

CONSPECTUS
Supramolecular coordination chemistry allows researchers to synthesize higher-order structures that approach the nanoscale dimensions of small enzymes. Frequently, such structures have highly symmetric macrocyclic square or cage shapes. To build functional structures that mimic the complex recognition, catalytic, and allosteric properties of enzymes, researchers must do more than synthesize highly symmetric nanoscale structures. They must also simultaneously incorporate different functionalities into these structures and learn how to regulate their relative arrangement with respect to each other. Designing such heteroligated coordination complexes remains a significant challenge for supramolecular chemists.
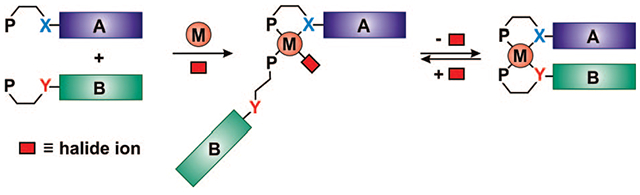
This Account focuses on the discovery and development of a novel supramolecular reaction known as the halide-induced ligand rearrangement (HILR) reaction. Two hemilabile ligands with different binding strengths combine with d8 transition metal precursors that contain halide ions. The reaction spontaneously results in heteroligated complexes and is highly modular and general. Indeed, it not only can be used to prepare tweezer complexes but also allows for the rapid and quantitative formation of heteroligated macrocyclic triple-decker/step and rectangular box complexes from a variety of different ligands and transition metal ions. The relative arrangement between functional groups A and B in these structures can be regulated in situ using small ancillary ligands such as halides, CO, and nitriles.
Based on this reaction, zinc- and magnesium-porphyrin moieties can be incorporated into heteroligated macrocyclic or tweezer scaffolds. These examples demonstrate the convergent and cofacial assembly of functional sites that are known to be involved in numerous processes in enzymes. They also show how the relative spatial and lateral distances of these sites can be varied, in many cases reversibly. Researchers can use such complexes to study a wide range of enzymatic processes, including catalysis, molecular recognition, electron transfer, and allosteric signal transfer.
Graphical Abstract
Introduction
Over the past few years, coordination chemistry-based synthetic methodologies have become increasingly attractive for the design of supramolecular complexes.1-11 Two of the most commonly used methods for preparing supramolecular coordination complexes are the directional bonding5-10 and symmetry interaction2,3,11 approaches. These methods rely on rigid ligands with strategically located functional groups and metal complexes with available coordination sites to form the desired structures. These approaches often provide access to highly symmetric structures with rigid, well-defined cavities that impart unique chemical reactivity, with respect to catalysis12-18 and molecular recognition.19-22 Our group has focused on developing a synthetic methodology known as the weak-link approach (WLA),1,4,23 which uses flexible hemilabile ligands 1 (Scheme 1) along with simple transition metal precursors to access structurally flexible supramolecular structures. Traditionally, the WLA has provided access to symmetric, homoligated supramolecular complexes, which are capable of adopting two different conformations: closed (2) and open (3) (Scheme 1). The ability to toggle between these two different conformations in situ via the addition or removal of chemical stimuli has enabled our group to design the first examples of supramolecular allosteric enzyme mimics,24-29 which when properly designed can provide a means of signal amplification in the context of chemical sensing.30,31
SCHEME 1.

Formation of Supramolecular Macrocycles via the WLA
A significant advance in the field of supramolecular chemistry would be the development of methodologies that expand synthetic capabilities from homoligated structures with two accessible states (4 and 5, Scheme 2A) to heteroligated32 architectures capable of undergoing multiple in situ transformations (6–8, Scheme 2B), providing control over the interactions of two different functionalities, A and B. The ability to target heteroligated structures containing two unique ligands with similar coordination motifs33-39 represents a significant challenge, with most methods involving multiple, often low yielding, steps with major product separation and isolation difficulties.
SCHEME 2.

(A) Homoligated Tweezer Complexes Formed via the Conventional WLA and (B) Heteroligated Tweezer Complexes Formed via the WLA and HILR
Recently, our group has discovered and developed a reaction known as the halide-induced ligand rearrangement (HILR), which allows one to access such complexes (Scheme 3).40,41 Therefore, these complexes can be chemically modified in situ to form closed (6), semiopen (7), and open (8) forms (Scheme 2B). The reaction allows for the cofacial arrangement of two different ligands in which the interactions between these ligands can be adjusted by the addition of small molecule chemical regulators. Indeed, we have shown that this capability can be used to realize a new class of allosteric enzyme mimics where a reactive pocket can be formed and destroyed through reactions that occur at the allosteric regulatory site, which is the metal hinge in this case (Scheme 2A).27
SCHEME 3.

The Halide-Induced Ligand Rearrangement (HILR)
Over the past several years, we have evaluated the generality of this rearrangement process by (1) using a wide variety of ligands with significantly different physical and chemical properties (i.e., size, shape, electronics, sterics, and symmetry) and (2) studying the effect of using different transition metal ions for the assembly of these complexes. Herein, we describe the development of several high-yielding ligand rearrangement processes and their ability to provide access to supramolecular assemblies that can incorporate many different functional sites, which can be used in molecular recognition, detection, catalysis, and electron transfer. The examples presented illustrate the high degree of selectivity and molecular diversity afforded by the HILR. Moreover, the supramolecular products obtained show how one can use external chemical stimuli to effectively regulate structural changes within these complexes.
The Halide-Induced Ligand Rearrangement (HILR)
Macrocyclic Complexes.
Our initial discovery of this rearrangement reaction originated from studying a series of ligands that contain both thioether–phosphine (PS) and ether–phosphine (PO) hemilabile coordination domains.40 Upon addition of the appropriate amounts of the desired hemilabile ligand and [Rh(NBD)Cl]2 (NBD = 2,5-norbornadiene) in CH2Cl2, the initial homoligated complexes 12a,b (Scheme 4) are formed. Interestingly, the 31P{1H} NMR spectroscopic resonances corresponding to the homoligated PS and PO moieties gradually disappear over several hours (time varies for each ligand set) with a concomitant appearance of a pair of doublet of doublets at ~δ 73 and 32, corresponding to complexes 13a,b, indicating the formation of the heteroligated macrocycles. Although the phenyl (13a) and biphenyl (13b) macrocycles were insoluble in common organic solvents (i.e., CH2Cl2 and THF), crystals of 13a, isolated as a precipitate, allowed for a single-crystal X-ray diffraction study (Figure 1A), which confirms the formation of the heteroligated product and is consistent with the solution structure deduced from spectroscopic data.
SCHEME 4.

The HILR for Macrocyclic Complexes
FIGURE 1.

Stick representations for the crystal structures of 13a (A) and 15a (B). Color scheme: Rh (pink), C (gray), P (orange), S (yellow), O (red), and Cl (green).
A key attribute of the complexes formed using this reaction is the combination of both S and O coordination about the RhI metal center. Abstraction of the Cl− ligand bound to the RhI centers of 13a,b results in the corresponding condensed macrocyclic products 14a,b. Since the Rh─O bonds are weaker than the Rh─S bonds in 14a,b, they can be cleaved selectively using Cl− to form 13a,b, CO to form 15a,b, or monodentate N-donors (pyridines and nitriles) to form 16a,b, leaving the Rh─S bonds intact. The ability to selectively cleave the Rh─O bonds in 14a,b without disrupting the Rh─S bonds not only allows one to easily regulate the aryl–aryl distance in this class of complexes (the vertical component) and therefore the size of the cavity of the resulting structure but also provides an orthogonal means to tailor the resulting structures via modification of the coordination environment at the structural site (the lateral component). For example, when Cl− or monodentate N-donors are used, the P─Rh─P geometry adopts a cis configuration (13a,b or 16a,b, respectively), whereas addition of CO (1 atm) converts the P─Rh─P geometry from cis to trans (15a,b), as confirmed by X-ray crystallography (Figure 1B). While the Rh─O bonds in 14a,b can be selectively cleaved to yield the semiopen complexes 15a,b or 16a,b, the fully open complexes 17a,b can be formed via the addition of Cl−/CO (1 atm) to the corresponding closed complexes 14a,b or by the addition of CO (1 atm) to 13a,b. The study also showed that the supramolecular ligand rearrangement process is induced by halide ions (Cl−, Br−, and I−), but not by several weakly or noncoordinating anions (BF4−, PF6− and B(ArF)4− (B(ArF)4− = B[3,5-(CF3)2(C6H3)]4−)).40
We have shown that the HILR also works with larger polydentate structures.40 For example, a 1,3,5-triphenylbenzene ligand containing two PS coordination domains and one PO coordination domain was synthesized and used to prepare trimetallic RhI complex 19 (Scheme 5). 31P{1H} NMR data show this complex also forms in a stepwise manner via the initial homoligated complex 18. Complex 19 undergoes a pattern of reactivity similar to the two-dimensional complexes 13a,b, resulting in fully closed complex 20 upon abstraction of chloride with AgBF4 (one homoligated Rh(κ2-PS)2 site in 18-20 remains unchanged). The observation of the HILR in trimetallic complexes was the first indication of the versatility of the rearrangement to generate multimetallic structures.
SCHEME 5.

The HILR for a Three-Dimensional Complex
Tweezer Complexes.
While the macrocyclic complexes described in the previous section provide a convenient approach for regulating the degree of cooperativity between two functional groups, they do not allow for the creation or total destruction of a reactive pocket, as observed in tweezer structures (Scheme 2). Such a phenomenon has been shown to have a significant effect on the rate of catalysis and enantioselectivity for a bimetallic epoxide ring-opening reaction.27 Indeed, the design of such tweezer complexes could become a general strategy for preparing new supramolecular sensors and allosteric enzyme mimics, a topic covered in a previous Account.23 In principle, the design of heteroligated tweezer complexes would allow for a gradual in situ switching among the following conformations: (1) those that possess a well-defined cavity incorporating two groups A and B in close proximity (6, Scheme 2B), (2) highly flexible open structures where groups A and B interact to a lesser degree or do not interact at all (8, Scheme 2B), and (3) intermediate structures in which only one arm is flexible (7, Scheme 2B). Furthermore, our group has previously demonstrated that moving from a macrocyclic- to a tweezer-based system often provides coordination complexes that are more soluble in common organic solvents.27 Additionally, ligands containing one hemilabile coordination domain are typically easier to synthesize than those that contain two, an important consideration as the complexity of the target supramolecular systems increases.
In order to glean more information regarding the effect of electronics and sterics on this reaction, a series of thioether–phosphine tweezer ligands were prepared and studied in the context of the HILR (21a–f, Scheme 6).41 Upon reaction of the appropriate combination of thioether–phosphine ligands 21a–f with phenyl-based tweezer ligands 22a,b and [Rh(NBD)Cl]2, the corresponding heteroligated RhCl(κ2-PS)(κ1-PO) complexes 25a–f formed in quantitative yield over a 4–18 h period as indicated by 31P{1H} NMR spectroscopy and X-ray crystallography (Figure 2). 31P{1H} NMR spectroscopy shows that the reaction proceeds via a process that is analogous to the one for the corresponding macrocyclic structures, in that the initial homoligated cationic tweezer products 23a–f and 24a,b form and react with one another over time to yield the desired heteroligated tweezer complexes 25a–f. The half-lives (t1/2) measured for each ligand combination indicate that the reaction is accelerated for ligands containing electron-deficient substituents on the aromatic groups appended to the thioether–phosphine moieties. Similar to the macrocyclic complexes, when noncoordinating, nonhalide counterions are used (e.g., BF4−), the rearrangement process is not observed. Interestingly, the reaction still proceeds in the absence of an ether moiety (22a) and in the presence of sterically demanding ligands (21e,f), illustrating the potential for incorporating a variety of different ligands and functional groups around the RhI metal center.
SCHEME 6.

Heteroligated Tweezer Complexes Formed via the HILR
FIGURE 2.

Stick representation for the crystal structure of 25e. Color scheme: Rh (pink), C (gray), P (orange), S (yellow), O (red), and Cl (green).
Compounds 25b–f (but not 25a where X = CH2) can be converted to the corresponding cationic complexes 26b–f via abstraction of Cl− using a stoichiometric amount of Na[B(ArF)4]. This transformation can be accomplished reversibly via the successive addition and abstraction of Cl−. Addition of CO (1 atm) to a solution of complexes 26b–f results in the quantitative formation of the corresponding cationic products 27b–f and also induces a change in geometry of the P─Rh─P coordination from cis to trans. The fully open complexes 28a–f can be generated via (1) the addition of CO (1 atm) to solutions of 25a–f, (2) the addition of Cl− to solutions of 27a–f, or (3) the addition of Cl− and CO to solutions of 26b–f. As these data illustrate, once the tweezer complexes 26b–f have been formed, they can be converted in situ to different structures, allowing for the facile control of the aryl–aryl interactions via small molecule reactions at the RhI structural site.
Triple-Decker/Step Complexes.
When the WLA is used to synthesize tweezer and macrocycle complexes that behave as abiotic allosteric enzyme mimics, the design of complexes capable of facilitating a bimetallic reaction has become a necessity. Indeed, we have shown that one can construct reactive pockets in molecules that facilitate catalytic acyl transfer25,30,31 and epoxide ring opening reactions.27,28 Small molecules that change the conformation or destroy these pockets significantly affect the rate and in certain cases entantioselectivities of the reactions.25,27-31 Since there are a relatively few documented bimetallic/multimetallic42-44 catalytic processes compared with reactions catalyzed by a monometallic species, it would be advantageous to design complexes whereby a catalyst or functional group can be activated or deactivated via steric blocking. In principle, a triple-decker type structure would allow for regulation of the interactions around an active catalytic site via the addition or removal of the appropriate chemical effector molecules.
In this regard, we have discovered that upon addition of both a suitable symmetric thioether–phosphine (or ether–phosphine) hemilabile ligand (i.e., 29a,b, Scheme 7) typically used to prepare macrocycles and an ether–phosphine (or thioether–phosphine) hemilabile ligand (i.e., 30a–c) typically used to form tweezer complexes to a solution containing [Rh(COD)Cl]2, the corresponding heteroligated complexes 31a–c form in quantitative yield as indicated by 31P{1H} NMR spectroscopy.45 These structures form via a process analogous to that presented for the macrocycle- and tweezer-based complexes.40,41 Similar to the analogous RhI tweezer complexes, the Cl− ligands can be abstracted from 31a–c to form the condensed complexes 32a–c. Significantly, this transformation can be effected reversibly, thereby providing a convenient way to regulate the sterics around the central aromatic group.
SCHEME 7.

Triple-Decker/Step Complexes Prepared via the HILR
The solid state and solution data for complex 31a (Scheme 7, Figure 3A) both illustrate the lack of close interaction between the aromatic groups of the tweezer ligands and the aromatic group of the central bifunctional hemilabile ligand. For complex 32a, the crystallographic data illustrate that the three aromatic groups are not aligned cofacially and form a step-like complex (Figure 3B). Although the complex adopts this orientation in the solid state, 2D NOESY NMR data indicate that the complexes are quite fluxional in solution and the outer aromatic groups can move back and forth freely, with the average structure best described as a triple-decker structure.
FIGURE 3.

Stick representation for the crystal structure of 31a (A) and 32a (B). Color scheme: Rh (pink), C (gray), P (orange), S (yellow), O (red), and Cl (green).
Heteroligated PtII Complexes
While RhI has proven useful for targeting otherwise inaccessible heteroligated supramolecular coordination complexes, the resulting compounds often must be handled and manipulated under an inert atmosphere. This requirement precludes using these complexes under conditions that would closely resemble those found in Nature. We hypothesized that PtII, which is also a d8 metal known to support phosphorus–sulfur hemilabile ligands,46 may also exhibit similar reactivity as the RhI system in the context of supramolecular ligand rearrangements. Furthermore, it is well established that isoelectronic and structurally related PtII complexes are not as susceptible to degradation under ambient conditions,47 which makes this transition metal an attractive candidate for the synthesis of new heteroligated coordination complexes.
While the heteroligated RhI analogues were formed using a combination of thioether– and ether–phosphine hemilabile ligands, the PtII analogues form via the participation of both alkyl- and arylthioether ligands (Scheme 8)48 Although the cationic complexes 36 and 39 can be isolated via a one-pot procedure, analysis of this process via 31P{1H} NMR spectroscopy illustrates that semiopen complexes (e.g., 35a) form initially, and Cl− can be abstracted using Na+ and Ag+ salts to give closed complexes 36 and 39. Furthermore, X-ray crystallographic analyses of single crystals of 35a and 36 confirm the presence of the heteroligated environment around the PtII metal center (Figure 4).
SCHEME 8.

Heteroligated PtII Complexes
FIGURE 4.

Stick representation for the crystal structures of 35a (A) and 36 (B). Color scheme: Pt (purple), C (gray), P (orange), S (yellow), and Cl (green).
Since halides (either Cl− or I−) can be used to cleave the relatively weak Pt─Saryl bonds in 36 to form 35b and 37, one can easily regulate the interactions between the groups appended to both S atoms. These heteroligated, thioether-based PtII complexes show a reactivity pattern different from the RhI complexes mentioned above. For example, acetonitrile is not a strong enough binder to cleave either Pt─S bond in complex 36, while it does break the Rh─O bond in 14a,b (Scheme 4). Thus, by choosing the appropriate metal and hemilabile ligands for the heteroligated rearrangement, one can tailor the small molecule reactivity with respect to the metal center, and systems can be designed that exhibit reactivities at regulatory and functional (catalytic) sites in the allosteric enzyme mimics that are orthogonal.
Structures Prepared via the HILR with Greater Complexity and Function
Assembly of Salen-Based Multimetallic Box Structures.
Our preliminary experiments during the development of the HILR indicated that the addition of pyridyl and nitrile ligands act to selectively break the Rh─O moieties in the macrocycles while leaving the Rh─S bonds intact (Scheme 4). The ability to target macrocyclic structures containing heteroligated coordination environments has led us to investigate the potential for using these macrocycles as building blocks for the preparation of large, supramolecular architectures. In principle, if a macrocyclic complex and a suitable bifunctional ligand are reacted in a 1:1 ratio, it is possible to quantitatively and rapidly assemble multimetallic box-type structures where the bifunctional ligands are aligned cofacially.
Upon the addition of bifunctional bridging ligands 41a–c to a solution of the closed heteroligated macrocycle 40 in a 1:1 ratio, the desired multimetallic structures 42a–c form after 10 min as indicated by 31P{1H} NMR spectroscopy (Scheme 9)49 The chemical shifts and coupling constants indicate that the Rh─O bonds are quantitatively cleaved and the P─Rh─P coordination environment retains the original cis configuration. A single-crystal X-ray diffraction study of 42a confirmed the local coordination environment about each RhI metal center and also illustrated that each 1,4-dicyanobenzene ligand is held in a cofacial orientation by the two heteroligated macrocycles (Figure 5). Importantly, while the area of the rectangle formed by the four RhI metal centers is 110 Å2 for 42a, the inherent ability to tailor complexes formed via the HILR and the WLA, in principle, will allow one to systematically vary the cavity size of the resulting structures via modification of the building blocks used for assembly.
SCHEME 9.

Multimetallic Box Complexes Formed via the HILR
FIGURE 5.

Stick representation for the crystal structure of 42a: (A) side view; (B) top view. The phenyl groups on the phosphines in the crystal structure have been omitted for clarity. Color scheme: Rh (pink), C (gray), P (orange), S (yellow), O (red), and N (blue).
Heteroligated Cofacial Porphyrin Complexes
Macrocyclic Complexes.
Since Collman et al. presented their initial work on the synthesis of cofacial porphyrin complexes,50 chemists have studied the properties of variants of such complexes for over 25 years and have generated many sophisticated and potentially useful species.17,51-53 Typically, these complexes are prepared via tedious multistep procedures, using rigid scaffolds that restrict the overall structural flexibility of the targeted complex. As a result, it is difficult to carry out comprehensive studies that address the consequences of changing the porphyrin–porphyrin distance, their orientation with respect to each other, and the metal centers that reside within them.54,55 We hypothesized that the HILR could provide access to cofacial porphyrin structures that not only enabled the systematic placement of different metals within the cofacial assembly but also would allow for significant control over the porphyrin orientations and interporphyrin distances.
Our initial attempts to synthesize these complexes were performed in CH2Cl2 at room temperature. Unfortunately, these conditions were not suitable for preparing the desired heteroligated complexes. By systematically varying the reaction conditions, we discovered that complexes 46 and 47 can be prepared in near quantitative yield by sonicating a THF solution containing ligands 43 (or 44), 45, and [Rh(COD)Cl]2 for 1 h (Scheme 10)56 Analysis of X-ray quality single crystals of 46 grown in the presence of DABCO show a solid state structure consistent with the one we assigned based upon solution spectroscopic data (Figure 6).
SCHEME 10.

Heteroligated Cofacial Porphyrin Macrocyclic Complexes Formed by the HILR
FIGURE 6.

Stick representation for the crystal structure of 46⊂DABCO: (A) side view; (B) top view. Color scheme: Rh (pink), Zn (purple), Mg (light violet), C (gray), P (orange), S (yellow), O (red), N (blue), and Cl (green).
The neutral complexes 46 and 47 can be converted into the closed cationic macrocycles 48 and 49 upon sonication with 2 equiv of Na[B(ArF)4] in CH2Cl2. Since the Rh─O bonds in macrocycles 48 and 49 are weaker than the Rh─S bonds, they can be quantitatively cleaved using CO (1 atm), resulting in the formation of the semiopen assemblies 50 and 51. Importantly, this transformation is accompanied by a change in the P─Rh─P geometry from cis to trans, which serves to modify the alignment of each porphyrin. The fully open complexes 52 and 53 can be generated via two different routes. First, adding 2 equiv of (n-Bu)4NCl followed by the introduction of CO (1 atm) to solutions of 48 or 49 results in the quantitative formation of the fully open assemblies 52 or 53, respectively. Alternatively, 52 or 53 can be directly prepared via introduction of CO (1 atm) to solutions of the semiopen assemblies 46 or 47, respectively. These transformations illustrate how one can (1) selectively access four different cofacial assemblies from a single macrocycle and (2) rapidly access heteroligated cofacial porphyrin systems in which each porphyrin ligand contains a different metal.
Tweezer Complexes.
While the macrocyclic cofacial porphyrin complexes synthesized to date have illustrated the ability for the HILR to provide access to porphyrin structures capable of in situ modification, a potential limitation of complexes 46 and 47 is their low solubility in common organic solvents, which, in turn, makes reversible in situ reactions difficult. As we mentioned earlier, one possible route to overcome limitations with respect to solubility is to design tweezer complexes, which have been shown to be significantly more soluble in many organic solvents.27 Our group has designed the analogous cofacial porphyrin tweezer complexes, which contain a single RhI regulatory site and can undergo significant geometrical distortions in situ when triggered by external chemical stimuli. Compared with the macrocyclic analogues, the tweezer complexes are all highly soluble in organic solvents (i.e., CH2Cl2, THF) owing, in part, to the incorporation of an extra mesityl group on the porphyrin ring and the decrease in overall charge and molecular weight. Furthermore, the increased solubility of these complexes allows them to undergo reversible reactions with small-molecule ligands without undesired precipitation.
Using protocols analogous to the ones developed for the macrocyclic complexes 46 and 47, the heteroligated complexes 57 and 58 were prepared in quantitative yield via the HILR using 2 equiv of 54 and 55 (or 56) and 1 equiv of the RhI precursor, [Rh(COE)2Cl]2 (Scheme 11).57 The closed cationic complexes 59 and 60 can be prepared quantitatively via abstraction of Cl− with Na[B(ArF)4]. Importantly, since tweezer complexes 57–60 are much more soluble in organic solvent compared with their macrocyclic analogues, the RhI hinge site can be addressed reversibly in situ with selected small-molecule ligands and elemental anions. For example, the addition of (n-Bu)4NCl (1 equiv) to a CH2Cl2 solution of complex 59 or 60 results in their quantitative conversion to complexes 57 or 58, respectively.
SCHEME 11.

Heteroligated Porphyrin Tweezer Complexes Prepared via the HILR
Upon addition of CO (1 atm) to separate solutions of 59 and 60 in CD2Cl2, respectively, complexes 61 and 62 are formed in quantitative yield. The fully open complexes 63 and 64 can be generated via two different routes, which both involve displacement of the thioether moieties bound to the RhI metal centers. For instance, introducing CO (1 atm) to a CD2Cl2 solution of complexes 57 or 58 yields the fully open, highly flexible tweezer complexes 63 or 64 quantitatively. These complexes also can be generated in situ from the closed complexes 59 or 60 upon the addition of a stoichiometric amount of (n-Bu)4NCl and CO (1 atm).
Conclusions and Outlook
Our efforts thus far demonstrate the power of using coordination chemistry, namely, the WLA and the HILR, for the preparation of unique and otherwise inaccessible supramolecular complexes. These heteroligated complexes can be readily accessed in quantitative yield using a wide variety of hemilabile ligands with varying electronic and steric properties when reacted with simple d8 transition metal precursors. It is anticipated that this new reaction will allow for the preparation of novel supramolecular complexes that can act in the context of allosteric catalysis/sensing and for the design of new architectures capable of mimicking the properties of enzyme active sites. For example, one can target tweezer complexes that contain both a catalyst and cocatalyst (i.e., porphyrin and imidazole/cysteine, respectively) that can be aligned cofacially, whereby the cocatalyst can activate a metal center for catalysis and potentially allow for the regulation of the catalytic rate by controlling the distance and relative alignment between the cocatalyst and the catalytic center. Additionally, it is now possible to target triple-decker complexes that contain two sterically demanding blocking ligands, which can crowd a catalytically active metal center and regulate a catalytic reaction via the addition or removal of small molecule effectors.
Acknowledgments
C.A.M. acknowledges the NSF, AFOSR, ARO, and DDRE for their generous support of this research and is grateful to the NIH for a Director’s Pioneer Award and the DoD for a NSSEF Fellowship.
Biography
Christopher G. Oliveri was born and raised in Long Island, New York. He earned a B.S. degree in chemistry (2002) and a M.S. degree in bioinorganic chemistry (2003) working under the supervision of Professor Stephen Koch at Stony Brook University. In 2003, he joined the graduate program at Northwestern University where he worked towards developing novel coordination chemistry-based cofacial porphyrin complexes under the auspices of Professors Chad A. Mirkin and SonBinh T. Nguyen. In 2007, he completed the Ph.D. program and is currently a Senior Chemist at ExxonMobil in the Process Research Laboratory in Annandale, New Jersey.
Pirmin A. Ulmann was born in Schaffhausen, Switzerland. He received his diploma in chemistry from ETH Zurich in 2004 under the guidance of Professor Erick M. Carreira. After a stint in process upscaling for a drug candidate at Merck Eprova AG, Schaffhausen, he joined the graduate program at Northwestern University, where he is currently pursuing his Ph.D. under the direction of Professor Chad A. Mirkin in the field of supramolecular coordination chemistry.
Michael J. Wiester attended the University of Texas at Austin (UT). While at UT, he did undergraduate research with Professor Richard Jones in the area of organometallic chemistry and earned his B.S. in chemistry in 2003. He joined the graduate program at Northwestern University where he is currently pursuing his Ph.D. in chemistry under the supervision of Professor Chad A. Mirkin.
Chad A. Mirkin received a B.S. degree from Dickinson College in 1986 and a Ph.D. degree in chemistry from the Pennsylvania State University in 1989. He was an NSF Postdoctoral Fellow at the Massachusetts Institute of Technology prior to becoming a chemistry professor at Northwestern University in 1991. He is currently the Director of the International Institute for Nanotechnology, the George B. Rathmann Professor of Chemistry, Professor of Medicine, and Professor of Materials Science and Engineering. He is author of over 335 manuscripts and over 350 patent applications (71 issued). He is the founder of two companies, Nanosphere and NanoInk, which are commercializing nanotechnology applications in the life science and semiconductor industries.
REFERENCES
- 1.Farrell JR; Mirkin CA; Guzei IA; Liable-Sands LM; Rheingold AL The Weak-Link Approach to the Synthesis of Inorganic Macrocyles. Angew. Chem., Int. Ed 1998, 37, 465–467. [DOI] [PubMed] [Google Scholar]
- 2.Caulder DL; Raymond RN The Rational Design of High Symmetry Coordination Clusters. J. Chem. Soc., Dalton Trans 1999, 1185–1200. [Google Scholar]
- 3.Caulder DL; Raymond RN Supermolecules by Design. Acc. Chem. Res 1999, 32, 975–982. [Google Scholar]
- 4.Holliday BJ; Mirkin CA Strategies for the Construction of Supramolecular Compounds through Coordination Chemistry. Angew. Chem., Int. Ed 2001, 40, 2022–2043. [PubMed] [Google Scholar]
- 5.Seidel SR; Stang PJ High-Symmetry Coordination Cages via Self-Assembly. Acc. Chem. Res 2002, 35, 972–983. [DOI] [PubMed] [Google Scholar]
- 6.Fujita M Self-Assembly of [2]Catenanes Containing Metals in Their Backbones. Acc. Chem. Res 1999, 32, 53–61. [Google Scholar]
- 7.Stang PJ; Olenyuk B Self-Assembly, Symmetry, and Molecular Architecture: Coordination as the Motif in the Rational Design of Supramolecular Metallacyclic Polygons and Polyhedra. Acc. Chem. Res 1997, 30, 502–518. [Google Scholar]
- 8.Leininger S; Olenyuk B; Stang PJ Self-Assembly of Discrete Cyclic Nanostructures Mediated by Transition Metals. Chem. Rev 2000, 100, 853–908. [DOI] [PubMed] [Google Scholar]
- 9.Benkstein KD; Hupp JT; Stern CL Molecular Rectangles Based on Rhenium(I) Coordination Chemistry. J. Am. Chem. Soc 1998, 120, 12982–12983. [DOI] [PubMed] [Google Scholar]
- 10.Lee SJ; Lin W Chiral Metallocycles: Rational Synthesis and Novel Applications. Acc. Chem. Res 2008, 41, 521–537. [DOI] [PubMed] [Google Scholar]
- 11.Albrecht M “Let’s Twist Again”-Double-Stranded, Triple-Stranded, and Circular Helicates. Chem. Rev 2001, 101, 3457–3497. [DOI] [PubMed] [Google Scholar]
- 12.Nishioka Y; Yamaguchi T; Yoshizawa M; Fujita M Unusual [2 + 4] and [2 + 2] Cycloadditions of Arenes in the Confined Cavity of Self-Assembled Cages. J. Am. Chem. Soc 2007, 129, 7000–7001. [DOI] [PubMed] [Google Scholar]
- 13.Yoshizawa M; Tamura M; Fujita M Diels-Alder in Aqueous Molecular Hosts: Unusual Regioselectivity and Efficient Catalysis. Science 2006, 312, 251–254. [DOI] [PubMed] [Google Scholar]
- 14.Pluth MD; Bergman RG; Raymond KN Acid Catalysis in Basic Solution: A Supramolecular Host Promotes Orthoformate Hydrolysis. Science 2007, 316, 85–88. [DOI] [PubMed] [Google Scholar]
- 15.Leung DH; Bergman RG; Raymond KN Highly Selective Supramolecular Catalyzed Allylic Alcohol Isomerization. J. Am. Chem. Soc 2007, 129, 2746–2747. [DOI] [PubMed] [Google Scholar]
- 16.Fiedler D; Leung DH; Bergman RG; Raymond KN Selective Molecular Recognition, C─H Bond Activation, and Catalysis in Nanoscale Reaction Vessels. Acc. Chem. Res 2005, 38, 351–360, and references cited therein. [DOI] [PubMed] [Google Scholar]
- 17.Merlau ML; del Pilar Mejia M; Nguyen ST; Hupp JT Artificial Enzymes Formed through Directed Assembly of Molecular Square Encapsulated Epoxidation Catalysts. Angew. Chem., Int. Ed 2001, 40, 4239–4242. [DOI] [PubMed] [Google Scholar]
- 18.Merlau M; Grande WJ; Nguyen ST; Hupp JT Enhanced Activity of Manganese(III) Porphyrin Epoxidation Catalysts through Supramolecular Complexation. J. Mol. Catal. A 2000, 156, 79–84. [Google Scholar]
- 19.Tashiro S; Tominaga M; Kawano M; Therrien B; Ozeki T; Fujita M Sequence-Selective Recognition of Peptides within the Single Binding Pocket of a Self-Assembled Coordination Cage. J. Am. Chem. Soc 2005, 127, 4546–4547. [DOI] [PubMed] [Google Scholar]
- 20.Yoshizawa M; Tamura M; Fujita M AND/OR Bimolecular Recognition. J. Am. Chem. Soc 2004, 126, 6846–6847. [DOI] [PubMed] [Google Scholar]
- 21.Fiedler D; Bergman RG; Raymond KN Stabilization of Reactive Organometallic Intermediates inside a Self-Assembled Nanoscale Host. Angew. Chem., Int. Ed 2006, 45, 745–748. [DOI] [PubMed] [Google Scholar]
- 22.Dong VM; Fiedler D; Carl B; Bergman RG; Raymond KN Molecular Recognition and Stabilization of Iminium Ions in Water. J. Am. Chem. Soc 2006, 128, 14464–14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gianneschi NC; Masar MS III; Mirkin CA Development of a Coordination Chemistry-Based Approach for Functional Supramolecular Structures. Acc. Chem. Res 2005, 38, 825–837. [DOI] [PubMed] [Google Scholar]
- 24.Kuwabara J; Stern CL; Mirkin CA A Coordination Chemistry Approach to a Multieffector Enzyme Mimic. J. Am. Chem. Soc 2007, 129, 10074–10075. [DOI] [PubMed] [Google Scholar]
- 25.Oliveri CG; Gianneschi NC; Nguyen ST; Mirkin CA; Stern CL; Wawrzak Z; Pink M Supramolecular Allosteric Cofacial Porphyrin Complexes. J. Am. Chem. Soc 2006, 128, 16286–16296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heo J; Mirkin CA Pseudo-Allosteric Recognition of Mandelic Acid with an Enantioselective Coordination Complex. Angew. Chem., Int. Ed 2006, 45, 941–944. [DOI] [PubMed] [Google Scholar]
- 27.Gianneschi NC; Cho S-H; Nguyen ST; Mirkin CA Reversibly Addressing an Allosteric Catalyst in Situ: Catalytic Molecular Tweezers. Angew. Chem., Int. Ed 2004, 43, 5503–5507. [DOI] [PubMed] [Google Scholar]
- 28.Gianneschi NC; Bertin PA; Nguyen ST; Mirkin CA; Zakharov LN; Rheingold AL A Supramolecular Approach to an Allosteric Catalyst. J. Am. Chem. Soc 2003, 125, 10508–10509. [DOI] [PubMed] [Google Scholar]
- 29.Yoon HJ; Heo J; Mirkin CA Allosteric Regulation of Phosphate Diester Transesterification Based upon a Dinuclear Zinc Catalyst Assembled via the Weak-Link Approach. J. Am. Chem. Soc 2007, 129, 14182–14183. [DOI] [PubMed] [Google Scholar]
- 30.Masar MS III; Gianneschi NC; Oliveri CG; Stern CL; Nguyen ST; Mirkin CA Allosterically Regulated Supramolecular Catalysis of Acyl Transfer Reactions for Signal Amplification and Detection of Small Molecules. J. Am. Chem. Soc 2007, 129, 10149–10158. [DOI] [PubMed] [Google Scholar]
- 31.Gianneschi NC; Nguyen ST; Mirkin CA Signal Amplification and Detection via a Supramolecular Allosteric Catalyst. J. Am. Chem. Soc 2005, 127, 1644–1645. [DOI] [PubMed] [Google Scholar]
- 32.In this paper, the term heteroligated refers to the simultaneous coordination of two different ligands of the same ligand class (i.e., phosphines, pyridines, etc.) to a metal.
- 33.Yoshizawa M; Nagao M; Kumazawa K; Fujita M Side Chain-Directed Complementary cis-Coordination of two Pyridines on Pd(II): Selective Multicomponent Assembly of Square-, Rectangular-, and Trigonal Prism-Shaped Molecules. J. Organomet. Chem 2005, 690, 5383–5388. [Google Scholar]
- 34.Baxter P; Lehn JM; DeCian A; Fischer J Multicomponent Self-Assembly: Spontaneous Formation of a Cyclindrical Complex from Five Ligands and Six Metal Ions. Angew. Chem., Int. Ed 1993, 32, 69–72. [Google Scholar]
- 35.Baxter PNW; Lehn JM; Baum G; Fenske D The Design and Generation of Inorganic Cylindrical Cage Architectures by Metal-Ion-Directed Multicomponent Self-Assembly. Chem.—Eur. J 1999, 5, 102–112. [Google Scholar]
- 36.Schmittel M; Kalsani V Functional, Discrete, Nanoscale Supramolecular Assemblies. Top. Curr. Chem 2005, 245, 1–53. [Google Scholar]
- 37.Kuil M; Soltner T; van Leeuwen PWNM; Reek JNH High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes. J. Am. Chem. Soc 2006, 128, 11344–11345. [DOI] [PubMed] [Google Scholar]
- 38.Pyle AM; Rehmann JP; Meshoyrer R; Kumar CV; Turro NJ; Barton JK Mixed-Ligand Complexes of Ruthenium(II) - Factors Governing Binding to DNA. J. Am. Chem. Soc 1989, 111, 3051–3058. [Google Scholar]
- 39.Bosnich B The Exciton Circular Dichroism and the Absolute Configurations of Molecules Containing Nonidentical Chromophores. The Cases of the Bis(o-phenanthroline)-2,2′-bipyridylruthenium(II) and Bis(2,2′-bipyridyl)-o-phenanthrolineruthenium(II) Ions. Inorg. Chem 1968, 7, 2379–2386. [Google Scholar]
- 40.Brown AM; Ovchinnikov MV; Stern CL; Mirkin CA Halide-Induced Supramolecular Ligand Rearrangement. J. Am. Chem. Soc 2004, 126, 14316–14317. [DOI] [PubMed] [Google Scholar]
- 41.Brown AM; Ovchinnikov MV; Mirkin CA Heteroligated Rh(I) Tweezer Complexes. Angew. Chem., Int. Ed 2005, 44, 4207–4209. [DOI] [PubMed] [Google Scholar]
- 42.Mackay LG; Wylie RS; Sanders JKM Catalytic Acyl Transfer by a Cyclic Porphyrin Trimer: Efficient Turnover without Product Inhibition. J. Am. Chem. Soc 1994, 116, 3141–3142. [Google Scholar]
- 43.Jacobsen EN Asymmetric Catalysis of Epoxide Ring-Opening Reactions. Acc. Chem. Res 2000, 33, 421–431. [DOI] [PubMed] [Google Scholar]
- 44.Shibasaki M, Yamamoto Y, Eds.; Multimetallic Catalysts in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- 45.Jeon YM; Heo J; Brown AM; Mirkin CA Triple-Decker Complexes Formed via the Weak Link Approach. Organometallics 2006, 25, 2729–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Zotto A; Mezzetti A; Rigo P; Bressan M; Morandini F; Morvillo A Synthesis and NMR-Studies of Palladium(II) and Platinum(II) Complexes with Hybrid Bidentate Ligands Ph2P(CH2)2SR. Inorg. Chim. Acta 1989, 158, 151–158. [Google Scholar]
- 47.Colladon M; Scarso A; Sgarbossa P; Michelin RA; Strukul G Regioselectivity and Diasteroselectivity in Pt(II)-Mediated “Green” Catalytic Epoxidation of Terminal Alkenes with Hydrogen Peroxide: Mechanistic Insight into a Peculiar Substrate Selectivity. J. Am. Chem. Soc 2007, 129, 7680–7689. [DOI] [PubMed] [Google Scholar]
- 48.Ulmann PA; Brown AM; Ovchinnikov MV; Mirkin CA; DiPasquale AG; Rheingold AL Spontaneous Formation of Heteroligated Pt(II) Complexes with Chelating Hemilabile Ligands. Chem.—Eur. J 2007, 13, 4529–4534. [DOI] [PubMed] [Google Scholar]
- 49.Brown AM; Ovchinnikov MV; Stern CL; Mirkin CA Tetrametallic Rectangular Box Complexes Assembled from Heteroligated Macrocycles. Chem. Commun 2006, 4386–4388. [DOI] [PubMed] [Google Scholar]
- 50.Collman JP; Chong AO; Jameson GB; Oakley RT; Rose E; Schmittou ER; Ibers JA Synthesis of “Face to Face” Porphyrin Dimers Linked by 5,15-Substituents: Potential Binuclear Multielectron Redox Catalysts. J. Am. Chem. Soc 1981, 103, 516–533. [Google Scholar]
- 51.Chang CJ; Loh Z-H; Shi C; Anson FC; Nocera DG Targeted Proton Delivery in the Catalyzed Reduction of Oxygen to Water by Bimetallic Pacman Porphyrins. J. Am. Chem. Soc 2004, 126, 10013–10020. [DOI] [PubMed] [Google Scholar]
- 52.Rosenthal J; Luckett TD; Hodgkiss JM; Nocera DG Photocatalytic Oxidation of Hydrocarbons by a Bis-iron(III)-μ-oxo Pacman Porphyrin Using O2 and Visible Light. J. Am. Chem. Soc 2006, 128, 6546–6547. [DOI] [PubMed] [Google Scholar]
- 53.Collman JP; Wagenknecht PS; Hutchison JE Cofacial Bis(metallo)diporphyrins as Potential Molecular Catalysts for Multielectron Reductions and Oxidations of Small Molecules. Angew. Chem., Int. Ed 1994, 106, 1620–1639. [Google Scholar]
- 54.Faure S; Stern C; Guilard R; Harvey PD Synthesis and Photophysical Properties of Meso-Substituted Bisporphyrins: Comparative Study of Phosphorescence Quenching for Dioxygen Sensing. Inorg. Chem 2005, 44, 9232–9241. [DOI] [PubMed] [Google Scholar]
- 55.Fukuzumi S; Okamoto K; Tokuda Y; Gros CP; Guilard R Dehydrogenation versus Oxygenation in Two-Electron and Four-Electron Reduction of Dioxygen by 9-Alkyl-10-methyl-9,10-dihydroacridines Catalyzed by Monomeric Cobalt Porphyrins and Cofacial Dicobalt Porphyrins in the Presence of Perchloric Acid. J. Am. Chem. Soc 2004, 126, 17059–17066. [DOI] [PubMed] [Google Scholar]
- 56.Oliveri CG; Heo J; Nguyen ST; Mirkin CA; Wawrzak Z A Convergent Coordination Chemistry-Based Approach to Dissymmetric Macrocyclic Cofacial Porphyrin Complexes. Inorg. Chem 2007, 46, 7716–7718. [DOI] [PubMed] [Google Scholar]
- 57.Oliveri CG; Nguyen ST; Mirkin CA A Highly Modular and Convergent Approach for the Synthesis of Stimulant-Responsive Heteroligated Cofacial Porphyrin Tweezer Complexes. Inorg. Chem 2008, 47, 2755–2763. [DOI] [PubMed] [Google Scholar]