

Abstract
Insulin Resistance syndromes (IR's), are a group of genetic disorders caused due a functional defect in chromosome 19p13. It is an autosomal recessive condition. Donohue Syndrome was initially described by Donohue and Uchida in 1948 and 1954, a case of sisters born to parents with a first-degree consanguineous marriage. Infants presented with typical facial features that resembled the Leprechaun elves of Irish fairy tales. The following is a report of a rare case of dental complications of Severe Insulin Resistance Syndrome. An eight year old female child, with characteristic features of severe insulin resistance syndrome, reported to the Department of Pediatric and Preventive Dentistry, Pravara Institute of Medical Sciences, Loni, presenting with cariously destructed molars and a previous history of dental treatment under local anaesthesia. Given her condition, it was decided to reduce the multiple appointments, to one appointment with all procedures done under general anaesthesia. The following case report discusses the advantages, disadvantages and post operative complications faced when forming a treatment strategy for Severe Insulin Resistance Syndrome.
Keywords: Anesthetic considerations, HAIR-AN syndrome, leprecaunism, Rabson–Mendenhall syndrome, severe insulin resistance syndromes
INTRODUCTION
Insulin resistance syndromes (IRs) are a group of genetic disorders caused due a functional defect in chromosome 19p13. It is an autosomal recessive condition. Depending on the severity of the mutation, there are many variations of this syndrome, of these five main variations have been described in literature. It ranges from Donohue syndrome (DS) (a complete loss insulin receptor [IR] function) to milder phenotypes such as Rabson–Mendenhall Syndrome (RMS) and Type A insulin resistance.[1]
DS was initially described by Donohue and Uchida in 1948 and 1954, a case of sisters born to parents with a first-degree consanguineous marriage.[1,2,3] Infants presented with typical facial features that resembled the leprechaun elves of Irish fairy tales. Some of the important and notable facial features included flat nasal bridge, thick lips, low-set large ears, and hypertelorism of the orbits. Clinical features include lipoatrophy, acanthosis nigricans, organomegaly, hypertrichosis, hirsutism, and postprandial hyperglycemia among many others.[2,3] Neurological abnormalities include delayed milestones and severe growth retardation. Endocrine anomalies include hyperplasia of the islet cells of Langerhans with altered carbohydrate metabolism and either ovaries with varying stages of maturation of multiple cysts or abnormal penile enlargement.[2]
It has a prevalence of one in four million live births, and it affects children born to first cousins. In India, there have been four such reported cases and worldwide, the number of reported cases is under 100.[4,5,6,7]
The milder phenotypes, namely RMS and Type A insulin resistance have similar features to the above condition. The prognosis of this condition depends on the severity of the mutation. In the case of DS, the survival rate is only up to 2–3 years, and in the case of RMS, patients live well into the third decade of life. However, since Type A insulin resistance is the mildest form, it gets diagnosed after puberty, and it is manageable with appropriate medication.[8,9,10]
Recently, along with oral hypoglycemics such as metformin and glitazone recombinant insulin-like growth factor-1 (IGF-1) and growth hormone therapy, the quality of life seems to have improved for patients with the milder phenotypes. DS being the most severe phenotype has no reported survival post 3 years of age.[11,12,13]
In the case report below, we present a rare case of insulin resistant diabetes combined with the classic features of leprechaunism.
CASE REPORT
Patient history and general examination
An 8-year-old female patient reported to the department of pediatric and preventive dentistry with a chief complaint of severe pain, with upper and lower right back quadrants of the jaws. The pain was relieved on medication for about 1 month, after which she complained of night pain for a week. The patients' earlier medical records indicated that she was diagnosed with DS since birth. Before starting her physical examination, parental consent was obtained.
Her general features included elfin facies, upturned nose, low-set ears, hypertelorism of the orbits, acanthosis nigricans, skin tags, hirsutism, and enlargement of the secondary sex organs [Figures 1 and 2]. Upper and lower extremities showed extreme skin roughness [Figures 3 and 4]. Her intraoral features included crowded and dysplastic dentition, narrow maxillary arch, anterior open bite, palatally placed 12 and 22, and hyperplastic gingival among visible features [Figure 5]. Macroglossia was seen with enlarged filiform papillae and vertical furrows [Figure 1]. She had carious 16, 26, 36, and 46. There was a previous history of root canal treatment with 36. She was extremely apprehensive on oral examination and showed delayed understanding (mild mental retardation).
Figure 1.
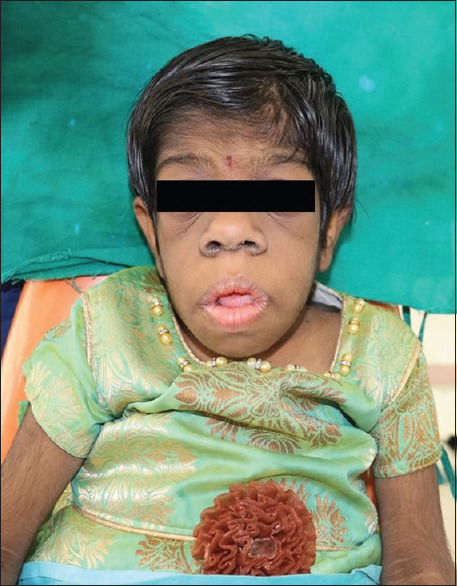
Macroglossia and elfin features
Figure 2.
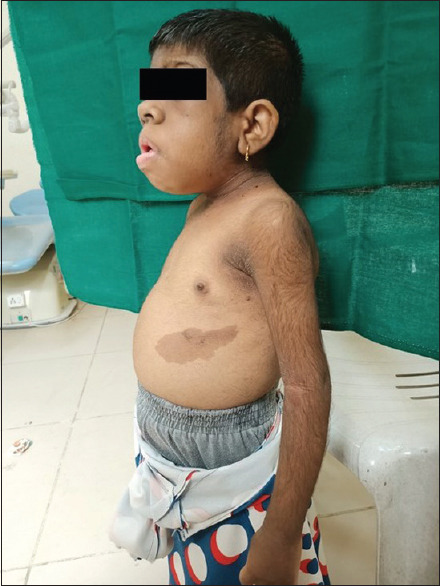
Marked acanthosis nigricans and hirsutism, distended abdomen
Figure 3.
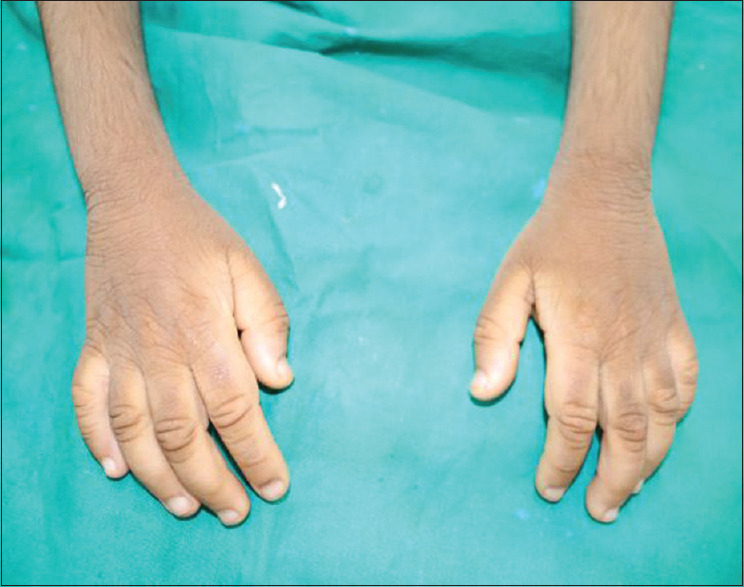
Rough skin (upper extermities)
Figure 4.
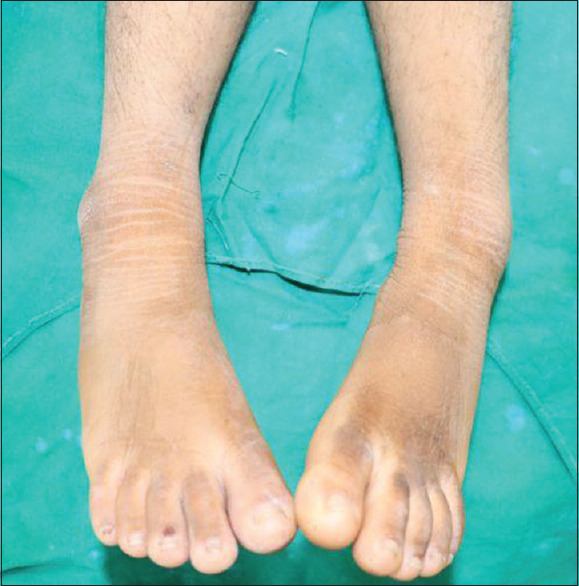
Rough skin (lower extremities)
Figure 5.
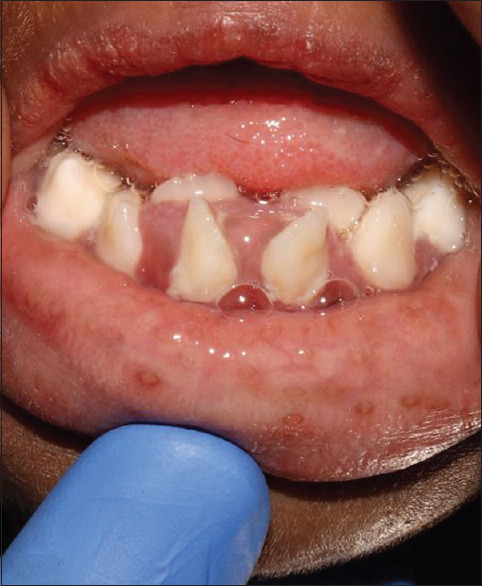
Crowding of teeth, hyperplastic gingiva
She was then referred to the department of pediatrics for a general examination as well as medical fitness. It became evident that she had organomegaly as well as a distended abdomen pointing downward along with the other features of severe insulin resistance syndrome (IRS). Blood investigations revealed had very low hemoglobin (Hb) (7.9 mg/dl) and hyperglycemia (fasting: 200–300 mg/dl, postprandial: 300–400 mg/dl). She was already on a combination of metformin and pioglitazone (one tablet twice a day). She was considered medically unfit to be treated on the chair as she was of the American Society of Anesthesiologists (ASA) Category 4. It was also very likely that she would either undergo hyperglycemic shock (high sugar) or syncopal shock (low Hb). This, along with the delayed mental faculties, would not be manageable on the dental chair, and therefore, it was decided that she would be taken under general anesthesia for dental treatment.
It is important to note that her genetic tests were never carried out due to poor socioeconomic background. Her diagnosis of DS was made in very early infancy due to her symptoms.
The patient was admitted to the pediatric wards 2 days prior to the decided date of the procedure to make sure that appropriate care was provided both before and after the procedure. It was decided to start her on insulin injections (injection glargine 8 units postnight meal) to keep her sugar under check (≥200 mg/dl).
Preanesthetic evaluation
On examination, she was found to have uncontrolled sugar (≤200 mg/dl), very low Hb (7.9 mg/dl), hyponatremia (126 mEq/l) as well as cervical lymphadenopathy. She soon developed upper respiratory tract infection, and thus, the procedure had to be postponed till the patient was optimized to meet the criteria to undergo treatment under general anesthesia.
Achievement of optimization of the patient was done by starting her on injection Actrapid (1.2 ml diluted in 39cc saline) and glargine (8 units post dinner). Alongside, she was also kept on her regular dose of oral hypoglycemics. She was also given a blood transfusion to bring up her Hb levels. Once the optimization was complete, the patient was taken under general anesthesia at the earliest.
Anesthetic considerations
Our patient, due to her condition, would be considered under ASA Category IV which is defined as a patient having a severe systemic condition that is a constant threat to life. Once the patient was taken under general anesthesia, it became clearly evident intraoral features, especially such as macroglossia, narrow high arched palate, comparatively long maxillary insicors, poor visibility of the uvula (according to Mallampati Classification, Class IV), and poor compliance of mandibular space, etc., would make this a difficult airway to negotiate.
Since the procedure would be performed intraorally, nasal intubation was tried. It was not successful; there was profuse bleeding and dropping of saturation levels. One of the reasons for difficulty with nasal intubation could be midfacial instability as well as active upper respiratory infection. It was thus decided to proceed with orotracheal intubation. Once the patient was confirmed stable, the intraoral dental procedures were finally carried out.
Dental procedures
The dental procedures that were carried out were oral prophylaxis, restorations, and extractions of severely carious teeth. Owing to the patient's condition, it was imperative to reduce the bacterial load as much as possible. The patient was given local anesthesia in spite of being under general anesthesia to reduce postoperative pain. She was given very controlled amounts to maintain stability. Not only were the carious teeth extracted but also the palatally placed teeth 12 and 22 were subsequently removed in an attempt to relieve the crowding [Figure 6]. The carious teeth on extraction showed the presence of granulomatous tissue. Post extraction, the sockets were thoroughly curetted and cleaned and irrigated with betadine. They were closed using resorbable Vicryl sutures.
Figure 6.
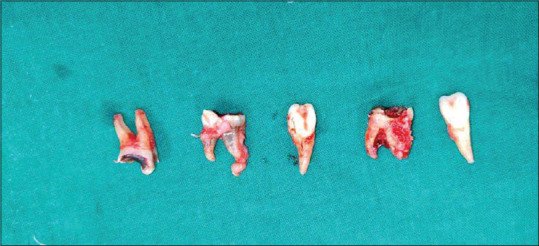
Postoperative picture, extraction of the offending teeth
Postoperative care
The patient was kept in postoperative care on the surgery floor for about 3–4 h. Her vital signs and sugar levels were kept closely monitored. The patient had suffered a mucosal injury during intubation, the result of which was bronchospasm of the left lung. She was thus moved to the pediatric intensive care unit where she was kept under vigilant monitoring. The patient was kept on electrolytes and the insulin drip along with injection glargine to maintain her electrolyte and sugar levels. The patient was kept under sedation for the remainder of the day.
The next day, the patient had gained complete consciousness, though she did experience postoperative lethargy. She was brought back to her regular routine by that very afternoon. Her insulin drip was stopped entirely. She was kept on nebulization with asthalin and budecort for 2 days. She was also started on postoperative antibiotics (injection augmentin 400 mg intravenous [IV] TDS and injection metronidazole 120 mg IV TDS). She was not started on anti-inflammatories as there was a risk of gastrointestinal (GI) bleed.
During her stay at the hospital, postoperatively, her wound healing sites as well as suture sites were carefully monitored for pain and swelling. Instead of anti-inflammatories, she was given topical local anesthetics to reduce the pain. When the patient was finally deemed stable, she was started on Syrup Ibuclin Junior twice a day. Due to her immune-compromised state and uncontrolled sugar rate, it was decided by the pediatricians as well as the pedodontists on the team to discharge the patient to avoid any more risk of hospital-acquired infections.
DISCUSSION
From the above report, it is evident that our patient had a less severe phenotype of the Insulin Receptor (INSR) mutation. One of the most important findings, in our patient, was her uncontrolled sugar level. IR 1 serves as a docking station for recruiting and activating other enzymes that ultimately mediates insulin's effects.[8] Abnormalities in any of the insulin signaling pathway molecules from the IR to the final effectors might be the reason for the pathogenesis of the severe IRS (SIRS). IR mutations have been found in all patients with leprechaunism, RMS, and Type A IRS.[9] Such patients exhibit a significant reduction in IR number, on the cell surface in spite of the presence of adequate cell receptors. Thus, this evidence suggests that there may be a defect before the insertion of the receptor into the plasma membrane.[9]
Insulin exerts its dominant effect on homeostasis through actions on carbohydrate, protein as well as lipid metabolism. There are also actions of insulin that are tissue specific on specific organs. Tissue-specific actions include potassium metabolism, cellular differentiation in adipocytes, stimulation of ovarian production of androgens, and the renal retention of sodium. These are reflected by the specific symptoms and characteristic features of this syndrome. A clear example of this would be the characteristic symptom “acanthosis nigricans” [Figure 2]. It results from long-term exposure of keratinocytes to insulin. Keratinocytes have IGF-1 receptors. Stimulation of IGF receptors due to prolonged exposure to insulin is one of the main etiologies of acanthosis nigricans. Another classic example is pineal gland hyperplasia. One of the main features to diagnose pineal gland hypoplasia is over excretion of the metabolite melatonin. Melatonin is secreted by the pineal gland and is responsible for antigonadotropic property; therefore, the overgrowth of secondary sexual characters is observed. Coming to a few of the dental abnormalities, Bar et al.[13] reported that children with diabetes mellitus (DM) exhibited accelerated tooth eruption, corresponding to later stages of tooth eruption corresponding only to the later stages of tooth eruption involving only the extra alveolar phases. Bar et al.[13] and others also reported that children with DM presented with more severe gingival inflammation. The reason for this is that hyperglycemia leads to deposition of advanced glycation end products which leads to phenotypic alteration of macrophages and other cells via a specific cell receptor. The alteration in phenotype leads to overproduction of pro-inflammatory cytokines which are then responsible for severe gingival inflammation that contributes to the reduction of quality as well as quantity of bone around the erupting tooth and shortens the eruption distance through the bone.[10,11,12,13,14]
Treatment of this condition is not very definitive and extremely challenging. There is no definitive treatment regimen documented in the literature, aside from managing the sugar level and insulin resistance. The first line of treatment is the administration of the tablet metformin, which is a biguanide that suppresses hepatic glucose output in combination with pioglitazones (or rosiglitazone) which is a thioglitazone. It binds avidly peroxisome proliferator-activated receptors to promote adipogenesis and fatty acid uptake.[11]
In recent years, recombinant DNA therapy has been theorized and tried for this condition. Therapy with IGF-1 (rh-IGF-1) alone or with its binding protein IGFBP-3 has been known to have a positive effect on IRS cases. This may improve glucose disposal by signaling via the IGF-1 receptor. It also has on the islet cells of the pancreas, thus having an effect on insulin secretion. This indicates that this line of treatment would be better as the first line of treatment.[11,12,13]
In 2008, Fukunaga et al.[10] reported a case of leprechaunism who lived beyond 2 years due to long-term treatment with rh IGF-1 therapy. According to their observations, few of their major adverse effects included macrodontia, crowding, and abnormal craniofacial growth pattern. However, it showed improved glucose metabolism and prevented postnatal growth retardation. Another line of treatment is therapy with recombinant methionyl human leptin (r-met-hu-leptin).[10,11] Cochran et al. have reported that r-met-hu-leptin in combination with metformin (200 mg) and pioglitazone (2 mg) showed 40%–60% decrease in fasting serum glucose and a marked improvement in glycosylated Hb levels in two siblings with RMS who underwent treatment for 10 months. According to a very recent case report by Plamper et al.,14 a new drug, mecasermin in combination with IGF-1 therapy has shown good results, the only adverse effect being lymphoid hypertrophy.[10]
Clinical features of our patient included acanthosis nigricans, hypertrichosis, typical facial features, Type I DM, and dental prematurity along with swollen and thick gingiva. Another very important feature is our patient's age which was 8 years, and as discussed earlier, patients with leprechaunism do not survive past 2 years of age unless started on early gene therapy. All of these findings corelate with RMS. Furthermore, since RMS has the associated dental findings, it is the most likely diagnosis for our patient. The other syndromes associated with the INSR mutation related to our patient's diagnosis are DS and Type A IRS (HAIR-AN).[10,11,12,13]. Comparative evaluation of characteristics between Donohue, IRS, and RMS is given in Table 1.
Table 1.
Comparison of features for Donohue, Rabson–Mendenhall, and Type A insulin resistance syndrome[10,14]
| Features | Donohue syndrome | Rabson–Mendenhall syndrome | Type A IRS |
|---|---|---|---|
| Skin | Hypertrichosis, acanthosis nigricans, hyperkeratosis, thick skin, dry skin, minimal subcutaneous fat | Hypertrichosis, acanthosis nigricans, hyperkeratosis, thick skin, hirsutism | Acanthosis nigricans, hirsutism |
| Organs | Organomegaly, nephrocalcinosis, renal dysfunction, enlarged polycystic ovaries, rectal prolapse, cholestasis | Organomegaly, nephrocalcinosis, renal dysfunction, enlarged polycystic ovaries, rectal prolapse, peripheral | Polycystic Ovarian Disorder |
| Biochemistry | Hyperinsulinemia, elevated plasma insulin, hyperglycemia, decreased IGF-1, IGFBP-3 | Hyperinsulinemia, decreased plasma insulin levels, risk of diabetic ketoacidosis, low LDL, high HDL, hypercalciuria | Extereme resistance to insulin, androgenism, increased testosterone |
| Dental findings | Macrodontia, crowding, macroglossia (could be because of prolonged therapy with IGF-1*) | Face shows vertical growth, increased gonial angle, macrodontia, crowding, macroglossia, thick and hyperplastic gingiva, dental prematurity, dysplastic dentition | Normal facial features, but high risk of caries and periodontal infections due to Type I DM |
| Neurological findings | Developmental/severe mental retardation | Minor delay in the development of mental faculties | No impairment |
| Life expectancy | Usually 2 years (except ref Fukunaga*) | Usually second to third decade of life microvascular complications | - |
IRS: Insulin resistance syndrome, LDL: Low-density lipoprotein, HDL: High-density lipoprotein, IGF: Insulin-like growth factor, IGFBP: IGF-binding protein 3, DM: Diabetes mellitus
It is imperative to note that children with DS have the most severe form of mutation and thus do not survive over 2 years. Only continuous treatment with recombinant IGF-1 and oral hypoglycemics makes it possible for them to rarely survive past 2 years.
Our patient has undergone no such treatment and is only on oral hypoglycemics as discussed earlier. It is thus evident that she may have a less severe phenotype of the mutation, which is also known as RMS. This can be further corroborated by her intraoral features which are characteristic to this syndrome as well as her mental faculties only being mildly delayed, as opposed to complete retardation as frequently seen with DS.
Our patient was a case of full-mouth rehabilitation and had slightly delayed mental faculties. Since she was at a constant risk of either hypoglycemic and hyperglycemic shock or syncopal shock, it was decided to take her under general anesthesia to perform all her treatment procedures. Preoperative as well as postoperative records were maintained by taking intraoral as well as extraoral pictures and radiographs.
Given the patient's intraoral and facial features and her line of treatment, nasotracheal approach would be tried. After a failed attempt at a nasotracheal approach, the orotracheal approach was considered. It was classed under Class V of the intubation difficulty scale.[9,15]
The patient was put under using a combination of propofol and sevoflurane instead of the standard thiopental and ketamine combination. Our patient's condition puts her at a severe risk of cardiovascular hypotension and saturation depression. Cardiovascular depression is one of the most common adverse effects of importance in our case. Ketamine, unlike other anesthetics, causes tachycardia and peripheral vasoconstriction due to the liberation of catecholamines. With our patient's condition, ketamine can cause myocardial dysfunction. Since sevoflurane has minimum airway reactivity, it is used very frequently in pediatric anesthesia cases.
The patient's treatment procedures consisted mainly of extractions. This was because she had DM Type I along with severe insulin resistance. This condition, as discussed earlier, causes multiple complications, such as high risk of caries, early eruption of permanent teeth, and as a result, severe crowding. It also causes increased risk of postoperative infections.
Postoperatively, the patient was only started on a regime of antibiotics to prevent postoperative infections. She was not given any analgesics since she was at a risk of GI bleed. She was given topical lignocaine hydrochloride jelly to manage the pain.
CONCLUSION
Although SIRS are extremely rare occurrences, they can be very lethal. Upon survival, there can be multiple complications and very poor prognosis. It is essential to manage any and all manageable symptoms effectively and efficiently. It is of paramount importance to find out and concentrate on the root cause by genetic testing and a confirmatory analysis of symptoms. Although the mutation in the INSR gene has been affecting people for decades, literature is yet to provide a clear diagnostic as well as a treatment protocol for this condition. With newer advancements in medical sciences and allied fields, life expectancy has increased and improved significantly. There is also a marked reduction in complications following the survival of infancy.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given his consent for images and other clinical information to be reported in the journal. The guardian understands that names and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Summitt RL, Favara BE. Leprechaunism (Donohue's syndrome): A case report. J Pediatr. 1969;74:601–10. doi: 10.1016/s0022-3476(69)80044-2. [DOI] [PubMed] [Google Scholar]
- 2.Clark DR, Edwards HE. Dysendocrinism. J Pediatr. 1948;32:739–48. [PubMed] [Google Scholar]
- 3.Donohue WL, Uchida I. Leprechaunism: A euphemism for a rare familial disorder. J Pediatr. 1954;45:505–19. doi: 10.1016/s0022-3476(54)80113-2. [DOI] [PubMed] [Google Scholar]
- 4.Hassan I, Altaf H, Yaseen A. Rabson-mendenhall syndrome. Indian J Dermatol. 2014;59:633. doi: 10.4103/0019-5154.143579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohanan S, Chandrashekar L, Semple RK, Thappa DM, Parameswaran N, Negi VS, et al. Rabson-mendenhall syndrome with recurrent cerebral infarcts caused by a novel INSR mutation. Int J Dermatol. 2013;52:182–5. doi: 10.1111/j.1365-4632.2012.05665.x. [DOI] [PubMed] [Google Scholar]
- 6.Sinnarajah K, Dayasiri MB, Dissanayake ND, Kudagammana ST, Jayaweera AH. Rabson mendenhall syndrome caused by a novel missense mutation. Int J Pediatr Endocrinol. 2016;2016:21. doi: 10.1186/s13633-016-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sen S, Roy A, Gangopadhyay A, Halder C. Leprechaunism-A case report. Indian J Dermatol. 2012;57:499–501. doi: 10.4103/0019-5154.103077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E, et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling. Cell. 1985;40:747–58. doi: 10.1016/0092-8674(85)90334-4. [DOI] [PubMed] [Google Scholar]
- 9.Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E, et al. The human insulin receptor cDNA: The structural basis for hormone-activated transmembrane signalling [Internet] Cell. 1985;Vol. 40:747–58. doi: 10.1016/0092-8674(85)90334-4. Available from: http://dx.doi.org/10.1016/0092-8674(85)90334-4 . [DOI] [PubMed] [Google Scholar]
- 10.Fukunaga T, Murakami T, Tanaka H, Miyawaki S, Yamashiro T, Takano-Yamamoto T. Dental and craniofacial characteristics in a patient with leprechaunism treated with insulin-like growth factor-I. Angle Orthod. 2008;78:745–51. doi: 10.2319/0003-3219(2008)078[0745:DACCIA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 11.de Kerdanet M, Caron-Debarle M, Nivot S, Gaillot T, Lascols O, Fremont B, et al. Ten-year improvement of insulin resistance and growth with recombinant human insulin-like growth factor 1 in a patient with insulin receptor mutations resulting in leprechaunism. Diabetes Metab. 2015;41:331–7. doi: 10.1016/j.diabet.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM, et al. The syndromes of insulin resistance and acanthosis nigricans.Insulin-receptor disorders in man. N Engl J Med. 1976;294:739–45. doi: 10.1056/NEJM197604012941401. [DOI] [PubMed] [Google Scholar]
- 13.Bar RS, Muggeo M, Kahn CR, Gorden P, Roth J. Characterization of insulin receptors in patients with the syndromes of insulin resistance and acanthosis nigricans. Diabetologia. 1980;18:209–16. doi: 10.1007/BF00251918. [DOI] [PubMed] [Google Scholar]
- 14.Plamper M, Gohlke B, Schreiner F, Woelfle J. Mecasermin in insulin receptor-related severe insulin resistance syndromes: Case report and review of the literature. Int J Mol Sci. 2018;19:1268. doi: 10.3390/ijms19051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirkwood A, Stuart G, Harding L. Donohue syndrome: A review of literature, case series, and anesthetic considerations. Paediatr Anaesth. 2018;28:23–7. doi: 10.1111/pan.13273. [DOI] [PubMed] [Google Scholar]