

Abstract
Most cells respond to viral infections by activating innate immune pathways that lead to the induction of antiviral restriction factors. One such factor, viperin, was discovered almost two decades ago based on its induction during viral infection. Since then, viperin has been shown to possess activity against numerous viruses via multiple proposed mechanisms. Most recently, however, viperin was demonstrated to catalyze the conversion of cytidine triphosphate (CTP) to 3′-deoxy-3′,4′-didehydro-CTP (ddhCTP), a previously unknown ribonucleotide. Incorporation of ddhCTP causes premature termination of RNA synthesis by the RNA-dependent RNA polymerase of some viruses. To date, production of ddhCTP by viperin represents the only activity of viperin that links its enzymatic activity directly to an antiviral mechanism in human cells. This review examines the multiple antiviral mechanisms and biological functions attributed to viperin.
Keywords: viperin, RSAD2, radical SAM protein, interferon, RNA-dependent RNA polymerase, chain terminator
1. INTRODUCTION
Viruses are ubiquitous in nature. They can be found freely disseminated in nearly every ecosystem and in intimate association with the cells they infect and require for replication. The multicellularity of higher metazoans required them to develop mechanisms that protect all cells within the whole organism. Most eukaryotic host cells possess the machinery to detect invading viruses by recognizing different viral molecular signatures as nonself. These cellular pathways are critical not only for determining the fate of the infected cell but also for communicating the presence of localized infection to surrounding cells, so as to prepare for the onslaught of subsequent infections with progeny viruses. The high degree of diversity within the virome requires cells to possess a variety of such sensors that span the repertoire of viral species. Nonetheless, production of virus-specific antiviral effectors by the host cell is genetically expensive, energetically demanding, and time-consuming, as exemplified by the adaptive immune responses.
Viral recognition by many of these sensing mechanisms ultimately converges into the shared and remarkably effective pathways that form the basis of innate immunity. The interferon (IFN) system, named for its ability to interfere with viral replication, is one such broad cellular response against microbes. This system is present in essentially all nucleated mammalian cells and, when activated, results in the induction of hundreds of interferon-stimulated genes (ISGs), many of which possess direct antiviral properties. First identified almost 20 years ago, viperin is one such IFN-inducible protein that can interfere with the replication of diverse viruses. Its homology to FE-S cluster–containing enzymes along with the multiple hypothesized mechanisms associated with its antiviral activity has confounded the field ever since its discovery. Despite the high conservation of viperin throughout evolution and its undoubtedly important role as an antiviral effector, a solid molecular basis for its antiviral activity had been lacking until recently. The recent demonstration that the enzymatic activity of viperin generates an antiviral ribonucleotide that can act as a chain terminator of viral RNA synthesis by some viral polymerases has provided a mechanism for the antiviral action of viperin (1) and perhaps will contribute to the establishment of a partially unifying mechanism for the viperin-mediated inhibition of virus multiplication. The goals of this review are to describe the reported roles of viperin as an IFN-inducible antiviral protein and to interpret the many previously postulated antiviral mechanisms of viperin in the context of this newly recognized enzymatic activity.
2. VIPERIN AS A CELLULAR PROTEIN
2.1. Discovery of Viperin
Viperin was initially identified in 1997 using differential display PCR while searching for transcripts that accumulate in fibroblasts infected with human cytomegalovirus (HCMV) (2). Accumulation of these transcripts, named cytomegalovirus inducible gene 5 (cig5) and cig33, depended on viral entry but not on viral replication, suggesting the involvement of an IFN response induced by a molecular signature present in the virions. Subsequently, these two complementary DNA fragments were shown to correspond to a single transcript, which was cloned as an IFN-inducible gene in human macrophages (3). The full human transcript encodes a 361-amino acid protein of about 42 kDa in size that is homologous to BEST5, an IFN-inducible protein expressed during rat osteoblast differentiation (4), and to Vig-1, the fish ortholog found to be induced in rhabdovirus-infected rainbow trout leukocytes (5). The murine ortholog, mvig, had also been found to be induced by vesicular stomatitis virus (VSV) and pseudorabies virus in mouse splenocytes (6).
The induction of the cig5 transcripts upon IFN treatment suggested antiviral properties, which were demonstrated by diminished HCMV replication in cells constitutively expressing cig5 (3). These features, along with its intracellular localization to the endoplasmic reticulum (ER), resulted in the renaming of this restriction factor to viperin for virus inhibitory protein, endoplasmic reticulum associated, interferon inducible (3). Nonetheless, differences in the intracellular localization of viperin between cell types and in different contexts have shed light on other cellular roles for this enigmatic protein. Since its discovery, functional viperin orthologs have been identified in both invertebrates, such as mollusks (7) and lancelets (8), and vertebrate species including fish (9), birds (10), and reptiles (11). These comparative studies demonstrate that viperin-mediated antiviral activity is an ancient and conserved response against viral infections. Based on these studies, viperin is one of the most well-studied ISGs due to its potency and broad-spectrum antiviral activity across taxa.
2.2. Genetics and Cellular Features of Viperin
In humans, the VIPERIN/RSAD2 gene is located in the short arm of chromosome 2 and found adjacent to and inverted with respect to the CMPK2 gene, which encodes cytidylate monophosphate kinase 2 (CMPK2) (Figure 1a). This genomic organization is present in all vertebrates, but both genes can be found to be fused in some lower organisms (1, 8). The CMPK2 gene encodes for a nucleoside kinase, which localizes to the mitochondria (12) and preferentially phosphorylates cytidine diphosphate (CDP) and uridine diphosphate (UDP) to yield their triphosphorylated forms cytidine triphosphate (CTP) and uridine triphosphate (UTP), respectively (1). Both viperin and CMPK2 are cotranscribed upon induction of IFN signaling (13, 14), consistent with their functional cooperation, as is discussed further in Section 3 (1). The basal and IFN-induced expression of both viperin and CMPK2 is negatively regulated by a multi-exonic nuclear-localized long non-coding RNA (lncRNA) known as lncRNA-CMPK2. This lncRNA was identified based on its IFN-dependent induction and named after the protein-coding CMPK2 gene (13) (Figure 1a). While the precise mechanism for lncRNA-CMPK2-mediated regulation of viperin and CMPK2 levels remains unknown, it functions at the transcriptional level without altering the stability of the target messenger RNAs (mRNAs) (13).
Figure 1.
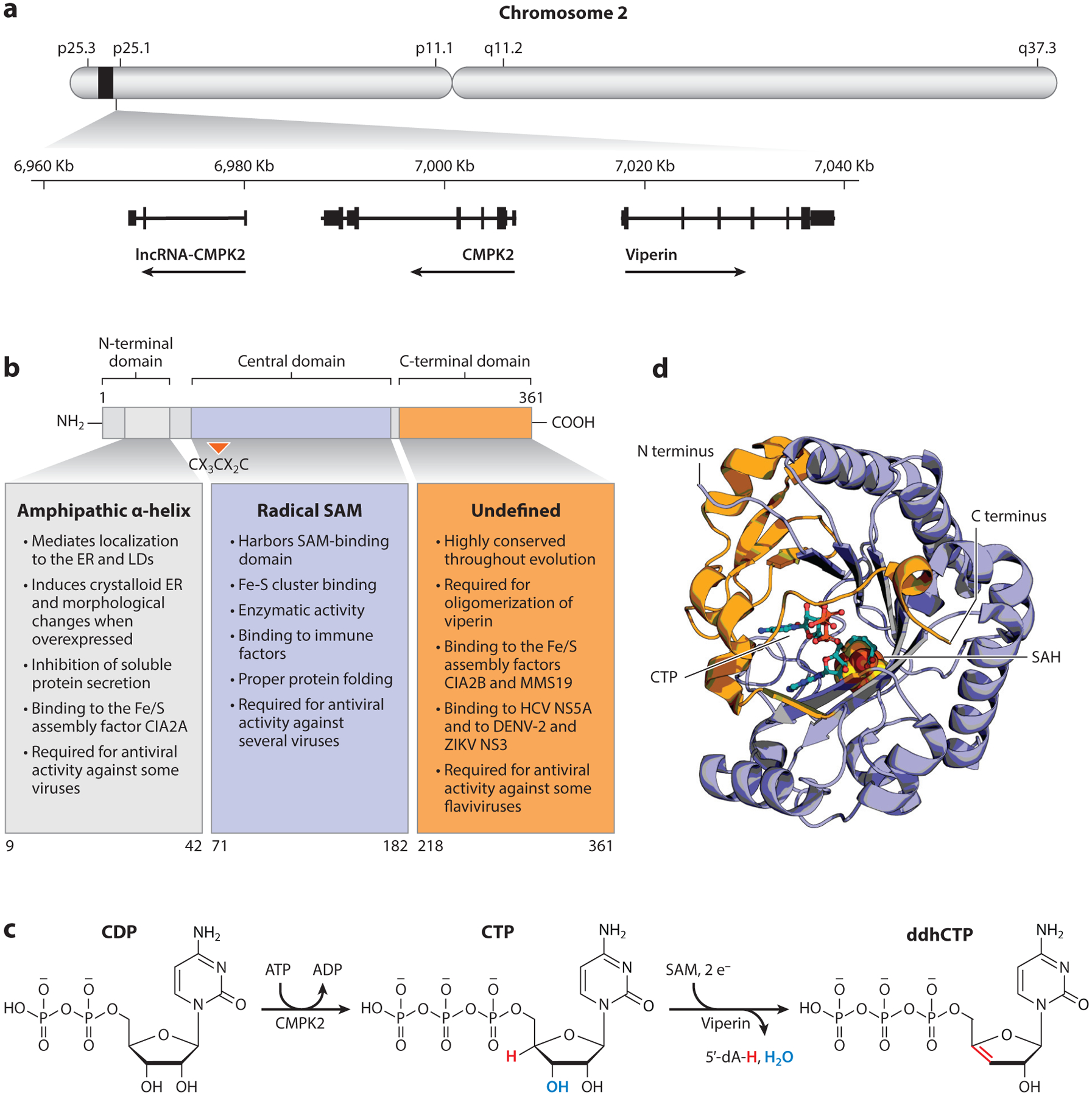
Molecular and biochemical properties of viperin. (a) Genetic organization of VIPERIN. The VIPERIN gene is located in the short arm of chromosome 2 and found adjacent to and inverted with respect to CMPK2. The gene lncRNA-CMPK2 is located adjacent to CMPK2 and acts as a negative regulator for CMPK2 levels. (b) Previously described domains of the viperin protein. The three predicted domains of viperin and their reported roles are shown. (c) Production of ddhCTP. The mitochondrial kinase CMPK2 catalyzes the phosphorylation of CDP to produce CTP, which can then be used as a substrate by viperin to produce ddhCTP. For a detailed mechanistic proposal for viperin catalysis, see Reference 1. (d) Crystal structure of mouse viperin in complex with SAH and its substrate CTP showing the RS and C-terminal domains are not isolable (6Q2P). Abbreviations: CDP, cytidine diphosphate; CIA, cytosolic iron-sulfur protein assembly; CTP, cytidine triphosphate; ddhCTP, 3′-deoxy-3′,4′-didehydro-cytidine triphosphate; DENV-2, dengue virus serotype-2; ER, endoplasmic reticulum; HCV, hepatitis C virus; LD, lipid droplet; RS, radical S-adenosyl-L-methionine; SAH, S-adenosylhomocysteine; SAM, S-adenosyl-L-methionine; ZIKV, Zika virus.
Sequence analyses of eukaryotic viperin have suggested three distinct domains that relate to its function (Figure 1b). The first 70 amino acids of human viperin constitute the N-terminal domain; this is the least conserved region when sequences from mammals and fish are compared. This domain contains a leucine zipper motif and was initially hypothesized to facilitate protein-protein interactions (3), but evidence for the functionality of this domain in such events remains to be elucidated. It was later demonstrated that the first 42 amino acid residues harbor an amphipathic α-helix that is both necessary and sufficient to localize viperin to the cytosolic face of the ER and to lipid droplets (LDs) (15, 16). Amphipathic α-helices are characterized by the presence of a polar cytosolic-exposed side and a hydrophobic side that dips into the hydrophobic phase of cellular membranes and induces curvature. Accordingly, overexpression of viperin has been shown to induce dramatic distortions in ER morphology and to inhibit the secretion of soluble proteins that originate from this organelle (16, 17). The localization of viperin to LDs has been associated with its ability to bind to viral proteins during infection and to associate with innate immune signaling components and enhance type I IFN synthesis (18–20). As we discuss later in Section 4.3, the N-terminal amphipathic α-helix is critical for many of the antiviral properties of viperin, likely due to its essential role in determining intracellular localization. Additionally, viperin has been localized to mitochondria in cells of brown adipose tissue (21) and transiently during HCMV infection in human foreskin fibroblasts, where it regulates β-oxidation of fatty acids and energy production (22, 23).
The central domain of viperin (residues 71–182, human numbering) displays significant homology to the MoaA/NifB/PQQE motif present in the radical S-adenosyl-L-methionine superfamily of enzymes (3, 5). This motif gives viperin the alternate name of radical S-adenosyl-L-methionine (SAM) domain-containing 2 (RSAD2). Radical SAM (RS) enzymes are broadly distributed in all kingdoms of life and are characterized by the presence of an almost invariant RS motif, CX3CX2C (residues 83–90) and the use of SAM as a cofactor. The three conserved cysteine residues coordinate a redox-active [4Fe-4S] cluster. Despite the recognition of this motif since the discovery of viperin, it was not until almost a decade later that biochemical evidence demonstrated RS activity (24, 25). Furthermore, the role of the Fe-S cluster in viperin-mediated catalysis was only recently described (1, 26). The RS motif is important for its antiviral action against multiple viruses, for regulation of fatty acid metabolism and other cellular functions, and for its enzymatic activity, which is discussed in more detail in Section 4. It also has been suggested to contribute to the stability and folding of the tertiary structure of viperin (26, 27).
Lastly, a defined role for the C-terminal domain remains to be described, but it appears to be important for its antiviral functions against members of the Flaviviridae (19, 20, 28). This domain is highly conserved in evolution and necessary for the interaction of viperin with the cytosolic Fe-S protein assembly factor cytosolic iron-sulfur protein assembly factor CIAO1, which is thought to facilitate capacity of viperin to bind iron (29).
2.3. Defining Viperin’s Structure and Substrate
The crystal structure of mouse viperin revealed how the central and C-terminal domains of viperin combine to yield an active antiviral protein (26). Most previous studies assumed that the central and C-terminal domains of viperin operated independently. However, the structure of viperin demonstrates that these domains form a partial α6β6 triosephosphate isomerase (TIM) barrel that harbors the active site [4Fe-4S] cluster, anchored by the RS motif. Importantly, this structure showed that these domains cannot be separated to form stable, isolable products. Like all other members of the RS superfamily, the [4Fe-4S] cluster anchors the SAM cofactor to facilitate generation of the highly reactive 5′-deoxy-5′-adenosyl radical (5′dA•). In the structural report by Fenwick and colleagues (26), it was noted that viperin displays structural similarity to the RS protein MoaA, a radical SAM enzyme that catalyzes the conversion of guanosine triphosphate (GTP) to (8S)-3′,8-cyclo-7,8-dihydroguanosine-5′-triphosphate involved in molybdenum cofactor biosynthesis (30, 31). Based on its structure, it was proposed that viperin might modify nucleotides or polyphosphate-containing molecules.
Subsequently, Gizzi et al. (1) reported the biochemical characterization of viperin as a 3′-deoxy-3′,4′-didehydro-cytidine triphosphate (ddhNTP) synthase that mediates the conversion of CTP to 3′-deoxy-3′,4′-didehydro-CTP (ddhCTP) (Figure 1c). This discovery was made by the simple, yet elegant, strategy of searching for viperin homologs that exist as gene fusions with other biochemically characterized species. This approach led to the identification of a fusion protein from the bacterium Lacinutrix mariniflava containing an N terminus with a predicted cytidylate monophosphate kinase—highly similar to human CMPK2—fused at its C terminus to a coding sequence with high homology to human viperin (32). Human CMPK2 catalyzes the conversion of CDP to CTP, indicating that CTP or a related nucleotide could be a substrate for viperin (Figure 1c). By screening a diverse set of nucleotides and deoxynucleotides, CTP was confirmed to be the substrate of viperin and further validated by showing direct deuterium transfer from the 4′-position of CTP to the 5′-position of 5′-dA—a hallmark of substrate-dependent catalysis by RS enzymes (33). The structure of ddhCTP was confirmed by two-dimensional nuclear magnetic resonance (NMR) techniques, correlation spectroscopy and heteronuclear single-quantum correlation, as well as 31P NMR. Interestingly, one would predict ddhCTP to be an unstable molecule due to the proximity of the double bond to the ribose ether. Contrary to this expectation, however, ddhCTP is in fact stable for more than a week in aqueous buffer at pH 7.5 and room temperature. Consistent with available data, a provisional mechanism for the viperin-catalyzed reaction was proposed to begin with homolytic cleavage of SAM to generate the 5′-dA•, which is typical of RS enzymes, followed by radical abstraction of a hydrogen atom from the 4′-position of the ribose of CTP. Similar to the situation observed in ribonucleotide reductase chemistry (34), the positioning of the 4′-radical would allow for loss of the 3′-hydroxyl group with general acid assistance. The resulting resonance-stabilized radical cation could then be reduced by one electron to yield ddhCTP (Figure 1c). The source of the electron in the last step is currently unclear; however, ketyl radicals are potent oxidants with potentials in the range of +2 V, making this step thermodynamically favorable (35). Similar to other RS enzyme reactions, the electron could be derived from a reduced Fe-S cluster, a prediction that would require viperin to utilize two electrons to complete each turnover: one to generate the 5′-dA• and another to reduce the penultimate product (36–38). A recent publication from the Ealick lab (39) used crystallography to show that, indeed, viperin selectively binds CTP, and its 4′-hydrogen atom is optimally oriented to react with the 5′-dA• during viperin catalysis (6Q2P in Figure 1d).
2.4. Viperin-Like Enzymes
The primary sequence of viperin is highly conserved in eukaryotic species. However, there is clear sequence divergence between mammalian viperins and distant viperin-like proteins, such as those from nonchordate eukaryotes or more distantly related proteins from bacteria and archaea. A major question is do these distant viperin-like proteins possess biochemical activities and perform biological functions similar to the mammalian viperins? Initial examination of several viperin-like proteins from nonmammalian species suggested that these proteins may carry out divergent chemistry unrelated to ddhNTP synthase activities. Honarmand Ebrahimi et al. (40) reported that the viperin-related protein from the thermophilic fungus Thielavia terrestris can couple the 5′-dA moiety of SAM to a UDP-glucose substrate in vitro, suggesting that viperin may be conjugating these two compounds. However, complete elucidation of the reaction product was not conducted. Shortly thereafter, Chakravarti and colleagues (41) reported that the viperin-like proteins from the fungus Trichoderma virens and the archaea Methanogenium limitans are similarly capable of coupling the 5′-dA moiety of SAM to isopentenyl pyrophosphate (IPP) (41). However, Chakravarti et al. (41) also confirmed the ability of these enzymes to convert CTP to ddhCTP, as had been previously reported by Gizzi et al. (1) for the mammalian viperin. Whether IPP is a biological substrate of viperin and the functional consequences of its conversion to its adenylated form, AIPP, remain to be elucidated.
A recent comprehensive study of viperin-like proteins clarified some of these discrepancies in substrate specificity. Viperin homologs from the fungus T. virens, the cnidarian Nematostella vectensis, the bacteria Lacinutrix mariniflava and Shewanella baltica, and the archaea Methanogenium limitans were shown to catalyze the ddhNTP synthase reaction, albeit with varying yet highly selective nucleotide triphosphate discernment (A.S. Gizzi, S.C. Almo & T.L. Grove, unpublished article). Structural characterization of viperin for both the T. virens with SAM and UTP and the N. vectensis protein with SAM and CTP showed that these enzymes bind to their respective substrates in a similar fashion as murine viperin (39). This report also identifies a highly conserved NΦHX4CX3CX2C sequence motif (where Φ represents Trp, Tyr, or Phe and X represents any amino acid) containing the catalytic machinery responsible for the ddhNTP synthase activity. Together, these results clarified the activity of viperin and viperin-like proteins and demonstrated the evolutionary conservation of the ddhNTP synthase activity and the likely broader phylogenetic role of viperin in innate immunity.
3. INDUCTION OF VIPERIN DURING VIRAL INFECTIONS
Initially identified as an IFN-inducible gene, the regulation of viperin expression follows both expected and unexpected patterns based on magnitude, kinetics of induction, and dependence on IFN signaling (Figure 2). Additionally, viperin has been shown to be constitutively expressed at high levels in some tissues such as the liver, the heart, adipose tissue, and some immune cells (21, 42). Discovery and characterization of novel components of the innate immunity will continue to provide insights into the mechanisms that contribute to the temporal and spatial regulation of viperin expression.
Figure 2.
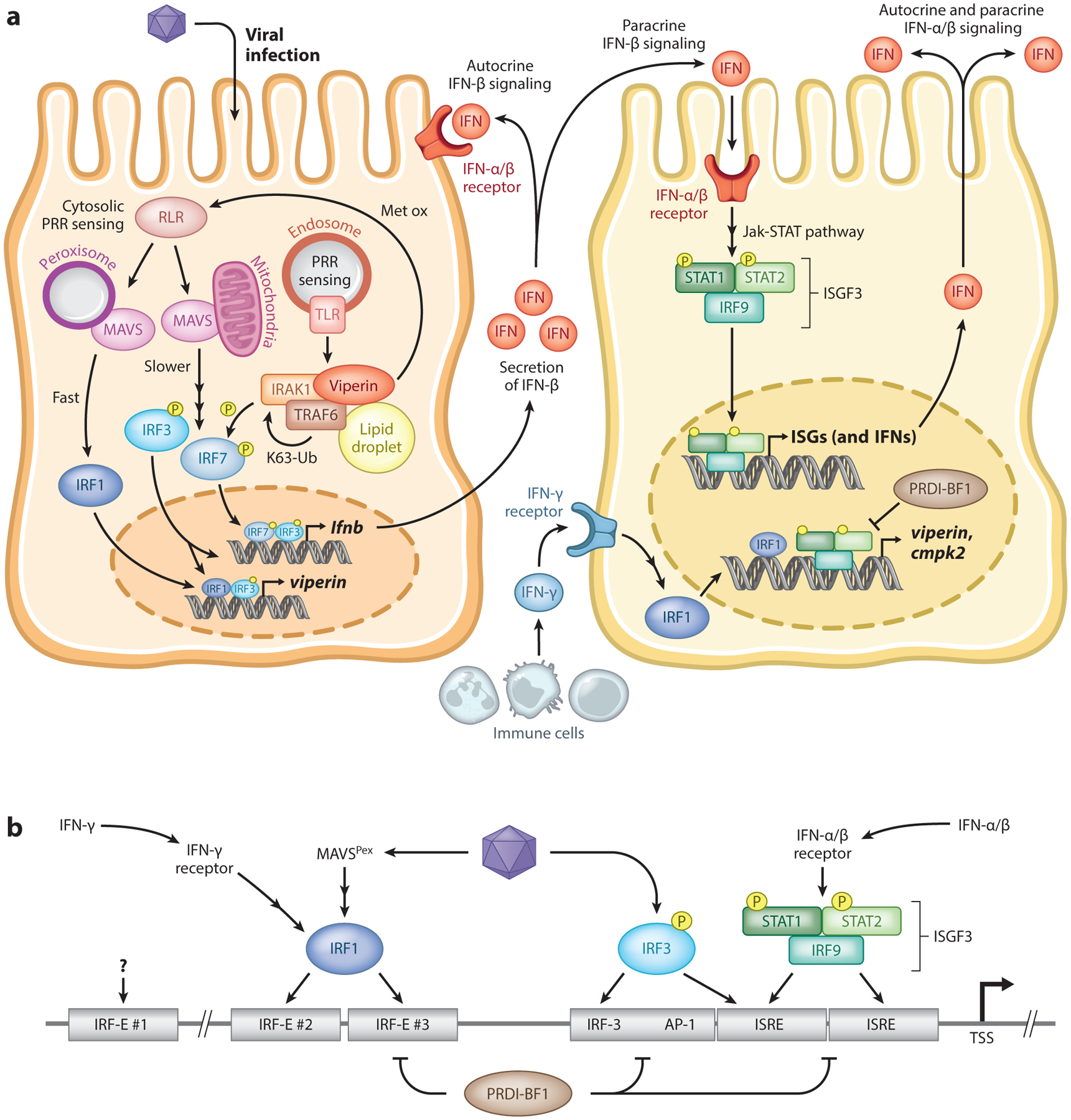
IFN-dependent and -independent pathways leading to induction of viperin upon viral infection. (a) Upon viral infection, sensing of viral nucleic acids by PRRs leads to the activation of downstream signaling factors that result in the induction of IFNs and, in some cases, viperin. The induction of IFN-β can occur to one of several mechanisms, including those dependent on endosomal TLRs and mitochondrial MAVS, that culminate in the activation of the transcription factors IRF3 and IRF7 that bind to the IFNB promoter and induce its expression. The direct induction of viperin can occur through peroxisomal MAVS and downstream activation of IRF1 or by IRF3. Viperin itself can increase IFNB induction by promoting TRAF6-dependent ubiquitination of IRAK1 and phosphorylation of IRF7. IFN-β is secreted and signals both in autocrine and paracrine manners upon binding to its receptor. Downstream activation of the Jak-STAT pathway results in the formation of the heterotrimeric complex ISGF3, which translocates to the nucleus and binds to the promoter of ISGs, including that of viperin, and other IFNs. The transcription factor PRDI-BF1 can act as a negative regulator by competing with ISGF3 for promoter binding. Additionally, IFN-γ secreted primarily by immune cells can activate IRF1 and directly induce viperin expression. (b) In binding of transcription factors to the VIPERIN promoter, the promoter contains two adjacent ISRE sites immediately upstream of the TSS for ISGF3 complex binding. The transcription factor IRF3 can directly bind to the ISREs and also to an upstream sequence. Activation of IRF1 by either IFN-γ signaling or peroxisomal MAVS can bind to two IRF-binding elements, IRF-E #2 and #3. A further upstream IRF-E sequence exists but does not appear to be involved in IRF1 binding. Abbreviations: AP-1, activator protein 1; IFN, interferon; IRAK1, interleukin-1 receptor-associated kinase 1; IRF, interferon regulatory factor; IRF-E, interferon regulatory factor element; ISGF3, interferon-stimulated gene factor 3; ISRE, interferon-stimulated response element; Jak-STAT, Janus kinase signal transducer and activator of transcription protein; MAVS, mitochondrial antiviral signaling; P, phosphorylated; PRDI-BF1, positive regulatory domain I binding factor 1; PRR, pattern recognition receptor; RLR, retinoic acid-inducible gene I (RIG-I)-like receptor; STAT, signal transducer and activator of transcription protein; TLR, Toll-like receptor; TRAF6, tumor necrosis factor receptor–associated factor 6; TSS, transcription start site.
3.1. Interferon-Dependent Induction of Viperin
As a classic IFN-inducible gene, viperin is expressed only minimally in most cells but is greatly induced upon IFN signaling (Figure 2a). In the context of viral and microbial infections, recognition of pathogen-associated molecular patterns (PAMPs) as nonself by cellular pattern recognition receptors (PRRs) results in the induction of inflammatory cytokines and type I (IFN-α/β) and type III (IFN-λ) IFN genes. Discrete molecular signatures derived from the microbe determine the specific PRR that is engaged and, therefore, the downstream pathway that is activated. Viperin expression can be induced by recognition of different PAMPs present in diverse DNA and RNA viruses (43) and bacteria (18, 44). This induction can be readily recapitulated after stimulation with synthetic analogs of double-stranded RNA (43) or B-form DNA (45), as well as the bacterial cell wall component lipopolysaccharide (LPS) (43, 46). Regardless of the PAMP recognized and the PRR activated, these pathways all ultimately converge in the activation of the transcription factors NF-κB and interferon regulatory factor 3 (IRF3), which translocate to the nucleus to bind to the promoters of IFN genes and induce their transcription (47). Once produced, IFNs are secreted and signal in both paracrine and autocrine manners upon binding to their specific heterodimeric receptors IFNAR or IFNLR for type I or type III IFNs, respectively, both of which can induce viperin expression (3, 48, 49). The dimerization of IFNAR or IFNLR subunits activates the Janus kinase signal transducer and activator of transcription protein (Jak-STAT) signal transduction pathway and ultimately induces the formation of the heterotrimeric ISG factor 3 (ISGF3) complex that directly binds to interferon-stimulated response elements (ISREs) within the promoter of ISGs to drive their transcription.
The VIPERIN promoter contains two sequential ISRE sites immediately upstream of the transcription start site. ISGF3 binds directly to these sites and promotes transcriptional activation (43, 50) (Figure 2b). Additionally, the transcription factor promyelocytic leukemia (PML) zinc finger (PLZF) is critical for IFN-α-dependent expression of viperin through interactions with the nuclear proteins PML and histone deacetylase 1 (HDAC1) and subsequent activation of the viperin promoter (51). The binding of ISGF3 to the viperin promoter has been shown to be negatively regulated through competition by positive regulatory domain I binding factor 1 (PRDI-BF1) (43), a repressor of IFN-β expression (52). It is also likely that IFN-inducible lncRNA-CMPK2 represses the expression of viperin through additional genomic interactions (13). These host factors may act as negative feedback regulators to modulate levels of viperin postinduction and limit the deleterious effects of its accumulation. Once transcribed, the viperin transcript is relatively stable [half-life of ~6–9 h (13)] but can be degraded by the endoribonucleolytic action of RNase MRP/RNase P (53). Thus, viperin mRNA levels are tightly regulated through both control of transcriptional activation and mRNA decay.
3.2. Interferon-Independent Induction of Viperin
Cell-type- and virus-specific features contribute to differences in the genetic regulation of viperin. While virus-induced upregulation of viperin is strongly dependent on functional IFN signaling (43, 54), there is accumulating evidence for IFN-independent induction of viperin expression directly through IRF3, activator protein 1 (AP-1), and IRF1 (6, 55, 56). The transcription factor IRF3, which is commonly activated through phosphorylation as a consequence of signaling cascades that result upon PAMP-PRR engagement, can directly induce the expression of some ISGs, including viperin (55). Indeed, initial studies showed that transcripts of fish ortholog vig-1 could be induced during viral infection even in the presence of the protein synthesis inhibitor cycloheximide (5). These findings suggested that IFN synthesis, and thus signaling, is dispensable for induction of vig-1.
Since then, accumulating evidence continues to highlight distinct mechanisms for induction of viperin during viral infections. For example, although the induction of viperin during HCMV infection relies primarily on the heterotrimeric ISGF3 complex, IRF3 can bind directly to the VIPERIN promoter and stimulate its transcription independently of IFN signaling (2, 3, 55, 57, 58). However, the diversity of the virome adds an additional layer of complexity as exemplified by viruses such as sindbis virus and Japanese encephalitis virus (JEV), which induce viperin by mechanisms dependent or independent of IFN signaling, respectively (56). Variation in requirements for viperin expression may not be conserved within a virus family, as chikungunya virus induces viperin expression through activation of the mitochondrial antiviral signaling (MAVS) adapter and downstream IRF3 independent of IFN secretion (59). IFN-independent activation of viperin was also reported in murine dendritic cells (DCs) infected with VSV; viperin could be induced in cells in which IFN signaling was blocked with neutralizing antibodies against IFN-α/β (6). Later evidence indicated that induction of VIPERIN by VSV is dependent on binding of the transcription factor IRF1 to two proximal IRF-elements (IRF-Es) within its promoter (60).
3.3. Biphasic Induction of Viperin
Notably, the magnitude and kinetics of viperin induction differ in almost every scenario reported to date. IFN-independent induction of viperin, while rapid, is modest compared with the delayed but stronger IFN signaling-dependent response. The key to these differences appears to revolve around the intracellular localization of the adapter MAVS and the downstream pathway activated. Detection of many RNA viruses by cytosolic retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) leads to activation of MAVS, a protein localized primarily to the outer mitochondrial membrane (reviewed in 61), and downstream signaling factors to induce transcription of type I IFNs. However, MAVS can also signal from peroxisomes, and peroxisomal MAVS has been proposed to drive distinct antiviral programs (62). In the current model, activation of mitochondrial MAVS leads to IRF3-dependent induction of the type I IFNs and subsequent IFN-α/β signaling, while peroxisomal MAVS is responsible for rapid but transient induction of some ISGs directly through activation of IRF1 in addition to IRF3 (62, 63) (Figure 2a). Additionally, the peroxosimal MAVS pathway that promotes IRF1-dependent responses is selectively responsible for induction of IFN-λ and signaling thereof (63). IFN-λ expression is used predominantly, if not exclusively, in epithelial cell types at anatomic barriers where it appears to establish a more rapid and specialized antiviral program, yet less potent and inflammatory (47, 64, 65). Thus, IRF1-dependent viperin expression in nonepithelial cells is likely induced by a distinct mechanism(s). Consistent with this proposal is the finding that MAVS, IRF3, and IFN signaling are not required for IRF1-dependent constitutive or early antiviral programs in hepatocytes (66, 67) where viperin is highly expressed at basal levels (21).
The VIPERIN promoter contains functional binding sites for both IRF1 and IRF3; thus, differences in temporal viperin induction can be explained by the intrinsic nature of the cell type and identity of antiviral program activated (Figure 2b). The apparent mechanistic redundancy in the induction of viperin likely contributes to requirements for inhibition of viral replication in both the cell infected and surrounding uninfected cells. Additionally, cell-type-specific biphasic induction of viperin could be related to distinct functions during acute versus chronic infections (47, 68) and to provide means to circumvent the many mechanisms that viruses employ to dampen these antiviral responses.
4. VIPERIN AS A MULTIFUNCTIONAL ANTIVIRAL FACTOR
4.1. Proposed Mechanisms of Action for Viperin
Since its discovery, viperin has been—and continues to be—one of the most studied ISGs due to its potent and broad-spectrum antiviral activities across species and cell types. During its initial characterization, viperin was not only induced by viral infection to inhibit HCMV replication (2, 3, 5) but also relocalized during HCMV infection. These findings suggested that HCMV and perhaps other viruses have evolved ways to counteract and repurpose the natural functions of viperin (3, 69). Viperin has been demonstrated to be a restriction factor for many diverse families of viruses in different cell types (Table 1), suggesting that viperin can inhibit viral replication through multiple mechanisms (Figure 3). It is important to note, however, that many of the studies on the antiviral effects of viperin have overexpressed viperin or used cells that lack endogenous expression of viperin [e.g., human embryonic kidney (HEK) 293T and HeLa cells (1, 3, 70)] and perhaps important regulators of viperin activity, which may contribute to apparently contradictory evidence in the literature. Furthermore, the overexpression of truncated or mutated versions of viperin to access the role of the distinct domains has always assumed that these can function independently, yet accumulating evidence argues that this is not the case (26, 27, 71).
Table 1.
Human viruses reported to be sensitive to the antiviral effects of viperin
| Family | Virus | Experimental evidence | Proposed domain(s) requireda | Proposed mechanism(s) of action | Reference(s) |
|---|---|---|---|---|---|
| Herpesviridae (dsDNA group) | HCMV | Overexpression of viperin, knockdown of viperin, co-IPs, microscopy | RS domain | Antiviral effects only if expressed prior to infection through an unknown mechanism; proviral: the HCMV vMIA protein binds to viperin and localizes it to the mitochondria where it binds to host proteins, altering cellular metabolism that favors HCMV replication; replacing the N terminus for a mitochondrial targeting signal is sufficient | 3, 22, 23 |
| KSHV | Overexpression of viperin, knockdown of viperin, co-IP, pharmacological disruption of lipid droplets, in vitro oxidation assays | Controversial—authors proposed protein methionines are substrates for viperin despite published biochemical evidence to the contrary | Proviral: binds to and promotes methionine oxidation of the KSHV helicase ORF44, which in turn enhances its stability | 69 | |
| Flaviviridae (+ssRNA group) | Hepatitis C virus | Overexpression of viperin, knockdown of viperin, replicons, FRET analysis, in vitro primer extension assays with purified RdRp | RS and aromatic amino acids on C terminus required when using replicon colony formation assays; C terminus required for binding to VAP-A and NS5A; role for the N terminus is controversial | Binds to NS5A through its C terminus and to the host factor VAP-A in replication complexes, which may interfere with viral replication; chain termination of RNA replication through ddhCTP-dependent inhibition of RdRp | 1, 20, 75, 78 |
| West Nile virus | Overexpression of viperin, replicons, Viperin−/− mice, in vitro primer extension assays with purified RdRp | RS domain and N terminus (only partially) | Inhibition is seen in virus-like particles and subgenomic replicons; likely chain termination of RNA replication through ddhCTP-dependent inhibition of RdRp | 1, 76, 94 | |
| Dengue virus type 2 | Overexpression of viperin, knockdown of viperin, replicons, FRET analysis, in vitro primer extension assays with purified RdRp | Controversial—RS domain required when using VLP; only C terminus required during infection | Inhibition is seen in VLPs, subgenomic replicons, and infections; interacts with capsid and NS3; likely chain termination of RNA replication through ddhCTP-dependent inhibition of RdRp | 1, 19, 76 | |
| Zika virus | Overexpression of viperin, viperin knockout cells, microscopy, co-IPs, in vitro primer extension assays with purified RdRp, addition of exogenous ddhCTP to cells | C-terminal domain (especially the last four amino acids); N-terminal and RS domains are indispensable | Likely interferes with RNA replication; interacts with NS3 and promotes its degradation; chain termination of RNA replication through ddhCTP-dependent inhibition of RdRp | 1, 28, 77, 79, 95 | |
| Tick-borne encephalitis virus | Overexpression of viperin, depletion of CIAO1, Fe55 incorporation, SAM depletion, flotation assays, microscopy, co-IPs | RS domain and incorporation of Fe are required; aromatic residues in C terminus are required for maturation of viperin and Fe incorporation | Inhibits viral +ssRNA synthesis through an RS-dependent mechanism; interacts with structural and nonstructural proteins and promotes degradation of NS3; induces release of noninfectious capsids and may prevent virion maturation by promoting Golgi-independent secretion of capsids | 29, 77, 82 | |
| JEV | Overexpression of viperin in presence of proteasome inhibitor | ND | Viperin is normally degraded in JEV-infected cells; inhibition of the proteasome rescues the antiviral properties of viperin by an unknown mechanism | 56 | |
| Langat virus | Flotation assays, microscopy, Viperin−/− mice | ND | Unknown; proposed to be through induction of secretion of noninfectious capsids; cell-type-specific effects in vivo | 82, 96 | |
| Picornaviridae (+ssRNA group) | Human rhinovirus | Viperin knockdown | ND | Unknown; unlikely to be dependent on ddhCTP production | 1, 97 |
| Enterovirus A71 | Overexpression of viperin, knockdown of viperin, co-IP | N-terminal domain | Binding to viral protein 2C at the ER through its N-terminal domain | 98 | |
| Togaviridae (−ssRNA group) | Chikungunya virus | Overexpression of viperin, Viperin−/− mice, colocalization with nsP2 at the ER | N-terminal domain is sufficient; intact RS domain is required in full-length viperin | Unknown | 81 |
| Sindbis virus | Viperin knockdown | ND | Unknown | 56, 99 | |
| Rhabdoviridae (−ssRNA group) | Rabies virus | Overexpression of viperin | RS domain | Reduction of cholesterol and sphingomyelin at the plasma membrane; likely inhibition of virion budding through alteration of lipid rafts | 86 |
| Vesicular stomatitis virus | Knockdown of viperin | ND | Unknown; proposed to be due to viperin-mediated promotion of RIG-I oxidation and stabilization | 69 | |
| Orthomyxoviridae (−ssRNA group) | Influenza A virus | Overexpression of murine viperin in human cells; however, no effect observed in Viperin−/− mice | ND | Disruption of lipid rafts inhibits virus release from the plasma membrane | 70, 100 |
| Arenaviridae (−ssRNA group) | Junín mammarenavirus | Overexpression of viperin and coimmunoprecipitations | N-terminal domain | Binds to viral N protein through its N-terminal domain at LDs and inhibits mRNA synthesis | 101 |
| Peribunyaviridae (−ssRNA group) | Bunyamwera orthobunyavirus | Overexpression of viperin | RS domain (only domain tested) | Unknown | 102 |
| Paramyxoviridae (−ssRNA group) | Respiratory syncytial virus | Overexpression of viperin in cell culture and in chinchillas, microscopy | ND | Unknown; may interfere with virus filament formation and cell-to-cell spread | 103, 104 |
| Measles virus | Overexpression of viperin | N terminus, RS domain, C terminus | Inhibits virus release; no effects on titers of cell-associated virus | 85 | |
| Retroviridae (ssRNA-RT group) | Human immunodeficiency virus-1 | Knockdown of viperin, overexpression of viperin, microscopy | RS domain; N-terminal and C-terminal domains are indispensable | Disruption of lipid rafts and inhibition of virion release from the plasma membrane; intact RS domain is required for redistribution of viperin to sites of virion release | 84 |
Abbreviations: CIA, cytosolic iron-sulfur protein assembly; co-IP, coimmunoprecipitation; ddhCTP, 3′-deoxy-3′, 4′-didehydro-cytidine triphosphate; dsDNA, double-stranded DNA; ER, endoplasmic reticulum; FRET, fluorescence resonance energy transfer; HCMV, human cytomegalovirus; JEV, Japanese encephalitis virus; KSHV, Kaposi’s sarcoma-associated herpesvirus; LD, lipid droplet; mRNA, messenger RNA; ND, not determined; NS3, nonstructural protein 3; NS5A, nonstructural protein 5A; nsP2, nonstructural protein 2; RdRp, RNA-dependent RNA polymerase; RIG-I, retinoic acid-inducible gene I; RS, radical S-adenosyl-L-methionine; RT, reverse transcriptase; SAM, S-adenosyl-L-methionine; ssRNA, single-stranded RNA; VAP-A, vesicle-associated membrane protein-associated protein-A; VLP, virus-like particle; vMIA, viral mitochondrial inhibitor of apoptosis.
Biochemical evidence indicates that neither the RS nor the C-terminal domain can operate independently, and their functions cannot be separated.
Figure 3.
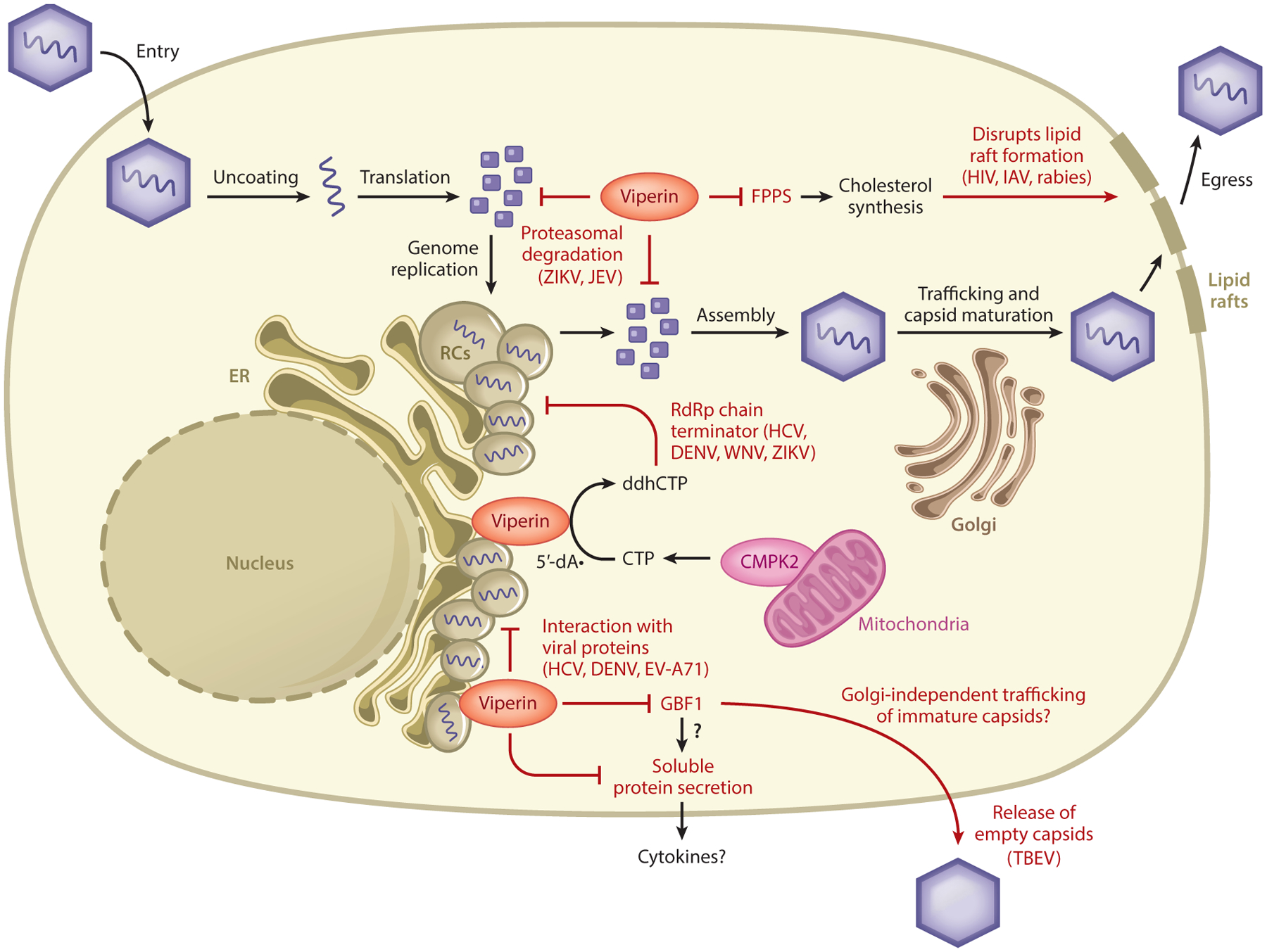
Reported inhibitory mechanisms of viral infections by viperin. Viperin localizes on the cytosolic face of the ER and to LDs, both of which often serve as platforms for viral RCs. Viperin catalyzes the conversion of CTP, possibly produced by CMPK2, to ddhCTP, which serves as a chain terminator during flavivirus replication. Viperin can also promote viral protein degradation, bind to proteins from several different viruses, and interfere with Golgi-dependent trafficking of soluble proteins and promote the release of immature capsids through sequestration of the host factor GBF1. Viperin can inhibit cholesterol synthesis through binding to FPPS, resulting in disruption of lipid rafts at the plasma membrane used by some viruses during their egress. Abbreviations: CMPK2, cytidylate monophosphate kinase 2; CTP, cytidine triphosphate; ddhCTP, 3′-deoxy-3′,4′-didehydro-cytidine triphosphate; DENV, dengue virus; ER, endoplasmic reticulum; EV-A71, enterovirus A71; FPPS, farnesyl diphosphate synthase; GBF1, Golgi-specific brefeldin A-resistance guanine nucleotide exchange factor 1; HCV, hepatitis C virus; HIV, human immunodeficiency virus; IAV, influenza A virus; JEV, Japanese encephalitis virus; LD, lipid droplet; RC, replication complex; TBEV, tick-borne encephalitis virus; WNV, West Nile virus; ZIKV, Zika virus.
4.2. Viperin as an Antiviral Ribonucleoside Synthase
Viperin can produce ddhCTP when provided with CTP both in vitro and in a cellular environment (1). A direct correlation between induction of viperin and ddhCTP production has been shown in immortalized murine macrophages (RAW264.7) treated with IFN-α. Remarkably, even when ddhCTP reaches ~43% of the total concentration of the CTP pool (~350 μM ddhCTP to ~800 μM CTP), the overall levels of CTP remain stable, indicating that the cellular mechanisms exist to preserve the nucleotide pool (i.e., increased ribonucleotide synthesis). Furthermore, although HEK 293T cells do not naturally express viperin, upon being transient transfected with a native viperin-expressing vector, these cells produce ~75 μM ddhCTP compared with undetectable levels in control cells.
It is noteworthy that the viperin constructs with C-terminal tags were completely inactive in vivo. One intriguing observation is that coexpression of CMPK2 with viperin resulted in an approximately fourfold increase in ddhCTP production. In vitro analyses revealed that CMPK2 preferentially phosphorylates dinucleotides in the order CDP > UDP, leading to a synergistic model in which CMPK2 increases the local concentration of CTP for viperin to use as substrate, enhancing ddhCTP production during viral infection (1). These data indicate that CMPK2 is a misannotated gene.
The resemblance of ddhCTP to known polymerase chain terminators provided mechanistic insights into the role of ddhCTP. It was demonstrated through the use of primed-template assays that ddhCTP is incorporated into the nascent RNA and acts as a chain terminator for the RNA-dependent RNA polymerase (RdRp) of multiple members of the Flaviviridae family (Table 1). The antiviral effects of this natural nucleoside were shown when Zika virus (ZIKV) titers were reduced in cells pretreated with synthetic ddhC nucleoside, which is capable of crossing the plasma membrane and can be metabolically converted into ddhCTP by host cell resident kinases (1).
Additional questions remain to be addressed. For example, not all viral RdRps appear to be sensitive to ddhCTP, as exhibited by the observation that the RdRps of human rhinovirus type C and poliovirus are not inhibited by ddhCTP (1). Additionally, viperin and CMPK2 localize to the cytoplasmic face of the ER and the mitochondria, respectively, and whether these reactions are somehow compartmentalized remains to be determined. One possibility is their transient localization to mitochondrial-associated ER membranes (MAMs)—domains known to facilitate the exchange of ions and larger molecules between these two organelles—as has been suggested for viperin (72, 73).
Viperin is expressed and ddhCTP is produced in response to viral infections, but how is this response turned off once the threat has subsided? A recent report by Yuan et al. (74) may afford some insight, as they have shown that primary epithelial cells can produce viperin mRNA in response to both IFN and viral infections, with undetectable amounts of viperin as accessed by immunoblotting techniques. They demonstrated that the lack of viperin protein accumulation was dependent on Lys197-acetylation by the histone acetyltransferase HAT1. This modification, in turn, recruits the ubiquitin conjugation factor E4 A (UBE4A) ubiquitin ligase to polyubiquitinate viperin at Lys206, which targets it for proteasomal degradation. As such, interfering peptides against UBE4A rescues the levels of viperin protein production in epithelium and renders mice more resistant to viral infections (74). To date, viperin degradation has received only modest attention, and it will be important to understand the breadth of this mechanism for modulating viperin levels and ddhCTP production and whether different viruses specifically target viperin for degradation as described for JEV (56).
4.3. Viperin as a Regulator of Secretion and Lipid Rafts
An inhibitory role for viperin in the secretion of soluble proteins from the ER was reported a decade ago (16). This property was attributed to the N-terminal amphipathic α-helix of viperin, which is also necessary for directing viperin to the ER and LDs (15, 16). Because members of the Flaviviridae replicate in replication complexes (RCs) derived from ER membranes and LDs and are sensitive to viperin activity (28, 75–77), the inhibitory effects of viperin on the replication of these viruses represented a potential mechanism for the action of this ISG. However, conflicting results have confounded a unifying mechanism for restriction of these and other viruses. For example, the N and C termini of viperin were found by two independent groups to be important for its antiviral effects against hepatitis C virus (HCV) through its ability to localize to ER-derived membranes and interact with viral proteins in RCs (20, 75). In contrast, another group reported that the C terminus exerted the antiviral properties of viperin against HCV and that the N terminus was dispensable (78). An essential role for the C-terminal domain was also shown for ZIKV and dengue virus (DENV), and the antiviral activity against the three flaviviruses was attributed to direct binding of viperin to viral proteins (19, 28, 79). Additionally, evidence suggesting that the N and C termini of viperin-harboring binding sites for binding of factors involved in maturation of the Fe-S cluster (71) further highlights the limitations associated with truncation mutants of viperin.
Discrepancies have also been reported for the role of the RS domain of viperin during flavivirus infection (19, 20, 75, 76), which is required for production of ddhCTP and RdRp-mediated inhibition of genome replication by flaviviruses (1). Notably, it has been suggested that cysteine-to-alanine mutations, which remove the Fe-S cluster, lead to folding defects that render viperin unstable and prone to aggregation (27). Thus, it is possible that the lack of antiviral effects reported when SAM-binding-defective derivatives of viperin are overexpressed could be related to inherent structural aberrations. Similarly, it was previously suggested that residues within the proposed C-terminal domain of viperin are required for substrate recognition (80). For some viruses such as chikungunya, however, the N-terminal, and not the SAM, domain was found to be both necessary and sufficient to inhibit viral replication and might reflect effects related almost exclusively to changes in ER function (81). More recently, the regulator of Golgi-dependent protein trafficking Golgi brefeldin A-resistant guanine nucleotide exchange factor 1 (GBF1) was identified as a binding partner of viperin, and it was proposed that this interaction could influence intracellular trafficking of membrane-associated viral particles from the ER (82).
Viperin has also been shown to inhibit release of some viruses from the plasma membrane. The first example was influenza A virus (IAV), when it was demonstrated that induction of murine viperin in HeLa cells resulted in inhibition of IAV budding from lipid rafts on the plasma membrane (70). Mechanistically, viperin was shown to bind to and inhibit the activity of farnesyl diphosphate synthase (FPPS), a key enzyme in the synthesis of isoprenoid-derived lipids, including cholesterol, altering the composition and fluidity of plasma membrane domains necessary for the budding of some enveloped viruses (70, 83). It was recently postulated that geranyl pyrophosphate and farnesyl pyrophosphate, two intermediates in the isoprenoid pathway, could serve as substrates of human viperin and that its C terminus may play a role in substrate recognition (80). However, this is highly unlikely based on recent biochemical and structural data of viperin and viperin-like proteins (39). Viperin-induced effects on lipid raft composition and inhibition of virus budding were also shown for human immunodeficiency virus (HIV) and rabies virus, as well as likely for Measles virus (84–86). In HIV-infected macrophages, the induction of viperin disrupts lipid rafts and viperin localization shifted from the ER to virus budding compartments labeled with CD81 in a manner that depends on its radical SAM domain (84). Whether production of ddhCTP affects the activity of enzymes in the isoprenoid pathway, and thus lipid synthesis, remains to be elucidated. Alternatively, inhibition of viral RNA synthesis by ddhCTP could affect the rate of capsid maturation and, as a consequence, decrease the efficiency and rate of viral egress.
4.4. Viperin as a Regulator of β-Oxidation and Thermogenesis
In the initial studies on the role of viperin during viral infections, viperin was shown to transiently redistribute to mitochondria in HCMV-infected cells, and viperin was also shown to enhance HCMV multiplication (23). These studies identified the HCMV-encoded viral mitochondrial inhibitor of apoptosis (vMIA) as the binding partner for viperin that mediates its translocation to mitochondria, where it blocks fatty acid β-oxidation through an interaction with the β subunit of the mitochondrial trifunctional protein complex (23). Inhibition of fatty acid catabolism results in decreased levels of adenosine triphosphate and changes in the actin cytoskeleton, which, in turn, activates the adenosine monophosphate–activated protein kinase and a signaling pathway that ultimately increases lipogenesis and accumulation of LDs (22, 23). While these responses are induced by HCMV to enhance its replication, simply directing functional viperin to the mitochondria alone is sufficient to induce these metabolic changes in the absence of HCMV infection (22, 23). Thus, it is likely that the interaction of viperin with mitochondrial proteins and the metabolic consequences thereof may occur in certain contexts. It is worth noting that proviral roles of viperin have been documented only for herpesviruses: HCMV and Kaposi’s sarcoma-associated herpesvirus (KSHV) (22, 69). The associated mechanisms may therefore represent an exception to the rule that can be accessed by herpesviruses because of their enormous proteomes.
From a metabolic standpoint, viperin was recently shown to be constitutively expressed in several tissues, including the liver, some immune cells, the heart, and adipose tissue (21, 42). Viperin was found to localize to the mitochondria in the brown adipose tissue of mice, causing downregulation of thermogenesis through inhibition of β-oxidation in these highly metabolic tissues (21). Thus, it appears that viperin can naturally regulate fatty acid metabolism in some cell types and that this can be repurposed by certain viruses—such as HCMV—in cell types where viperin is poorly expressed and localized to the ER. Notably, in endothelial cells, fatty acid oxidation is important for the synthesis of deoxynucleoside triphosphates (dNTPs), which are produced by reduction of ribonucleotides, and DNA replication (87). While no evidence for a role in the synthesis of ribonucleotides was found in these cells, likely due to compensatory synthesis by salvage pathways (87), a role of viperin and CMPK2 in intrinsic production of ribonucleotides and a relationship to fatty acid metabolism in other scenarios remain a possibility.
4.5. Viperin as an Enhancer of Antiviral Responses
In addition to its many roles as a direct inhibitor of viral replication and egress, viperin has been linked to modulation of innate and adaptive immune responses. The initial insights relating immune roles for viperin came from the characterization of immune pathways in mice engineered to lack its expression (18, 88). These studies demonstrated that viperin facilitates innate immune signaling in plasmacytoid dendritic cells (pDCs), leading to enhanced production of type I IFNs (18, 89). In these specialized immune cells, viperin acts as a scaffold for the recruitment and activation of immune signaling components downstream of the viral nucleic acid sensors Toll-like receptor 7 (TLR7) and TLR9 at ER-derived LDs (18, 89). Mechanistically, viperin is required for tumor necrosis factor receptor–associated factor 6 (TRAF6)-mediated lysine 63 (K63) polyubiquitination of the kinase interleukin-1 receptor-associated kinase 1 (IRAK1), which is responsible for the phosphorylation of IRF7 and subsequent transcriptional induction of type I IFNs (89). As such, the ability of viperin to facilitate the assembly of immune components on specialized organelles—namely endosomes and LDs—as signal-propagation complexes is important for the augmentation of IFN production and links the intracellular location of viperin with one of its functions (18). A more recent study showed that the interaction of both TRAF6 and IRAK1 with viperin increases SAM cleavage approximately tenfold in transfected HEK 293T cells. Interestingly, while these components are important for enhancing the production of ddhCTP by viperin, this enzymatic activity was found not to be required for activation of IRAK1 (89). Instead, the latter relies on stabilization of viperin through cofactor-induced structural changes in its structure (89). Thus, it appears that the ability of SAM to stabilize viperin when it complexes with IRAK1 and TRAF6 is important for promoting the enzymatic activity of viperin and concomitantly the activation of IRAK1 but that production of ddhCTP is not necessary for this to occur.
While the effects of viperin are specific to endosomal nucleic acid sensors and viperin is dispensable for the induction of IFNs by cytosolic PAMPs and for the induction of proinflammatory cytokines in pDCs (18), this may not be the case for all cells. A recent report showed that viperin catalyzes the oxidation of methionine residues in several helicases, including the cytosolic viral RNA sensor RIG-I, in a manner that may depend on its SAM domain (69). These post-translational modifications were shown to increase the stability and signaling activity of RIG-I and thus affect the magnitude of IFN-β induction and control of viral infections in mouse embryonic fibroblasts (69). Nonetheless, these enhancement roles for viperin during innate immune sensing are in disagreement with a report suggesting that viperin acts as a negative regulator of IFN-β synthesis in bone marrow macrophages through binding to MAVS at mitochondrial-associated ER membranes (73). Differences in cell types, PAMPs engaged, and/or the specific assays used are likely responsible for these discrepancies.
5. CMPK2 ROLES IN INFLAMMATION AND INNATE IMMUNITY
5.1. CMPK2 Regulates Inflammasome Activation
The activity of human CMPK2 was first reported as a nucleoside monophosphate kinase that preferentially catalyzes the phosphorylation of deoxycytidine monophosphate and deoxyuridine monophosphate to their respective diphosphates. Based on this activity and its localization to the mitochondria, it was suggested that the primary role of CMPK2 was to function as part of the mitochondrial nucleotide salvage pathway (12, 14). We recently demonstrated that, contrary to previous reports, human CMPK2 is misannotated and has activity only for pyrimidine diphosphates (CDP or UDP), which are converted to their respective triphosphates (1). The CMPK2 protein is predicted to have two domains: an N-terminal domain of unknown function and a C-terminal thymidylate kinase domain that likely harbors the catalytic site. The N-terminal domain contains no known sequence homology to any protein other than other eukaryotic CMPK2 proteins, and its biological role has not been examined. CMPK2 contains orthologs in bacterial and archaeal species, which lack the N-terminal domain present in higher eukaryotes and are suggestive of broad evolutionary functional conservation for only the C-terminal domain.
Recent reports suggest that CMPK2 may play a role in inflammation by activating the inflammasome, the hallmarks of which are the cellular release of cytokines such as IFN-β, TNF-α, and IL-1β. Zhong et al. (90) recently reported that the activity of CMPK2 is required for proper inflammasome activation, and RNA interference (RNAi)-mediated silencing of CMPK2 in mouse-derived bone marrow macrophages results in an approximately fivefold decrease in IL-1β. Furthermore, expression of wild-type CMPK2, but not of the catalytically inactive D330A variant, rescues this phenotype. While these authors were unaware at the time of publication that CMPK2 actually converts CDP to CTP, they suggested that the role of CMPK2 in inflammasome activation is to maintain the mitochondrial dNTP pool required for de novo mitochondrial DNA synthesis by increasing the levels of 2′-deoxycytidine diphosphate, the initially proposed CMPK2 substrate. It is interesting to note that several studies show increased levels of viperin mRNA in inflammasome-activated cells (43, 91). Thus, it is tempting to speculate that CMPK2 and viperin act in concert to modulate the inflammatory response: CMPK2 applies the gas, and viperin applies the brakes.
5.2. CMPK2 as an Antiviral Factor
CMPK2 and viperin are both induced by IFN signaling, but the effects of CMPK2 on viral restriction dependent or independent of viperin have not been extensively investigated. A recent report describes a role for CMPK2 in restriction of HIV replication (14). In this case, CD4+ T cells isolated from HIV-infected patients stimulated with IFN-α show induction of CMPK2, in addition to common ISGs, including viperin. Although it was noted that both viperin and CMPK2 were upregulated by IFN, covariate analysis of the ISG expression profiles suggested independent regulation and likely independent roles in viral restriction. Indeed, CMPK2, but not viperin, is induced in acute monocytic leukemia cells when stimulated with IFN, and an approximately tenfold increase in HIV titer is seen in cells where CMPK2 has been silenced by RNAi. These results lead to the possibility that viperin and CMPK2 can work together or individually to restrict viral replication in different cellular contexts. How the activity of CMPK2 could lead to restriction of virus is not apparent from its biochemical function, and further studies are needed to understand this phenomenon.
6. TOWARD A UNIFYING MECHANISM FOR VIPERIN ACTION?
Can most, if not all, of the reported effects of viperin be explained on the basis of its enzymatic activity? Perhaps. The antiviral effects by viperin reported to date can be generally placed into four categories: (a) inhibition of viral RNA replication, (b) perturbation of the secretory pathway, (c) direct binding to viral proteins, and (d) dysregulation of lipid raft formation by altering lipid metabolism (Figure 3). To date, two DNA viruses, HCMV and KSHV, appear to have repurposed cellular roles for viperin to their benefit during viral replication (Table 1).
In contrast, the antiviral effects of viperin are strongly linked to its localization to the ER. For example, the amphipathic α-helix in the N terminus of viperin when overexpressed can induce morphological changes to the ER that have been linked to its ability to alter protein secretion (16). The ER and LDs are also sites of viral replication and assembly for multiple viruses; thus, a role of viperin in inhibiting the replication of such viruses is not surprising. Furthermore, the interactions between viperin and proteins of viruses such as HCV and DENV have been determined only by fluorescence resonance energy transfer analyses (19, 20), which can be confounded by the high levels of proteins expressed within the same cell. Although these interactions are plausible, the RdRp of these viruses is sensitive to ddhCTP, and even a 1% incorporation of ddhCTP into the nascent RNA chain is likely sufficient to cause premature chain termination (1).
Additionally, several lines of evidence point to a role for the ER in proteasome-dependent protein degradation—another pathway reported to be involved in viperin’s antiviral properties. Notably, the viruses reported to be affected by viperin that fall in these categories are viruses that require membranes of the ER for their replication and/or are sensitive to the antiviral effects of ddhCTP. Lastly, the antiviral effects of viperin against viruses such as IAV, HIV, and rabies virus are related to fatty acid metabolism, disruption of lipid rafts, and inhibition of egress (70, 84, 86). Although currently unknown, it is possible that viperin’s enzymatic activity is linked to lipid metabolism through either ddhCTP production or an as-yet-unidentified metabolic reaction. Further studies on viperin’s enzymatic activity, functions of ddhCTP, and their interplay with other metabolic pathways are necessary to obtain a complete picture of the role of viperin as a broad-spectrum antiviral effector protein.
7. CONCLUSION
The recent biochemical characterization of viperin as a catalyst that produces a natural antiviral ribonucleoside during viral infections has impacted the existing literature; however, many aspects of viperin’s broad-spectrum effector activity remain to be explained (Table 1). Is ddhCTP-mediated chain termination specific to the RdRps of flaviviruses? If so, is the ability of viperin to inhibit replication and/or egress of other viruses related to its enzymatic activity and ddhCTP production? Are these molecules able to cross organelle membranes and regulate molecular and metabolic pathways? A recent study showed that viperin can inhibit RNA synthesis by bacteriophage T7 DNA-dependent RNA polymerase in mammalian cells, which supports the evolutionary conservation of viperin and viperin-like proteins in antiviral immunity (92). Importantly, viperin did not inhibit RNA polymerase II–dependent RNA synthesis from a cytomegalovirus promoter (92), highlighting the specificity of viperin for some polymerases. This inhibition of T7 polymerase function by viperin appears to be independent of its cytoplasmic localization (92), leaving a potential direct role for ddhCTP against viruses that replicate in the nucleus an open question.
Lastly, viperin has been shown to be induced by microbial constituents and inhibit the infection of some bacteria such as Shigella flexneri and Listeria monocytogenes (93). For S. flexneri, the current model is linked to viperin-induced changes in cellular cholesterol levels that impair bacterial entry (93). Similar effects on cholesterol levels have been linked to viperin’s antiviral activity against several viruses, including IAV, HIV, and rabies virus (70, 86). Whether these observations reflect broader indirect mechanisms of viperin function based on ddhCTP production remains an unanswered question. Are the production of ddhCTP and the regulation of lipid synthesis by viperin two independent processes, or are they coupled? And what determines these differences in the context of an infection? Although CMPK2 is often coinduced with viperin, during some scenarios their regulation is independent from each other (1, 14). Does coinduction of CMPK2 favor ddhCTP production by viperin, whereas their downregulation favors viperin’s role in regulating lipogenesis? Further research is needed to address the lack of complete alignment in the literature and the potential link between viperin’s enzymatic production of the antiviral ribonucleoside and regulation of metabolic pathways involved in the viral replication cycle of viruses insensitive to ddhCTP-dependent termination of RdRp-catalyzed RNA synthesis.
While viperin contributes to viral restriction and innate immunity through a wide range of mechanisms, its roles in cellular function and development are likely to be equally complex and diverse. We speculate that these physiological cellular functions may be broadly coopted by pathogens to afford selective advantages. For instance, the correlation between viperin’s localization to the mitochondria during thermogenesis in adipose tissue and the analogous requirement for the mitochondrial localization of viperin for HCMV viral replication is conspicuous. Perhaps the known and other yet-to-be-described functions of viperin could aid in the discovery of novel mechanisms utilized by viruses to evade the host immune response and enable new therapeutic strategies.
SUMMARY POINTS.
Viperin is a highly conserved protein from the radical S-adenosyl-L-methionine superfamily of enzymes and is induced during viral infections through interferon-dependent and -independent pathways.
Viperin exerts antiviral activity against many different viruses through apparent multifunctional mechanisms.
Recently, viperin was found to function as a 3′-deoxy-3′, 4′-didehydro-cytidine triphosphate synthase that uses cytidine triphosphate (CTP) as a substrate to produce 3′-deoxy-3′, 4′-didehydro-CTP (ddhCTP), the first antiviral ribonucleoside encoded by the human genome to be described.
The VIPERIN gene is frequently coinduced with CMPK2, a gene that encodes for a mitochondrial kinase responsible for the conversion of cytidine diphosphate to CTP and thus likely provides viperin with substrate availability.
The product of viperin’s enzymatic reaction, ddhCTP, acts as a premature chain terminator and inhibits the RNA-dependent RNA polymerase of flaviviruses.
Expression of full-length, active viperin inhibits synthesis of RNA by bacteriophage T7 polymerase in mammalian cells, while not affecting RNA synthesis of the native RNA polymerase II.
Viperin has also been implicated in regulation of lipid synthesis, soluble protein secretion, and augmentation of innate immune pathways. Whether these properties are related to its enzymatic activity remains to be explored.
Further work is needed to fully understand the stated multifunctional antiviral properties of viperin and unify the mechanisms exerted by this enigmatic enzyme.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Gizzi AS, Grove TL, Arnold JJ, Jose J, Jangra RK, et al. 2018. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558:610–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu H, Cong JP, Shenk T. 1997. Use of differential display analysis to assess the effect of human cytomegalovirus infection on the accumulation of cellular RNAs: induction of interferon-responsive RNAs. PNAS 94:13985–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chin K-C, Cresswell P. 2001. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. PNAS 98:15125–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grewal TS, Genever PG, Brabbs AC, Birch M, Skerry TM. 2000. Best5: a novel interferon-inducible gene expressed during bone formation. FASEB J. 14:523–31 [DOI] [PubMed] [Google Scholar]
- 5.Boudinot P, Massin P, Blanco M, Riffault S, Benmansour A. 1999. vig-1, a new fish gene induced by the rhabdovirus glycoprotein, has a virus-induced homologue in humans and shares conserved motifs with the MoaA family. J. Virol 73:1846–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudinot P, Riffault S, Salhi S, Carrat C, Sedlik C, et al. 2000. Vesicular stomatitis virus and pseudorabies virus induce a vig1/cig5 homologue in mouse dendritic cells via different pathways. J. Gen. Virol 81:2675–82 [DOI] [PubMed] [Google Scholar]
- 7.Green TJ, Speck P, Geng L, Raftos D, Beard MR, Helbig KJ. 2015. Oyster viperin retains direct antiviral activity and its transcription occurs via a signalling pathway involving a heat-stable haemolymph protein. J. Gen. Virol 96:3587–97 [DOI] [PubMed] [Google Scholar]
- 8.Lei M, Liu H, Liu S, Zhang Y, Zhang S. 2015. Identification and functional characterization of viperin of amphioxus Branchiostoma japonicum: implications for ancient origin of viperin-mediated antiviral response. Dev. Comp. Immunol 53:293–302 [DOI] [PubMed] [Google Scholar]
- 9.Eslamloo K, Ghorbani A, Xue X, Inkpen SM, Larijani M, Rise ML. 2019. Characterization and transcript expression analyses of Atlantic cod viperin. Front. Immunol 10:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goossens KE, Karpala AJ, Rohringer A, Ward A, Bean AG. 2015. Characterisation of chicken viperin. Mol. Immunol 63:373–80 [DOI] [PubMed] [Google Scholar]
- 11.Milic NL, Davis S, Carr JM, Isberg S, Beard MR, Helbig KJ. 2015. Sequence analysis and characterisation of virally induced viperin in the saltwater crocodile (Crocodylus porosus). Dev. Comp. Immunol 51:108–15 [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Johansson M, Karlsson A. 2008. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem 283:1563–71 [DOI] [PubMed] [Google Scholar]
- 13.Kambara H, Niazi F, Kostadinova L, Moonka DK, Siegel CT, et al. 2014. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res. 42:10668–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.El-Diwany R, Soliman M, Sugawara S, Breitwieser F, Skaist A, et al. 2018. CMPK2 and BCL-G are associated with type 1 interferon–induced HIV restriction in humans. Sci. Adv 4:eaat0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinson ER, Cresswell P. 2009. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic α-helix. PNAS 106:20452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinson ER, Cresswell P. 2009. The N-terminal amphipathic α-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J. Biol. Chem 284:4705–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinbusch MMF, Caron MMJ, Surtel DAM, van den Akker GGH, van Dijk PJ, et al. 2019. The antiviral protein viperin regulates chondrogenic differentiation via CXCL10 protein secretion. J. Biol. Chem 294:5121–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitoh T, Satoh T, Yamamoto N, Uematsu S, Takeuchi O, et al. 2011. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34:352–63 [DOI] [PubMed] [Google Scholar]
- 19.Helbig KJ, Carr JM, Calvert JK, Wati S, Clarke JN, et al. 2013. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLOS Neglected Trop. Dis 7:e2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Helbig KJ, Eyre NS, Yip E, Narayana S, Li K, et al. 2011. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 54:1506–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eom J, Kim JJ, Yoon SG, Jeong H, Son S, et al. 2019. Intrinsic expression of viperin regulates thermogenesis in adipose tissues. PNAS 116:17419–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo JY, Cresswell P. 2013. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLOS Pathog. 9:e1003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seo JY, Yaneva R, Hinson ER, Cresswell P. 2011. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332:1093–97 [DOI] [PubMed] [Google Scholar]
- 24.Duschene KS, Broderick JB. 2010. The antiviral protein viperin is a radical SAM enzyme. FEBS Lett. 584:1263–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaveta G, Shi J, Chow VT, Song J. 2010. Structural characterization reveals that viperin is a radical S-adenosyl-L-methionine (SAM) enzyme. Biochem. Biophys. Res. Commun 391:1390–95 [DOI] [PubMed] [Google Scholar]
- 26.Fenwick MK, Li Y, Cresswell P, Modis Y, Ealick SE. 2017. Structural studies of viperin, an antiviral radical SAM enzyme. PNAS 114:6806–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haldar S, Paul S, Joshi N, Dasgupta A, Chattopadhyay K. 2012. The presence of the iron-sulfur motif is important for the conformational stability of the antiviral protein, Viperin. PLOS ONE 7:e31797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van der Hoek KH, Eyre NS, Shue B, Khantisitthiporn O, Glab-Ampi K, et al. 2017. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep 7:4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Upadhyay AS, Vonderstein K, Pichlmair A, Stehling O, Bennett KL, et al. 2014. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell. Microbiol 16:834–48 [DOI] [PubMed] [Google Scholar]
- 30.Hanzelmann P, Schindelin H. 2006. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. PNAS 103:6829–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanzelmann P, Schindelin H. 2004. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. PNAS 101:12870–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. 1999. Detecting protein function and protein-protein interactions from genome sequences. Science 285:751–53 [DOI] [PubMed] [Google Scholar]
- 33.Holliday GL, Akiva E, Meng EC, Brown SD, Calhoun S, et al. 2018. Atlas of the radical SAM superfamily: divergent evolution of function using a “plug and play” domain. Methods Enzymol. 606:1–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minnihan EC, Nocera DG, Stubbe J. 2013. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res 46:2524–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarz HA, Dodson RW. 1989. Reduction potentials of CO2− and the alcohol radicals. J. Phys. Chem 93:409–14 [Google Scholar]
- 36.Silakov A, Grove TL, Radle MI, Bauerle MR, Green MT, et al. 2014. Characterization of a cross-linked protein nucleic acid substrate radical in the reaction catalyzed by RlmN. J. Am. Chem. Soc 136:8221–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grove TL, Radle MI, Krebs C, Booker SJ. 2011. Cfr and RlmN contain a single [4Fe-4S] cluster, which directs two distinct reactivities for S-adenosylmethionine: methyl transfer by SN2 displacement and radical generation. J. Am. Chem. Soc 133:19586–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grove TL, Livada J, Schwalm EL, Green MT, Booker SJ, Silakov A. 2013. A substrate radical intermediate in catalysis by the antibiotic resistance protein Cfr. Nat. Chem. Biol 9:422–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fenwick MK, Su D, Dong M, Lin H, Ealick SE. 2020. Structural basis of substrate selectivity of viperin. Biochemistry 59:652–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Honarmand Ebrahimi K, Carr SB, McCullagh J, Wickens J, Rees NH, et al. 2017. The radical-SAM enzyme Viperin catalyzes reductive addition of a 5′-deoxyadenosyl radical to UDP-glucose in vitro. FEBS Lett. 591:2394–405 [DOI] [PubMed] [Google Scholar]
- 41.Chakravarti A, Selvadurai K, Shahoei R, Lee H, Fatma S, et al. 2018. Reconstitution and substrate specificity for isopentenyl pyrophosphate of the antiviral radical SAM enzyme viperin. J. Biol. Chem 293:14122–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eom J, Yoo J, Kim JJ, Lee JB, Choi W, et al. 2018. Viperin deficiency promotes polarization of macrophages and secretion of M1 and M2 cytokines. Immune Netw. 18:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Severa M, Coccia EM, Fitzgerald KA. 2006. Toll-like receptor-dependent and -independent viperin gene expression and counter-regulation by PRDI-binding factor-1/BLIMP1. J. Biol. Chem 281:26188–95 [DOI] [PubMed] [Google Scholar]
- 44.Weiss G, Rasmussen S, Zeuthen LH, Nielsen BN, Jarmer H, et al. 2010. Lactobacillus acidophilus induces virus immune defence genes in murine dendritic cells by a Toll-like receptor-2-dependent mechanism. Immunology 131:268–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo F, Liu H, Yang S, Fang Y, Zhao Z, et al. 2019. Nonreceptor tyrosine kinase c-Abl- and Arg-mediated IRF3 phosphorylation regulates innate immune responses by promoting type I IFN production. J. Immunol 202:2254–65 [DOI] [PubMed] [Google Scholar]
- 46.Olofsson PS, Jatta K, Wagsater D, Gredmark S, Hedin U, et al. 2005. The antiviral cytomegalovirus inducible gene 5/viperin is expressed in atherosclerosis and regulated by proinflammatory agents. Arterioscler. Thromb. Vasc. Biol 25:e113–16 [DOI] [PubMed] [Google Scholar]
- 47.Lazear HM, Schoggins JW, Diamond MS. 2019. Shared and distinct functions of type I and type III interferons. Immunity 50:907–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, et al. 2018. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLOS Pathog. 14:e1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol 81:7749–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duschene KS, Broderick JB. 2012. Viperin: a radical response to viral infection. Biomol. Concepts 3:255–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu D, Holko M, Sadler AJ, Scott B, Higashiyama S, et al. 2009. Promyelocytic leukemia zinc finger protein regulates interferon-mediated innate immunity. Immunity 30:802–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller AD, Maniatis T. 1991. Identification and characterization of a novel repressor of beta-interferon gene expression. Genes Dev. 5:868–79 [DOI] [PubMed] [Google Scholar]
- 53.Mattijssen S, Hinson ER, Onnekink C, Hermanns P, Zabel B, et al. 2011. Viperin mRNA is a novel target for the human RNase MRP/RNase P endoribonuclease. Cell. Mol. Life Sci 68:2469–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rivieccio MA, Suh H-S, Zhao Y, Zhao M-L, Chin KC, et al. 2006. TLR3 ligation activates an antiviral response in human fetal astrocytes: a role for viperin/cig5. J. Immunol 177:4735–41 [DOI] [PubMed] [Google Scholar]
- 55.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, et al. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol 76:5532–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chan Y-L, Chang T-H, Liao C-L, Lin Y-L. 2008. The cellular antiviral protein viperin is attenuated by proteasome-mediated protein degradation in Japanese encephalitis virus-infected cells. J. Virol 82:10455–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeFilippis VR, Robinson B, Keck TM, Hansen SG, Nelson JA, Früh KJ. 2006. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J. Virol 80:1032–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ashley CL, Abendroth A, McSharry BP, Slobedman B. 2019. Interferon-independent upregulation of interferon-stimulated genes during human cytomegalovirus infection is dependent on IRF3 expression. Viruses 11:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White LK, Sali T, Alvarado D, Gatti E, Pierre P, et al. 2011. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J. Virol 85:606–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stirnweiss A, Ksienzyk A, Klages K, Rand U, Grashoff M, et al. 2010. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J. Immunol 184:5179–85 [DOI] [PubMed] [Google Scholar]
- 61.Vazquez C, Horner SM. 2015. MAVS coordination of antiviral innate immunity. J. Virol 89:6974–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, et al. 2010. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141:668–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Odendall C, Dixit E, Stavru F, Bierne H, Franz KM, et al. 2014. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol 15:717–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLOS Pathog. 4:e1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, et al. 2019. Differential activation of the transcription factor IRF1 underlies the distinct immune responses elicited by type I and type III interferons. Immunity 51:451–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamane D, Feng H, Rivera-Serrano EE, Selitsky SR, Hirai-Yuki A, et al. 2019. Basal expression of interferon regulatory factor 1 drives intrinsic hepatocyte resistance to multiple RNA viruses. Nat. Microbiol 4:1096–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Panda D, Gjinaj E, Bachu M, Squire E, Novatt H, et al. 2019. IRF1 maintains optimal constitutive expression of antiviral genes and regulates the early antiviral response. Front. Immunol 10:1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hinson ER, Joshi NS, Chen JH, Rahner C, Jung YW, et al. 2010. Viperin is highly induced in neutrophils and macrophages during acute and chronic lymphocytic choriomeningitis virus infection. J. Immunol 184:5723–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bai L, Dong J, Liu Z, Rao Y, Feng P, Lan K. 2019. Viperin catalyzes methionine oxidation to promote protein expression and function of helicases. Sci. Adv 5:eaax1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang X, Hinson ER, Cresswell P. 2007. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2:96–105 [DOI] [PubMed] [Google Scholar]
- 71.Upadhyay AS, Stehling O, Panayiotou C, Rösser R, Lill R, Överby AK. 2017. Cellular requirements for iron–sulfur cluster insertion into the antiviral radical SAM protein viperin. J. Biol. Chem 292:13879–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seo JY, Yaneva R, Cresswell P. 2011. Viperin: a multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe 10:534–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hee JS, Cresswell P. 2017. Viperin interaction with mitochondrial antiviral signaling protein (MAVS) limits viperin-mediated inhibition of the interferon response in macrophages. PLOS ONE 12:e0172236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuan Y, Miao Y, Qian L, Zhang Y, Liu C, et al. 2019. Targeting UBE4A revives viperin protein in epithelium to enhance host antiviral defense. Mol. Cell 77:734–47 [DOI] [PubMed] [Google Scholar]
- 75.Jiang D, Guo H, Xu C, Chang J, Gu B, et al. 2008. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol 82:1665–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang D, Weidner JM, Qing M, Pan X-B, Guo H, et al. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and dengue virus infections. J. Virol 84:8332–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Panayiotou C, Lindqvist R, Kurhade C, Vonderstein K, Pasto J, et al. 2018. Viperin restricts Zika virus and tick-borne encephalitis virus replication by targeting NS3 for proteasomal degradation. J. Virol 92:e02054–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang S, Wu X, Pan T, Song W, Wang Y, et al. 2012. Viperin inhibits hepatitis C virus replication by interfering with binding of NS5A to host protein hVAP-33. J. Gen. Virol 93:83–92 [DOI] [PubMed] [Google Scholar]
- 79.Vanwalscappel B, Gadea G, Despres P. 2019. A viperin mutant bearing the K358R substitution lost its anti-ZIKA virus activity. Int. J. Mol. Sci 20:1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mikulecky P, Andreeva E, Amara P, Weissenhorn W, Nicolet Y, Macheboeuf P. 2018. Human viperin catalyzes the modification of GPP and FPP potentially affecting cholesterol synthesis. FEBS Lett. 592:199–208 [DOI] [PubMed] [Google Scholar]
- 81.Teng TS, Foo SS, Simamarta D, Lum FM, Teo TH, et al. 2012. Viperin restricts chikungunya virus replication and pathology. J. Clin. Invest 122:4447–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vonderstein K, Nilsson E, Hubel P, Nygård Skalman L, Upadhyay A, et al. 2018. Viperin targets flavivirus virulence by inducing assembly of noninfectious capsid particles. J. Virol 92:e01751–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Makins C, Ghosh S, Roman-Melendez GD, Malec PA, Kennedy RT, Marsh EN. 2016. Does viperin function as a radical S-adenosyl-l-methionine-dependent enzyme in regulating farnesylpyrophosphate synthase expression and activity? J. Biol. Chem 291:26806–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nasr N, Maddocks S, Turville SG, Harman AN, Woolger N, et al. 2012. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 120:778–88 [DOI] [PubMed] [Google Scholar]
- 85.Kurokawa C, Iankov ID, Galanis E. 2019. A key anti-viral protein, RSAD2/VIPERIN, restricts the release of measles virus from infected cells. Virus Res. 263:145–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang H-B, Lu Z-L, Wei X-K, Zhong T-Z, Zhong Y-Z, et al. 2016. Viperin inhibits rabies virus replication via reduced cholesterol and sphingomyelin and is regulated upstream by TLR4. Sci. Rep 6:30529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, et al. 2015. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520:192–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qiu L-Q, Cresswell P, Chin K-C. 2009. Viperin is required for optimal Th2 responses and T-cell receptor–mediated activation of NF-κB and AP-1. Blood 113:3520–29 [DOI] [PubMed] [Google Scholar]
- 89.Dumbrepatil AB, Ghosh S, Zegalia KA, Malec PA, Hoff JD, et al. 2019. Viperin interacts with the kinase IRAK1 and the E3 ubiquitin ligase TRAF6, coupling innate immune signaling to antiviral ribonucleotide synthesis. J. Biol. Chem 294:6888–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, et al. 2018. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560:198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Das A, Yang C-S, Arifuzzaman S, Kim S, Kim SY, et al. 2018. High-resolution mapping and dynamics of the transcriptome, transcription factors, and transcription co-factor networks in classically and alternatively activated macrophages. Front. Immunol 9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dukhovny A, Shlomai A, Sklan EH. 2018. The antiviral protein Viperin suppresses T7 promoter dependent RNA synthesis-possible implications for its antiviral activity. Sci. Rep 8:8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Helbig KJ, Teh MY, Crosse KM, Monson EA, Smith M, et al. 2019. The interferon stimulated gene viperin, restricts Shigella. flexneri in vitro. Sci. Rep 9:15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szretter KJ, Brien JD, Thackray LB, Virgin HW, Cresswell P, Diamond MS. 2011. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol 85:11557–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vanwalscappel B, Tada T, Landau NR. 2018. Toll-like receptor agonist R848 blocks Zika virus replication by inducing the antiviral protein viperin. Virology 522:199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lindqvist R, Kurhade C, Gilthorpe JD, Överby AK. 2018. Cell-type- and region-specific restriction of neurotropic flavivirus infection by viperin. J. Neuroinflamm 15:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Proud D, Turner RB, Winther B, Wiehler S, Tiesman JP, et al. 2008. Gene expression profiles during in vivo human rhinovirus infection: insights into the host response. Am. J. Respir. Crit. Care Med 178:962–68 [DOI] [PubMed] [Google Scholar]
- 98.Wei C, Zheng C, Sun J, Luo D, Tang Y, et al. 2018. Viperin inhibits enterovirus A71 replication by interacting with viral 2C protein. Viruses 11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y, Burke CW, Ryman KD, Klimstra WB. 2007. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol 81:11246–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tan KS, Olfat F, Phoon MC, Hsu JP, Howe JLC, et al. 2012. In vivo and in vitro studies on the antiviral activities of viperin against influenza H1N1 virus infection. J. Gen. Virol 93:1269–77 [DOI] [PubMed] [Google Scholar]
- 101.Pena Carcamo JR, Morell ML, Vazquez CA, Vatansever S, Upadhyay AS, et al. 2018. The interplay between viperin antiviral activity, lipid droplets and Junín mammarenavirus multiplication. Virology 514:216–29 [DOI] [PubMed] [Google Scholar]
- 102.Carlton-Smith C, Elliott RM. 2012. Viperin, MTAP44, and protein kinase R contribute to the interferon-induced inhibition of Bunyamwera Orthobunyavirus replication. J. Virol 86:11548–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McGillivary G, Jordan ZB, Peeples ME, Bakaletz LO. 2013. Replication of respiratory syncytial virus is inhibited by the host defense molecule viperin. J. Innate Immunity 5:60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jumat MR, Huong TN, Ravi LI, Stanford R, Tan BH, Sugrue RJ. 2015. Viperin protein expression inhibits the late stage of respiratory syncytial virus morphogenesis. Antivir. Res 114:11–20 [DOI] [PubMed] [Google Scholar]