expression data of the different FACS gates. 2,275 genes were found in the EC RNA data. Fractions were between 0.055 and 0.063 out of total RNA, demonstrating EC predominance. We next performed Kruskal Wallis tests to check whether there was a statistically significant difference in the fraction of non-EC across the four different FACS gates. Kruskal Wallis test on the sum of these genes across the different FACS gates was insignificant p-value = 0.089, indicating that there was no difference in fraction of EC along the different sorted populations (Figure S1C). Similarly, 219 genes were found in the label free MS data. Their fractions out of the total identified proteins were 0.073-0.080. Kruskal Wallis p-value = 0.235 (Figure S1C).
Dataset filtering and processing
For gene expression out of 28,727 identified genes, we considered for further analysis (pathway enrichment, phylogenetic tree, PTR definition) only genes with a mean TPM of the four replicates above 5 in at least one of the four zones (PN, PP, PC, CV). In order to provide an overview of the whole transcriptome zonation (Figures 2A and 2B), the TPM cut-off was maintained at 1 and the expression profiles were confirmed by qRT-PCR (Figure 2C).
Of 5,015 detected proteins, only those quantified in at least two replicates in one of the zones were retained for imputation. The label-free quantification (LFQ) intensities were imputed with a constant (the minimal LFQ of each sample) for missing values. To normalize the variability among the experimental replicates, the values of each pool across the four zones were normalized to their mean. Finally, where protein abundance was required, the normalized value was multiplied for the mean LFQ of all the 16 samples before normalization.
Of 19,607 detected phospho-sites (p-site) corresponding to 3,447 proteins, only class I (localization probability > 0.75) were considered for analysis. Normalized ratio exported from MaxQuant was used for quantification. As described for proteins, only p-sites quantified in at least two replicates in one of the zones were retained for imputation. The sample/spike ratios were first log2 transformed and then imputed from normal distribution with Perseus v1.6.14.0 (Tyanova et al., 2016b). Finally, normalization between replicates was performed as described for protein analysis.
In order to compare multiple datasets, transcripts, proteins and p-sites were matched with their corresponding ENSEMBL gene ID.
Zonation pattern definition
To define if the expression of a specific transcript, protein or p-sites was zonated across zones, we performed Kruskal-Wallis (KW) test, followed by Benjamini–Hochberg (BH) procedure to correct for multiple hypotheses. Transcripts/proteins/p-sites with KW p-value <0.05 and BH FDR <0.25 were considered as zonated.
A zonation score was expressed by Center-of-Mass (CoM) as described (Ben-Moshe et al., 2019). We defined sorting gate PN as gate1, PP as 2, PP as 3 and CV as 4. Thus, for each pool consisting of the 4 zones, taking mRNA as example,
To calculate for the overall CoM, the mean of each zone was used.
The patterns of the zonation - portal, central, vessel and sinusoidal - for zonated genes were defined as follows: Considering the continuum of the vasculature, two gates were combined. Portal area combined PN and PP, central area combined CV and PC, vessel area combined PN and CV and sinusoidal area combined PP and PC. Thus, each of these areas contained 8 samples. The mean value for each combined area was calculated and the pattern was defined according to which area expressed the maximum mean value. To avoid the confoundment generated by combination of portal and central gates that are not neighboring gates, thus masking the difference of these gates, we applied a further filtration for vessel and sinusoid pattern. As extreme low and high CoM is indicative for portal or central zonation, the genes CoM <2.2 or >2.8 were excluded for vessel and sinusoidal pattern.
To generate heat maps of the expression profiles, the expression was normalized to the maximum value across all 16 samples. Genes were sorted by their Center-of-Mass, except for the heat maps for vessel/sinusoid patterns, where log2 fold change of vessel area to sinusoid area was used to order the genes. In order to compare the zonation profiles of different data sets (transcript, protein p-peptides), each zonation profile was represented by percentage of maximum expression. Graph was generated by Gitools v2.3.1 (Perez-Llamas and Lopez-Bigas, 2011).
Correlation of spatial sort RNAseq and published scRNAseq
For the comparison of spatial sorting RNAseq with previous scRNAseq of L-EC (Halpern et al., 2018), we matched the two datasets by gene name. Comparison was calculated on the mean fractions of the different repeats in each FACS gate and the mean fractions of all cells assigned to the same liver lobule layer in the scRNAseq data. Zonation FDR on Kruskal-Wallis test was recalculated for number of genes common to both datasets (n=13,070 genes). We next filtered for zonated genes (FDR qval <0.25 in both datasets), whose dynamic range was greater than 1.2. Dynamic range was defined for each dataset as the ratio between the maximal and minimal expressions of the genes across the different liver lobule layers. To avoid noisy genes, we only considered genes with fraction higher than 5x10-6 in at least one lobule layer. This filter yielded 2,463 highly expressed zonated genes in the spatial sorting dataset and 76 in the scRNAseq, with common 48 genes intersecting. We then calculated the Center-of-Mass (CoM) for these 48 genes in each of the data sets and calculated the Pearson correlation (rPearson = 0.873, p-val = 5.746x10-16).
Correlation of protein and mRNA abundance
To compare protein and mRNA abundance, we matched the two data sets by their Gene ID. We obtained 4,169 protein-mRNA pairs and protein-to-transcript ratio (PTR) was calculated by building the ratio between average protein and RNA abundance across the zones (Figure 3B; Table S5). The PTR values of the protein-mRNA pairs followed a Gaussian distribution. High PTR or low PTR genes were defined as PTR > median+SD or PTR < median-SD (Figure 3C).
Comparison of RNA, protein and p-peptides zonation
To compare the zonation of protein and mRNA, data sets were matched by Gene ID (Table S5). The 4 CoM values corresponding to the 4 replicates was calculated for each data set and unpaired two-sided Student's t test was performed between the 4 CoM relative to mRNA and the 4 CoM relative to protein. The extent of zonation shift was determined by calculating the ΔCoM for each protein-mRNA pair. Finally, the comparison is visualized by plotting the -log10 p-value against the ΔCoM, where genes with p <0.05 and ΔCoM > 0.1 is considering to be differentially zonated between protein and mRNA (Figure 4D). The same approach was applied to compare the zonation between p-peptide and protein (Figure 4G).
Pathway and protein domain analysis
Pathway or protein domain analysis for each indicated subset of genes was performed with STRING (Szklarczyk et al., 2019) v11.0, searching against KEGG (Kanehisa and Goto, 2000) or SMART (Letunic and Bork, 2018) database (Table S4).
Selected pathways with FDR < 0.01 were represented as dot plot using ggplot2 package v3.3.1 (Wickham, 2016) in RStudio v1.2.5042. Circle size was proportional to gene count and color indicating the -log10 FDR. Pathways were sorted accordingly to the median CoM (Figure S4B), log2 fold change (FC) (Figure S4D) and PTR (Figure 3D) of the genes belonging to each pathway. SMART protein domains were represented as bar graph sorted by median CoM with the bar color indicating the FDR range (Figure 4I).
Protein network analysis
Protein-protein interaction networks for both high and low PTR proteins were obtained with STRING v11.0 (Szklarczyk et al., 2019). Network parameters, including the source and the target node defining the direction of the interaction and the combined interaction score defining the weight of the network edge, were imported into Gephi v0.9.2 (Bastian et al., 2009) for the network visualization. Networks were visualized as ForceAtlas 2 with the node size proportional to the LFQ value and edge thickness to the combined interaction score. Nodes and edges relative to specific pathways were colored as indicated in the figures (Figures 3E and 3F; Table S4).
Phylogenetic tree analysis of L-EC kinome and phosphatome
Phylogenic analysis was performed with CORAL (Metz et al., 2018) or CORALp (Min et al., 2019) for the L-EC kinome and phosphatome, respectively (Figure S3). The circle size was proportional to the expression indicated by the mean TPM across zones. The color represented the overall zonation score (CoM) from portal (blue) to central (red). In case of multiple p-peptides corresponding to the same gene, the one with the lowest Kruskal Wallis p-value was selected for the analysis (Figure 5F).
Differential gene expression analysis
To investigate the gene regulation induced by the receptor tyrosine kinase Tie1 blockade, L-EC was spatially sorted from C57/B6 mice 2 hours after treatment with anti-Tie1 antibody, and processed for RNAseq as described above. To obtain the differentially expressed genes in each zone, the obtained gene counts from Tie1 treated samples and control samples were analysed with the DESeq2 (Yousif et al., 2020) (Table S7). Gene regulation was visualized as volcano plot by plotting the -log10 q-val against the log2 fold change with the EnhancedVolcano (Blighe et al., 2020) v1.4.0 package in RStudio (Figure 6A). To compare the regulatory effect of Tie1 on portal and central, genes that were significantly regulated in portal or in central, i.e., q-value < 0.05 in at least of one of the two DEseq2 output, were selected and their -log10 q-value was visualized as density plot using ggplot2 package. The difference was determined by Wilcoxon matched-pairs signed rank test (p-value < 2.2 x10-16).
Wnt9b promoter analysis
Searching for putative transcription factor binding sites in the promoter region of Wnt9b was performed by the Search Motif Tool from the Eukaryotic Promoter Database (SIB) (Dreos et al., 2015). The search was based on the library from Transcription Factor Motifs (JASPAR CORE 2018 vertebrates), retrieved from -1000 to 1000 bp relative to the Wnt9b transcription start site with a p-value cut-off at 0.001. Motif was selected for Foxo1 or STAT3, respectively. The retrieved putative binding sites for FoxO1 and STAT3 were indicated as shown in Figure S7F.
Phosphosite motif analysis
15-mer sequences were extracted from -7 to +7 position flanking the detected phosphosites. Motif analysis was performed using PhosphoSitePlus (Hornbeck et al., 2015) v6.5.9.2. Sequences for p-S, p-T, p-Y were loaded separately as foreground, searched against the corresponding background. Proline-directed, acidic, basic or other motif categories were classified as previously described (Villén et al., 2007). The sequences that could not be assigned to any aforementioned motif were designated as none (Figure S4E). To visualize the consensus sequences flanking the phosphosites for each pattern, sequence logos were generated by PhosphoSitePlus v6.5.9.2. The sequences belonging to each category of each phosphosite were loaded separately as foreground and searched against the respective background of the specific phosphosite, using PST production algorithm (Figure S4F).
Quantification and statistical analysis
Kruskal-Wallis tests followed by Benjamini–Hochberg correction were performed when comparing multiple groups. Wilcoxon matched-pairs signed rank test or unpaired two-sided Student's t tests were performed when comparing two groups as indicated in the figure legends. N=4 pools (each 30 mice) were used for RNAseq, label free proteome and phosphoproteome analysis. Mouse number was indicated by data point in animal experiments, as indicated in Figures 6, 7, and S7. ∗, p-value <0.05; ∗∗, p-value < 0.01; ∗∗∗, p-value < 0.001; ∗∗∗∗, p-value < 0.0001. Data are expressed as mean ± SD where applied except in Figure S1C (median with IQR) and S1D (mean ± SEM). Flow cytometry data was analysed with FlowJo v10. Statistics and graphics were performed with GraphPad Prism v8 and Rstudio v1.2.5042. Heat maps were generated with Gitools v2.3.1. Networks were visualized with Gephi v0.9.2. Cartoons were created with Biorender.com.
Acknowledgments
We thank the MS-based Protein Analysis Unit and the High-Throughput Sequencing Unit of the Genomics and Proteomics, the Flow cytometry, the Light Microscopy Core Facilities, and the Laboratory Animal Facility at the German Cancer Research Center (DKFZ) as well as the Proteomics Core Facility at the Center for Molecular Biology (ZMBH) at Heidelberg University for providing excellent support. We thank Silvia Calderazzo (DKFZ Division of Biostatistics) for biostatistical consulting.
This work was supported by grants from the Deutsche Forschungsgemeinschaft (projects A1 and A2 within CRC1324 “Wnt Signaling” [project number 331,351,713] to H.G.A. and M.B.; projects A4 and C3 within CRC-TR209 “Liver Cancer” [project number 314905040 to H.G.A. and M.H.]; project C5 within CRC1366 “Vascular Control of Organ Function” [project number 39404578 to H.G.A.]), the European Research Council Advanced Grant “AngioMature” [project 787181 to H.G.A.], and the State of Baden-Württemberg Foundation special program “Angioformatics Single Cell Platform” [to H.G.A.].
Author contributions
D.I., J.S., and H.G.A. conceived and designed the study. D.I., J.S., K.H.L., M.J., G.W., M.K., P.A.V., M.R., and C.S. performed the experiments. D.I., J.S., S.B.-M., and S.R.K. analyzed the data. M.S., T.R., C.S., D.H., I.S., M.B., S.C., M.H., and S.I. provided scientific input, reagents, as well as technical and infrastructural support. D.I., J.S., and H.G.A. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.devcel.2021.05.001.
Contributor Information
Donato Inverso, Email: inverso@angioscience.de.
Hellmut G. Augustin, Email: augustin@angioscience.de.
Supplemental information
References
- Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian, M., Heymann, S., and Jacomy, M. (2009). Gephi: an open source software for exploring and manipulating networks. Third international AAAI conference on weblogs and social media 8, pp. 361–362.
- Ben-Moshe S., Shapira Y., Moor A.E., Manco R., Veg T., Bahar Halpern K., Itzkovitz S. Spatial sorting enables comprehensive characterization of liver zonation. Nat. Metab. 2019;1:899–911. doi: 10.1038/s42255-019-0109-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blighe, K., Rana, S., and Lewis, M. (2020). EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. R package version 1.8.0, 10.18129/B9.bioc.EnhancedVolcano.
- Cao Z., Ye T., Sun Y., Ji G., Shido K., Chen Y., Luo L., Na F., Li X., Huang Z. Targeting the vascular and perivascular niches as a regenerative therapy for lung and liver fibrosis. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aai8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobie R., Wilson-Kanamori J.R., Henderson B.E.P., Smith J.R., Matchett K.P., Portman J.R., Wallenborg K., Picelli S., Zagorska A., Pendem S.V. Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 2019;29:1832–1847.e8. doi: 10.1016/j.celrep.2019.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreos R., Ambrosini G., Périer R.C., Bucher P. The Eukaryotic Promoter Database: expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res. 2015;43:D92–D96. doi: 10.1093/nar/gku1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan M., Wang H., Gaur U., Little P.J., Xu J., Zheng W. FOXO signaling pathways as therapeutic targets in cancer. Int. J. Biol. Sci. 2017;13:815–827. doi: 10.7150/ijbs.20052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausto N., Campbell J.S., Riehle K.J. Liver regeneration. Hepatology. 2006;43(Supplement 1):S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- Fíla J., Honys D. Enrichment techniques employed in phosphoproteomics. Amino Acids. 2012;43:1025–1047. doi: 10.1007/s00726-011-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebhardt R. Metabolic zonation of the liver: regulation and implications for liver function. Pharmacol. Ther. 1992;53:275–354. doi: 10.1016/0163-7258(92)90055-5. [DOI] [PubMed] [Google Scholar]
- Geissmann F., Cameron T.O., Sidobre S., Manlongat N., Kronenberg M., Briskin M.J., Dustin M.L., Littman D.R. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gola A., Dorrington M.G., Speranza E., Sala C., Shih R.M., Radtke A.J., Wong H.S., Baptista A.P., Hernandez J.M., Castellani G. Commensal-driven immune zonation of the liver promotes host defence. Nature. 2021;589:131–136. doi: 10.1038/s41586-020-2977-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti L.G., Inverso D., Sironi L., Di Lucia P., Fioravanti J., Ganzer L., Fiocchi A., Vacca M., Aiolfi R., Sammicheli S. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell. 2015;161:486–500. doi: 10.1016/j.cell.2015.03.005. [DOI] [PubMed] [Google Scholar]
- Halpern K.B., Shenhav R., Massalha H., Toth B., Egozi A., Massasa E.E., Medgalia C., David E., Giladi A., Moor A.E. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 2018;36:962–970. doi: 10.1038/nbt.4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern K.B., Shenhav R., Matcovitch-Natan O., Toth B., Lemze D., Golan M., Massasa E.E., Baydatch S., Landen S., Moor A.E. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature. 2017;542:352–356. doi: 10.1038/nature21065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Wang Y., Pu W., Huang X., Qiu L., Li Y., Yu W., Zhao H., Liu X., He L. Lineage tracing reveals the bipotency of SOX9+ hepatocytes during liver regeneration. Stem Cell Rep. 2019;12:624–638. doi: 10.1016/j.stemcr.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser J., Mayo A., Keren L., Alon U. Central dogma rates and the trade-off between precision and economy in gene expression. Nat. Commun. 2019;10:68. doi: 10.1038/s41467-018-07391-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Srivastava K., Wieland M., Runge A., Mogler C., Besemfelder E., Terhardt D., Vogel M.J., Cao L., Korn C. Endothelial cell-derived angiopoietin-2 controls liver regeneration as a spatiotemporal rheostat. Science. 2014;343:416–419. doi: 10.1126/science.1244880. [DOI] [PubMed] [Google Scholar]
- Huch M., Dorrell C., Boj S.F., van Es J.H., Li V.S., van de Wetering M., Sato T., Hamer K., Sasaki N., Finegold M.J. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494:247–250. doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R., Beausoleil S.A., Villén J., Haas W., Sowa M.E., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh J., Chand A., Gough D., Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer. 2019;19:82–96. doi: 10.1038/s41568-018-0090-8. [DOI] [PubMed] [Google Scholar]
- Inverso D., Iannacone M. Spatiotemporal dynamics of effector CD8+ T cell responses within the liver. J. Leukoc. Biol. 2016;99:51–55. doi: 10.1189/jlb.4MR0415-150R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., Allen B., Korhonen E.A., Nitschké M., Yang H.W., Baluk P., Saharinen P., Alitalo K., Daly C., Thurston G., McDonald D.M. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J. Clin. Invest. 2016;126:3511–3525. doi: 10.1172/JCI84871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018;46:D493–D496. doi: 10.1093/nar/gkx922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Martinez-Ledesma E., Zhang C., Gao F., Zheng S., Ding J., Wu S., Nguyen N., Clifford S.C., Wen P.Y. Tie2-FGFR1 interaction induces adaptive PI3K inhibitor resistance by upregulating Aurora A/PLK1/CDK1 signaling in glioblastoma. Cancer Res. 2019;79:5088–5101. doi: 10.1158/0008-5472.CAN-19-0325. [DOI] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lorenz L., Axnick J., Buschmann T., Henning C., Urner S., Fang S., Nurmi H., Eichhorst N., Holtmeier R., Bódis K. Mechanosensing by β1 integrin induces angiocrine signals for liver growth and survival. Nature. 2018;562:128–132. doi: 10.1038/s41586-018-0522-3. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–12. [Google Scholar]
- Marx V. A dream of single-cell proteomics. Nat. Methods. 2019;16:809–812. doi: 10.1038/s41592-019-0540-6. [DOI] [PubMed] [Google Scholar]
- McDonald B., McAvoy E.F., Lam F., Gill V., de la Motte C., Savani R.C., Kubes P. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008;205:915–927. doi: 10.1084/jem.20071765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara H.A., Cai Y., Wagle M.V., Sontani Y., Roots C.M., Miosge L.A., O'Connor J.H., Sutton H.J., Ganusov V.V., Heath W.R. Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aaj1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mederacke I., Dapito D.H., Affò S., Uchinami H., Schwabe R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015;10:305–315. doi: 10.1038/nprot.2015.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergner J., Frejno M., List M., Papacek M., Chen X., Chaudhary A., Samaras P., Richter S., Shikata H., Messerer M. Mass-spectrometry-based draft of the Arabidopsis proteome. Nature. 2020;579:409–414. doi: 10.1038/s41586-020-2094-2. [DOI] [PubMed] [Google Scholar]
- Metz K.S., Deoudes E.M., Berginski M.E., Jimenez-Ruiz I., Aksoy B.A., Hammerbacher J., Gomez S.M., Phanstiel D.H. Coral: clear and customizable visualization of human kinome data. Cell Syst. 2018;7:347–350.e1. doi: 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min A., Deoudes E., Bond M.L., Davis E.S., Phanstiel D.H. CoralP: flexible visualization of the human phosphatome. J. Open Source Softw. 2019;4:1837. doi: 10.21105/joss.01837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell C., Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- Morse M.A., Sun W., Kim R., He A.R., Abada P.B., Mynderse M., Finn R.S. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 2019;25:912–920. doi: 10.1158/1078-0432.CCR-18-1254. [DOI] [PubMed] [Google Scholar]
- Niklason L., Dai G. Arterial venous differentiation for vascular bioengineering. Annu. Rev. Biomed. Eng. 2018;20:431–447. doi: 10.1146/annurev-bioeng-062117-121231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J.H., Kollipara R., Chu G., Ji H., Xiao Y., Ding Z., Miao L., Tothova Z., Horner J.W., Carrasco D.R. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Llamas C., Lopez-Bigas N. Gitools: analysis and visualisation of genomic data using interactive heat-maps. PLOS One. 2011;6 doi: 10.1371/journal.pone.0019541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potel C.M., Lin M.H., Heck A.J.R., Lemeer S. Defeating major contaminants in Fe3+- immobilized metal ion affinity chromatography (IMAC) phosphopeptide enrichment. Mol. Cell. Proteomics. 2018;17:1028–1034. doi: 10.1074/mcp.TIR117.000518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X., Tompkins K., Batts L.E., Puri M., Baldwin H.S. Abnormal embryonic lymphatic vessel development in Tie1 hypomorphic mice. Development. 2010;137:1285–1295. doi: 10.1242/dev.043380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha A.S., Vidal V., Mertz M., Kendall T.J., Charlet A., Okamoto H., Schedl A. The angiocrine factor Rspondin3 is a key determinant of liver zonation. Cell Rep. 2015;13:1757–1764. doi: 10.1016/j.celrep.2015.10.049. [DOI] [PubMed] [Google Scholar]
- Salih D.A., Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr. Opin. Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Sharma A., Seow J.J.W., Dutertre C.-A., Pai R., Blériot C., Mishra A., Wong R.M.M., Singh G.S.N., Sudhagar S., Khalilnezhad S. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183:377–394.e21. doi: 10.1016/j.cell.2020.08.040. [DOI] [PubMed] [Google Scholar]
- Sharma K., D'Souza R.C., Tyanova S., Schaab C., Wiśniewski J.R., Cox J., Mann M. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havlis J., Olsen J.V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Singhal M., Gengenbacher N., La Porta S., Gehrs S., Shi J., Kamiyama M., Bodenmiller D.M., Fischl A., Schieb B., Besemfelder E. Preclinical validation of a novel metastasis-inhibiting Tie1 function-blocking antibody. EMBO Mol. Med. 2020;12:e11164. doi: 10.15252/emmm.201911164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Kaisho T., Yoshida N., Takeda J., Kishimoto T., Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J. Immunol. 1998;161:4652–4660. [PubMed] [Google Scholar]
- Tanaka M., Iwakiri Y. The hepatic lymphatic vascular system: structure, function, markers, and lymphangiogenesis. Cell. Mol. Gastroenterol. Hepatol. 2016;2:733–749. doi: 10.1016/j.jcmgh.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Van Hoof D., Muñoz J., Braam S.R., Pinkse M.W., Linding R., Heck A.J., Mummery C.L., Krijgsveld J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell. 2009;5:214–226. doi: 10.1016/j.stem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Villén J., Beausoleil S.A., Gerber S.A., Gygi S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Zhao L., Fish M., Logan C.Y., Nusse R. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature. 2015;524:180–185. doi: 10.1038/nature14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Nakayama M., Pitulescu M.E., Schmidt T.S., Bochenek M.L., Sakakibara A., Adams S., Davy A., Deutsch U., Lüthi U. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- Wickham H. Springer-Verlag; 2016. Ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Xiong X., Kuang H., Ansari S., Liu T., Gong J., Wang S., Zhao X.-Y., Ji Y., Li C., Guo L. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell. 2019;75:644–660.e5. doi: 10.1016/j.molcel.2019.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M., Xu H.H., Lin Y., Sun X., Wang L.J., Fang Z.P., Su X.H., Liang X.-J., Hu Y., Liu Z.M. LECT2, a ligand for Tie1, plays a crucial role in liver fibrogenesis. Cell. 2019;178:1478–1492.e20. doi: 10.1016/j.cell.2019.07.021. [DOI] [PubMed] [Google Scholar]
- Yousif A., Drou N., Rowe J., Khalfan M., Gunsalus K.C. NASQAR: a web-based platform for high-throughput sequencing data analysis and visualization. BMC Bioinformatics. 2020;21:267. doi: 10.1186/s12859-020-03577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq FASTAQ files generated during this study are available at the Gene Expression Omnibus (GEO) repository (accession number GEO: GSE155797). The proteomic and phosphoproteomic thermo.raw files are available at the PRIDE database (accession number PRIDE: PXD020760 and PRIDE: PXD020805). All data are also available from the corresponding authors on reasonable request.