

Abstract
DNA-functionalized gold nanoparticles can be used to induce the formation and control the unit cell parameters of highly ordered face-centered cubic crystal lattices. Nanoparticle spacing increases linearly with longer DNA interconnect length, yielding maximum unit cell parameters of 77 nm and 0.52% inorganic-filled space for the DNA constructs studied. In general, we show that longer DNA connections result in a decrease in the overall crystallinity and order of the lattice due to greater conformational flexibility.
In 1996, our group introduced the concept of programmable materials synthesis based upon polyvalent oligonucleotide gold nanoparticle conjugates and complementary oligonucleotide linker molecules.1 In a parallel effort, the Alivisatos group developed methods of aligning monovalent nanoparticle conjugates on DNA templates to create small, periodic nanoparticle clusters.2 These approaches have generated significant interest from researchers looking to harness the unique physical and chemical properties of such hybrid materials for therapeutic,3,4 spectroscopic5–8 and materials applications.9–12 The polyvalent conjugates, ranging in core diameter from 5 to 250 nm, are easily prepared and modified,13 and they possess a host of interesting properties, including intense surface plasmon resonances,5,14 cooperative binding,15 programmable recognition,16,17 and catalytic activity,18 making them an interesting and flexible synthon for complex materials.19–23
When the concept of programmable materials synthesis from oligonucleotide nanoparticle conjugates was first introduced, a goal was to be able to use DNA to direct the three-dimensional assembly of nanoparticles into discrete preconceived structures.1 Early demonstrations focused primarily on the formation of amorphous polymers, where rough placement and periodicity of the particles was controllable.1,24 Later advances showed that crystallization might be possible and suggested that it might be viable to form different crystal structures from one type of nanoparticle building block but different sequences of DNA.25,26 Recently, both our group and the Gang group independently discovered different but related routes for using DNA linkers to effect the crystallization of polyvalent oligonucleotide gold nanoparticle conjugates (DNA–AuNPs).27,28 Both groups have shown that with different types of linkers, one can form either face-centered cubic (FCC) or body-centered cubic (BCC) unit cells with unit cell edge lengths as long as 39 nanometers (nm) and as little as 3% inorganic matter by volume.27–29 In addition to controlling the type of crystal structure, one should, in principle, be able to systematically adjust the lattice constants of these structures by varying the length of oligonucleotide interconnects. Such adjustments would allow for a versatile method of modulating this novel crystalline material with highly tailorable architectural parameters using a single type of DNA–AuNP and the appropriate oligonucleotide linkers (Scheme 1A). In this work, we explore that possibility and the consequences of changing oligonucleotide linker length on both the unit cell lattice parameters and the overall crystallinity of the resulting structures.
Scheme 1.
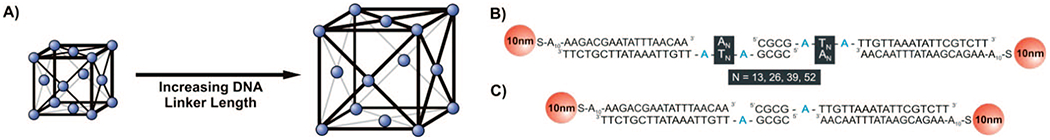
Materials Sequence and Designa
a (A) Illustration of the three-dimensional arrangement of particles forming a face-centered cubic lattice showing the effect of increasing linker length on lattice parameters, (B) sequences of oligonucleotides used to form the various N = X linkers ranging from 13 duplex spacers up to 52; (C) sequences of oligonucleotides used to form the X-linker system
The inorganic building block for all experiments presented herein is a 10 nm AuNP functionalized with ~ 58 ± 5 hexylthiol terminated 28-mer oligonucleotides (see Supporting Information for detailed methods). To form the crystal structures, linker strands were designed, each with a 3′ end sequence complementary to the Au-bound DNA, a poly(dT) sequence of varying length flanked on both ends by an adenine flexor (to allow DNA strand flexibility) and a self-complementary 5′ GCGC end. The poly(dT) sequences were 13, 26, 39 and 52 bases in length (Scheme 1B). To make the particle interconnects more rigid, these poly(dT) sequences were hybridized to complementary poly(dA) sequences of equivalent lengths. A fifth linker was synthesized without a poly(dT) sequence and only one flexor (X-linker, Scheme 1C). Each duplex linker sequence was then added to a different solution of DNA-AuNPs in a 40 to 1 ratio, transferred to a quartz capillary, and heated to 45 °C. The temperature was lowered at a rate of 0.1 °C/min until the samples reached 35 °C, at which time they were characterized by small-angle X-ray scattering (SAXS, Figure 1A). Previous work has shown that lowering the temperature of the solution containing the aggregates to approximately 5 °C below the melting transition (Tm) of the DNA interconnects and annealing at this temperature typically yields the most ordered FCC structures.27
Figure 1.
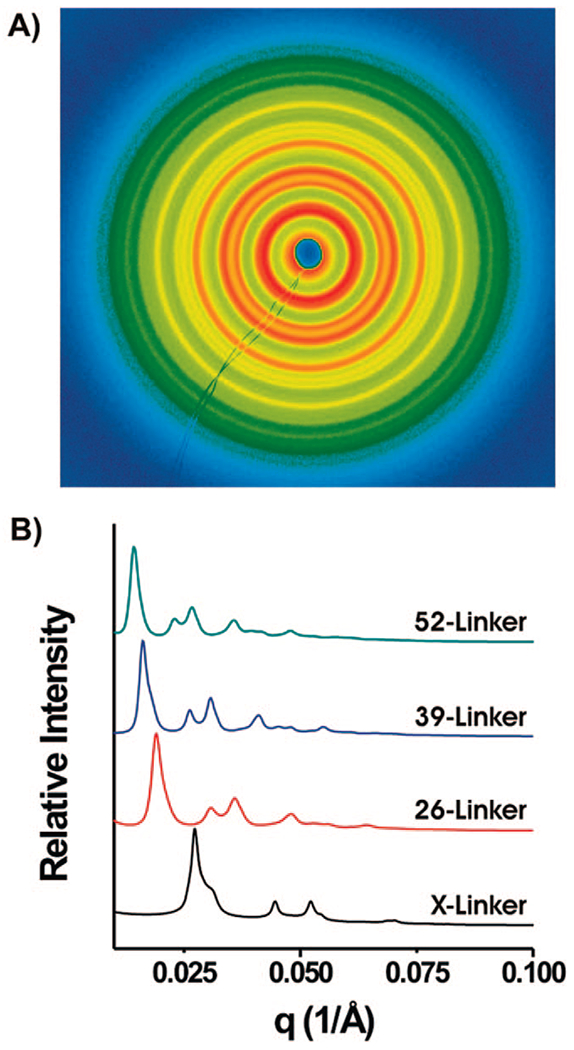
2-Dimensional and 1-Dimensional SAXS data. (A) 2-D data collected for the N = 39 linker system displayed in false colors. (B) 1-D line profiles of the 2-D data taken for the X-linker, N = 26, 39, and 52 systems demonstrating that the q* peak shifts to smaller values of q with increasing linker length. The 1-Dimensional data for the N = 13 bp linker was not included in part B, as this structure could not be annealed at 35 °C due to the low melting temperature of the A13/T13 duplex region. When N = 13 aggregates were formed at room temperature and subsequently imaged, they did show an FCC structure, but not of the quality of the structures formed with the other linkers (Figure 2, Supporting Figures 1B and 2B).
The 1-dimensional SAXS data (azimuthally averaged intensity versus scattering vector q, Figure 1B) allows one to easily observe the effect of increasing linker length on these crystalline structures. For all experiments, q = (4π sin θ)/λ (in 1/Å), where θ and λ are the scattering angle and wavelength of the X-ray used, respectively. The strong intensity of the first peak (moving out radially from the center) followed by a smaller shoulder and two peaks of similar intensity with their positions relative to that of the first peak (q*) such that q/q* = 1, √4/3, √8/3, √11/3, etc. (Figure 2) is indicative of a face-centered cubic arrangement. The 2-dimensional images for all N = X linker length FCC structures can be found in Supporting Figure 1A–F. As the length of DNA strands separating the particles gets larger, the scattering peaks shift to smaller q values (Figure 1B) as expected, since q is inversely proportional to the interparticle spacing (d-spacing) and values of q are given as 1/Å.
Figure 2.

Correlating linker length with crystal quality. The orange trace represents the modeled SAXS scattering for a perfectly ordered FCC crystal. The blue traces are the experimental data for the various length linkers and the values above the arrows are the theoretical q/q* peak locations. The strong correlation between simulated and experimental spectra confirms the presence of highly ordered FCC crystal structures.
In analyzing the SAXS data, the position of the first order peak of the FCC unit cell (q*) equals 2π/d111, where d111 is the d-spacing of the 111 plane of the FCC crystal. The X-linker (Figure 1B, black trace) shows the closest interparticle spacing and also the clearest delineation of the second peak (shoulder of the larger first peak). As the particle spacing increases, this shoulder can no longer be distinguished from the dominant first peak, (Figure 1B, red, blue, and green traces). However, increasing interparticle distance results in higher order scattering peaks observable at larger q values (Figure 1B, red, blue and green traces, Supporting Figure 2). These peaks are predicted by modeling27 (Figure 2) but until now have never been observed for oligonucleotide gold nanoparticle crystals. It is important to note that the existence of higher order peaks at longer linker lengths does not necessarily indicate larger or better quality FCC crystal structures. The appearance of these peaks is a result of the smaller q* values seen with increased interparticle distances. Intensities of peaks at higher q values are weaker because the detector does not efficiently measure scattering at q values larger than 0.06 Å−1 for the 10.3 nm gold particles used in this work. For the range of linker lengths studied, this decrease in intensity results in the obscuring of fifth order and higher (X-linker) and ninth order and higher (52-linker) peaks (Figure 2 and Supporting Figures 1A–F). Therefore, crystal quality cannot be determined by the number of peaks, but rather by the peak widths, where narrow peaks indicate greater constraint of AuNP position. The data clearly show that shorter linker lengths produce more ordered crystals (Figure 2 and Table 1).
Table 1.
Trends in FCC Crystal Parametersa
| name | nearest neighbor distance (nm) |
unit cell edge length (nm) |
percent filled space (% Au) |
aggregate diameter (nm) |
Δq/q* (×102) |
|---|---|---|---|---|---|
| X-linker | 28.1 | 39.8 | 3.74 | 957 | 1.24 |
| 13-linker | 32.4 | 45.9 | 2.44 | 586 | 2.41 |
| 26-linker | 40.3 | 57.0 | 1.27 | 769 | 2.74 |
| 39-linker | 47.7 | 67.5 | 0.77 | 1270 | 2.72 |
| 52-linker | 54.2 | 76.6 | 0.52 | 1100 | 4.04 |
Shown are calculations illustrating the effect of DNA linker length on AuNP nearest neighbor distances, unit cell edge length, the percent of the unit cell filled by in organic matter, aggregate diameter, and overall crystal order expressed as a function of the full width at half-maximum of the q* peak (Δq/q*).
To determine the extent to which DNA linker strands control the interparticle spacing of the crystals, we plotted the total number of DNA bases between particles versus gold nanoparticle nearest neighbor spacing as measured by SAXS. The nearest neighbor spacing can be calculated with eq 1:
| (1) |
where D is the nearest neighbor spacing in Å. When these nearest neighbor spacings are plotted versus number of bases for the linkers, a linear trend is observed with an R2 value of 0.987. The data follow eq 2:
| (2) |
where D is the gold nanoparticle nearest neighbor distance in nm and x is the total number of bases between the gold nanoparticles. The y-intercept of 11.1 nm can be attributed to the combined gold nanoparticle radii (~10.3 nm) and the two hexyl-thiol tethering moieties (~0.8 nm). The linearity of this equation allows one to easily design oligonucleotide linker sequences to form crystals with desired nanoparticle spacings (Figure 3). From these measured interparticle distances, it is possible to express a structure’s density as a percentage of space filled by the inorganic gold component using eq. 3:
| (3) |
where r is the radius of the gold nanoparticles in nm and a corresponds to the unit cell edge length in nm, calculated as √2 times D (the nearest neighbor distance). From our calculations, crystals with as little as 0.52% inorganic-filled space (in the case of the N = 52 linker) have been formed (Table 1). Moreover, we determined the size of the crystalline domains using the Debye-Scherrer formula (eq 4):
| (4) |
where t is the diameter of the crystal in Å, λ is the wavelength of scattered light in Å, θ is the diffraction angle associated with the q* peak and B is the angular full width at half-maximum of q*. From this analysis, the average diameters of the crystalline domains were determined to be approximately 957, 586, 769, 1270 and 1100 nm for the X-, 13-, 26-, 39-, and 52-linker systems, respectively (Table 1).
Figure 3.

Interparticle spacing as a function of total DNA length. A plot of the gold nearest neighbor distances as a function of the total number of bases used to separate them.
An attempt was made to extend the series to an N = 65 linker; however, the aggregate structures did not have a clearly defined FCC unit cell (Supporting Figures 1F and 2F). Interestingly, although previous data from others have indicated that longer linker lengths lead to higher order crystals,28 our data exhibit the opposite trend. The shorter lengths of duplex DNA provide the gold nanoparticles with less flexible interconnects (demonstrated by narrower peaks in the 1D spectra), as duplex DNA is known to be rigid up to approximately 50 nm.30 Thus as the linker lengths grow, the nanoparticles become less spatially constrained relative to a perfect FCC lattice, leading to a broadening of the peaks observed for longer linkers as indicated by the Δq/q* values, (Table 1 and Figure 2).
In conclusion, through these experiments, we have shown that the use of duplex DNA as a programmable linker allows one to direct the interparticle spacing of bioinorganic nanomaterials with a high degree of order. In addition to allowing one to program crystal structures with specific gold–gold particle distances and a desired percentage of inorganic-filled space, it should be possible to modify eq. 2 for particles of different sizes by adjusting the y-intercept value to account for an increase or decrease in the radii of nanoparticle building blocks. Finally, the rules established herein for predicting lattice parameters and controlling general materials architecture should be extendable to assemblies composed of other DNA-modified inorganic, organic, and biological synthons.
Supplementary Material
Acknowledgment.
C.A.M. acknowledges the NSF-NSEC, the AFOSR, and the NCI CCNE for the support of this work. He also is grateful for a NIH Director’s Pioneer Award. H.D.H. acknowledges the U.S. Department of Homeland Security (DHS) for a Graduate Fellowship under the DHS Scholarship and Fellowship Program. Portions of this work were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by E.I. DuPont de Nemours & Co., The Dow Chemical Company, and the State of Illinois. Use of the APS was supported by U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Supporting Information Available: Text giving details for the preparation of polyvalent DNA-AuNPs and the crystalline materials and the SAXS experimental parameters and figures showing all 1- and 2-dimensional SAXS images. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Mirkin CA; Letsinger RL; Mucic RC; Storhoff JJ Nature 1996, 382, 607–609. [DOI] [PubMed] [Google Scholar]
- (2).Alivisatos AP; Johnsson KP; Peng XG; Wilson TE; Loweth CJ; Bruchez MP; Schultz PG Nature 1996, 382, 609–611. [DOI] [PubMed] [Google Scholar]
- (3).Rosi NL; Giljohann DA; Thaxton CS; Lytton-Jean AKR; Han MS; Mirkin CA Science 2006, 312, 1027–1030. [DOI] [PubMed] [Google Scholar]
- (4).Seferos DS; Giljohann DA; Hill HD; Prigodich AE; Mirkin CA J. Am. Chem. Soc 2007, 129, 15477–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Daniel MC; Astruc D Chem. Rev 2004, 104, 293–346. [DOI] [PubMed] [Google Scholar]
- (6).Liu JW; Lu YJ Am. Chem. Soc 2003, 125, 6642–6643. [DOI] [PubMed] [Google Scholar]
- (7).Ozin GA, Arsenault AC Nanochemistry: A Chemical Approach to Nanomaterials; Royal Society of Chemistry: Cambridge, U.K., 2005. [Google Scholar]
- (8).Liu JW; Lu Y Angew. Chem., Int. Ed 2006, 45, 90–94. [Google Scholar]
- (9).Niemeyer CM Angew. Chem., Int. Ed 2001, 40, 4128–4158. [DOI] [PubMed] [Google Scholar]
- (10).Katz E; Willner I Angew. Chem., Int. Ed 2004, 43, 6042–6108. [DOI] [PubMed] [Google Scholar]
- (11).Niemeyer CM; Simon U Eur. J. Inorg. Chem 2005, 3641–3655. [Google Scholar]
- (12).Dillenback LM; Goodrich GP; Keating CD Nano. Lett 2006, 6, 16–23. [DOI] [PubMed] [Google Scholar]
- (13).Hurst SJ; Lytton-Jean AKR; Mirkin CA Anal. Chem 2006, 78, 8313–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Park SY; Stroud D Phys. Rev. B 2003, 67, 212202. [Google Scholar]
- (15).Lytton-Jean AKR; Mirkin CA J. Am. Chem. Soc 2005, 127, 12754–12755. [DOI] [PubMed] [Google Scholar]
- (16).Storhoff JJ; Mirkin CA Chem. Rev 1999, 99, 1849–1862. [DOI] [PubMed] [Google Scholar]
- (17).Nykypanchuk D; Maye MM; van der Lelie D; Gang O Langmuir 2007, 23, 6305–6314. [DOI] [PubMed] [Google Scholar]
- (18).Taton TA; Mirkin CA; Letsinger RL Science 2000, 289, 1757–1760. [DOI] [PubMed] [Google Scholar]
- (19).Baiancaniello PL; Kim AJ; Crocker JC Phys. Rev. Lett 2005, 94, 058302. [DOI] [PubMed] [Google Scholar]
- (20).Park SY; Stroud D Phys. Rev. B 2003, 68, 224201. [Google Scholar]
- (21).Park SY; Lee JS; Georganopoulou D; Mirkin CA; Schatz GC J. Phys. Chem. B 2006, 110, 12673–12681. [DOI] [PubMed] [Google Scholar]
- (22).Strable E; Johnson JE; Finn MG Nano Lett. 2004, 4, 1385–1389. [Google Scholar]
- (23).Kim AJ; Biancaniello PL; Crocker JC Chem. Phys. Lett 2006, 22, 1991–2001. [DOI] [PubMed] [Google Scholar]
- (24).Storhoff JJ; Lazarides AA; Mucic RC; Mirkin CA; Letsinger RL; Schatz GC J. Am. Chem. Soc 2000, 122, 4640–4650. [Google Scholar]
- (25).Park S-J; Lazarides AA; Mirkin CA; Letsinger RL Angew. Chem., Int. Ed 2001, 40, 2909–2912. [PubMed] [Google Scholar]
- (26).Park SJ; Lazarides AA; Storhoff JJ; Pesce L; Mirkin CA J. Phys. Chem. B 2004, 108, 12375–12380. [Google Scholar]
- (27).Park SY; Lytton-Jean AKR; Lee B; Weigand S; Schatz GC; Mirkin CA Nature 2008, 451, 553–556. [DOI] [PubMed] [Google Scholar]
- (28).Nykypanchuk D; Maye MM; van der Lelie D; Gang O Nature 2008, 451, 549–552. [DOI] [PubMed] [Google Scholar]
- (29).Xiong H; van der Lelie D; Gang OJ Am. Chem. Soc 2008, 130, 2442–2443. [DOI] [PubMed] [Google Scholar]
- (30).Schellman JA; Harvey C H Biophys. Chem 1995, 55, 95–114. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.